Week 04 HW
Part A. Conceptual Questions
Answer any NINE of the following questions from Shuguang Zhang:
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Meat is roughly 20% protein by weight, so 0.2*500g=100g of protein. This is the only amino acid in meat, as carbs are sugars, and fats are triglycerides (fatty acids + glycerol).
1 g = 6.02217364335E+23 dalton
100 g = 6.022173643E+25 daltons
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Humans eat beef but don’t become a cow because the stomach metabolizes complex proteins and cells down to base level molecules and amino acids.
3. Why are there only 20 natural amino acids?
For evolutionary reasons probably. Much like how human language has a finite set of phonemes which allow us to express infinitely more higher-level syllables, words, and concepts and sentences - biology has a base grammar of 20 units. This has proven to be enough - 64 codons map onto 20 amino acids plus stop signals. There may have been more but this is evidently evolutionarily optimal as it is now.
4. Can you make other non-natural amino acids? Design some new amino acids.
β-amino acids are interesting - usually the amino group is attached onto the α-carbon, but here they are attached on the β-carbon. Due to this, proteases (enzymes which support digestion) are highly ineffective against β-peptides.
Others I googled:
- Fluoroleucine — leucine with fluorine substituted in; more hydrophobic and metabolically stable
- Azidohomoalanine — methionine analog with an azide group, useful for click chemistry bioconjugation
5. Where did amino acids come from before enzymes that make them, and before life started?
- Meterorites that naturally carry amino acids
- Miller-Urey experiment showed amino acids could form spontaneously from simple molecules + an electric arc
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Left-handed. Natural α-helices are right-handed because they’re built from L-amino acids. D-amino acids are the mirror image, so the resulting helix is the mirror image too — left-handed.
7. Can you discover additional helices in proteins?
Yes — beyond the common α-helix, proteins also contain 3₁₀-helices (3 residues per turn, tighter) and π-helices (4.4 residues per turn, rarer and wider). These are already known but underappreciated. Computational analysis of PDB structures keeps surfacing edge cases and unusual conformations that don’t fit neatly into existing categories.
8. Why are most molecular helices right-handed?
Because natural amino acids are L-enantiomers. The geometry of the L-α-carbon makes right-handed coiling energetically favorable — the side chains point outward without steric clashes in a right-handed helix. A left-handed helix built from L-amino acids would force side chains into the backbone, creating strain.
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheets have “sticky edges” — the backbone NH and C=O groups along the edge strands are unsatisfied hydrogen bond donors/acceptors. These can pair with the edge of another β-sheet. The driving forces are hydrogen bonding along the backbone and hydrophobic stacking between sheet faces. This makes lateral growth into large, ordered aggregates thermodynamically favorable.
10. Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
When proteins misfold or partially unfold, they expose their backbone, which can then hydrogen-bond with other misfolded proteins into cross-β structure (hydrogen bonds running perpendicular to the fibril axis). This structure is extremely stable — often more so than the native fold — so once nucleation starts, it propagates. Many proteins will form amyloid under the right conditions; some just do so more readily due to sequence composition or environmental stress.
Yes, amyloid fibrils can be used as materials — they’re stiff, stable, and self-assembling. Researchers have used them as scaffolds for nanomaterials, hydrogels, and functional coatings.
11. Design a β-sheet motif that forms a well-ordered structure.
Part B. Protein Analysis and Visualization
(1) Briefly describe the protein you selected and why you selected it.
Insulin. A 51-amino acid peptide hormone secreted by pancreatic β-cells that regulates blood glucose by signaling cells to take up glucose. I chose it because it’s small and well-studied, historically significant (first recombinantly produced therapeutic protein), and I’m curious how something so tiny has such a large physiological effect.
(2) Identify the amino acid sequence of your protein. How long is it? What is the most frequent amino acid? How many protein sequence homologs are there? Does your protein belong to any protein family?
51 amino acids total — two chains: A (21 aa) and B (30 aa), linked by two disulfide bonds. Most frequent: leucine (L) and cysteine (C), both at 6 occurrences. Many homologs — the insulin/IGF/relaxin superfamily includes IGF-1, IGF-2, relaxin, and insulin-like peptides across many organisms. Belongs to the insulin family (InterPro: Insulin/IGF/relaxin superfamily).
(3) Identify the structure page of your protein in RCSB. When was the structure solved? Is it a good quality structure? Are there any other molecules in the solved structure apart from protein? Does your protein belong to any structure classification family?
PDB: 4INS — human insulin hexamer, solved in 1989 at 1.5 Å resolution. Good quality structure. In addition to protein, the hexamer contains two zinc ions (Zn²⁺) coordinated by His B10 residues at the center, plus water molecules.
Why extra zinc ions? Insulin is stored as a zinc-stabilized hexamer in β-cells; once secreted, the hexamer dissociates into monomers and the zinc stays behind, so zinc is necessary for storage and secretion but not for receptor binding. Zinc deficiency is linked to impaired insulin secretion and increased type 2 diabetes risk.
In structural classification, insulin belongs to the “Insulin-like” fold under the all-α class.
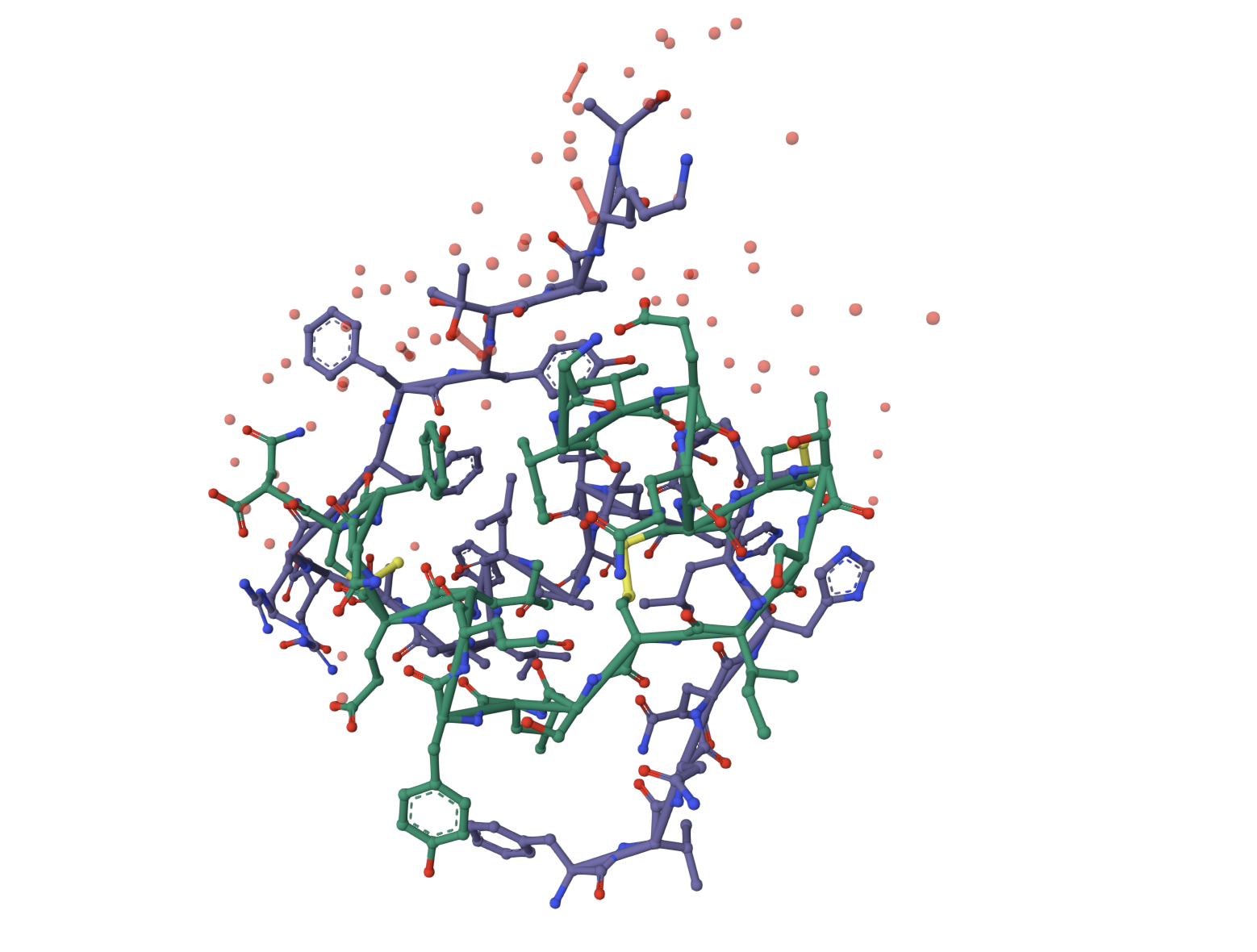
(4) Open the structure in 3D visualization software. Visualize as “cartoon”, “ribbon”, and “ball and stick”. Color by secondary structure — does it have more helices or sheets? Color by residue type — what can you tell about hydrophobic vs hydrophilic distribution? Visualize the surface — does it have any binding pockets?

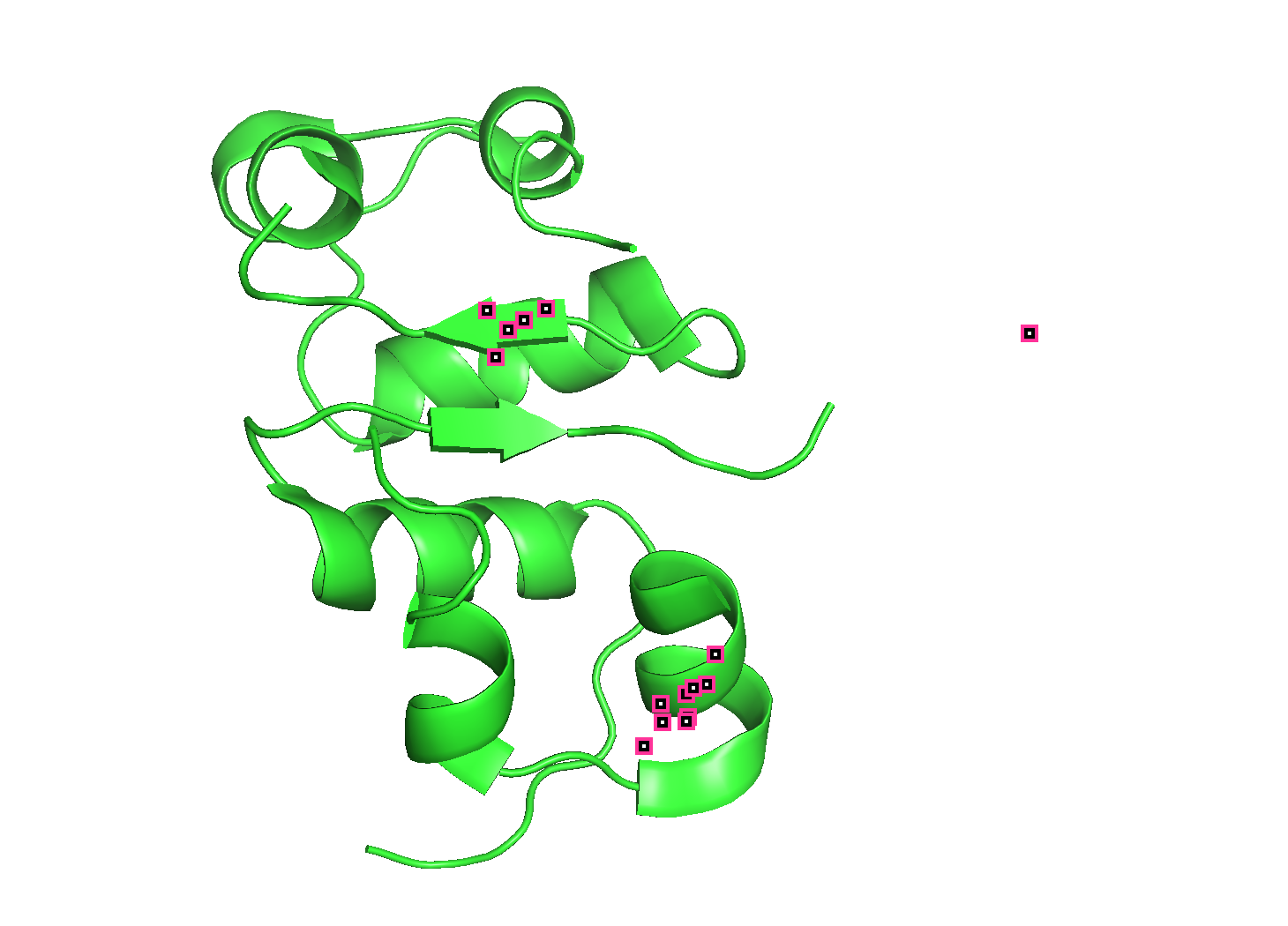
- Color by secondary structure — does it have more helices or sheets
It has helices
Red spirals = α-helices
Yellow flat arrow shapes = β-sheets
- Color by residue type — does it have more helices or sheets
PyMOL: util.cbag
 - green is helices. I don’t see any β-sheets.
- green is helices. I don’t see any β-sheets.
- Visualize the surface — does it have any binding pockets?
Red is helix, Yellow is sheet, Green is loop.
PyMOL: show surface
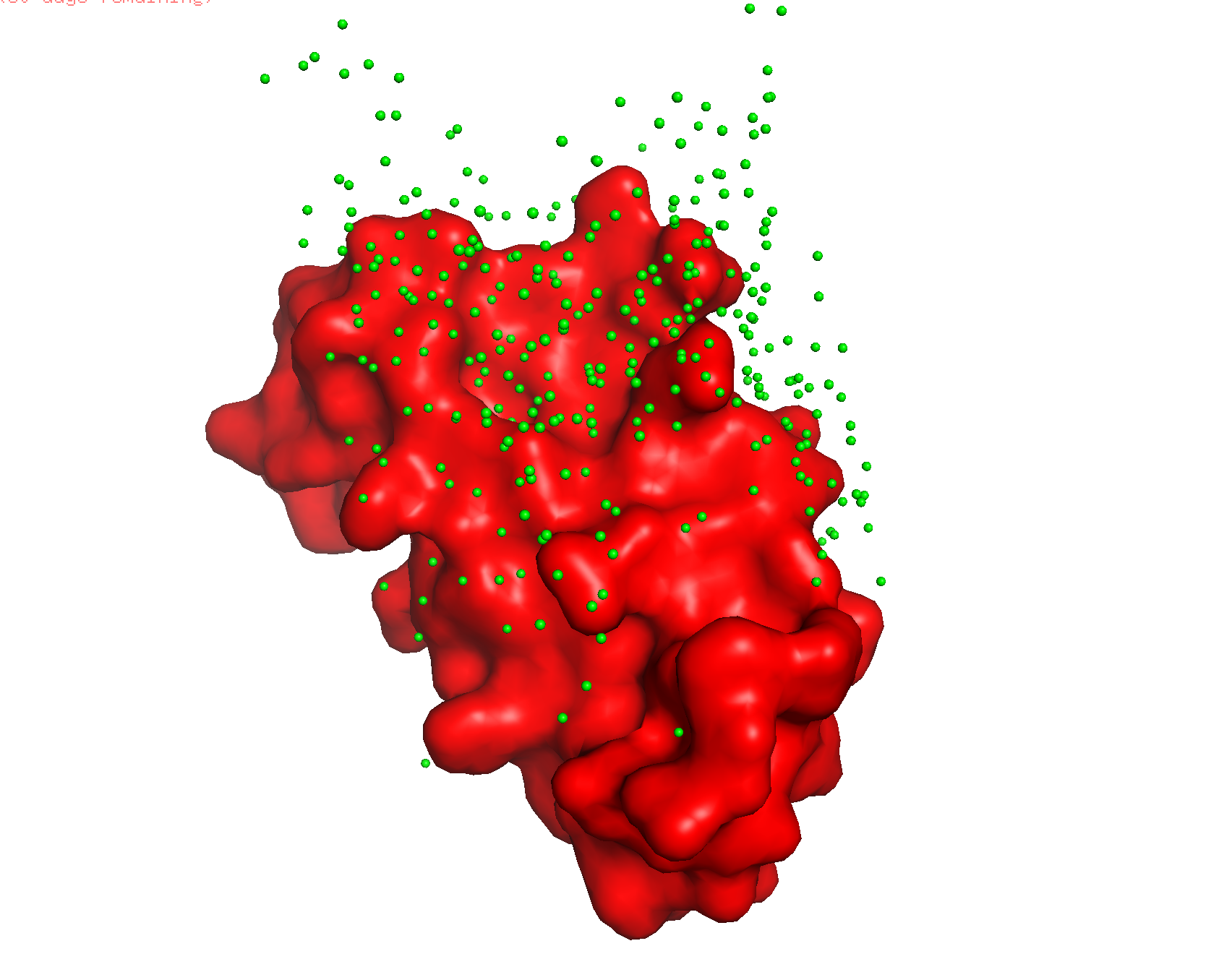
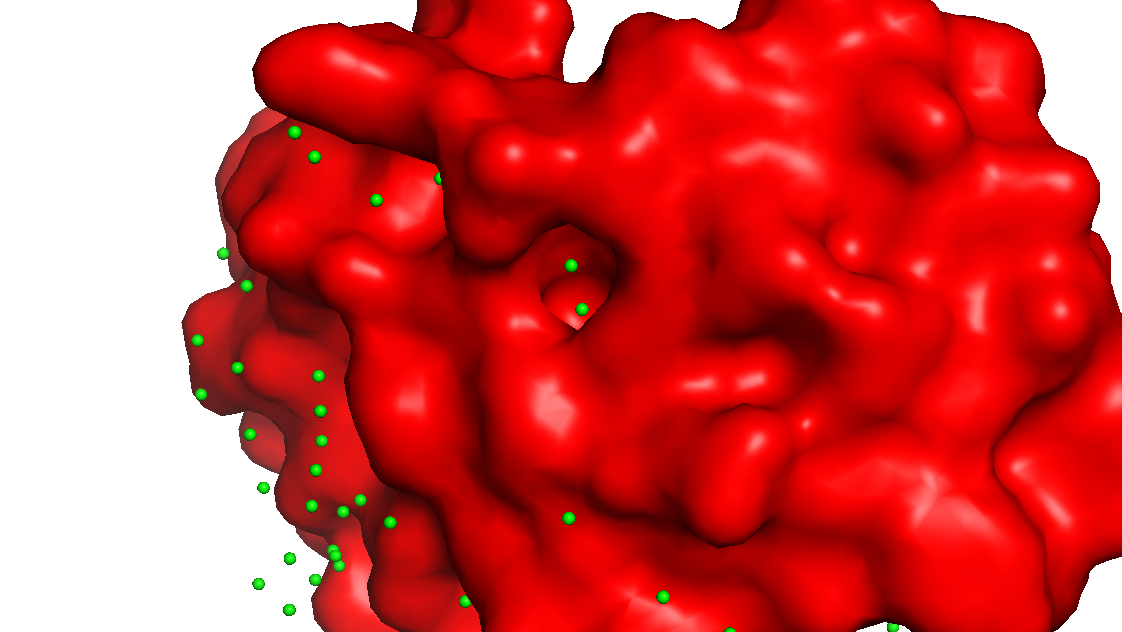
The surface doesn’t show a deep binding pocket — the receptor-binding interface is relatively flat.
Part C. Using ML-Based Protein Design Tools
4INS : GIVEQCCTSICSLYQLENYCNFVNQHLCGSHLVEALYLVCGERGFFYTPKT
C1. Protein Language Modeling
https://colab.research.google.com/drive/1Pu0Nmmpn-OjL_UDqrjnAhowjz1ZP1hKJ?usp=sharing
Deep Mutational Scans
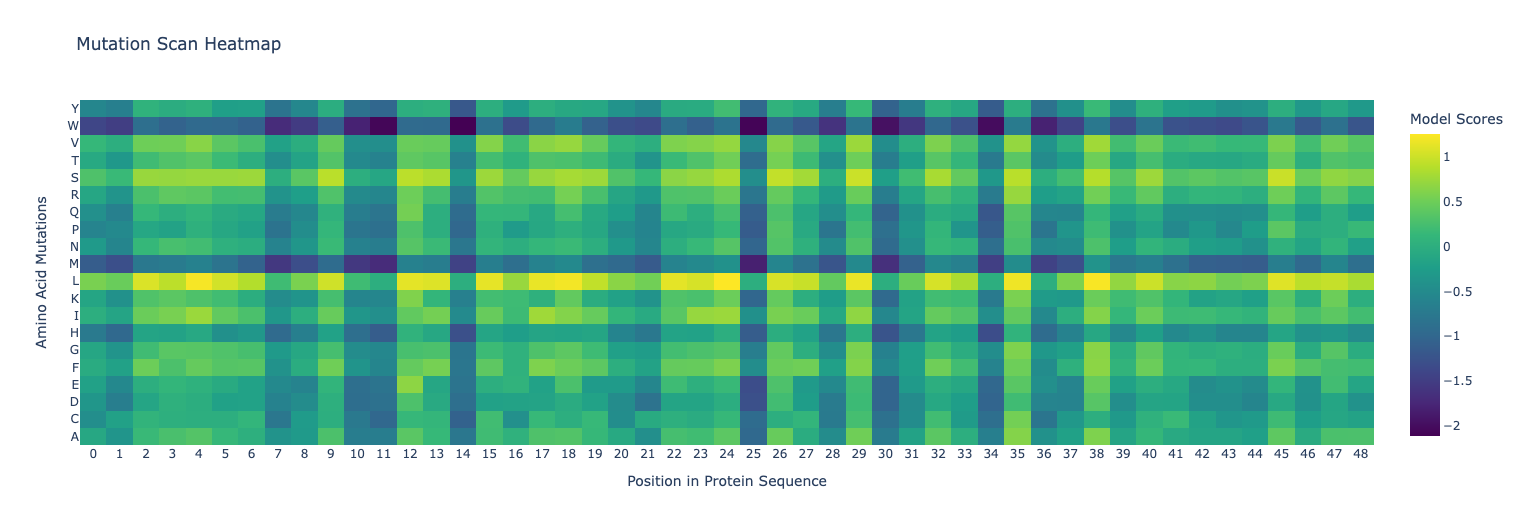
Use ESM2 to generate an unsupervised deep mutational scan. Explain any particular pattern (choose a residue and mutation that stands out). (Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
Latent Space Analysis
Use the provided sequence dataset to embed proteins in reduced dimensionality. Analyze the different formed neighborhoods. Place your protein in the resulting map and explain its position and similarity to its neighbors.
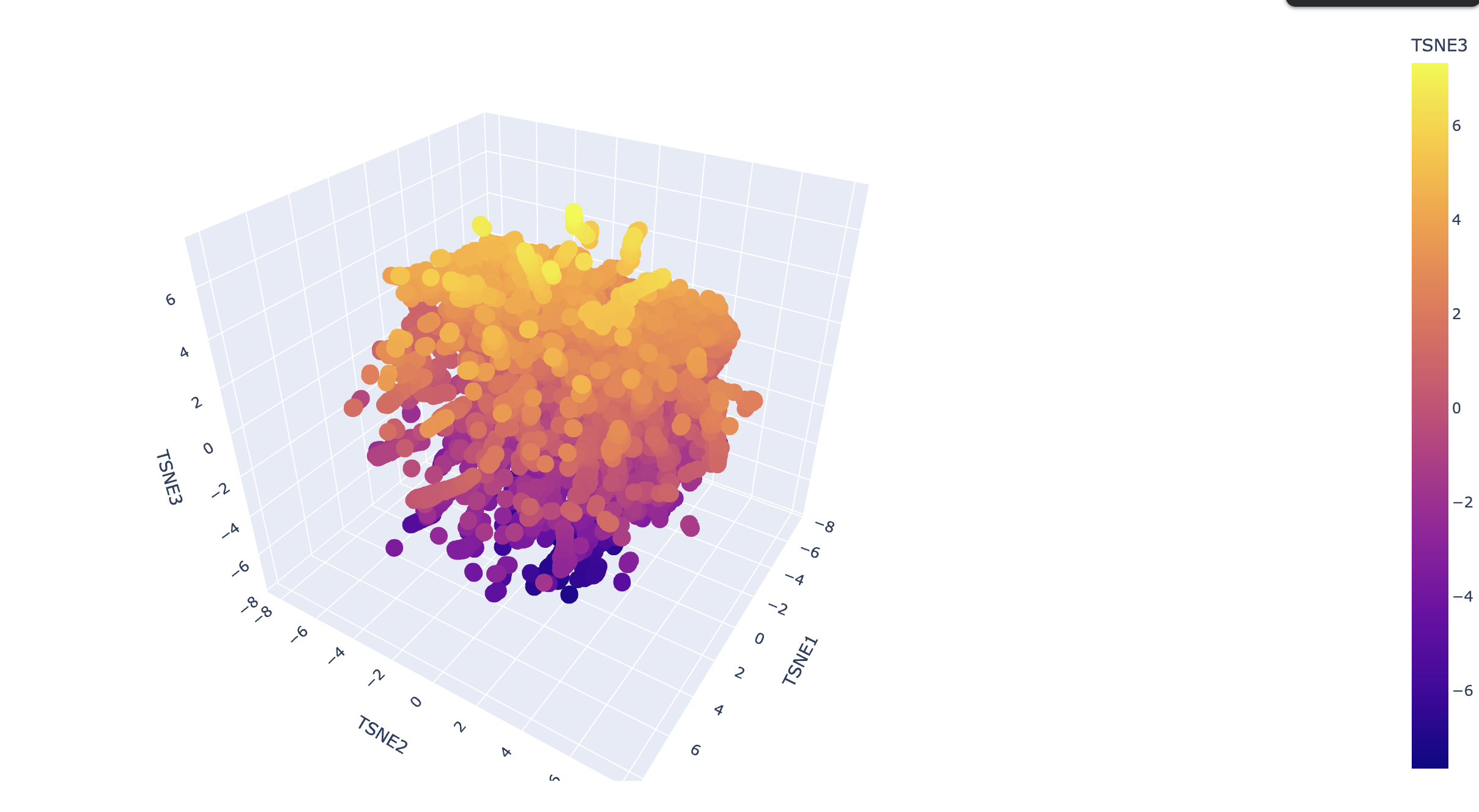
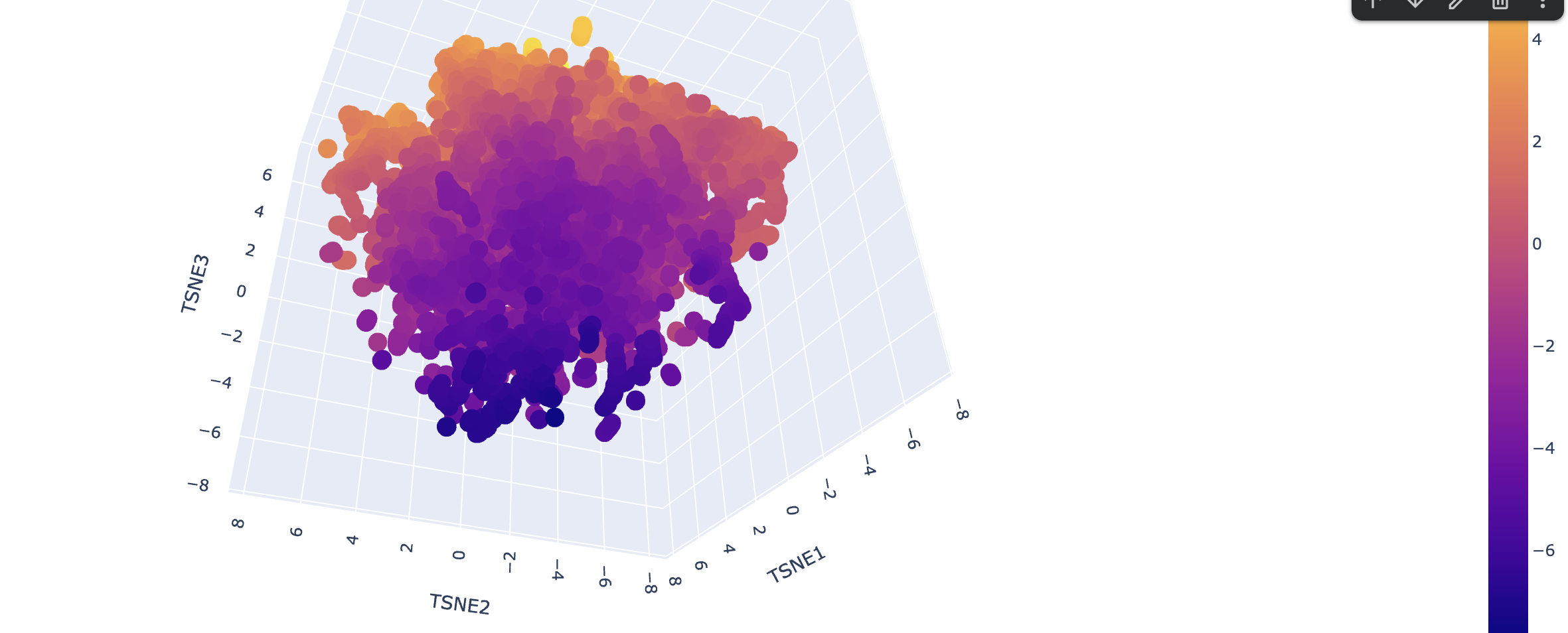
Explain its position and similarity to its neighbors
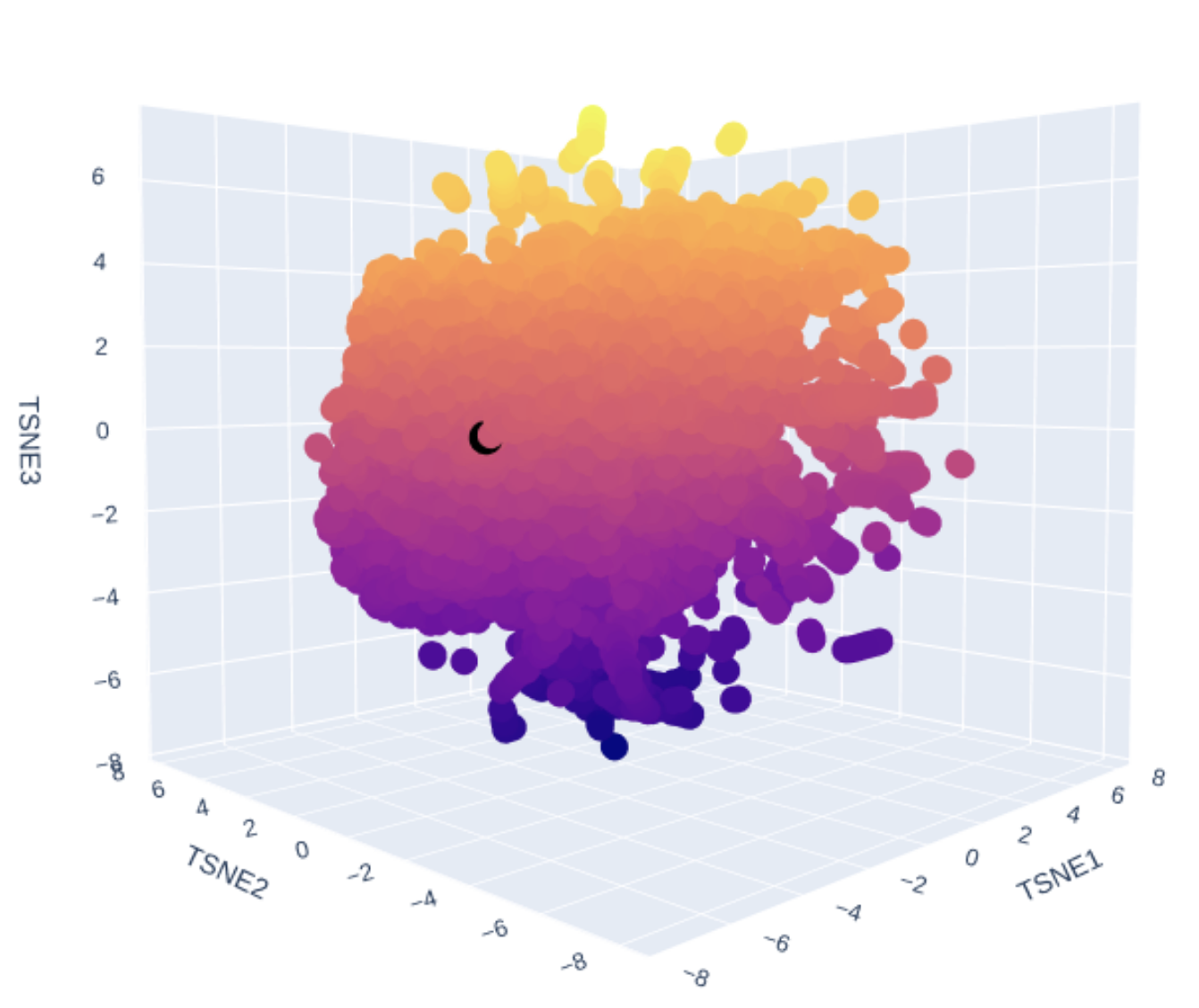
!!! TODO - I don’t know enough to describe it.
C2. Protein Folding
Fold your protein with ESMFold. Do the predicted coordinates match your original structure? Try changing the sequence — first some mutations, then large segments. Is your protein structure resilient to mutations?
!!! TODO - Cannot see the coordinates. Structure looks interesting.
Predicted folded:
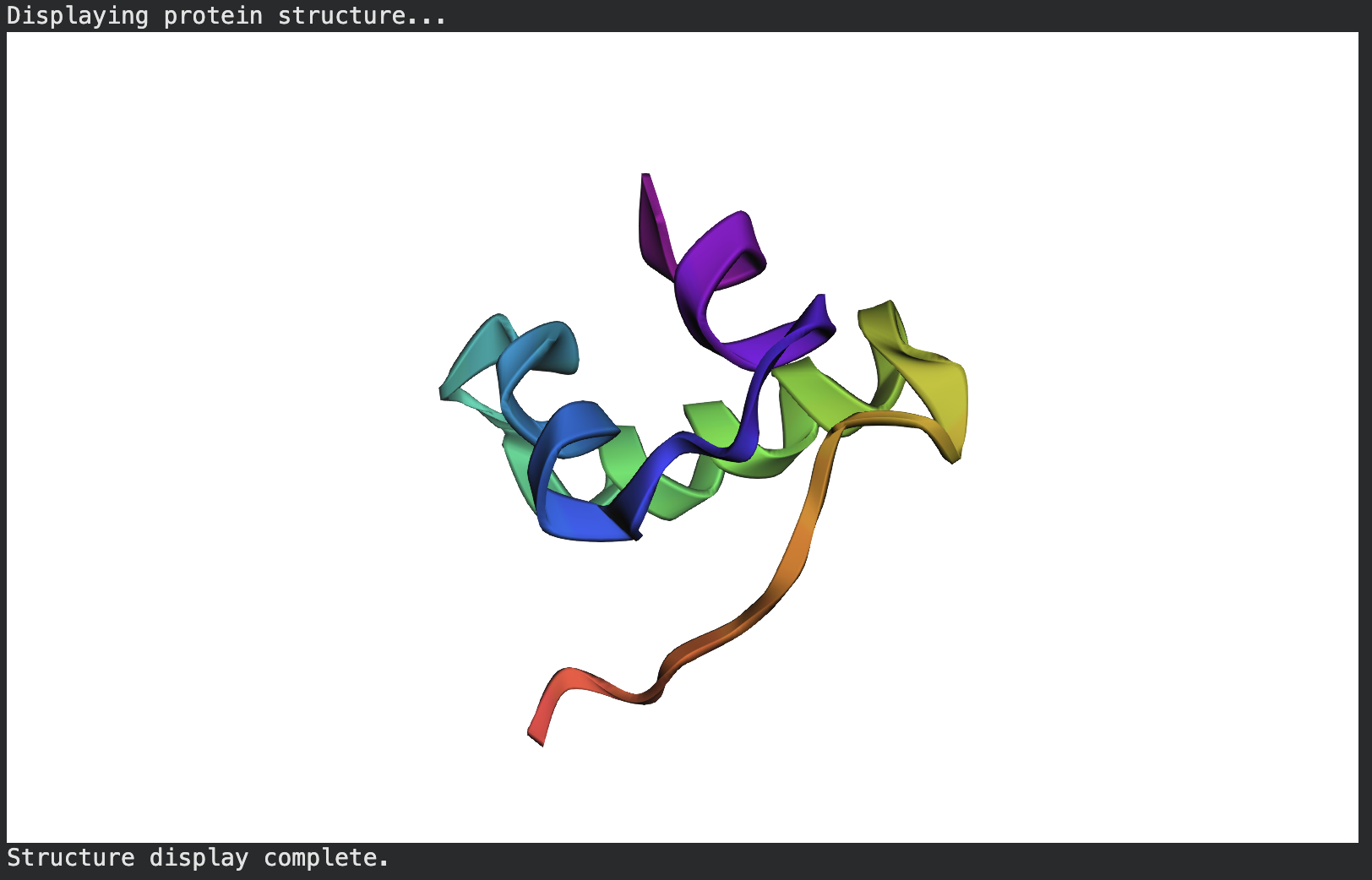
ptm0.413_r3_default.pdb - PDB file.
Original 4INS Ribbon Diagram:
C3. Protein Generation
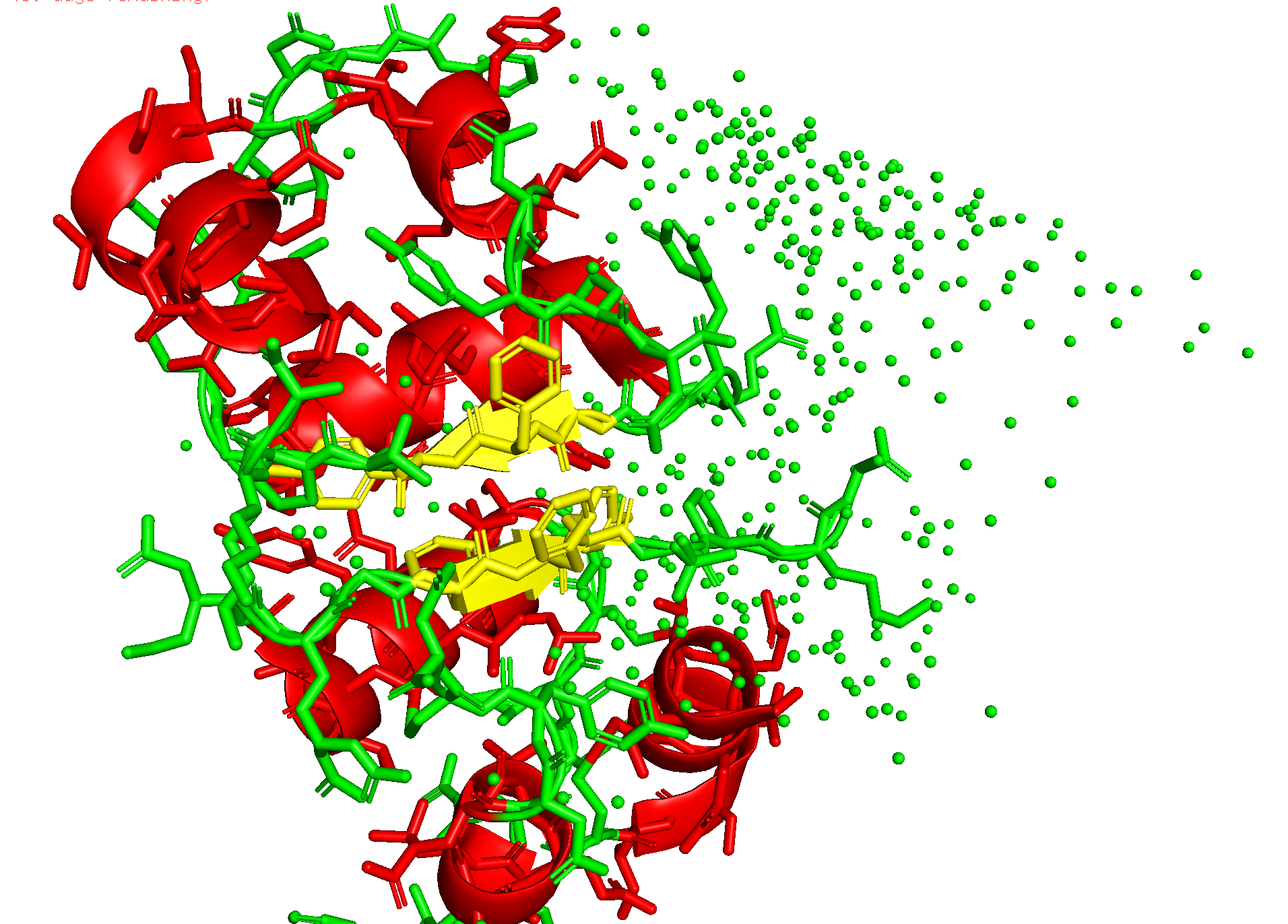
Use ProteinMPNN to inverse-fold your protein backbone. Analyze the predicted sequence probabilities and compare to the original. Input the predicted sequence into ESMFold and compare the predicted structure to your original.
.png)
Comparing:
ESMFold predicted structure vs. original:
Predicted:
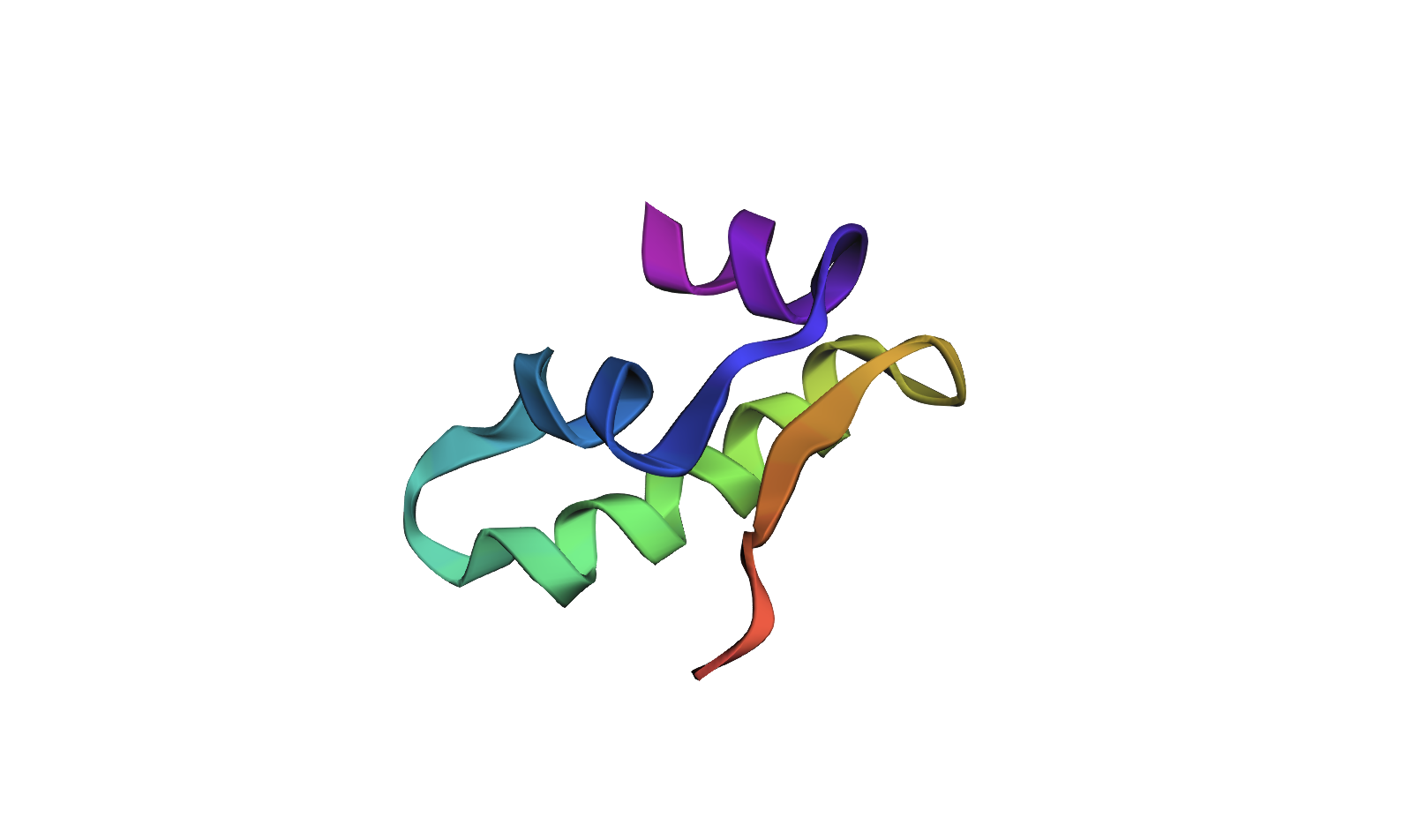
Original:
Part D. Group Brainstorm on Bacteriophage Engineering
NA - Sick