📄Committed Listener MOU
I am an HTGAA Committed Listener, my responsibilities are:
Watching class lectures and recitations
Participating in node reviews
Developing and documenting my homework
Actively communicating with other students and TAs on the forum
Allowing HTGAA and BioClub to share my work (with attribution)
Honestly reporting on my work, and appropriately attributing and citing the work of others (both human and non-human)
Following locally applicable health and safety guidance
Promoting a respectful environment free of harassment and discrimination
Signed by committing this file to my documentation page/repository,
Liam
08 March 2026
HW.1: Class assignment 1. Describe an application Identify a biological engineering tool or application you wish to develop and explain your motivation.
I would like to develop a way to make plants grow 100x faster. I find this a very interesting and ambitious question. Perhaps you reverse-engineer the genome, morphological development and constraints, proteins/enzymes/catalysts for growth. Perhaps you design a separate organism (two bacterium?) which produces biomass - a combination of a carbon sequester and a cellulose printer. Perhaps you attempt to design a minimal artificial cell, like a Xenobot / JCVI minimal cells - using new AI design software, you create a minimal genome/DNA, design your own morphological topology through simulation, which is compiled down to gene regulatory networks (GRN’s), transcription factors/thresholds, and DNA.
HW.1: Benchling & In-silico Gel Art Make a free account at benchling.com, Import the Lambda DNA. Simulate Restriction Enzyme Digestion with the following Enzymes: EcoRI HindIII BamHI KpnI EcoRV SacI SalI Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks. Benchling screenshots. Experimental design for Gel art. HW.2: Gel Art - Restriction Digests and Gel Electrophoresis In the wet-lab perform the lab experiment you designed in Part 1 and outlined in this week’s lab protocol “Gel Art: Restriction Digests and Gel Electrophoresis”.
https://2026a.htgaa.org/2026a/course-pages/weeks/week-03/index.html
HW.1: Python Script for Opentrons Artwork Review recitation materials and lab documentation. Design artwork using the GUI at opentrons-art.rcdonovan.com. Write a Python script using coordinates from the GUI via the “HTGAA26 Opentrons Colab”. Sign up for a robot time slot and run the script on the Opentrons robot. Submit Python file via provided form. HW.2: Post-Lab Reflection 2.1. Find and describe a published paper utilizing Opentrons or similar liquid handling automation tools. The paper I have found: Slowpoke: An Automated Golden Gate Cloning Workflow for Opentrons OT‑2 and Flex
Part A. Conceptual Questions Answer any NINE of the following questions from Shuguang Zhang:
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) Meat is roughly 20% protein by weight, so 0.2*500g=100g of protein. This is the only amino acid in meat, as carbs are sugars, and fats are triglycerides (fatty acids + glycerol).
Part A. SOD1 Binder Peptide Design Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? A Phusion HF PCR Master Mix is a pre-combined PCR reaction system optimised for a specific engineered DNA polymerase.
Phusion DNA polymerase — provides the catalytic activity which synthesises DNA, and includes a 3’→5’ exonuclease proofreading to reduce error Reaction buffer MgCl₂ — magnesium ions dNTPs — deoxynucelotide triphosphates: dATP, dCTP, dGTP, and dTTP Stabilizers/additives Water A typical setup only requires after adding:
https://2026a.htgaa.org/2026a/course-pages/weeks/week-07/index.html
Part 1: Intracellular Artificial Neural Networks (IANNs) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? They can compute continuous values, not just discrete ones.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal. A feedback-response mechanism, for example how the pancreas runs a sort of PID controller / control system for managing glucose levels in the body - through two hormones that move glucose levels in opposite directions - insulin and glucagon.
https://2026a.htgaa.org/2026a/course-pages/weeks/week-09/index.html
General homework questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Cell-free protein synthesis is technology where proteins are produced using lysed cell machinery (ribosomes, enzymes) rather than living cells.
https://2026a.htgaa.org/2026a/course-pages/weeks/week-10/index.html
Homework: Final Project Identify at least one aspect of your project to measure (protein mass, sequence, biomarker presence/quantity, etc.). For this homework, let’s say my project is to produce human insulin (INS_HUMAN) via recombinant synthesis (cell-based system - E. Coli).
The aspect I want to measure is purification/presence of the insulin protein.
https://2026a.htgaa.org/2026a/course-pages/weeks/week-11/index.html
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork Contribute at least one pixel to the global artwork experiment before editing ends. Discuss on the Discourse forum. I missed the deadline for this, sorry!
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents Provide 1–2 sentence descriptions of each component’s role in the cell-free reaction: E. coli Lysate: BL21 (DE3) Star Lysate (includes T7 RNA Polymerase) Provides the core machinery for translation and transcription of proteins.
Identify a biological engineering tool or application you wish to develop and explain your motivation.
I would like to develop a way to make plants grow 100x faster. I find this a very interesting and ambitious question. Perhaps you reverse-engineer the genome, morphological development and constraints, proteins/enzymes/catalysts for growth. Perhaps you design a separate organism (two bacterium?) which produces biomass - a combination of a carbon sequester and a cellulose printer. Perhaps you attempt to design a minimal artificial cell, like a Xenobot / JCVI minimal cells - using new AI design software, you create a minimal genome/DNA, design your own morphological topology through simulation, which is compiled down to gene regulatory networks (GRN’s), transcription factors/thresholds, and DNA.
Why? Because trees and plants are great. They are calming, they look beautiful, they are functionally useful. Originally I wanted to build my own house, and was wondering - why is wood so expensive? If we could grow wood more quickly and effectively, that would be useful. It would also be fun to rapidly green certain areas of the world to produce arable land - the Australian desert, for example.
2. Establish governance goals
Describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
Enhance biosecurity (prevent misuse and uncontrolled spread)
Prevent incidents
Restrict access to engineered strains, protocols, and enabling tools
Use genetic containment (kill-switches, auxotrophy, sterility)
Avoid traits that increase invasiveness or persistence outside intended settings
Help respond
Establish monitoring and reporting systems for unexpected dissemination
Prohibit open release until long-term impacts are understood
Prefer reversible or self-limiting designs over permanent alterations
Help respond
Post-deployment surveillance and remediation plans
Defined liability and responsibility for environmental harms
Equity, autonomy, and constructive use (ensure benefits are fairly distributed)
Minimizing burdens to stakeholders
Community consultation for land-use and deployment decisions
Avoid shifting risks onto local ecosystems or vulnerable populations
Feasibility without blocking research
Clear regulatory pathways that enable safe experimentation
Transparency and documentation to support responsible scaling
Promote beneficial applications
Prioritize reforestation, sustainable materials, and climate-positive outcomes
Discourage purely extractive or destabilizing commercial deployment
3. Design governance actions
Describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”)
Purpose: What is done now and what changes are you proposing?
Design: What is needed to make it “work”? (including the actor(s) involved - who must opt-in, fund, approve, or implement, etc)
Assumptions: What could you have wrong (incorrect assumptions, uncertainties)?
Risks of Failure & “Success”: How might this fail, including any unintended consequences of the “success” of your proposed actions?
Evaluate each action against objectives including:
Biosecurity enhancement
Lab safety
Environmental protection
Cost/burden minimization
Feasibility and research impact
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
3
3
2
• By preventing incidents
3
3
2
• By helping respond
2
2
3
Foster Lab Safety
3
2
1
• By preventing incident
3
2
1
• By helping respond
2
2
2
Protect the environment
3
2
3
• By preventing incidents
3
2
2
• By helping respond
2
1
3
Other considerations
• Minimizing costs and burdens to stakeholders
2
2
1
• Feasibility?
2
3
1
• Not impede research
1
2
1
• Promote constructive applications
3
2
3
5. Prioritize options
Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties.
For this, you can choose one or more relevant audiences for your recommendation, which could range from the very local (e.g. to MIT leadership or Cambridge Mayoral Office) to the national (e.g. to President Biden or the head of a Federal Agency) to the international (e.g. to the United Nations Office of the Secretary-General, or the leadership of a multinational firm or industry consortia). These could also be one of the “actor” groups in your matrix.
I would prioritise Containment-by-design + staged release. Given that there is immense uncertainty in how this project could be achieved, it is a waste of resources to consider other governance actions for now. Rapid iteration to reduce uncertainty is the path towards achievement. As part of this - a scalable safety protocol throughout this process facilitates rapid experimentation without risk of ruin, until the project can achieve milestones necessary for unlocking funding and revenue.
HW.2: Lecture prep for W2
Answer prep questions from three faculty members:
Homework Questions from Professor Jacobson:
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
Error rate refers to errors per nucleotide added per replication. An error could be a misincorporation (wrong base expressed for a pair), for example.
Error rate of polymerase synthesis is 1/1e7 (1:10^7).
The rate of errors in polymerase copying the human genome’s DNA is 1/1e7 * 3e9, which is nonzero.
Biology deals with the likely error through multiple levels of mitigation:
Proofreading during synthesis corrects errors
Mismatch repair after synthesis repairs errors
Redundancy and selection at multiple levels - DNA is double-stranded, cells exist in huge populations, misfolded proteins get degraded, defective RNAs are destroyed, faulty cells undergo apoptosis
Damage repair system
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Our assumptions:
Average Human Protein: 1036 bp.
~30,000 proteins observed in mammalian genome.
A protein of length L = 3L nucleotides (bases) + a stop codon in the genome
Coding is the process by which DNA is transcribed into mRNA (triplets / codons), and mRNA (codons) is translated into a linear chain of amino acids (polypeptides), which folds into 3D protein structures.
How many different ways are there to code for an average human protein, meaning how many different DNA encodings would compile (transcribe and translate) down to the same protein (chain of amino acids) of length 1036 bp?
Codons are 3 nucleotides, each which have a base (A,C,G,T). There are 64 possible triplet combinations (codons) using the four bases (A, U, G, C). Each codon encodes one amino acid. An amino acid can be encoded by multiple codons. For instance, codons GAA and GAG both specify glutamic acid and exhibit redundancy. This is referred to as degeneracy.
The degeneracy of an amino acid refers to the number of codons which encode it. ie. d(Leu)=6, meaning Leucine has 6 codons which encode it.
Average codon degeneracy across amino acids is roughly 3.
So to calculate the number of possible encodings for a protein of length L=5 amino acids, we compute the degeneracy of each amino acid, and compute their product to find the maximum number of permutations. ie. for a protein of L=5, average degeneracy d(*)=3, num_permutations=d(*) * d(*) * d(*) * d(*) * d(*) = d(*)^L = 3^L
So for an average human protein of L=1036 bp, the number of possible encodings could be 3^L = 3^1036.
There is an intractable number of possible encodings. However, functional “good” encodings are a tiny subset constrained by expression, folding, RNA processing, regulation, and host biology.
Homework Questions from Dr. LeProust:
What’s the most commonly used method for oligo synthesis currently?
solid-phase chemical synthesis with phosphoramidite chemistry
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Because direct phosphoramidite synthesis has a per-step yield <1.0, errors compound exponentially with length. P(success)=(1-e)^200 is improbable (e ~= 0.01)
Why can’t you make a 2000bp gene via direct oligo synthesis?
(1-e)^2000 is near impossible, due to errors accumulating from each synthetic cycle/step.
expected number of cleavage events scales ~linearly with cycle count and purine content
Misincorporations accumulate (wrong base addition)
Homework Question from George Church:
Choose ONE of the following three questions to answer; and please cite AI prompts or paper citations used, if any.
[Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
[Given slides #2 & 4 (AA:NA and NA:NA codes)] What code would you suggest for AA:AA interactions?
[(Advanced students)] Given the one paragraph abstracts for these real 2026 grant programs sketch a response to one of them or devise one of your own:
Out of the 20 amino acids needed, the body synthesizes 11-12, while the remaining 8-9, known as essential amino acids, must be obtained through diet.
This is not accurate to all animals, it seems? Counterexample: cats. Cats require taurine.
The Lysine Contingency was a genetic alteration Henry Wu performed in the dinosaur genome. The modification knocked out the ability of the dinosaurs to produce the amino acid Lysine.
This forced the dinosaurs to depend on lysine supplements provided by the park’s veterinary staff. In this way, dinosaurs could never escape from the park because they would never survive long without the food supplements.
Haha, I have to rewatch this film.
The way I would hack around this would be to introduce a substance containing the microbes that cows digest and feed it to the dinosaurs. These microbes synthesise the essential amino acids from nitrogen, thus mitigating the need for the dinosaurs to produce Lysine themselves, instead forming a symbiotic relationship with the microbes in their gut.
I don’t know what this question means, but it reminds me also of Liebig’s law - would the restriction of one amino acid necessarily debilitate the dinosaurs so they can’t escape, or is nature more nonlinear and complex than that?
LLM prompts used:
10 essential amino acids in all animals?
across all animals?
cows can synthesise most of their needed amino acids? how many which ones
how long can you survive without just one of the amnio acids ?
Make a free account at benchling.com, Import the Lambda DNA.
Simulate Restriction Enzyme Digestion with the following Enzymes: EcoRI HindIII BamHI KpnI EcoRV SacI SalI
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
Benchling screenshots.
Experimental design for Gel art.
HW.2: Gel Art - Restriction Digests and Gel Electrophoresis
In the wet-lab perform the lab experiment you designed in Part 1 and outlined in this week’s lab protocol “Gel Art: Restriction Digests and Gel Electrophoresis”.
N/A - no access to BioClub Tokyo Lab.
HW.3: DNA Design Challenge
3.1. Choose your protein.
In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose.
Describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
Proteins are translated from mRNA by tRNA’s. The tRNA’s “pair” with codons from the mRNA. A codon is a 3-base sequence which is then mapped onto a single amino acid. As we covered last week, there are 64 different codons (permutations of a string of 3 nuceleotide bases) which map down to only 20 amino acids. The degeneracy means we can swap out parts of the DNA/mRNA to express the same amino acids aka proteins. Why would we do this? Because mRNA codons are translated into amino acids by the available tRNA in the organism. Each tRNA matches a codon (or several synonymous codons, see wobble pairing at 3rd base). There is not a uniform concentration of tRNA for all codons. So some mRNA codons will translate more efficiently than others, because there is more tRNA.
To restate:
DNA encodes triplet codons.
mRNA is transcribed from DNA.
Ribosomes read mRNA in triplets.
tRNAs carrying amino acids base-pair with codons (binding with the tRNA’s complementary anticodon)
Translation rate is approximately proportional to local charged tRNA abundance and ribosomal processivity.
Multiple codons encode the same amino acid, yet different organisms use these synonymous codons at different frequencies (codon usage bias). If a gene from organism A is expressed in organism B without modification, the codon distribution may not match the tRNA pool of B.
You need to optimize codon usage in order to achieve (good) yields from your biomanufacturing process.
I choose Escherichia coli (E. coli) as the target host for optimization:
Takes less time
Cell division is faster
Well established protocols to isolate plasmid
Each cell has single chromosome
Single circular plasmid
Each replicated cell has exact copy of DNA
Easy method
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words how the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
Recombinant expression in a host organism like E. Coli.
Clone the coding sequence into an expression vector (a plasmid).
Promoter - T7 under lac control: binds the RNA polymerase
Ribosome binding site - Shine–Dalgarno AGGAGG: recruits ribosome
Coding sequence - see Miraculin DNA sequence above.
Antibiotic resistance gene - ampR: for selection of culture
Transform into E. Coli (transform the plasmid into host cells.)
Bacteria are given a heat shock.
Colonies grow.
Pick colonies.
Plate on ampicillin → only plasmid-containing cells survive.
Inoculate the liquid cultures (by introducing single colonies)
Induce expression (e.g., add IPTG if T7/lac system).
T7 RNA polymerase binds promoter
DNA is transcribed into mRNA
Ribosome binds RBS on mRNA.
tRNA translates into protein, stop at terminator.
tRNAs decode codons
Amino acids polymerize into polypeptide
Harvest. Cells are lysed. Protein is purified.
Lyse cells (sonication or chemical lysis).
Purify protein (e.g., His-tag + Ni-NTA affinity column).
Apparently E. coli is possible but non-ideal for a cysteine-rich, glycosylated plant secreted protein like miraculin.
3.5. [Optional] How does it work in nature/biological systems?
Describe how a single gene codes for multiple proteins at the transcriptional level. Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein!!! See example below.
HW.4: Twist DNA Synthesis Order
Steps to build a plasmid:
Import DNA into Benchling.
Add promoter, RBS, start/stop codons, 7x His Tag, and terminator
Export .fasta and import into Twist.
Order Twist clonal gene, using pTwist Amp High Copy vector.
Export .gb (genbank) file for plasmid.
Import plasmid .gb file into Benchling, open Info>Toplogy and set Circular.
HW.5: DNA Read/Write/Edit
DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
No idea. Possibly my basil plant.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would use long-read sequencing (1–100+ kb). Even though it is more expensive, it would provide greater accuracy.
The way that DNA sequencing works currenly is by taking DNA, lysing it, and then reassembling fragments based on probabilistic approaches. The “read length” refers to how large these fragments are in terms of base pairs. A fragment of length = 1 bp would be near useless, since there is no way to “place” it probabilistically within the greater genome. A fragment of length = 150bp map well because apparently the human genome is largely non-repetitive at that scale.
Short-read sequencing is a read of 50–600 bp. Long-read sequencing is 1-100 kb.
Technologies:
Polymerase-based sequencing
Enzymatic digest sequencing
Nanopore sequencing
DNA microarrays
DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
I have no idea.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Recombinant DNA synthesis
Oligonucleotide synthesis - can make complex motifs, extremely large DNA molecules (1kbp+)
DNA Edit
(i) What DNA would you want to edit and why?
I have no idea. Potentially plant DNA. I don’t know anything about what DNA plants have. I would like to figure out how to increase the growth speed, change the bark texture. Or even doing experiments on yeast. Perhaps I could figure out the enzymes/proteins and what DNA/genes code for it, and then edit that.
(ii) What technology or technologies would you use to perform these DNA edits and why?
Write a Python script using coordinates from the GUI via the “HTGAA26 Opentrons Colab”.
Sign up for a robot time slot and run the script on the Opentrons robot.
Submit Python file via provided form.
HW.2: Post-Lab Reflection
2.1. Find and describe a published paper utilizing Opentrons or similar liquid handling automation tools.
The paper I have found: Slowpoke: An Automated Golden Gate Cloning Workflow for Opentrons OT‑2 and Flex
Slowpoke is a tool which generates Opentron protocols for DNA assembly. DNA assembly is used to assemble larger strings of DNA than can be synthesised in one go, by joining together oligonucleotides.
It provides facilities to automate:
Golden Gate Cloning - automates the DNA assembly reaction setup, E. Coli transformation, and plating.
Colony PCR - automates colony PCR screening of resulting transformants.
Users provide for:
Golden Gate Cloning
Genetic toolkit map - e.g. MoClo YTK, STK plate layout
Custom parts map
Combination file
Colony PCR
Colony template positions
PCR deck maps
Reaction recipes.
The robot protocol automates the full pipeline of assembly, transformation, plating and colony PCR:
DNA and enzyme buffer extraction.
Golden gate reaction.
Transformation.
Plating.
Colony PCR.
It is compatible with multiple MoClo/Golden Gate toolkits (YTK, STK, and extensible to others).
Manual steps still required:
Colony picking - most labour-intensive step.
Sealing PCR plates in OT-2 thermocycler module.
Transferring PCR tubes to benchtop thermocycler.
Incubation, strain storage, and plasmid purification - still accounts for a lot of time.
2.2. Describe your intended automation use for your final project, including pseudocode, scripts, or implementation plans.
I intend to use a cloud lab platform to screen an array of biosensor constructs that I have designed, synthesised, and expressed using cell-free protein synthesis (CFPS).
HW.3: Final Project Ideas
Submit 1–3 slides with three individual project concept ideas.
Week 04 HW
Part A. Conceptual Questions
Answer any NINE of the following questions from Shuguang Zhang:
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Meat is roughly 20% protein by weight, so 0.2*500g=100g of protein. This is the only amino acid in meat, as carbs are sugars, and fats are triglycerides (fatty acids + glycerol).
1 g = 6.02217364335E+23 dalton
100 g = 6.022173643E+25 daltons
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Humans eat beef but don’t become a cow because the stomach metabolizes complex proteins and cells down to base level molecules and amino acids.
3. Why are there only 20 natural amino acids?
For evolutionary reasons probably. Much like how human language has a finite set of phonemes which allow us to express infinitely more higher-level syllables, words, and concepts and sentences - biology has a base grammar of 20 units. This has proven to be enough - 64 codons map onto 20 amino acids plus stop signals. There may have been more but this is evidently evolutionarily optimal as it is now.
4. Can you make other non-natural amino acids? Design some new amino acids.
β-amino acids are interesting - usually the amino group is attached onto the α-carbon, but here they are attached on the β-carbon. Due to this, proteases (enzymes which support digestion) are highly ineffective against β-peptides.
Others I googled:
Fluoroleucine — leucine with fluorine substituted in; more hydrophobic and metabolically stable
Azidohomoalanine — methionine analog with an azide group, useful for click chemistry bioconjugation
5. Where did amino acids come from before enzymes that make them, and before life started?
Meterorites that naturally carry amino acids
Miller-Urey experiment showed amino acids could form spontaneously from simple molecules + an electric arc
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Left-handed. Natural α-helices are right-handed because they’re built from L-amino acids. D-amino acids are the mirror image, so the resulting helix is the mirror image too — left-handed.
7. Can you discover additional helices in proteins?
Yes — beyond the common α-helix, proteins also contain 3₁₀-helices (3 residues per turn, tighter) and π-helices (4.4 residues per turn, rarer and wider). These are already known but underappreciated. Computational analysis of PDB structures keeps surfacing edge cases and unusual conformations that don’t fit neatly into existing categories.
8. Why are most molecular helices right-handed?
Because natural amino acids are L-enantiomers. The geometry of the L-α-carbon makes right-handed coiling energetically favorable — the side chains point outward without steric clashes in a right-handed helix. A left-handed helix built from L-amino acids would force side chains into the backbone, creating strain.
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheets have “sticky edges” — the backbone NH and C=O groups along the edge strands are unsatisfied hydrogen bond donors/acceptors. These can pair with the edge of another β-sheet. The driving forces are hydrogen bonding along the backbone and hydrophobic stacking between sheet faces. This makes lateral growth into large, ordered aggregates thermodynamically favorable.
10. Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
When proteins misfold or partially unfold, they expose their backbone, which can then hydrogen-bond with other misfolded proteins into cross-β structure (hydrogen bonds running perpendicular to the fibril axis). This structure is extremely stable — often more so than the native fold — so once nucleation starts, it propagates. Many proteins will form amyloid under the right conditions; some just do so more readily due to sequence composition or environmental stress.
Yes, amyloid fibrils can be used as materials — they’re stiff, stable, and self-assembling. Researchers have used them as scaffolds for nanomaterials, hydrogels, and functional coatings.
11. Design a β-sheet motif that forms a well-ordered structure.
Part B. Protein Analysis and Visualization
(1) Briefly describe the protein you selected and why you selected it.
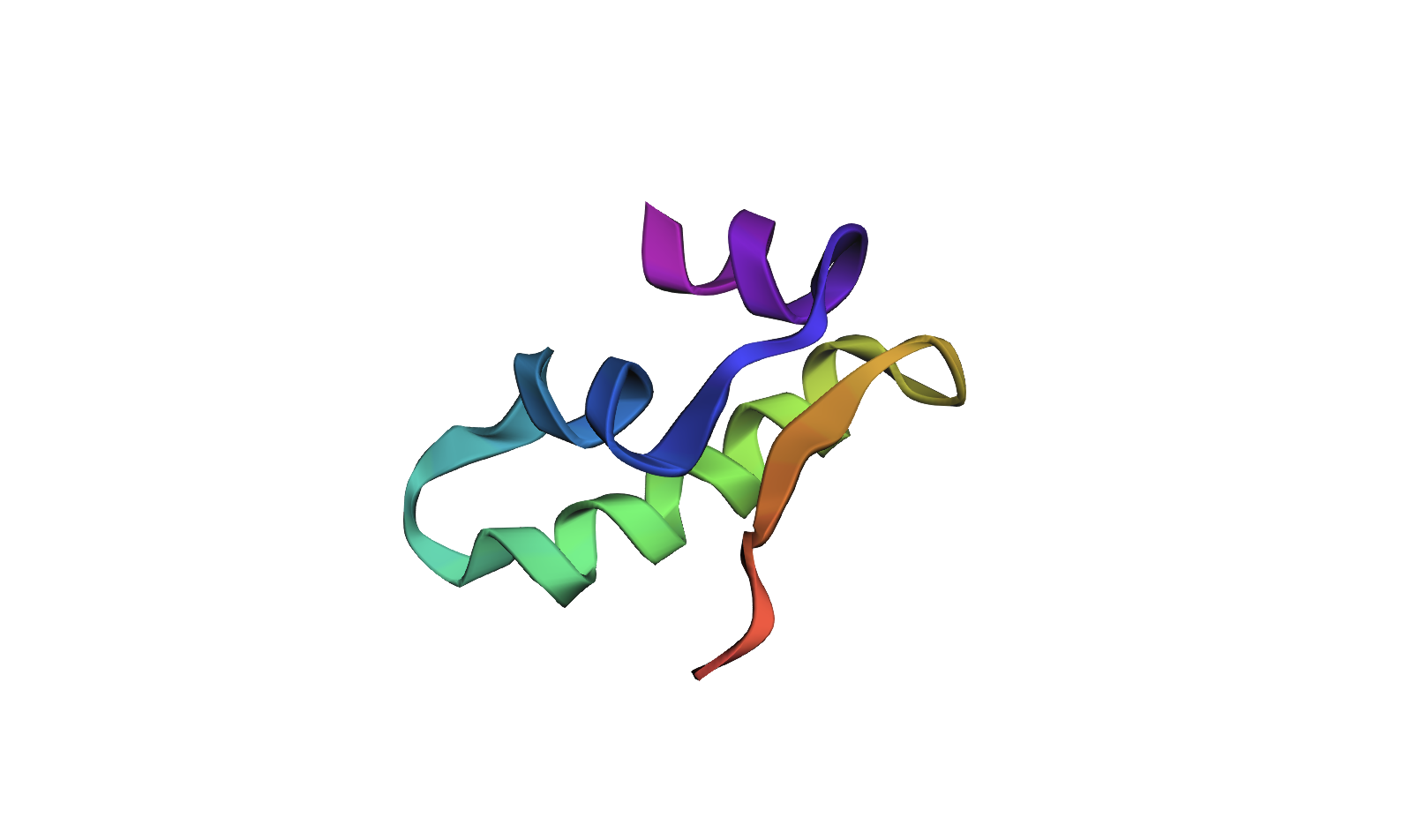
Insulin. A 51-amino acid peptide hormone secreted by pancreatic β-cells that regulates blood glucose by signaling cells to take up glucose. I chose it because it’s small and well-studied, historically significant (first recombinantly produced therapeutic protein), and I’m curious how something so tiny has such a large physiological effect.
(2) Identify the amino acid sequence of your protein. How long is it? What is the most frequent amino acid? How many protein sequence homologs are there? Does your protein belong to any protein family?
51 amino acids total — two chains: A (21 aa) and B (30 aa), linked by two disulfide bonds. Most frequent: leucine (L) and cysteine (C), both at 6 occurrences. Many homologs — the insulin/IGF/relaxin superfamily includes IGF-1, IGF-2, relaxin, and insulin-like peptides across many organisms. Belongs to the insulin family (InterPro: Insulin/IGF/relaxin superfamily).
(3) Identify the structure page of your protein in RCSB. When was the structure solved? Is it a good quality structure? Are there any other molecules in the solved structure apart from protein? Does your protein belong to any structure classification family?
PDB: 4INS — human insulin hexamer, solved in 1989 at 1.5 Å resolution. Good quality structure. In addition to protein, the hexamer contains two zinc ions (Zn²⁺) coordinated by His B10 residues at the center, plus water molecules.
Why extra zinc ions? Insulin is stored as a zinc-stabilized hexamer in β-cells; once secreted, the hexamer dissociates into monomers and the zinc stays behind, so zinc is necessary for storage and secretion but not for receptor binding. Zinc deficiency is linked to impaired insulin secretion and increased type 2 diabetes risk.
In structural classification, insulin belongs to the “Insulin-like” fold under the all-α class.
(4) Open the structure in 3D visualization software. Visualize as “cartoon”, “ribbon”, and “ball and stick”. Color by secondary structure — does it have more helices or sheets? Color by residue type — what can you tell about hydrophobic vs hydrophilic distribution? Visualize the surface — does it have any binding pockets?
Color by secondary structure — does it have more helices or sheets
It has helices
Red spirals = α-helices
Yellow flat arrow shapes = β-sheets
Color by residue type — does it have more helices or sheets
PyMOL: util.cbag
- green is helices. I don’t see any β-sheets.
Visualize the surface — does it have any binding pockets?
Red is helix, Yellow is sheet, Green is loop.
PyMOL: show surface
The surface doesn’t show a deep binding pocket — the receptor-binding interface is relatively flat.
Use ESM2 to generate an unsupervised deep mutational scan. Explain any particular pattern (choose a residue and mutation that stands out). (Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
Latent Space Analysis
Use the provided sequence dataset to embed proteins in reduced dimensionality. Analyze the different formed neighborhoods. Place your protein in the resulting map and explain its position and similarity to its neighbors.
Explain its position and similarity to its neighbors
!!! TODO - I don’t know enough to describe it.
C2. Protein Folding
Fold your protein with ESMFold. Do the predicted coordinates match your original structure? Try changing the sequence — first some mutations, then large segments. Is your protein structure resilient to mutations?
!!! TODO - Cannot see the coordinates. Structure looks interesting.
Use ProteinMPNN to inverse-fold your protein backbone. Analyze the predicted sequence probabilities and compare to the original. Input the predicted sequence into ESMFold and compare the predicted structure to your original.
Part D. Group Brainstorm on Bacteriophage Engineering
NA - Sick
Week 05 HW
Part A. SOD1 Binder Peptide Design
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Challenge: Design short peptides that bind mutant SOD1, then decide which ones are worth advancing toward therapy.
Models used:
PepMLM: target sequence-conditioned peptide generation via masked language modeling
Retrieve the human SOD1 sequence from UniProt (P00441), introduce the A4V mutation, and use the PepMLM Colab to generate four peptides of length 12 amino acids conditioned on the mutant sequence. Add the known binder FLYRWLPSRRGG for comparison. Record perplexity scores.
A4V mutant SOD1 sequence (deleted M at position 1, changed A→V at position 4):
A note on perplexity: A lower perplexity score means higher model confidence that the peptide satisfies the criteria for binding the target.
Part 2: Evaluate Binders with AlphaFold3
Submit each peptide + mutant SOD1 as separate chains to the AlphaFold Server. Record the ipTM score and describe where each peptide appears to bind — does it localize near the N-terminus (A4V site), the β-barrel, or the dimer interface? Is it surface-bound or partially buried? In a short paragraph, describe the ipTM values and whether any PepMLM-generated peptide matches or exceeds the known binder.
Peptide
Binding location
ipTM score
WRSPAVAVAHWE
None
0.28
WRVGWVGVELKE
None
0.35
WRSPAAXIEHKX
None
0.33
WRVYAAXIEWGK
None
0.34
Part 3: Evaluate Properties in the PeptiVerse
Using PeptiVerse, evaluate the therapeutic properties of each peptide against the A4V mutant SOD1 sequence. Check: predicted binding affinity, solubility, hemolysis probability, net charge (pH 7), and molecular weight.
Peptide
Solubility
Hemolysis
Binding Affinity
MW (Da)
Net Charge (pH 7)
WRSPAVAVAHWE
1.0
0.044 (Non)
5.361 (Weak)
1408.6
-0.14
WRVGWVGVELKE
1.0
0.117 (Non)
7.089 (Medium)
1457.7
-0.23
WRSPAAXIEHKX
1.0
0.011 (Non)
4.645 (Weak)
1158.5
0.85
WRVYAAXIEWGK
1.0
0.043 (Non)
6.724 (Weak)
1360.7
0.76
FLYRWLPSRRGG (known)
1.0
0.047 (Non)
5.962 (Weak)
1507.7
2.76
The best peptide to advance for wet lab validation would be WRVGWVGVELKE due to its relatively high binding affinity (7.089, Medium).
Part 4: Generate Optimized Peptides with moPPIt
Using the moPPIt Colab: paste your A4V mutant SOD1 sequence, choose specific residue indices to target (e.g. near position 4, the dimer interface, or another surface patch), set peptide length to 12 aa, and enable motif + affinity guidance. Briefly describe how the moPPIt peptides differ from your PepMLM peptides. How would you evaluate these before advancing to clinical studies?
Binder
Hemolysis
Solubility
Affinity
Motif
SVKTKCCTTYQS
0.964
0.917
6.576
0.890
DDTKKCSCIQTH
0.975
0.917
6.314
0.915
ENGETFQCTKKV
0.970
0.833
6.044
0.935
KKSKKAFVCCVC
0.963
0.667
8.172
0.614
For the long execution time and computational resources required, the main advantage of moPPIt over PepMLM (in this context) is the motif score — there was no option to check motif specificity in PeptiVerse. All other properties of the PepMLM-generated sequences were comparable to the moPPIt peptides.
Part B. BRD4 Drug Discovery Platform Tutorial
(Optional — skipped)
Part C. Final Project: L-Protein Mutants
High level summary: The objective of this assignment is to improve the stability and auto-folding of the lysis protein of a MS2-phage. This mechanism is key to the understanding of how phages can potentially solve antibiotic-resistance.
Chose option 3: generating random mutations in the lysis protein while avoiding loss-of-function or nonsense codons. A Python script (Colab) was used to load active mutations from experimental data and apply them randomly to unique positions.
AF2 Multimer was used to co-fold mutant sequence 1 with DnaJ. The plDDT score indicates low model confidence in the folding of the mutant L protein. Overall, the random mutation approach is very time-consuming for obtaining leads.
Week 06 HW
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
A Phusion HF PCR Master Mix is a pre-combined PCR reaction system optimised for a specific engineered DNA polymerase.
Phusion DNA polymerase — provides the catalytic activity which synthesises DNA, and includes a 3’→5’ exonuclease proofreading to reduce error
Reaction buffer
MgCl₂ — magnesium ions
dNTPs — deoxynucelotide triphosphates: dATP, dCTP, dGTP, and dTTP
Stabilizers/additives
Water
A typical setup only requires after adding:
Forward primer
Reverse primer
Template DNA
Additional water to reach final volume
What are some factors that determine primer annealing temperature during PCR?
Factors:
Primer melting temperature — dominant factor
GC content of primer
Primer length
Sequence features
Salt concentration in the reaction buffer
Template–primer mismatch tolerance
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR creates DNA fragments by enzymatic replication using primers that define the fragment boundaries.
Protocol: The reaction contains template DNA, forward and reverse primers, dNTPs, buffer, Mg²⁺, and a thermostable DNA polymerase (e.g. Phusion or Taq). The protocol cycles temperature: denaturation (~95 °C) separates strands, annealing (~50–65 °C) allows primers to bind, and extension (~72 °C) synthesizes new DNA. After ~25–35 cycles, the region between the primers is exponentially amplified, producing many linear copies of a precisely defined sequence.
Restriction enzyme digestion produces linear fragments by cutting DNA at specific recognition sequences using restriction endonucleases.
Protocol: The protocol involves incubating DNA with one or more enzymes in the appropriate buffer (often ~37 °C) for a set time. The enzyme recognizes a short sequence (typically 4–8 bp) and cleaves the phosphodiester backbone, generating fragments with defined ends (blunt or sticky). The resulting fragment sizes depend entirely on where those recognition sites exist in the DNA.
Conceptually, PCR synthesizes a fragment by copying between two designed boundaries, whereas restriction digestion extracts a fragment by cutting an existing molecule at predetermined sequence motifs.
To understand when both are useful, consider an objective: engineer E. coli to produce human insulin, which requires building a plasmid containing the insulin gene under a bacterial promoter.
3 difference scenarios for getting insulin:
DNA comes from a biological sample (e.g. human genomic DNA). The insulin gene is buried inside billions of unrelated bases, so PCR is used to isolate and amplify only that specific region using primers that define its boundaries. PCR is therefore used when the goal is to retrieve a specific gene from a complex DNA mixture.
DNA already exists in a plasmid (e.g. moving GFP from plasmid A into plasmid B). The fragment is already isolated, so restriction enzymes are used to cut DNA at specific recognition sequences, allowing the gene to be excised and inserted into another vector. Restriction digestion is therefore used when the task is to cut and rearrange existing DNA molecules.
DNA is chemically synthesized because the sequence is already known. The synthesized fragment may still be PCR-amplified if more copies are needed, and restriction enzymes (or similar assembly methods) are used to insert it into plasmids. In practice, PCR isolates or amplifies sequences, while restriction enzymes cut DNA molecules so fragments can be inserted, removed, or reorganized.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Desiderata: Linear DNA fragments whose terminal 20–40 bp regions are perfectly homologous to the neighboring fragment, unique, structurally stable, and present in a clean preparation so the Gibson enzymes can expose the overlaps, allow annealing, fill gaps, and ligate the final construct.
How does the plasmid DNA enter the E. coli cells during transformation?
A cell is boundaried by a lipid membrane wall. It is not a solid wall. It is more of a dense molecular fluid. Each phospholipid is held in place only by weak interactions (hydrophobic forces, van der Waals forces). Like an electron has no fixed static wall, rather a field it creates, a cell has no fixed solid wall, it is a highly dense molecular fluid.
Lipids constantly fluctuate, small gaps appear and disappear.
During transformation, the culture is treated to brief heat shock (~42 °C for ~30–60 s). The rapid temperature change causes a sudden increase in the lipid kinetic energy, resulting in transient disordering of phospholipid packing, resulting in transient aqueous pores in bilayer. Plasmid DNA molecules enter through these pores.
This is paired with a treatment of calcium ions, which neutralises negative charges on the DNA phosphate backbone and the membrane surface, and thus reduces electrostatic repulsion between DNA and cell envelope.
Describe another assembly method in detail (such as Golden Gate Assembly)
Explain the other method in 5–7 sentences plus diagrams (either handmade or online).
Design fragments with Type IIS sites and specific 4-bp overhangs. PCR amplify or synthesize fragments with those flanking sites. Mix fragments, plasmid backbone, Type IIS enzyme, ligase, and buffer. Run digestion–ligation thermal cycles. Transform assembled plasmid into bacteria.
Model this assembly method with Benchling or Asimov Kernel!
Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
They can compute continuous values, not just discrete ones.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
A feedback-response mechanism, for example how the pancreas runs a sort of PID controller / control system for managing glucose levels in the body - through two hormones that move glucose levels in opposite directions - insulin and glucagon.
Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Mycelium composites. You can use them to grow coffee cups, fashion (cloths).
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
You might want to genetically engineer fungi to grow coffee cups.
The advantage is that in fungi, the chassis is the product - filamentous growth means the organism shapes itself into the material. Bacteria are essentially isotropic point-particles in materials terms; hyphae are intrinsically networked, anisotropic, and 3D, which is the prerequisite for any morphogenetic program over a material.
Submit the Google Form with your draft Aim 1, final project summary, HTGAA industry council selections, and shared folder for DNA designs.
.
Review Part 3: DNA Design Challenge of the week 2 homework. Design at least 1 insert sequence and place it into the Benchling/Kernel/Other folder you shared in the Google Form above. Document the backbone vector it will be synthesized in on your website.
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis is technology where proteins are produced using lysed cell machinery (ribosomes, enzymes) rather than living cells.
The main advantages of CFPS:
Rapid, direct expression from DNA (plasmids). Bacterial colonies require tagging, growth and selection, whereas CFS are simpler and more straightforward.
Direct control over the reaction environment. CFS systems work more directly with primitive biomolecular elements, rather than encapsulated cells, so they are more straightforward to measure, instrument and perturb.
Describe the main components of a cell-free expression system and explain the role of each component.
This performs the core processes of protein production. Transcription and translation: DNA → mRNA → protein
DNA template. e.g. plasmid DNA, linear PCR
Energy system - supplies chemical energy (ATP, GTP) for transcription and translation.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Transcription and translation burn ATP and GTP very quickly. Without sufficient ATP/GTP production, the reaction rapidly stalls.
Consider a car. Igniting fuel will not propel a car forward, it will dispel energy uselessly. However attaching a piston and a chamber can direct that energy in one direction.
A cell is like a car with a piston. A cell-free system lacks such a system.
This is accomplished in a cell through the membrane. The cell membrane (composed of lipids) creates constrained geometry and controlled sequencing of interactions.
Describe a method you could use to ensure continuous ATP supply in your cell-free experiment..
Add phosphoenolpyruvate, PEP, plus pyruvate kinase.
Protocol: I would run a small optimization matrix rather than assume one energy condition is best. For example, I would test 10, 20, and 40 mM PEP with fixed 1.5 mM ATP and fixed pyruvate kinase, then measure protein yield at 0, 30, 60, 120, and 240 minutes. If yield stops early while ATP is low, I would increase PEP or pyruvate kinase. If yield stops despite ATP remaining, then the problem is probably not ATP supply but substrate depletion, pH drift, magnesium imbalance, mRNA degradation, or protein instability. This separates “energy failure” from other failure modes.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic: lacking a nucleus. Examples: E. Coli lysates.
Eurokaryotic: having a nucleus. Examples: Wheat Germ Extract, Rabbit Reticulocyte Lysate, Tobacco BY-2 Lysate
C-peptide still cleaved enzymatically post-translation.
The biosynthesis of insulin involves multiple intermediary steps:
preproinsulin: Insulin is synthesized as an inactive precursor molecule, a 110 amino acid-long protein called preproinsulin
proinsulin: Preproinsulin is translated directly into the rough endoplasmic reticulum (RER), where its signal peptide is removed by signal peptidase to form proinsulin
proinsulin:
As the proinsulin folds, opposite ends of the protein, called the “A-chain” and the “B-chain”, are fused together with three disulfide bonds.[26]
Folded proinsulin then transits through the Golgi apparatus and is packaged into specialized secretory vesicles, or granules.[26]
In the granule, proinsulin is cleaved by proprotein convertase 1/3 and proprotein convertase 2, removing the middle part of the protein, called the “C-peptide”.[26]
Finally, carboxypeptidase E removes two pairs of amino acids from the protein’s ends, resulting in active insulin
Preproinsulin contains a signal peptide. Signal peptides function to prompt a cell to translocate the protein, usually to the cellular membrane.
C-peptide is a connecting protein produced when the pancreas cleaves proinsulin to make mature insulin. C-peptide excision is required and is done by PC1/3 + PC2 in vivo — neither CFE system reproduces that, so both routes need an in vitro cleavage step (trypsin + carboxypeptidase B, ?apparently? standard industrial route).
prokaryotic = make it cheap and finish it on the bench; eukaryotic = watch it fold the way a β-cell folds it.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
(This question was super hard and I was lost. I am verbatim coping the response from the Claude LLM here because I really loved how first-principles the answer was.)
Concepts I became aware of:
membrane protein
fake wall - detergents, liposomes
concept of a “screen”
readout - another new term which fits into screen. in programming, we’d just call it a test.
Proteins are chains of amino acids that fold into 3D shapes. Most proteins are water-soluble: their surface is “hydrophilic” (water-loving), they float around in the watery interior of the cell, no problem.
Membrane proteins are different. They’re designed to live embedded in the cell’s outer wall — the lipid bilayer, which is a thin sheet of greasy fat molecules. The parts of the protein that sit inside that greasy sheet are themselves greasy (“hydrophobic”, water-hating). Think of a bolt designed to be installed through a wall: the threaded shaft is meant to be inside the wall material, only the head and tail stick out into air.
Now: in CFPS, there’s no wall. The protein gets manufactured into pure water. Its greasy mid-section is exposed to water, which it hates. It does what greasy things do in water — clumps up with other greasy bits (think oil droplets in vinegar). It misfolds, aggregates into junk, and you get no usable product.
So the central engineering problem for membrane-protein CFPS is: you have to provide a fake wall for the protein to embed into while it’s being made. Everything else in the protocol is in service of that.
The menu of fake walls (in rough order of complexity):
Detergents — soap molecules. They have a greasy tail and a water-loving head, so they can wrap around the greasy parts of the protein and keep it in solution. Cheap, easy. Downside: detergents are often harsh enough to denature the protein.
Liposomes — actual little lipid bubbles, basically tiny vesicles made of the same fat the real membrane is made of. The protein can insert into the bubble’s wall as it’s manufactured. More native, harder to work with.
Nanodiscs — a clever trick: a small flat patch of lipid bilayer (~10 nm across) held together by a belt of protein around the edge, like a coin made of fat with a metal rim. The membrane protein sits in the middle of the disc. Very clean, very defined, beloved by structural biologists. Most expensive.
SMALPs — a synthetic polymer (SMA) that does the nanodisc trick without needing the protein belt. Cheaper than nanodiscs.
The supporting cast. A real cell does more than just provide a wall. It has:
Translocons — machines built into the membrane that thread the protein into the wall as it’s being made (like a sewing machine guiding fabric through). Without these, the protein doesn’t insert correctly even if a wall is present. You can buy these as “inverted membrane vesicles” — little fragments of bacterial membrane with the translocons still in them — and dump them into the reaction.
Chaperones — helper proteins that prevent misfolding. Add as purified extras.
Redox environment — some membrane proteins have internal “staples” (disulfide bonds) that only form in an oxidizing environment. Standard cell juice is reducing (the opposite). You add chemicals to flip it.
How you’d actually run the experiment. You’re optimizing across maybe 5 variables (which wall, how much wall, with or without translocons, temperature, redox state). Too many combinations to try one at a time, so you do a screen: 48–96 small parallel reactions in a plate, each with a different combination, and a fast readout that tells you which ones worked.
The standard fast readout uses split-GFP: GFP is the green fluorescent protein, and you can split it into a big piece and a tiny tag (11 amino acids). Attach the tiny tag to your membrane protein. Add the big piece to the reaction. They only find each other and turn green if your membrane protein folded correctly and the tag is accessible. So fluorescence = success, no fluorescence = junk. Read the plate in a few minutes, identify the winning conditions, then scale those up.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
system starts strong, then fuel runs out or waste accumulates
ATP donor depleted, Amino acids consumed, NTP depletion, Phosphate buildup, Enzymes lose activity, Too much DNA overloads machinery
Process consistency
Troubleshooting is assumed to mean diagnosis here. Implementation of a solution is up to the experimetal designer.
Three possible reasons for low yield:
Poor folding. The protein may be successfully translated, but immediately misfolds, aggregates, or becomes insoluble. This is especially common for large proteins, membrane proteins, disulfide-rich proteins, or eukaryotic proteins expressed in bacterial extracts.
Troubleshooting strategy:
DNA degradation: incubate DNA in extract, Sample at 0, 15, 30, 60 min, Run DNA gel or qPCR across the gene
Energy depletion / reaction burnout. Cell-free systems consume ATP extremely rapidly.
Troubleshooting strategy:
Assume we measure yield somehow - eg. fluorescence (e.g. GFP), luciferase activity, SDS-PAGE band intensity, western blot, or mass spectrometry
Can diagnose energy depletion as yield declining abnormally (e.g. abruptly). Supplement fresh ATP mix midway.
if production resumes -> energy limitation confirmed
Weak Transcription initiation: measure mRNA by RT-qPCR, compare target template against a known-good positive-control template, keep coding region same, swap promoter/5’ region
if DNA intact but mRNA low = transcription problem
if positive control produces mRNA but target does not = target promoter/sequence design problem
if both fail = extract/polymerase/reaction chemistry problem
Useful synthetic cell.
Design an example of a useful synthetic minimal cell as follows:
Pick a function and describe it.
What would your synthetic cell do? What is the input and what is the output?
The synthetic cell would grow wood. The input is CO2 and photons and output is the structure it grows. Wood is a composite material made out of 40% cellulose (contained within cells), 30% lignin, and 30% hemicellulose.
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
No, since wood is made from cells. Cell-free structure could be created using crystalline approaches.
Could this function be realized by genetically modified natural cell?
Yes, but it would be more complex in the limit.
Describe the desired outcome of your synthetic cell operation.
Programming a tree to grow into a custom shape (a house) via morphological programming, growing a tree faster and more efficiently (without ancestral code which may no longer apply in current environment).
Design all components that would need to be part of your synthetic cell.
What would be the membrane made of?
Two lipids as in JVCI-syn3A - phosphatidylglycerol and a specific glycolipid.
What would you encapsulate inside? Enzymes, small molecules.
Carbon capture and fixation - CETCH cycle inspired design?
Energy production - cyanobacteria?
Cellulose extrusion - Komagataeibacter? Not sure if it’s possible to put a bacteria in a cell
Lignin production
There are many more things, I know. ;) These are the core ones that might be enough to answer the exercise.
Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
Bacterial systems lack a nucleus and Tx/Tl happens in same place (vs. in human cell - where transcription occurs in the nucleus, translation occurs outside the nucleus but still in the cell).
I’m not sure yet. It would not seem we need mammalian gene complexes or closures from different organelles.
How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
It needs to exchange:
light
CO2 / bicarbonate
water
ions
small molecule substrates
waste products
possibly output molecules
Channels:
Light - NA
CO2 - membrane-permeable
Bicarbonate - may need a bicarbonate transporter or channel
Water - membrane-permeable
Experimental details
List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
There are so many haha. And it would be the most adequate to describe this as an engineering design - ie. a hierarchy of encapsulation.
This monolith synthetic cell will have too much uncertainty to design-build-test and solve in one iteration. It will definitely require divide-and-conquer to break it down into testable subunits.
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment.
Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept.
Seasoning which turns rapidly digestible starch into slower digestible starch, which reduces glucose spikes.
How will the idea work, in more detail? Write 3-4 sentences or more.
This product is an enzyme powder sprinkled onto starchy food after cooking, especially rice, potato, pasta, corn, or sweet potato. The active enzyme is a starch-debranching enzyme such as pullulanase or isoamylase, which cuts branch points in amylopectin and creates more linear starch chains. When the food is then cooled, these linear chains pack together into retrograded resistant starch, which human digestive enzymes break down more slowly than normal cooked starch. The result is not that carbohydrates disappear, but that some rapidly digestible starch is shifted into a slower-digesting or resistant form, reducing the speed and size of the post-meal glucose rise.
What societal challenge or market need will this address?
This addresses the need for simple food-preparation tools that reduce post-meal glucose spikes without requiring people to fully change what they eat. It is especially relevant for people managing diabetes, insulin dosing, blood glucose variability, or low-GI diets. It would be used like a seasoning or cooking aid for high-starch foods, not as a replacement for insulin or medical treatment.
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
The cell-free limitation would be handled by treating the product as a freeze-dried biochemical system rather than a living organism. The enzyme would be produced in microbes, purified, then dried with stabilizers such as trehalose or other protein-protective excipients so it can survive storage as an inactive powder. Water from the food rehydrates the system and turns the enzyme back on, while buffer salts keep the local pH in the enzyme’s working range. Because there is no cell to repair damaged proteins or regenerate itself, the reaction is designed as a one-time use system: the enzyme acts during a short warm window, modifies the starch structure, then becomes inactive during later heating, digestion, or storage.
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/ .
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Scientific research and invention requires intelligence. AI systems provide intelligence in much greater amounts, for orders of magnitude lower cost than biological intelligence. AI systems require energy and data centers. Building data centers in space can be done in a different tradeoff space - powered by solar (at much higher solar efficiency due to lack of atmosphere), unconstrained by regulatory cost. However transporting hardware to space is expensive.
What if we could build parts of the data center in space using cell-based systems? If transporting a seed to space could grow a tree, potentially we could transport a seed which grows a data center (or even a rack for a server).
Transporting seeds to space that grow into objects offers exponential savings compared to sending a data center as a manufactured object. This is due to the cost economics of sending load into space - the relationship between the mass of the rocket’s fuel and the mass of the payload is exponential, dictated by the Tsiolkovsky rocket equation. You can also think about it in the inverse - reducing load makes the fuel cost exponentially cheaper.
This requires solving:
morphological programming - how to program a single cell to grow into a 3D structure
materials design - ie. what is this rack made out of? Something that could survive in space’s vacuum? self-sealing lichen/coral
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
Engineered photosynthetic cyanobacteria/lichen-like chassis genes controlling carbon fixation, extracellular polysaccharide secretion, melanin biosynthesis, mineralization, desiccation tolerance, and 3D morphogenesis.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
These enable 3D structure production in the same way a plant would, mineralisation provides the hard dead shell that would provide protection of the living inner cells from the vacuum of space.
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
Above in Q1
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
This experiment would be broken down:
solving morphological programming - growing a simple 3D shape from programming a single cell
building complex structures - instead of a shape made of a single material, growing a mineral layer and now the cell inner layer
testing photosynthesis - adding photosynthesis genes and building the mechnism for the organism to convert energy
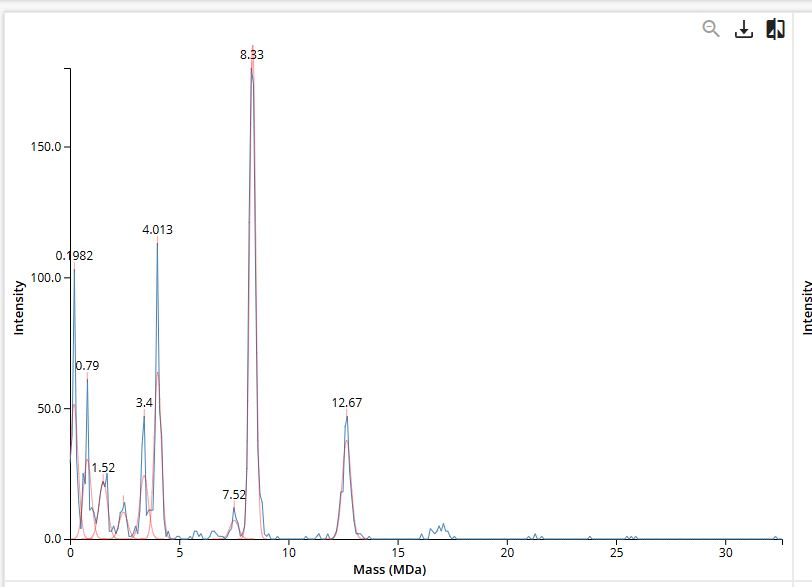
You don't know M, and you don't know which divisor goes with which peak.
The trick: adjacent peaks must differ by exactly 1 in the divisor
peak_27 ≈ M / 27
peak_28 ≈ M / 28
peak_29 ≈ M / 29
Determine the protein MW using the relationship between m/z, MW, and z.
m/z = (MW + z * mh) / z
MW = z * ((m/z) - mh)
MW = 27.002599690371586 * (m1 - mh)
= 27.002599690371586 * (1037.4423 - 1.00728)
= 27986.439950142267 Da
Calculate measurement accuracy by comparing experimental vs. theoretical weight.
# Two adjacent isotope peaks: same protein, same charge state z,
# but peak 2 has one extra neutron (so +1.003 Da in true mass)
m1 = (MW + z*mh) / z # lighter isotope
m2 = (MW + 1.003 + z*mh) / z # heavier isotope (+1 neutron)
# Subtract
delta_m = m2 - m1
= ((MW + 1.003 + z*mh) - (MW + z*mh)) / z
= 1.003 / z
# Rearrange
z = 1.003 / delta_m
Small note on units: m/z effectively is in Da — mass (Da) divided by charge (just an integer count) leaves you with Da on the x-axis; some textbooks call the unit “Thomson” (Th), but numerically it’s the same as Da.
Homework: Waters Part II — Secondary/Tertiary Structure
Explain the differences between native and denatured protein conformations. What happens when a protein unfolds? How is this determined with a mass spectrometer? What changes appear in the spectra between analyses?
A protein is one or more chains of amino acids. When DNA is read, it is assembled and stored in another form in mRNA, and later assembled by RNA into an amino acid chain.
These amino acids have inherent physical properties that results in various forces which twist the shape into a certain form. This process is referred to as protein folding and is a process of energy minimisation. When the folding reaches an equilibrium the protein is referred to as folded into its native form.
The 3D form of a protein is what confers its functionality. Proteins are in a sense 3D machines composed of chemical elements. Some proteins are merely static shapes, such as signalling molecules. Whereas others have a dynamic mechanical function, such as the ATP synthase.
How is this determined with a mass spectrometer? What changes appear in the spectra between analyses?
A mass spectrometer is a device which measures the mass-to-charge ratio of gaseus ions, which can be used to identify chemical substances.
Molecules are chemical compounds of atoms (ie. CO2 - one carbon, two oxygen atoms). An atomic element is composed of protons and neutrons in its nucleus, and electrons in its orbit. Ionisation is unpairing an electron from an atom.
A mass spectrometer ionises a substance, producing a charge (a free electron). For most small molecules and atoms (like Carbon-12), ionization predictably produces a single, stable charge state (typically +1).
A mass spectrometer outputs a plot of intensity (y) and $m/z$ mass-charge quotient (x). Mass can be used to characterise a specific atom, and charge is emitted when the substance is present and ionised (thus emitting an electron).
Thus a mass spectrometer can be used to map the presence of atoms and larger structures (molecules and proteins).
From what we know:
a denatured protein is highly charged, where each basic residue (amino acid) tends to become protonated.
a native protein has few charges, evenly spread across the surface.
The differences between the two runs:
Native — envelope at high m/z (~3000–4000), narrow (4–5 peaks), few charges. Compact part, few surface sites.
Denatured — envelope at low m/z (~1000–2000), broad (15–25 peaks), many charges. Sprawl, sites everywhere.
Mass itself — native shows the assembled mass: subunits stuck together, plus any bound ligand or metal. Denatured shows only individual subunit masses; the assembly has come apart and the cargo has fallen off.
Peak sharpness — denatured peaks are crisp; native peaks are fuzzier because the folded part drags along bound water, salt adducts, and some conformational wobble.
To detect proteins, typically a divide-and-conquer strategy is used. Proteins are cut using an enzyme (a protease) into smaller groups of residues (peptides), and then the spectral measurement of the mass-charge profiles of all the individual peptides is used to match against a signature of existing measured peptides.
Digestion: divide-and-conquer using enzyme.
Separation: separate using liquid chromatography (LC), peptides pass through specrometer gradually.
Ionisation: peptides are converted into charged, gas-phase ions.
This relies on a database of peptide fragments, whose usage is detailed below:
Peptide identification problem: given an unknown peptide — a chain of amino acids — identify which amino acids it contains and in what order, using only mass-charge measurements. One measurement of the intact peptide gives total mass but not order; many different orderings yield the same mass. Workaround: break many copies of the peptide at random points along the chain, producing sub-chains of every possible length, then measure the mass-charge of every sub-chain. The resulting set of mass-charges encodes the sequence, but noisily and incompletely — decoding the amino-acid order directly from that pattern is ill-posed because many candidate sequences fit any partial set of sub-chain masses. Reformulate as lookup against the known list of proteins in the organism, which fixes every peptide that could possibly exist (~10⁶ candidates). For each candidate, predict what its sub-chain mass-charges should be; filter the candidate set down to ~10¹ by demanding the intact-peptide mass match; score the survivors by how well their predicted sub-chain masses overlap with the observation; take the best. Bound the error rate by running the same pipeline against a catalog of fake (reversed) sequences and tuning the score threshold so fake matches stay below 1% of accepted matches — a calibrated null check standing in for ground truth.
In cases where the peptide sequence is unknown, de novo sequencing is performed.
From the native eGFP mass spectrum, discern the charge state of the peak at ~2800 m/z. What is the charge state? How can you tell?
Homework: Waters Part III — Peptide Mapping (Primary Structure)
Count the Lysines (K) and Arginines (R) in the eGFP sequence; circle or highlight them.
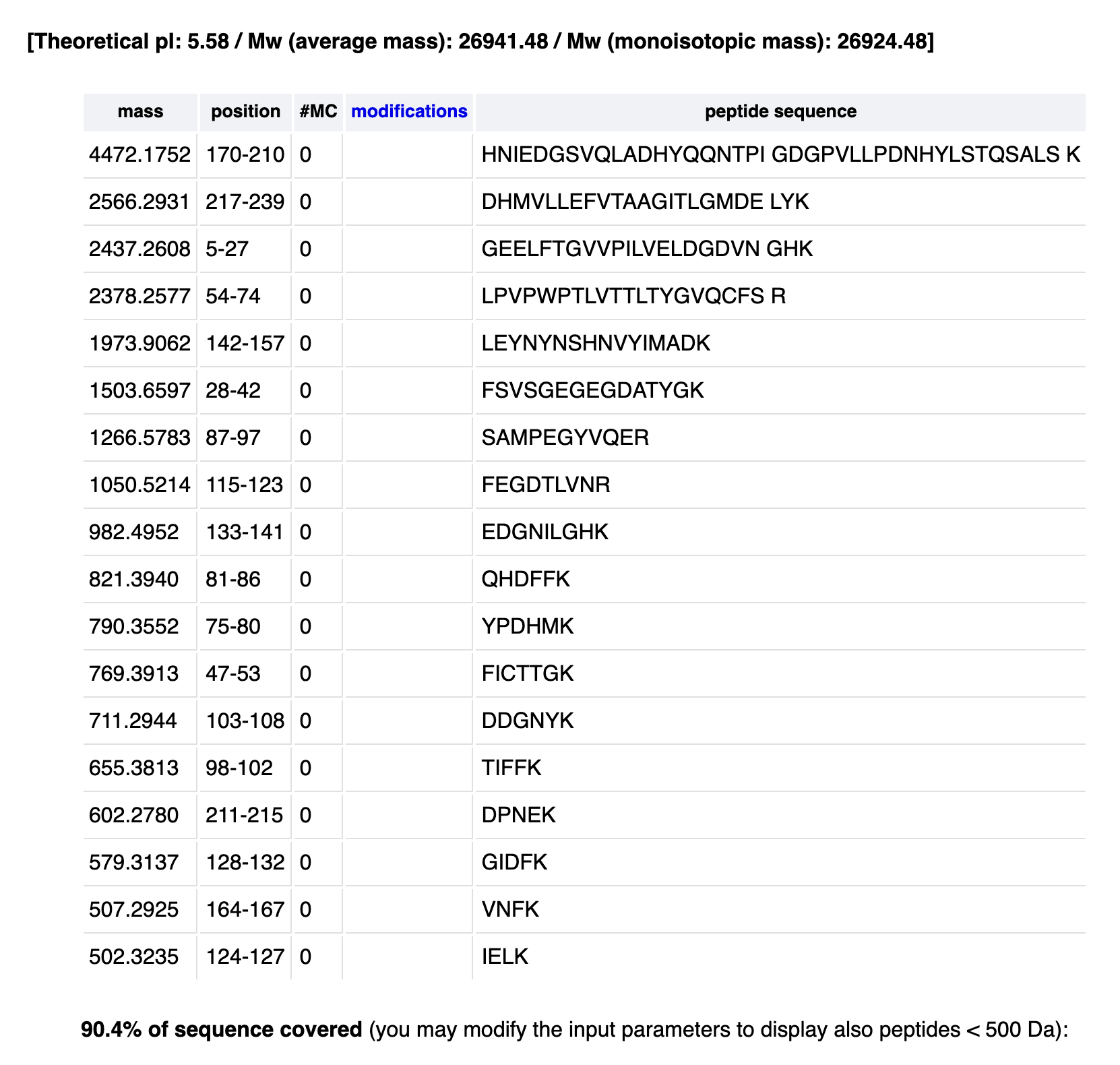
How many peptides will be generated from tryptic digestion?
Navigate to the ExPASy PeptideMass tool.
Copy/paste the eGFP sequence.
Replicate the parameters shown in Figure 4.
Report the number of peptides generated.
18
Based on the LC-MS peptide map data, count the chromatographic peaks between 0.5–6 minutes (>10% relative abundance).
23-25
Does the peak count match the predicted peptide number? Are there more or fewer peaks?
There are more peaks!
Identify the m/z of the peptide in Figure 5b. Determine the charge (z) of the most abundant charge state using isotope separation. Calculate the singly charged peptide mass [M+H]⁺.
z = (m2 - mH) / (m1 - m2)
m/z = (MW + z * mh) / z
MW = z * ((m/z) - mh)
mh = 1.00728
m = MW + z * mh
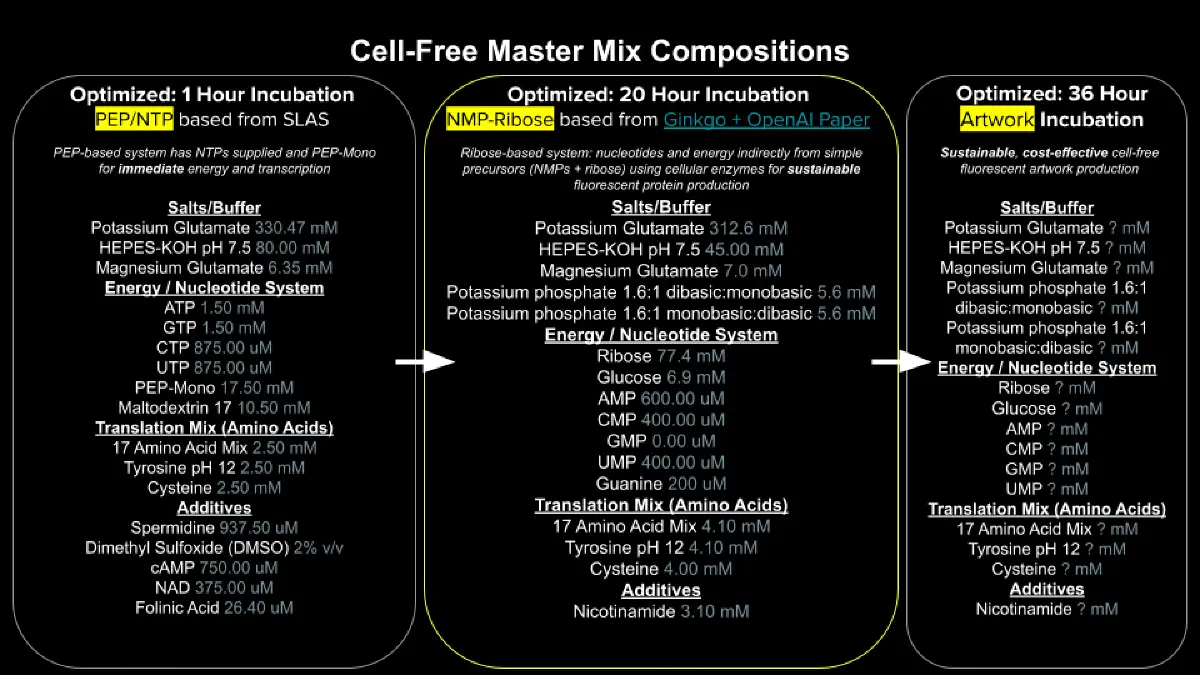
Potassium Glutamate — Provides K⁺ at high concentration (~100–200 mM) to support ribosome function and translation fidelity
HEPES-KOH pH 7.5 — Zwitterionic Good’s buffer that holds pH near the optimum for transcription/translation enzymes despite acid production
Magnesium Glutamate — Supplies Mg²⁺, the critical divalent cofactor for ribosome assembly, tRNA structure, aminoacyl-tRNA binding, peptidyl transferase, and RNA polymerase activity
Potassium phosphate monobasic (KH₂PO₄) / dibasic (K₂HPO₄) — Together act as a secondary phosphate buffer near pH 7 and supply inorganic phosphate that feeds the energy regeneration system
Ribose — Sugar substrate that gets phosphorylated to ribose-5-phosphate and then to PRPP, supplying the sugar-phosphate backbone for nucleotide salvage
Glucose — Primary carbon/energy fuel feeding glycolysis to regenerate ATP via substrate-level phosphorylation
AMP, CMP, GMP, UMP — Cheap nucleoside monophosphate inputs that kinases phosphorylate up to NTPs for transcription and translation
Guanine — Free base salvaged with PRPP into GMP, cheaply replenishing the heavily-consumed GTP pool used in translation
17 Amino Acid Mix — Provides 17 of the 20 proteinogenic amino acids as monomers that aminoacyl-tRNA synthetases load onto tRNAs for ribosomal polymerization
Additives: Nicotinamide
Inhibits NAD+ consuming enzymes. NAD⁺ is required at the GAPDH step of glycolysis, so once it’s gone, ATP regeneration from glucose stalls and translation dies.
Backfill: Nuclease Free Water
Nuclease-Free Water — Brings the reaction to final volume while avoiding contaminating RNases/DNases that would degrade the mRNA and DNA template.
water that’s been DEPC-treated and/or filtered, packaged sterile, and certified by the manufacturer to have no detectable RNase activity
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix (2–3 sentences).
(This is my best effort answer using Claude’s LLM!)
The whole design question of any CFPS recipe is: what’s your refueling strategy.
PEP-NTP — bring premium fuel and a pressurized recharge cartridge. PEP-NTP mix supplies pre-made high-energy substrates directly.
Glucose-NMP — bring crude oil and an onboard refinery. NMP-Ribose-Glucose mix uses glucose-fed glycolysis as a slower but sustained ATP regenerator.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
For each of the 6 fluorescent proteins used for collaborative painting, identify and explain at least one biophysical or functional property affecting cell-free expression or readout (1–2 sentences each):
Slow chromophore maturation (hours) plus low brightness means much of the protein made during a short CFPS reaction never becomes fluorescent inside the readout window
Combines fast maturation with comparatively low pKa for fluorescence (~5.5), making readout robust to the pH drift that occurs in glycolysis-fueled cell-free reactions as organic acids (lactate, pyruvate) accumulate
Blue FP with excitation ~403 nm and emission ~456 nm, which collides directly with NAD(P)H autofluorescence
Create a hypothesis for adjusting one or more reagents in the cell-free mastermix to improve a specific biophysical or functional property and maximize fluorescence over 36-hour incubation. Clearly state the protein, reagents, and expected effect.
Hypothesis: For expression of eGFP in BL21(DE3) Star E. coli lysate, increasing the ATP-regeneration capacity and stabilizing redox balance will extend productive translation time, improve chromophore maturation, and increase total fluorescence over a 36-hour incubation.
Protein: enhanced green fluorescent protein, eGFP.
Reagents to adjust: increase glucose or replace part of the glucose with maltodextrin as a slower-release carbon source
Expected effect: Maltodextrin should feed glycolysis more gradually than free glucose, reducing early fuel depletion and pH stress while sustaining ATP regeneration for longer
Begin composing master mix compositions here once assigned artwork wells are received.
Final phase: analyze fluorescence data to determine favorable reagent compositions (due one week after data return). Reaction composition per well: 6 μL Lysate, 10 μL 2X Optimized Master Mix, 2 μL assigned fluorescent protein DNA template, 2 μL custom reagent supplements (20 μL total).
Not sure where data was, sorry doing this HW late!!
Pippetting Units Moles (mol) measure the absolute amount of a substance Molarity (M) measures the concentration of that substance in a solution Moles (mol): A unit representing particles (atoms, molecules, etc.). Molarity (M): Concentration defined as moles of solute per liter of solution (mol/L). Conversions 1 L = 1000 mL = 1,000,000 μL 1 M = 1000 mM = 1,000,000 μM Pipette sizes P20, P200, P1000 - each fitting up to 20μL, 200μL and 1000μL (1mL) Equipment Pippette Eppendorf Tube PCR tube strip Reagents: dH2O - distilled water (purified) Gel loading dye - used for ??? Assays procedure to see if the thing is there or not thing can be a substance, chemical, entity, bacteria, etc. “see” could be measured qualitatively or quantitatively Serial dilutions What is this? It’s a geometric process which downsamples a concentration. This procedure conveys useful information in multiple areas: For measuring population counts using the human eye, you cannot count anything above 102, so a 1mL broth which might contain 107-10^9 populants can be downsampled to a 1μL broth. There is an innate assumption that the serial dilution process retains a uniform distribution of the original broth. For virology/immunology, you define strength by the last dilution that still works (neutralizes, infects, agglutinates) Dose–response curves - these are log-spaced. The serial dilution process is in a sense a geometric process (reduces by a ratio 1:10 each step, which progressively downscales in logarithmic sense). Serial dilution is how you map an unknown huge concentration into the measurable window of any detector How do you dilute? C1 * V1 = C2 * V2 rearrange: V1 = (C2*V2) / C1 V_water = V2 - V1 How do you do serial dilutions? Scenario: The stock concentration of a mystery substance (MS) is 5 M. Calculate how to dilute to 100 µM (0.1 mM): SerialDilute(1:499), SerialDilute(1:99) → Step 1: Dilute 5 M (5,000,000 µM) to 10,000 µM (500x dilution). Step 2: Dilute 10,000 µM to 100 µM (100x dilution). https://2026a.htgaa.org/2026a/course-pages/weeks/week-01/lab/index.html
Subsections of Labs
Week 1 Lab: Pipetting
Pippetting
Units
Moles (mol) measure the absolute amount of a substance
Molarity (M) measures the concentration of that substance in a solution
Moles (mol): A unit representing particles (atoms, molecules, etc.).
Molarity (M): Concentration defined as moles of solute per liter of solution (mol/L).
Conversions
1 L = 1000 mL = 1,000,000 μL
1 M = 1000 mM = 1,000,000 μM
Pipette sizes
P20, P200, P1000 - each fitting up to 20μL, 200μL and 1000μL (1mL)
Equipment
Pippette
Eppendorf Tube
PCR tube strip
Reagents:
dH2O - distilled water (purified)
Gel loading dye - used for ???
Assays
procedure to see if the thing is there or not
thing can be a substance, chemical, entity, bacteria, etc.
“see” could be measured qualitatively or quantitatively
It’s a geometric process which downsamples a concentration.
This procedure conveys useful information in multiple areas:
For measuring population counts using the human eye, you cannot count anything above 10^2, so a 1mL broth which might contain 10^7-10^9 populants can be downsampled to a 1μL broth. There is an innate assumption that the serial dilution process retains a uniform distribution of the original broth.
For virology/immunology, you define strength by the last dilution that still works (neutralizes, infects, agglutinates)
Dose–response curves - these are log-spaced. The serial dilution process is in a sense a geometric process (reduces by a ratio 1:10 each step, which progressively downscales in logarithmic sense).
Serial dilution is how you map an unknown huge concentration into the measurable window of any detector
How do you dilute?
C1 * V1 = C2 * V2
rearrange: V1 = (C2*V2) / C1
V_water = V2 - V1
How do you do serial dilutions?
Scenario: The stock concentration of a mystery substance (MS) is 5 M. Calculate how to dilute to 100 µM (0.1 mM):
SerialDilute(1:499), SerialDilute(1:99) →
Step 1: Dilute 5 M (5,000,000 µM) to 10,000 µM (500x dilution).
Step 2: Dilute 10,000 µM to 100 µM (100x dilution).
Slides Section 1: Abstract Trees are an extremely old technology — the design hasn’t meaningfully changed in over 10 million years, yet a premium tree still takes 20 years to grow and wood remains expensive. The deeper question behind “how do we grow trees 100× faster?” is the problem of morphology: how does a single seed, running purely local computation in each cell, self-assemble into a global 3D form? We can design a bridge in CAD, but we have no equivalent for designing an organism — no way to translate a target 3D shape into the per-cell program that grows it. This project takes an engineering-first approach: build the missing CAD-for-cells layer in silico first, then map it onto biological substrates. As validation, I built Morpheus, a voxel-based cell morphology simulator in which each cell runs the same short program, communicates only with neighbours via diffusing hormone gradients, and collectively grows a cylinder (a “cigar”) from a single seed cell. The longer-term aim is to compile these designs onto a real chassis — the JCVI-syn3.0 minimal cell — and use the same primitives to grow custom organoids, faster trees, and eventually plants engineered into specific 3D shapes such as a house frame or a portable dwelling-seed for space travel.
Trees are an extremely old technology — the design hasn’t meaningfully changed in over 10 million years, yet a premium tree still takes 20 years to grow and wood remains expensive. The deeper question behind “how do we grow trees 100× faster?” is the problem of morphology: how does a single seed, running purely local computation in each cell, self-assemble into a global 3D form? We can design a bridge in CAD, but we have no equivalent for designing an organism — no way to translate a target 3D shape into the per-cell program that grows it. This project takes an engineering-first approach: build the missing CAD-for-cells layer in silico first, then map it onto biological substrates. As validation, I built Morpheus, a voxel-based cell morphology simulator in which each cell runs the same short program, communicates only with neighbours via diffusing hormone gradients, and collectively grows a cylinder (a “cigar”) from a single seed cell. The longer-term aim is to compile these designs onto a real chassis — the JCVI-syn3.0 minimal cell — and use the same primitives to grow custom organoids, faster trees, and eventually plants engineered into specific 3D shapes such as a house frame or a portable dwelling-seed for space travel.
Section 2: Project Aims
Aim
Description
Aim 1: Experimental
Build an in silico voxel-based morphology simulator in which a single seed cell, running a local-only program with hormone-gradient communication, self-assembles into a target 3D shape (cylinder / “cigar”). Demonstrate that global form can emerge from purely local rules using primitives — point → 2D circle → 3D cylinder — and release the runtime as open source (github.com/liamzebedee/morpheus).
Aim 2: Development
Compile the simulated cell program onto a real biological chassis. Use JCVI-syn3.0 as the minimal cell, encode the local program as a synthetic gene-regulatory network (toggles, oscillators, feed-forward loops), implement positional sensing via diffusing hormone analogues (auxin-like morphogens), and verify a 1D → 2D → 3D shape ladder in vivo. Candidate hosts include E. coli and mycelium for early scaffolds.
Aim 3: Visionary
Establish the missing science of “CAD for cells”: a programming model that maps a target 3D form onto the local code each cell must run. If realized, this opens a new branch of the tech tree — designing organoids, organisms, and cell-grown materials directly. Concrete applications: trees that grow 100× faster, medicine grown in fruit (e.g. insulin pods), portable dwellings grown from a seed, plants whose geometry is engineered for apartment blocks or for Mars.
Section 3: Background
Literature Context
Two works frame the project. Beal, Lu & Weiss (2011), “Automatic Compilation from High-Level Biologically-Oriented Programming Language to Genetic Regulatory Networks” — establishes that high-level descriptions of cellular behaviour can, in principle, be compiled into concrete GRN constructs, and surveys the GRN motifs (toggles, repressilators, feed-forward loops, AND/OR/NOT gates) that earlier synthetic-biology landmarks built from real cells: Gardner, Cantor & Collins (2000) on the genetic toggle switch; Elowitz & Leibler (2000) on the repressilator; Atkinson et al. (2003) on rationally designed circuits; Alon (2003) on network motifs; and François & Hakim (2004) on morphogen-gradient compartmentation. Trewavas (2014), “Plant Behavior and Intelligence” / the physics-and-computation literature on plants — frames plant tropism as sensing → processing → actuating, where a plant performs spatial integration via distributed sensing and acts via directional growth. Both lines support the project’s central claim: that the tools and motifs to encode local cell programs already exist, but the compiler from desired 3D form down to per-cell code does not.
Innovation
The novel contribution is treating morphology as a programming problem and shipping a working in silico substrate for it. Rather than studying one organism’s developmental biology, Morpheus is a general voxel-based runtime where every cell runs the same program against local state and hormone gradients — closer to a CAD tool than a biology paper. The shape-decomposition approach (point → circle → cylinder, built from inheritable local state, emit/read gradients, and programmed cell death as scaffolding) is a concrete proposal for a primitive set in this design language. Combined with the observation that LLMs are now intelligent enough to discover the per-cell code given a target shape and a set of primitives, this reframes morphology from a biology-first problem into an engineering-first one.
Significance
Wood is expensive, but that’s the surface. Beneath it: we have no science for designing organisms, organoids, or cell-grown materials, and no theory of how the genome encodes 3D form. Every existing engineering discipline rests on divide-and-conquer — break the design into glued-together sub-parts — and biology stubbornly does not work that way: every cell runs the same program, all communication is local, and global structure is emergent. Cracking that gap would matter far beyond trees: it underlies how genotype maps to phenotype, why simulating tissue is currently intractable (one week of compute simulates ten minutes of a single JCVI-syn3.0 cell), and why drug design still requires human and animal trials instead of in-silico verification. A working theory unlocks faster trees, drug-bearing plants, cell-grown structures with no glue or joints, and self-assembling factories light enough for space travel.
Ethical Considerations
The capacity to design organisms with engineered 3D form raises clear dual-use concerns: non-maleficence (engineered organisms must not become invasive or disrupt ecosystems), beneficence (the technology should expand access — cheaper wood, cheaper medicine — rather than concentrate it), justice (benefits like grown-housing or grown-medicine must reach beyond well-resourced labs), and responsibility (designers are accountable for downstream effects of self-replicating, self-assembling systems they release).
Concretely: the project stays in silico through Aim 1, with no environmental release. Any in vivo work in Aim 2 uses contained chassis (JCVI-syn3.0 is intentionally fragile and unable to survive outside lab media) and follows standard BSL-1/2 containment. Unintended consequences considered include horizontal gene transfer, ecological escape of engineered plants, and the displacement risk to forestry-dependent economies if grown-on-demand wood matures faster than transition policy. Mitigations include kill-switches in any chassis, restricting in vivo work to non-reproducing cell-free or auxotrophic strains, and publishing the simulator and design language openly so that scrutiny and dual-use review aren’t bottlenecked behind a single lab.
Section 4: Experimental Design
Detailed Experimental Plan
Define the target shape. Pick a minimal 3D form that exercises self-assembly (a vertical cylinder, the “cigar”). Week 1.
Decompose the shape into primitives. Cylinder = (a) seed point, (b) radial 2D circle of given radius, (c) extrusion along +z up to a target height. Week 1.
Specify the cell substrate. Voxel-based 3D grid; one cell per voxel; cells have local state, can divide into a free neighbour cell, can emit and read scalar hormone gradients, and can undergo programmed death. Week 1.
Build the simulator runtime (Morpheus). Python; deterministic per-tick update; gradient diffusion via a cheap PDE-like relaxation; render with a 3D viewer. Open-sourced at github.com/liamzebedee/morpheus. Weeks 2–3.
Implement primitive 1: seed → axis. Seed cell sets is_axis=True with axis_potential=1.0; while potential > threshold, replicate +z and pass axis_potential * DECAY to child. Yields a 1D vertical line. Week 3.
Implement primitive 2: axis → radial circle. Axis cells emit a radial hormone gradient g_radial; any cell sensing g_radial < RADIUS_THRESHOLD flips inside=True and replicates outward in ±x, ±y. Yields a 2D disc per slice. Week 3.
Compose into a cylinder. Run primitives 1 and 2 simultaneously; verify the result is a vertical cylinder of correct height and radius. Week 4.
Tune parameters. Sweep DECAY, STOP_THRESHOLD, RADIUS_THRESHOLD against target dimensions; record sensitivity. Week 4.
Document the cell-program API. Lock down replicate_toward, emit_gradient, read_gradient, child_state, programmed death, inherited vs. read-only state. Week 5.
Catalogue tactics. Decompose into reusable patterns: clocks, oscillators, multi-scale staging, scaffold + cell-death “remove the formwork”. Week 5.
Map primitives onto biological constructs. For each runtime primitive, identify a real GRN equivalent: toggle (Gardner et al.), repressilator (Elowitz & Leibler), feed-forward loop (Alon), morphogen gradient (François & Hakim). Week 6.
Pick a chassis for in vivo Aim 2. JCVI-syn3.0 minimal cell as the base; E. coli as a pragmatic intermediate for early circuit testing. Week 6.
Design an in vivo 1D demo. Single-axis growth via a synthetic morphogen — diffusible peptide + receptor + AND-gate that triggers division along a polarity cue. Order parts via Twist; assemble with Gibson. Future.
Stage to 2D and 3D demos. Add a second orthogonal morphogen for radial growth; verify on solid media with fluorescent reporters at axis vs. radial cells; analyse via microscopy + image segmentation. Future.
Techniques Checklist
Category
Techniques
Fundamentals
Pipetting, Lab Safety, Bioethical Considerations
DNA
Construct Design, Sequencing, Editing, Restriction Enzyme Digestion, Gel Electrophoresis, DNA Purification, Databases
Chassis Selection (JCVI-syn3.0), Registry of Standard Biological Parts, Plasmid Prep, Bacterial Culturing, QC/Analysis, Bacterial Processing
Cell-Free Systems
Cell-Free Reactions, Freeze-Dried Systems, miniPCR Tools, Protein Purification
Assembly
Primer Design, PCR Reactions, Gibson Assembly
CRISPR
CRISPR/Cas9 (knockout sweep for essential cellulose-production genes in a minimal wood-producing organism)
Technique Deep-Dive
Chassis selection — JCVI-syn3.0 minimal cell. This project deliberately uses the minimal cell as its Aim 2 chassis because it strips the substrate down to the irreducible machinery for life, leaving the synthetic GRN and morphogen circuits as the dominant behavioural signal. JCVI’s recently released full-cell simulation model is a complementary asset: it allows the same per-cell program to be tested in silico at full biochemical fidelity before any wet-lab run. The trade-off is compute cost — current JCVI-syn3.0 simulation is roughly six days of wall-clock per fifteen-minute biological cell cycle — which justifies Morpheus operating at the abstract voxel level for early design iteration and reserving JCVI-syn3.0 for later validation only.
Construct design via synthetic GRNs. The cell program is encoded as a gene-regulatory network using established motifs: a toggle switch (Gardner, Cantor, Collins 2000) to lock cell identity post-differentiation; a repressilator (Elowitz & Leibler 2000) as an internal clock for staged development; a feed-forward loop (Alon 2003) for noise-robust thresholding of morphogen concentration; and a morphogen + reaction-diffusion module (François & Hakim 2004) for the spatial gradients that encode position. Constructs are assembled in Benchling, ordered as gene fragments via Twist, joined by Gibson assembly, and verified by sequencing and fluorescent-reporter readout. This deep-dive matters because the leap from voxel-program to wet-lab is, mechanistically, exactly this translation: every Morpheus primitive must map onto one of these GRN motifs to be physically realizable.
Industry Partners
JCVI (minimal cell + simulation), Ginkgo Bioworks (autonomous lab for circuit iteration), Twist Bioscience (DNA fragment supply), Opentrons (liquid handling for parallel circuit assays).
Section 5: Results & Validation
Validation Approach
Aim 1 is validated by demonstrating, in silico, that a single seed cell running a short local-only program self-assembles into a target 3D shape (a cylinder). The deliverable is a working open-source simulator (github.com/liamzebedee/morpheus) and a reproducible run that grows the cylinder from one cell using only inheritable state and diffusing hormone gradients — no global coordinator and no per-cell knowledge of absolute position.
Protocol
Initialize an empty voxel grid with one seed cell at the origin; mark it is_seed=True.
Each tick, every live cell executes the same Python program:
On seed step: set is_axis=True, axis_potential=1.0.
If is_axis and +z neighbour empty and axis_potential > STOP_THRESHOLD: replicate_toward('+z', child_state={is_axis: True, axis_potential: axis_potential * DECAY}).
If is_axis: emit_gradient('g_radial', 1.0).
If read_gradient('g_radial') < RADIUS_THRESHOLD: inside = True.
If inside and not yet expanded: replicate_toward each empty neighbour in ±x and ±y; mark has_grown_radial = True.
Diffuse g_radial across the grid each tick (relaxation step).
Run until no further divisions occur.
Render the final voxel field; measure resulting cylinder height and radius; compare to target.
Techniques Used
The validation exercises several synthetic-biology techniques in their in silico equivalents. Gradient emission and reading correspond directly to morphogen design (François & Hakim 2004): a scalar field broadcast by a class of cells and decoded by neighbours via a concentration threshold. Inherited child state implements asymmetric division with locked identity, the runtime analogue of a GRN toggle switch (Gardner, Cantor & Collins 2000). The decay-step counter on axis_potential plays the role of an internal clock — a count-down equivalent to a damped repressilator (Elowitz & Leibler 2000). And the inside flag’s threshold-and-latch behaviour is a feed-forward loop (Alon 2003) that filters transient gradient noise into a stable commit-to-divide decision. Each of these has a known wet-lab realization, which is what makes the simulation a credible blueprint for Aim 2 rather than a toy.
Data & Analysis
The simulator successfully grows a vertical cylinder from a single seed cell. With DECAY=0.9, STOP_THRESHOLD=0.37, RADIUS_THRESHOLD=1.5, the resulting structure contains roughly 120 cells: an axial column rises along +z until the inherited axis_potential decays below threshold (yielding a height of ~10 voxels, consistent with log(0.37)/log(0.9) ≈ 9.4), and at each axial level the radial hormone gradient produces a disc of radius ~1.5 voxels. The shape is stable across asynchronous update orders and across small perturbations to gradient diffusion, confirming that the global form is a function of the local rules rather than an artefact of update ordering. This is a direct validation that programming a cell with only local information can produce a globally specified 3D shape — the central claim of the project.
Challenges
The biggest unexpected challenge was conceptual rather than technical: writing local-only code is genuinely uncomfortable for an engineer trained on divide-and-conquer, because you cannot reach for a global coordinator or per-cell coordinates. Several early prototypes silently smuggled global state in via shared counters; rewriting them under the discipline of “every cell runs the same program against only its own state and what it can read locally” was the actual work. A second challenge is scale: the cylinder demo is ~120 cells, but a real leaf is ~10⁹ cells and a JCVI-syn3.0 cell takes ~6 days to simulate per 15-minute biological cell cycle, so the in silico-to-in vivo compilation will need much faster surrogate models or hybrid abstractions. Mitigations include hierarchical abstractions (simulate at the voxel level for design, drop to JCVI-fidelity only for spot-checks), and using LLMs to discover candidate cell programs given a target shape and a primitive library, which compresses the design search dramatically. A third anticipated challenge in Aim 2 is morphogen crosstalk in vivo — real diffusible peptides do not cleanly separate into orthogonal channels — which is why the proposed in vivo demos start with a 1D axis only, and only then add a second orthogonal morphogen.
Section 6: Additional Information
References
Gardner, T. S., Cantor, C. R., & Collins, J. J. (2000). Construction of a genetic toggle switch in Escherichia coli. Nature, 403(6767), 339–342.
Elowitz, M. B., & Leibler, S. (2000). A synthetic oscillatory network of transcriptional regulators (the repressilator). Nature, 403(6767), 335–338.
Atkinson, M. R., Savageau, M. A., Myers, J. T., & Ninfa, A. J. (2003). Development of genetic circuitry exhibiting toggle switch or oscillatory behavior in Escherichia coli. Cell, 113(5), 597–607.
Alon, U. (2003). Biological networks: the tinkerer as an engineer. Science, 301(5641), 1866–1867.
François, P., & Hakim, V. (2004). Design of genetic networks with specified functions by evolution in silico. PNAS, 101(2), 580–585.
Beal, J., Lu, T., & Weiss, R. (2011). Automatic compilation from high-level biologically-oriented programming language to genetic regulatory networks. PLoS ONE, 6(8), e22490.
Trewavas, A. (2014). Plant Behaviour and Intelligence. Oxford University Press.
J. Craig Venter Institute. JCVI-syn3.0 minimal cell and full-cell simulation model release (2025).
Source code for the morphology simulator: github.com/liamzebedee/morpheus
Supply List & Budget
Item
Quantity
Estimated Cost
Compute (laptop CPU + occasional GPU) for Morpheus simulation runs
1
$0 (existing)
LLM API credits for cell-program search & shape decomposition
ongoing
~$50
JCVI-syn3.0 minimal cell strain (Aim 2, future)
1 vial
~$500
Twist gene fragments for GRN constructs (toggle, repressilator, FFL, morphogen)
~8 fragments
~$1,200
Gibson assembly master mix
1 kit
~$300
Plasmid prep + sequencing reactions
~20
~$400
Fluorescent reporter parts (GFP/mCherry) from iGEM Registry
A Rust implementation of Radhika Nagpal’s MIT thesis, “Programmable Self-Assembly: Constructing Global Shape using Biologically-inspired Local Interactions and Origami Mathematics” https://github.com/liamzebedee/biogami
Morphology - final presentation
Cigars and tomatoes are both natural products, so in principle you should be able to grow a cigar and pick it off a tree — we just don’t know how to program a cell to grow into that shape. To study this I built an in silico 3D cell simulation that runs a programming language mapping onto real cell mechanics: hormones for signaling, internal state analogous to internal chemical concentrations, logic as found in GRN circuits, neighbour sensing, and the ability to divide. Using an LLM to search this language spec, I discovered a program that grows a single cell into a cylinder. The core idea is how global form arises from purely local interactions — every cell runs the same program with only a local view, no eyes — and it turns out it can be remarkably simple: grow out and grow up. A center cell emits a hormone whose concentration only reaches a certain radius, and cells are programmed to divide when the concentration exceeds a threshold (growing out). A second process grows up: cells inherit a biased chemical state carrying a signal that decays as the structure grows upward, encoding a preset height. There is still far more to discover — we still cannot take a plant, load it into a simulator, alter its developmental program, and reliably grow a new morphology in silico. Understanding how organisms decode and generate form from DNA is likely to be as foundational this century as molecules and atoms were in the early 20th, and now is the perfect time to figure it out.
cells are like programmable agents that serve as units for a rule-based behavior for the self-assembly of larger multicellular entities: tissues, organs, and organisms. In fact, a portion of the genes expressed in the context of a stable state S are cell-cell communication proteins, such as cytokines and extracellular matrix components, which mediate the interaction between cells to form a higher-level cell-cell interaction network. This cellular network in turn has a state space in which attractor states would represent tissue states, such as inflammatory states, regenerative states, and “tumor states.”

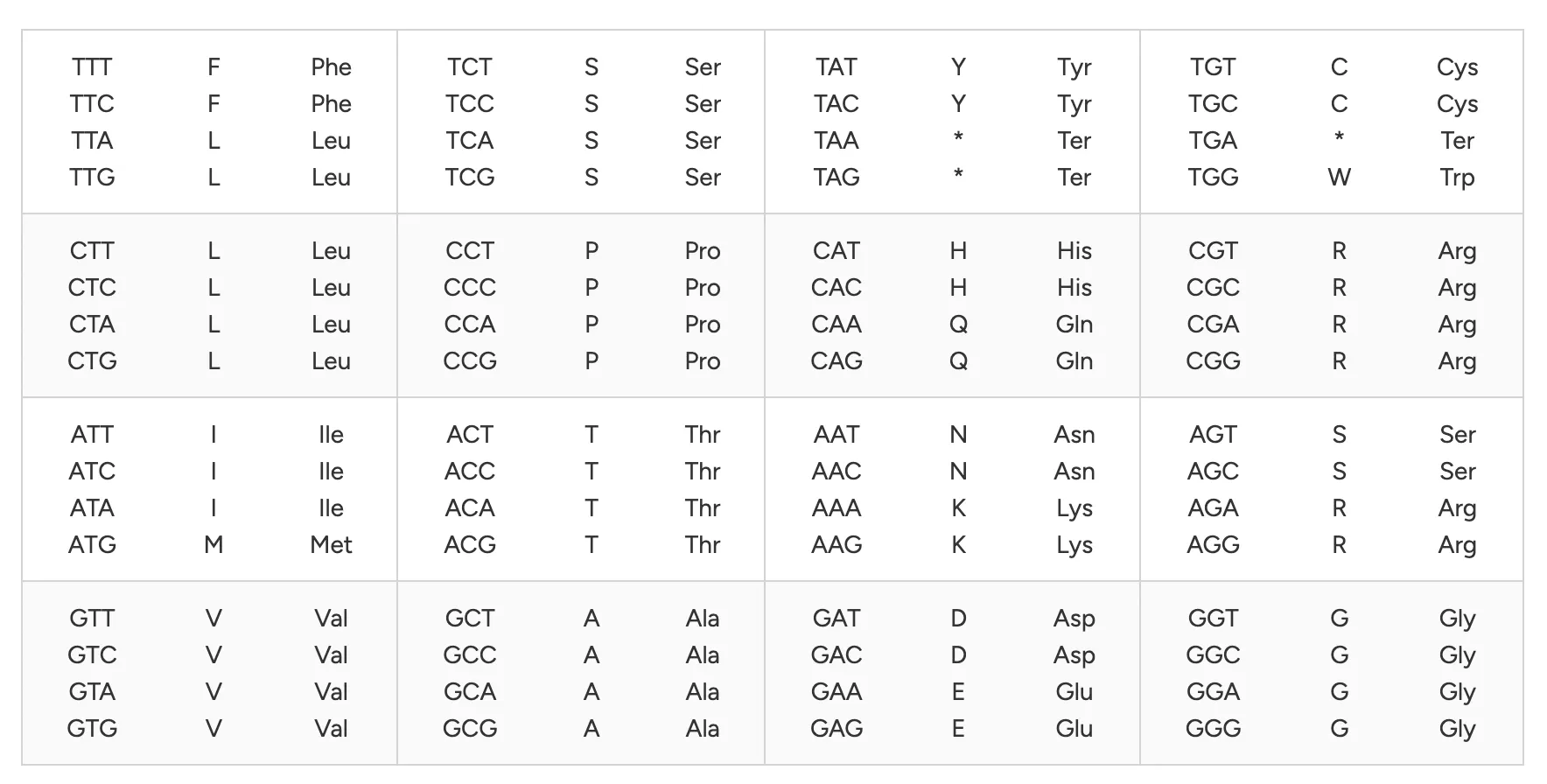
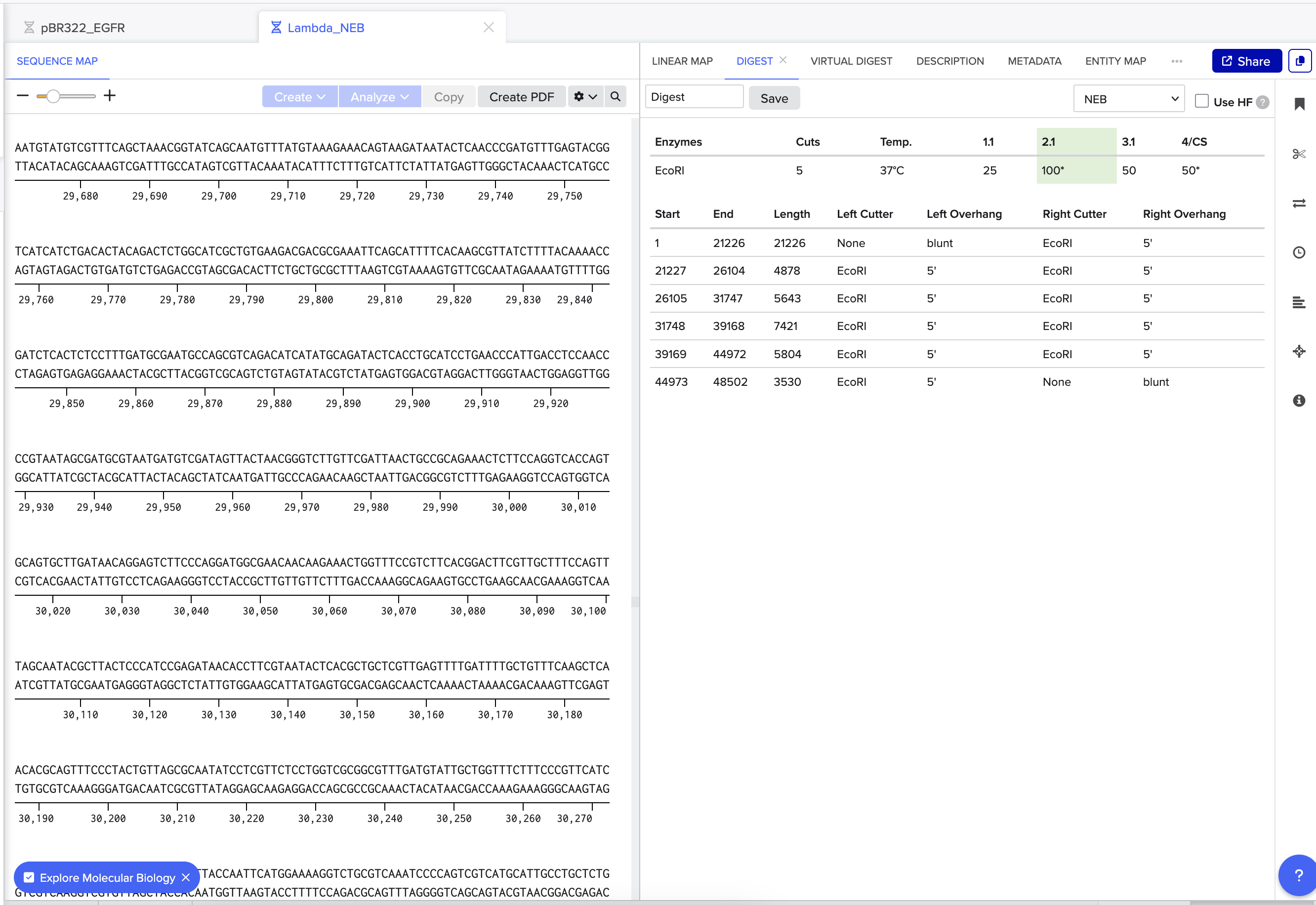

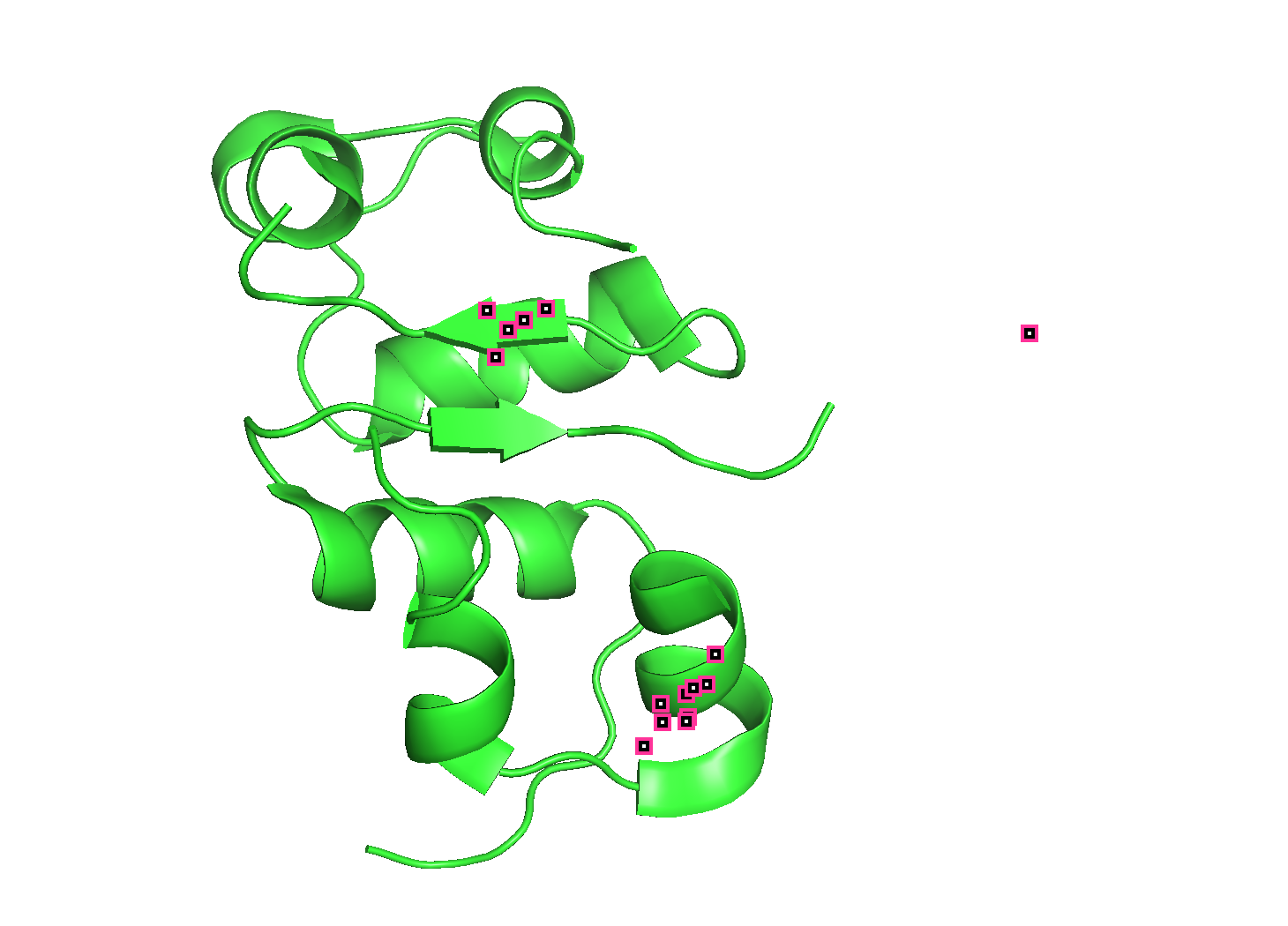
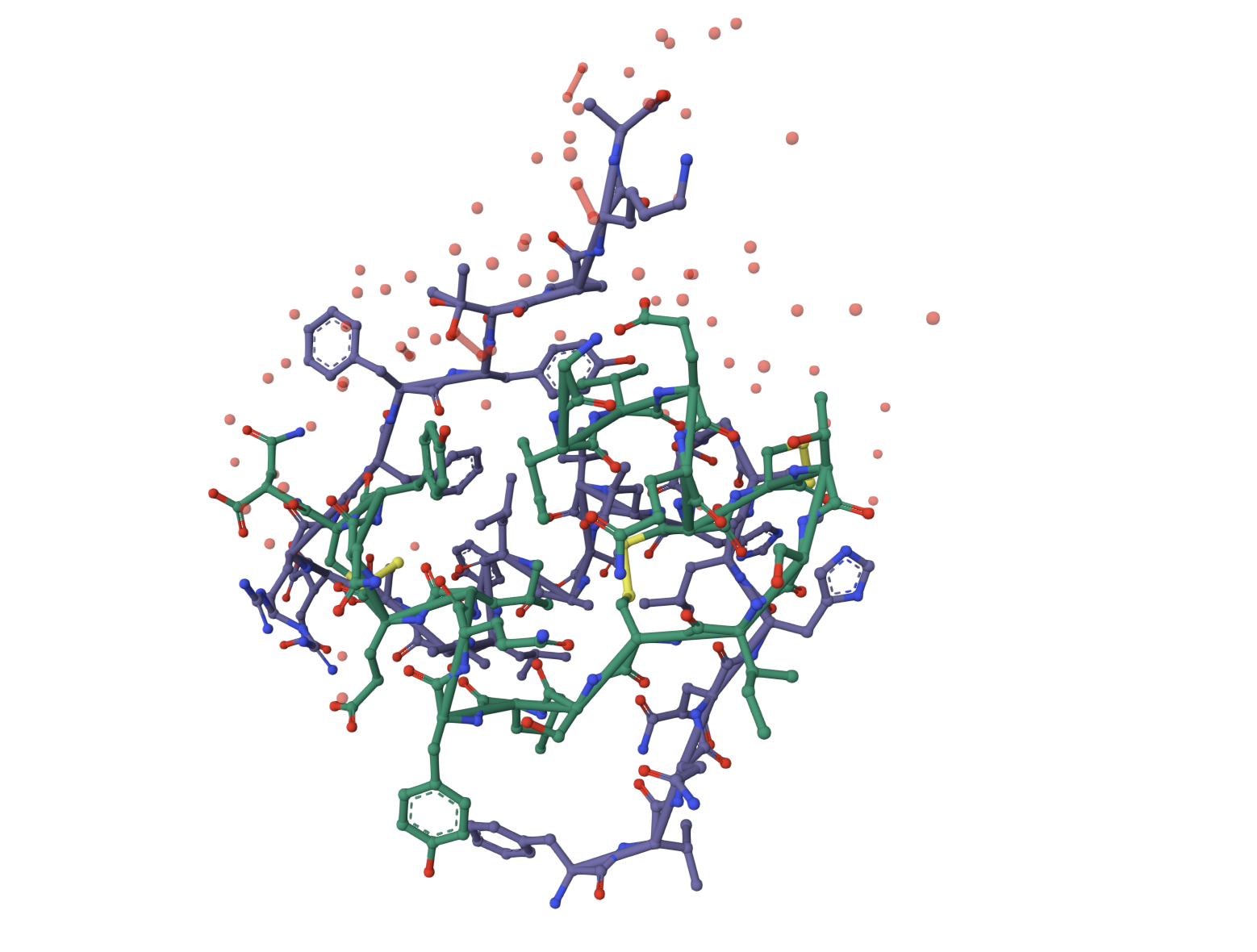
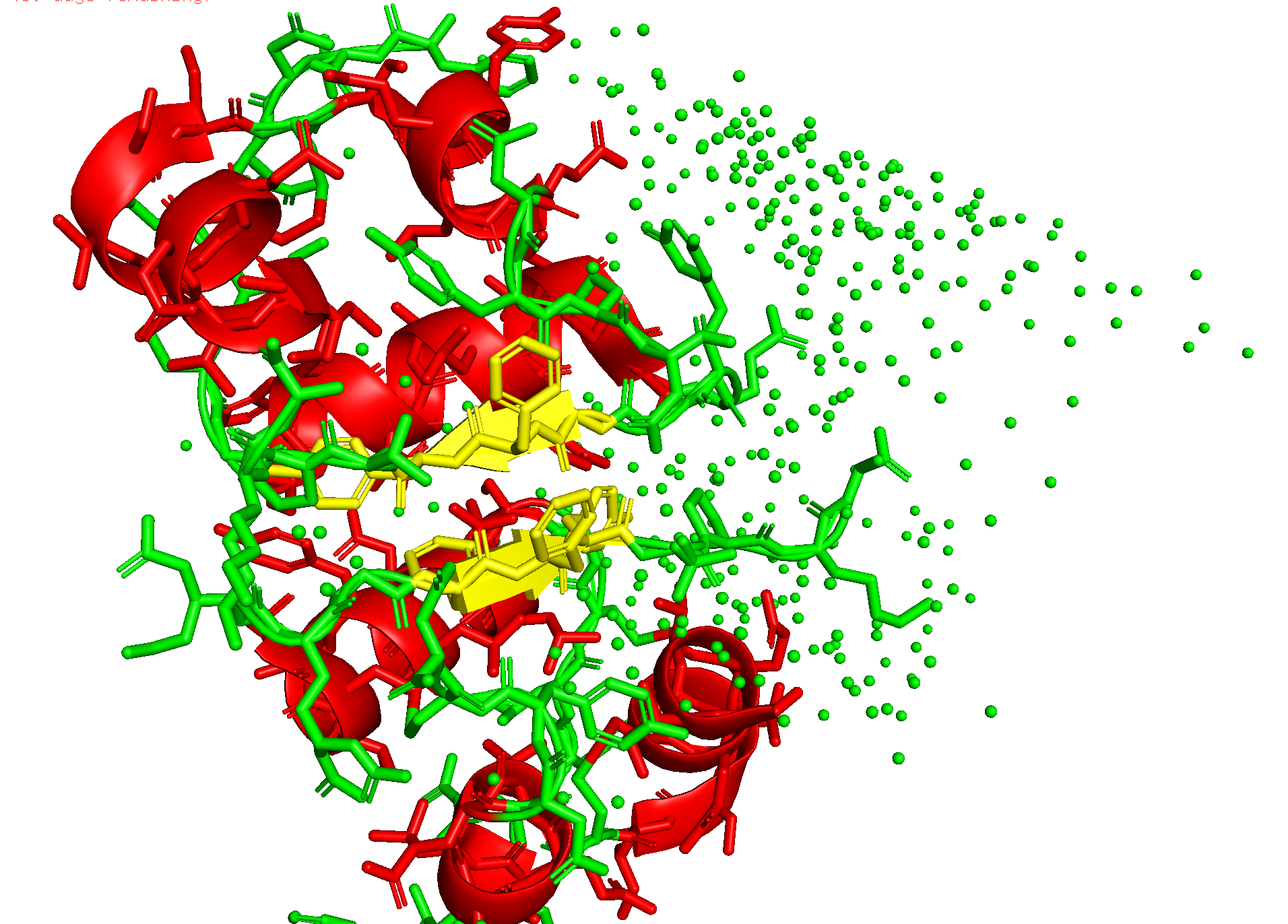
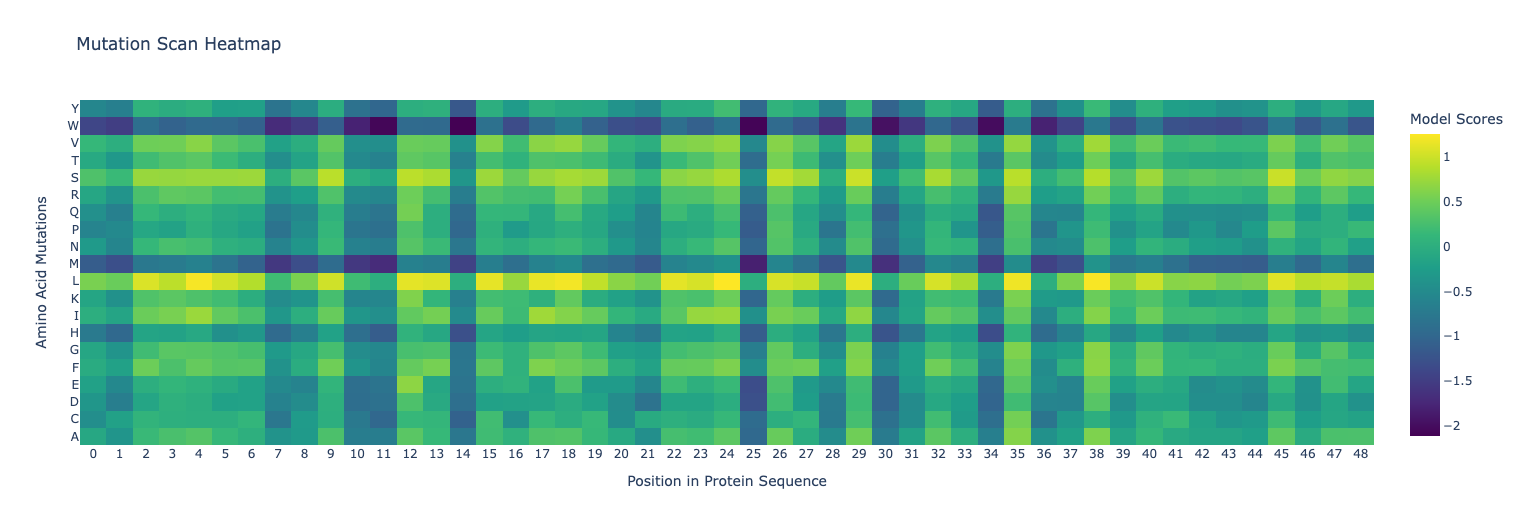
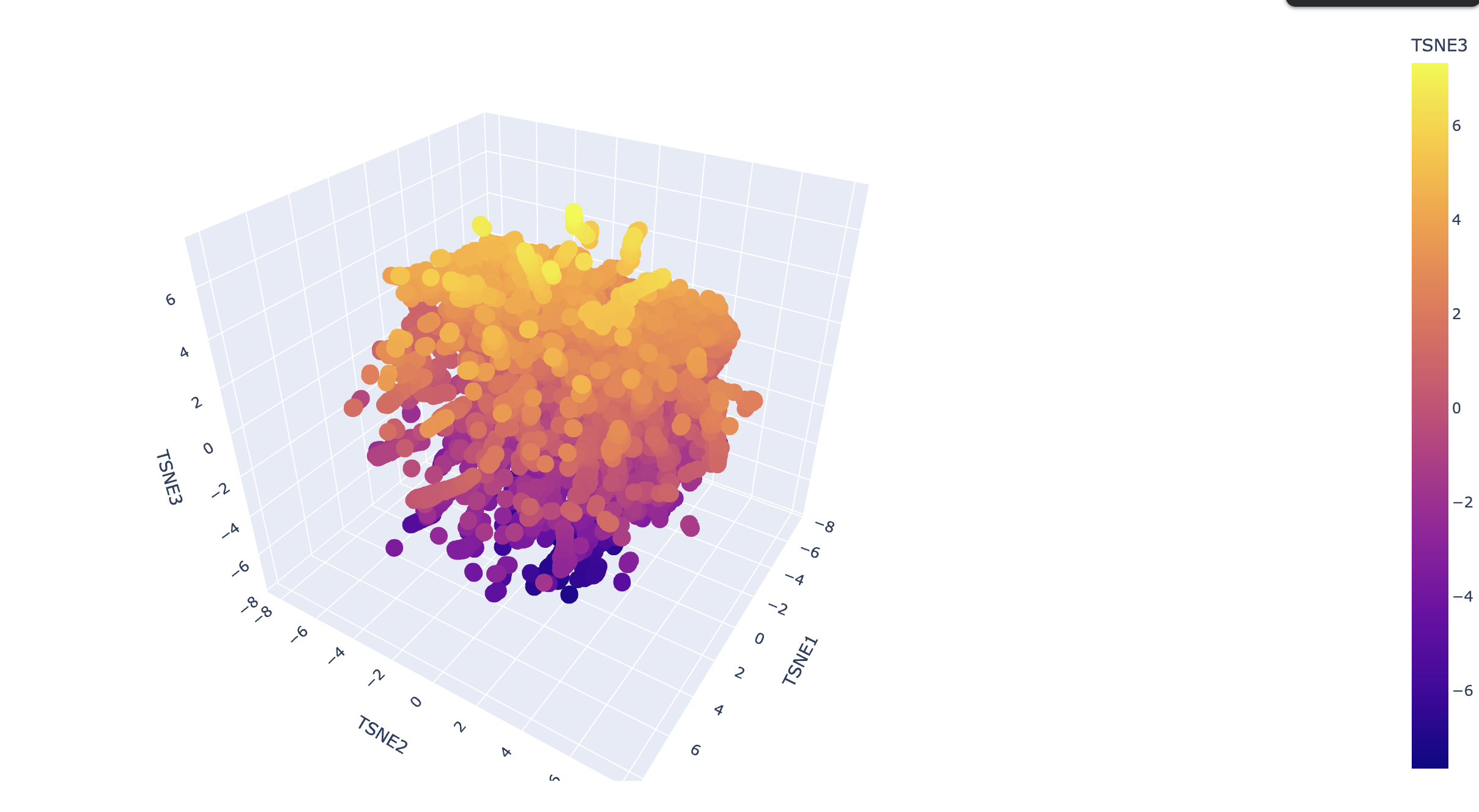
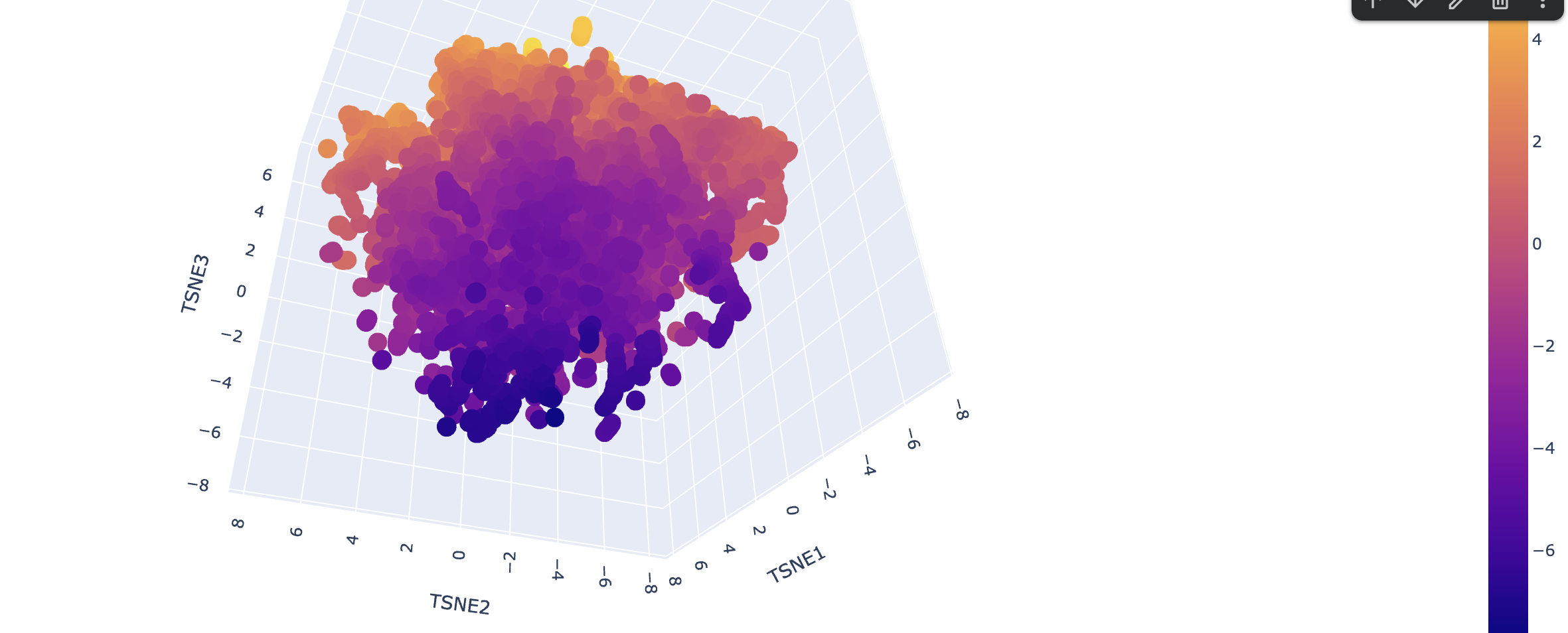
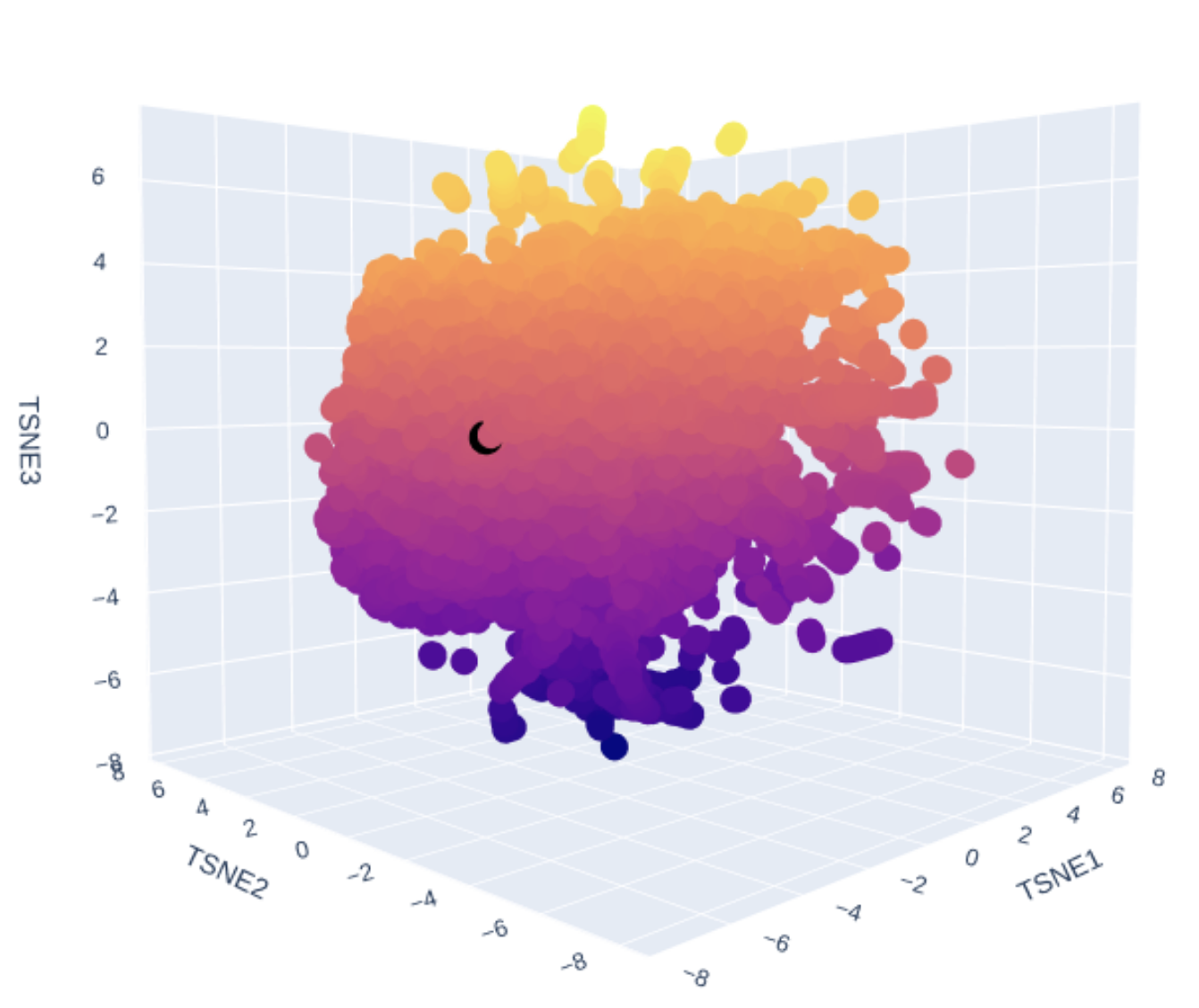
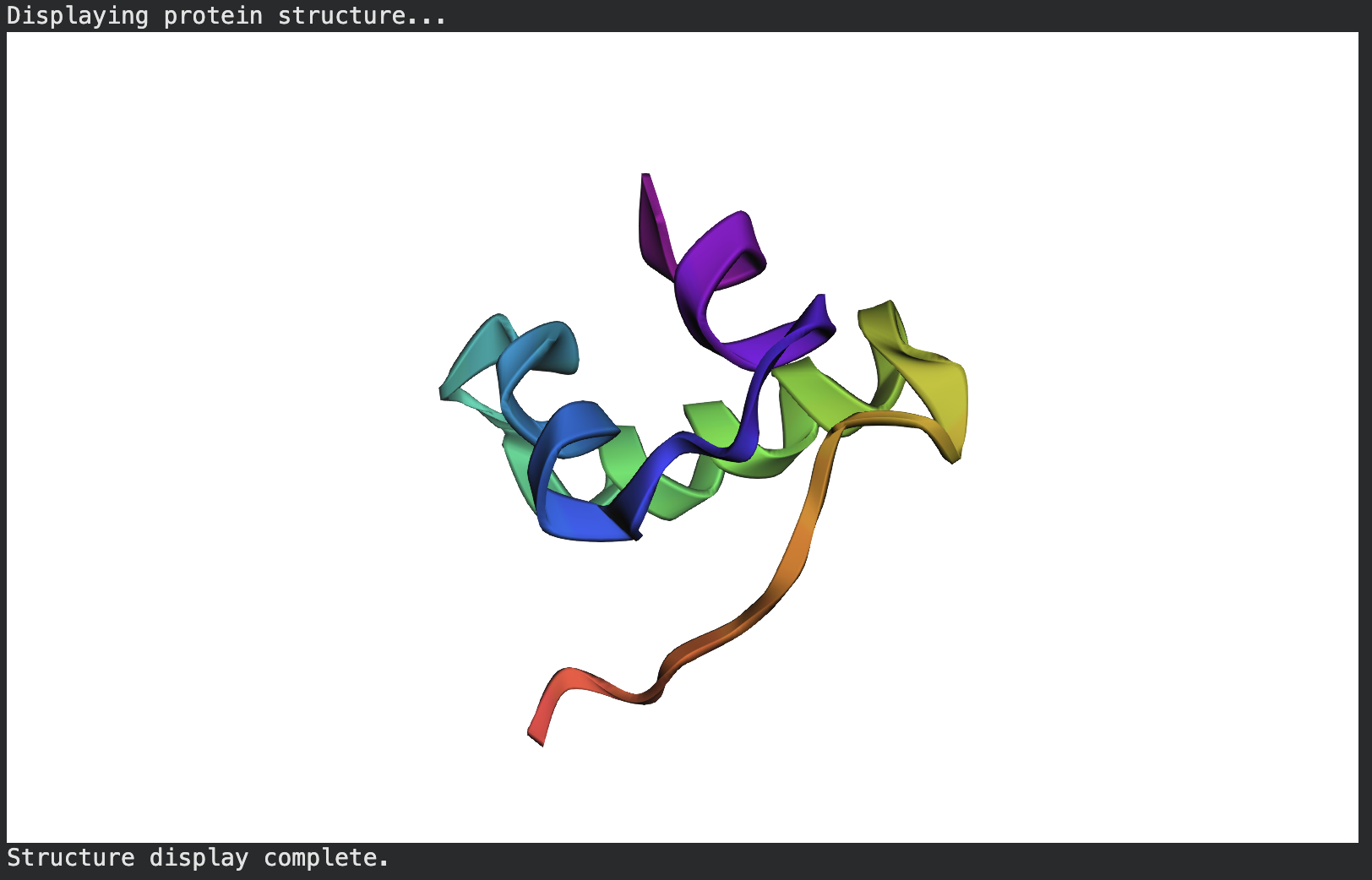
.png)