A Wearable or At-Home Androgen Biosensing Platform for PCOS Diagnosis and Management
I am interested in developing an easy-to-use biosensor/microfluidic kit that can detect and quantify androgen levels from blood or sweat to support the diagnosis and personalized management of Polycystic Ovary Syndrome (PCOS).
PCOS is a common hormonal disorder affecting 5-13% of reproductive-aged women and is characterized by androgen excess, irregular or absent ovulation, and polycystic ovaries. Symptoms can include severe acne, hirsutism, hair loss, and infertility, and the condition is associated with long-term risks such as diabetes, cardiovascular disease, and endometrial cancer.
Currently, one of the key diagnostic criteria for PCOS is elevated androgen levels, which are typically assessed through blood tests performed specifically on days 2-5 of the menstrual cycle. However, for many women with PCOS, menstrual cycles are irregular or absent, making it difficult to determine when testing should occur. As a result, diagnosis can be delayed or inconclusive.
A biosensing platform capable of continuously or repeatedly monitoring androgen levels throughout an entire cycle would provide a more accurate picture of hormonal dynamics. This could improve diagnostic reliability and enable more personalized treatment strategies by tracking hormonal responses to lifestyle changes or medications over time.
Governance and Policy Goals
1. Ensure Biological Safety and Security
Prevent misuse, unsafe operation, or unintended biological harm associated with the biosensing platform.
Sub-goals:
Restrict access to raw sensor calibration, modification protocols, or firmware to reduce the risk of misuse.
Incorporate built-in safety checks, clear user instructions, and warnings to minimize incorrect use or misinterpretation of results.
2. Protect User Privacy and Data Security
Protect sensitive hormonal and reproductive health data to prevent discrimination, stigma, or exploitation.
Sub-goals:
Minimize data collection to only what is strictly necessary for hormonal monitoring and trend analysis.
Ensure hormonal data is encrypted, anonymized, or processed locally when possible to reduce exposure.
3. Promote Equity and Accessibility in Women’s Health
Ensure the tool reduces existing barriers to diagnosis and care rather than reinforcing healthcare inequities.
Sub-goals:
Design the biosensing kit to be affordable and usable outside specialized clinical settings.
Validate the platform across diverse populations to reduce bias and improve diagnostic reliability for underrepresented groups.
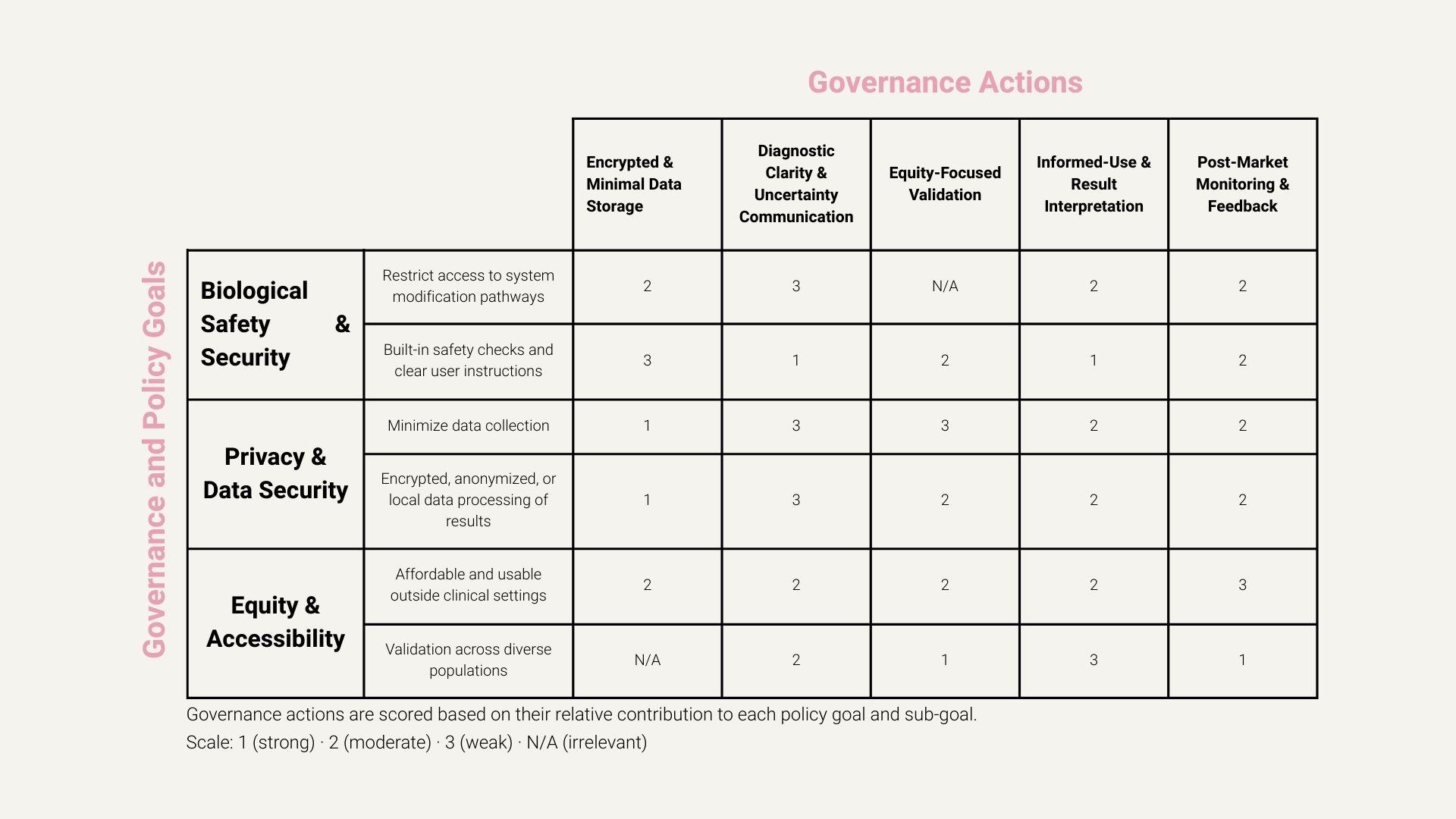
Governance Actions
1. Encrypted and Minimal Data Storage System
Actor: Companies (product developers), with regulatory oversight Ensure that hormonal data is encrypted and that only the minimum necessary data is collected or stored, preferably processed locally on-device.
2. Diagnostic Clarity and Uncertainty Communication
Actor: Companies + regulators Ensure that diagnostic outputs clearly distinguish between high-confidence diagnoses and ambiguous or borderline results. The tool should explicitly communicate diagnostic confidence and uncertainty, and automatically prompt users to seek further clinical evaluation when results fall outside validated confidence thresholds.
3. Equity-Focused Validation Across Diverse Populations
Actor: Academic researchers, funders, public health agencies Conduct and incentivize validation studies across diverse populations to reduce bias and improve diagnostic reliability for underrepresented groups.
4. Informed-Use and Result Interpretation Requirements
Actor: Companies + regulators Require a short, standardized informed-use process before first use that explains the scope of the tool, the meaning of diagnostic confidence and uncertainty, and appropriate user actions in response to different types of results.
5. Post-Market Monitoring and Feedback Loop
Actor: Regulators + companies Require ongoing, anonymized post-market monitoring of the tool after deployment, including reporting of false positives, false negatives, and aggregate user outcomes. This feedback should be used to identify unintended harms, performance gaps across populations, and opportunities for improvement over time.
Actions Ranking
Recommendation for Best Governance Action
Based on the scoring analysis, I would prioritize a combination of governance actions, with Encrypted and Minimal Data Storage as the primary action, alongside Informed-Use and Result Interpretation Requirements and Post-Market Monitoring and Feedback as complementary measures.
The highest priority should be the implementation of an encrypted and minimal data storage system. This action consistently scored strongest across privacy and data security sub-goals and provides a foundational safeguard against harm. Because hormonal and reproductive health data is highly sensitive, failures in data protection could lead to discrimination, stigma, or misuse at scale. Prioritizing data minimization and local or encrypted processing reduces these risks regardless of downstream user behavior or diagnostic accuracy. This action is also relatively feasible for companies to implement early and does not significantly postpone research or innovation.
As a second priority, I would emphasize informed-use and result interpretation requirements together with post-market monitoring. Informed-use mechanisms reduce misinterpretation and panic by helping users understand results and lower uncertainty, while post-market monitoring enables the detection of errors, biases, or unintended harms that emerge after deployment.
One key trade-off is the tension between data minimization and the need for a lot of data to support monitoring. There is also uncertainty around how effectively users will engage with and understand informed-use materials, which could limit their intended protective impact.
This recommendation is directed toward the research and development team responsible for building the biosensing platform, including academic labs and early-stage startups developing the technology. Overall, the scoring highlights that no single action is sufficient on its own. Instead, a comprehensive approach that prioritizes privacy infrastructure while supporting safe use and continuous evaluation offers the most ethically robust path forward.
Reflection on Ethical Concerns
An ethical concern that became especially salient to me this week is the risk associated with centralized access to sensitive hormonal and reproductive health data. Considering what could happen if a single actor gained access to aggregated user data highlighted how data breaches or misuse could cause large-scale harm, including discrimination or loss of trust. This concern reinforced the importance of prioritizing encrypted and minimal data storage, local data processing when possible, and ongoing post-market monitoring, rather than treating privacy as a secondary or purely technical consideration.
Lab Preparation
🦠 Complete Lab Specific Training in Person DONE 🦠 Complete Safety Training in Atlas DONE
Week 2 Lecture Prep
Questions from Professor Jacobson:
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
Answer: The error rate of polymerase is approximately 1 in 10⁶. The human genome is about 3 × 10⁹ bp, so that corresponds to roughly 3,000 potential errors per genome copy (and across many dividing cells, the total number of errors could be much higher). Biology deals with this discrepancy through multiple layers of error correction - using proofreading and repair mechanisms that reduce errors, such as the MutS repair system.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Answer: Most amino acids can be encoded by multiple codons, so there are an astronomically large number of possible DNA sequences that could encode the same protein, with many choices per amino acid compounding across the full length of the gene.
In practice, not all of these sequences work because certain codons are translated more efficiently than others, and some sequences can create unfavorable secondary structures or other issues. As a result, only a small subset of possible DNA sequences is functional and practical for expressing the desired protein.
Questions from Dr. LeProust:
What’s the most commonly used method for oligo synthesis currently?
Answer: The most commonly used method for oligo synthesis is chemical phosphoramidite DNA synthesis, which builds DNA sequences one nucleotide at a time through repeated cycles of coupling, protection, and deprotection on a solid support.
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Answer: It is difficult to synthesize oligos longer than 200nt via direct chemical synthesis because each nucleotide addition (chemical reaction) has an error rate, and these errors accumulate with every synthesis cycle, causing the correctness of the sequences to decrease as the length increases.
Why can’t you make a 2000bp gene via direct oligo synthesis?
Answer: A 2000bp gene cannot be made via direct oligo synthesis because synthesis errors accumulate with each nucleotide addition. As sequence length increases, the probability of producing a correct full-length sequence drops dramatically. Since direct oligo synthesis is already unreliable beyond 200nt, synthesizing a 2000bp sequence in a single run is effectively impossible. In addition, oligo synthesis produces single-stranded DNA, and generating a correct double-stranded gene of this length would further compound errors, making direct synthesis impractical.
Question from George Church:
Using Google & Prof. Church’s slide #4, what are the 10 essential amino acids in all animals, and how does this affect your view of the “Lysine Contingency”?
Answer: The 10 essential amino acids in animals are: Histidine, Isoleucine, Leucine, Lysine, Methionine, Phenylalanine, Threonine, Tryptophan, Valine, and Arginine.
I did not watch Jurassic Park, but according to what I read online, the “Lysine Contingency” was a genetic alteration Henry Wu performed in the dinosaurs’ genome to knock out their ability to produce lysine, forcing them to rely on lysine supplements provided by park staff in an attempt to imprison them in the park and prevent them from destroying the global ecosystem.
After checking where lysine comes from (I asked Gemini: “How do animals produce lysine?”), I found that animals cannot produce lysine at all and must obtain it through nutrition. This means that humans, animals, and dinosaurs would all need dietary lysine regardless. Based on this, I conclude that Henry Wu’s lysine contingency was useless as a containment strategy, and it is unclear what genetic mechanism he could have targeted to achieve this goal, since there is no direct lysine biosynthesis pathway in animals to knock out.
*You can find parts 1 and 2 in the week 2 lab section!
Part 3: DNA Design Challenge
My protein choice: Pro-resilin! Pro-resilin is a highly elastic, rubber-like structural protein found in insects that enables efficient energy storage for jumping, flight, and sound production. About seven years ago, my biotechnology teacher in high school showed us an article about a very cool leading professor in Israel, who was doing things that seemed almost impossible. I vividly remember one of the projects he was working on: scientists had identified a protein that enables a particular spider species to jump remarkably high relative to its body size, and this scientist wanted to give humans the same ability by creating shoes with soles made from some protein similar to Pro-resilin.
That moment has stayed with me until today. I think it would be very cool to look at the structure of this protein, try to understand what gives it its unique characteristics, and potentially think about what could be done with it.
I went to UniProt.org and searched for “Pro-resilin protein Drosophila melanogaster”
Here is the AA sequence:
*I chose the Drosophila melanogaster (fruit fly) resilin protein because its resilin is the best-characterized and most widely studied form of the protein.
Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence. To determine the nucleotide sequence corresponding to Pro-resilin in Drosophila melanogaster, I followed the link for the canonical transcript (Q9V7U0-1) on UniProt, which directed me to Ensembl Metazoa. There, I downloaded only the coding sequence (CDS) in FASTA format, excluding UTRs and introns, to obtain the exact DNA sequence that encodes the protein. The sequence is shown below:
There are several reasons why we need to optimize codon usage. Although multiple codons can encode the same amino acid, different organisms have preferences for certain codons over others - a phenomenon known as codon bias. This means that a DNA sequence that works efficiently in one organism may not be translated as efficiently in another. If rare or non-preferred codons are used, the ribosome may stall, leading to lower protein expression or incomplete translation. Therefore, it is important to adapt the codons in the sequence to match the preferred codon usage of the organism in which the protein will be expressed.
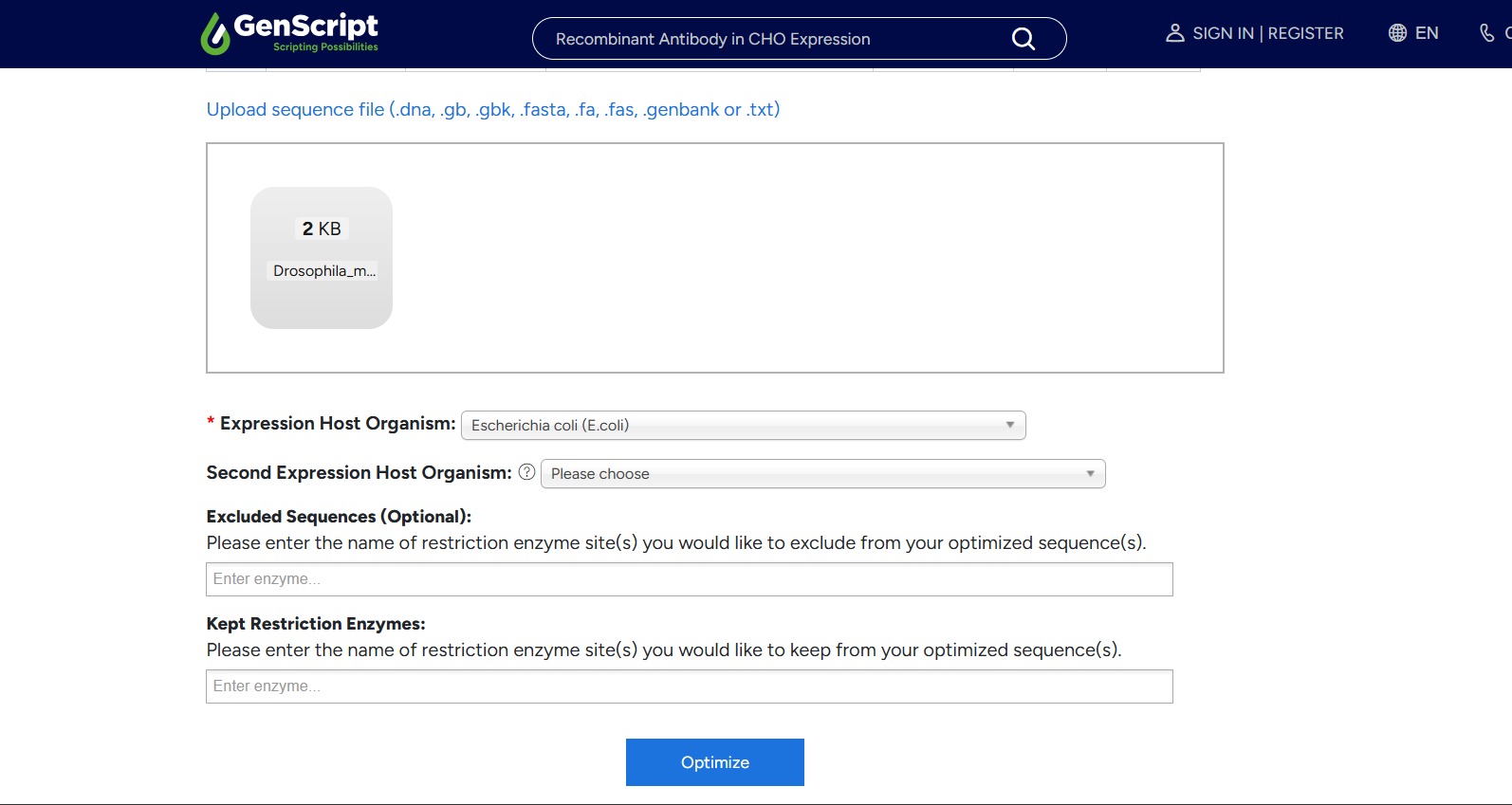
I chose to optimize the sequence for Escherichia coli because it is one of the most well-characterized and widely used organisms for recombinant protein expression. E. coli grows quickly, is easy to manipulate genetically, and has a large body of existing protocols and tools available. Since this is my first time going through the full synthetic gene design and expression workflow, I felt it was best to work with a well-established system before moving on to more complex organisms.
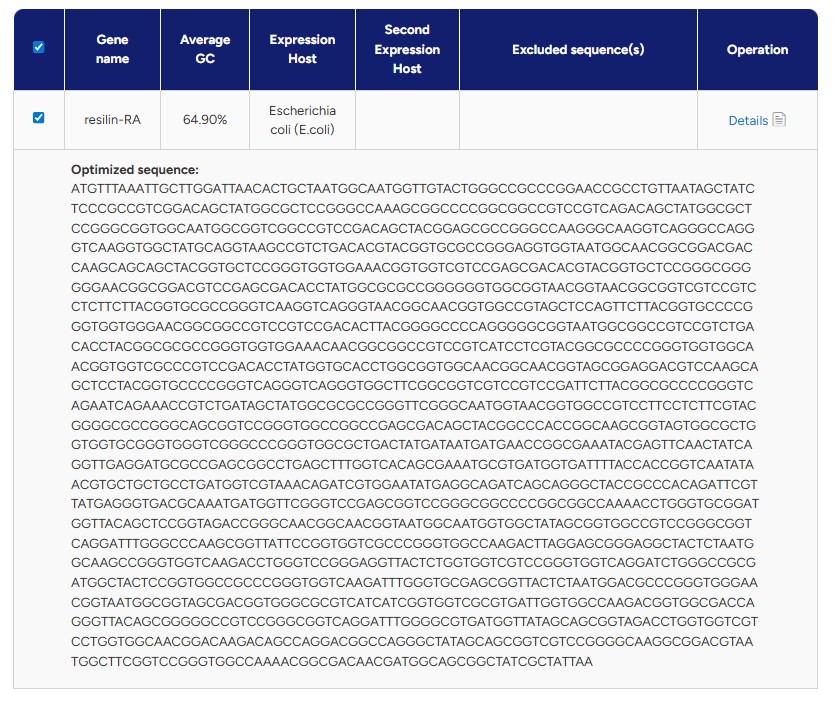
What technologies could be used to produce this protein from your DNA? One technology that can be used to produce this protein from my DNA is recombinant protein expression in bacteria, which is exactly the approach taken here. After obtaining and codon-optimizing the coding sequence (CDS), the gene is inserted into a plasmid vector that contains a promoter, ribosome binding site (RBS), terminator, and a selectable marker such as an antibiotic resistance gene. The plasmid is then transformed into E. coli cells, which are grown under selective conditions to ensure that only bacteria containing the correct vector survive.
Once inside the bacteria, the promoter drives transcription of the inserted DNA into mRNA. The ribosome then binds to the mRNA and translates the coding sequence into the corresponding protein. As the bacteria grow and divide, they produce increasing amounts of the recombinant protein. After sufficient expression, the protein can be purified. This results in the isolation of the desired protein.
Part 4: Prepare a Twist DNA Synthesis Order
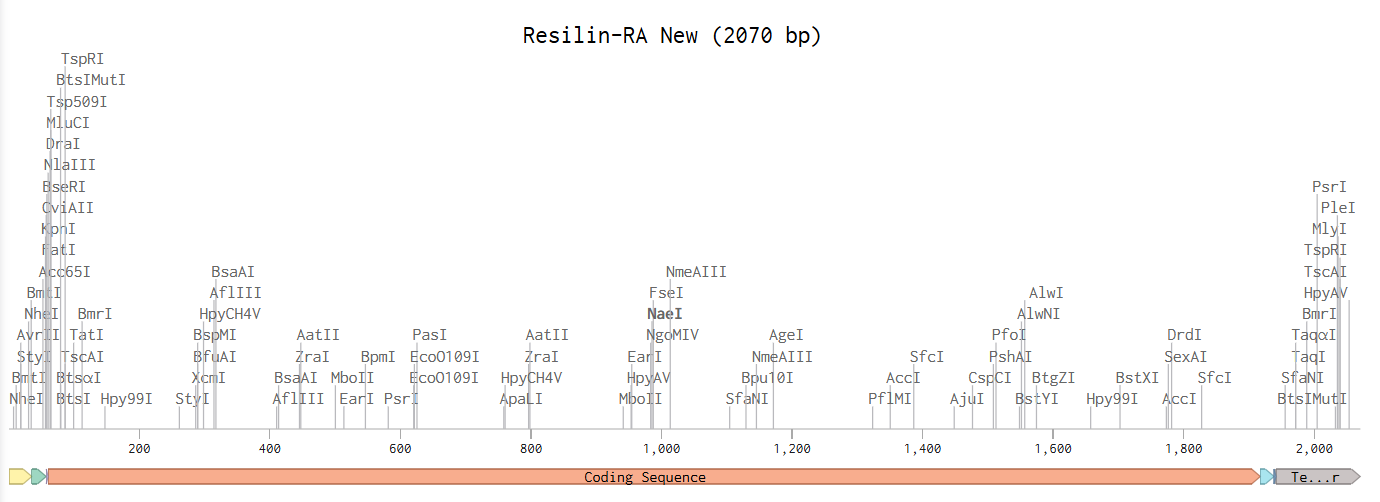
Linear Map After following all of the instructions on the website, I have obtained the following Linear Map:
When I proceeded to Twist, the platform flagged issues with my sequence and recommended re-optimization. After running their internal optimization, the system still classified the sequence as too complex, however, it did allow me to download the plasmid design.
Here it is:
Yayyyyy :)
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? I would like to sequence the gut microbiome of athletes before and after intense training, or before and after a traumatic injury (such as ligament tears or broken bones), to investigate whether specific bacterial compositions or functional genes correlate with recovery speed, inflammation levels, or injury resilience. I think it would be fascinating to explore whether the microbiome, often referred to as “the second brain”, plays an even greater role in recovery and overall health than we currently understand.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why To obtain a microbiome sample, I would probably rely on a non-invasive stool sample collection method, since from what I have learned, this is typically how researchers study the gut microbiome. For sequencing, I would probably choose next-generation sequencing (NGS), specifically shotgun metagenomic sequencing. NGS allows for a lot of DNA fragments to be sequenced in parallel, enabling comprehensive analysis of complex microbial communities. This approach would allow me not only to identify the bacterial species present, but also to analyze functional genes that may be associated with inflammation regulation, tissue repair, and recovery capacity.
Shotgun metagenomic sequencing using Illumina technology is considered a second-generation sequencing method because it enables massively parallel sequencing of millions of short DNA fragments simultaneously. Unlike first-generation Sanger sequencing, which reads one fragment at a time, second-generation platforms generate high-throughput short reads using sequencing-by-synthesis chemistry.
The input for this method is the total DNA extracted from a stool sample. The extracted DNA is fragmented into smaller pieces, typically a few hundred base pairs long. Synthetic adapter sequences are ligated to both ends of each fragment, allowing them to bind to the sequencing flow cell. The fragments are then amplified by PCR to generate sufficient material, creating a sequencing library ready for analysis.
In Illumina sequencing, DNA fragments bind to a flow cell and undergo cluster amplification. During sequencing-by-synthesis, fluorescently labeled nucleotides are incorporated one base at a time to synthesize the complementary strand. After each incorporation, a camera detects the emitted fluorescence signal, and the color corresponds to a specific base (A, T, C, or G). This process repeats in cycles, allowing the machine to determine the DNA sequence through base calling.
The output consists of millions of short DNA reads, typically stored in FASTQ format. Each read includes both the nucleotide sequence and a quality score (a measure of confidence in each base call) for each base. These reads can then be computationally analyzed to determine microbial composition and identify functional genes within the microbiome.
5.2 DNA Write
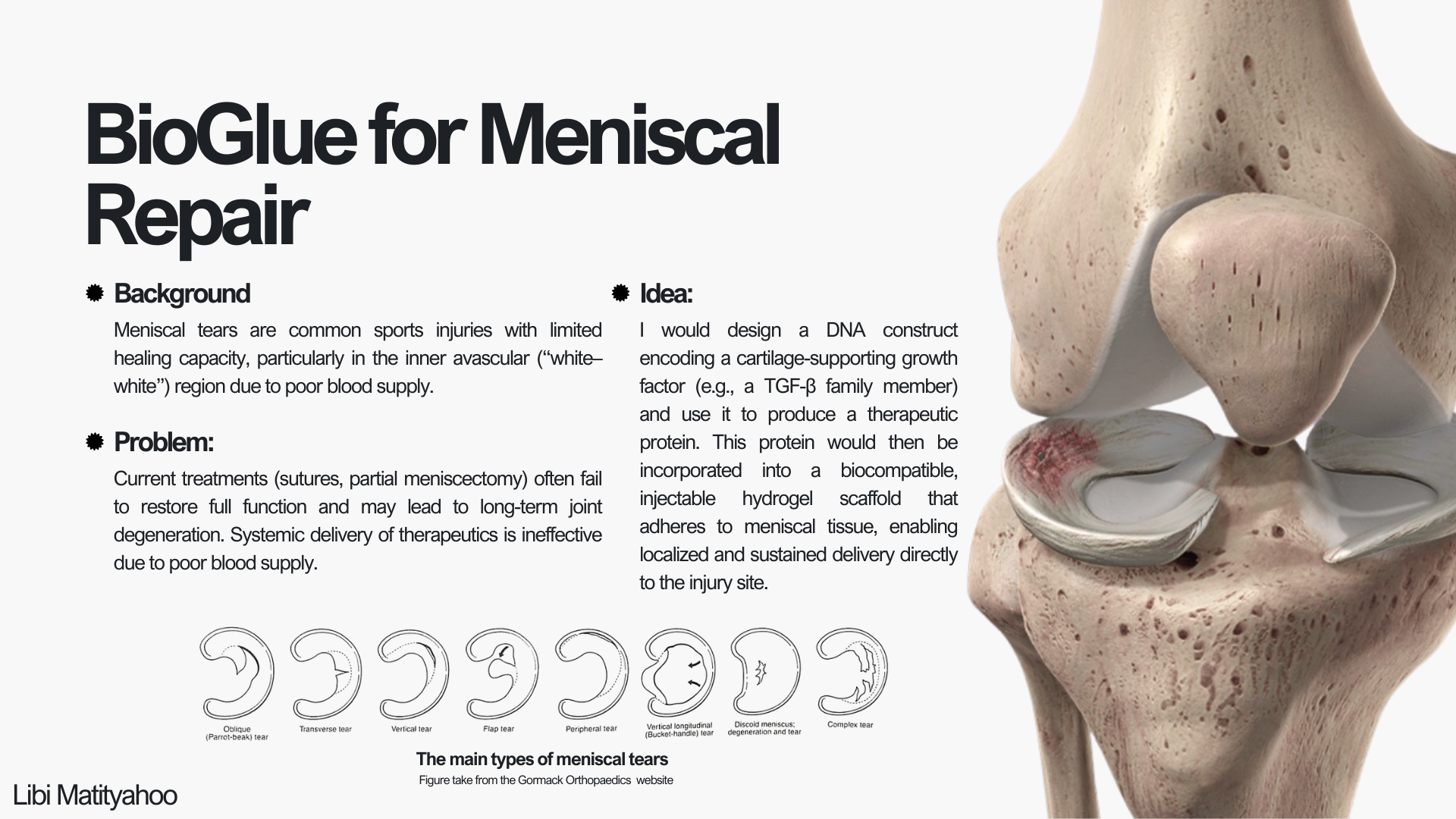
(i) What DNA would you want to synthesize (e.g., write) and why? I would like to synthesize DNA that can be transcribed and translated into a therapeutic protein “paste” (or hydrogel-like material) to support healing of joint cartilage, and specifically the meniscus. Since there isn’t a sufficient solution for meniscal tears today, and the effects of surgeries can be lifelong, I think that creating something like this could be game-changing. One of the main challenges with meniscus healing is that blood supply is not consistent across the tissue (especially toward the inner region), which makes delivery of therapeutics through the bloodstream difficult. Because of that, an externally applied material that can consistently deliver needed proteins or signaling molecules directly to the injury site might help the meniscus heal better and faster. More specifically, I would start by synthesizing a construct encoding a cartilage-supporting growth factor, such as members of the TGF-β superfamily (Fortier et al., 2011), or a small engineered protein designed to promote extracellular matrix production. This construct could eventually be incorporated into a biomaterial scaffold for localized and sustained protein delivery.
(ii) What technology or technologies would you use to perform this DNA synthesis and why? The DNA would be synthesized using solid-phase phosphoramidite synthesis, which builds short oligonucleotides one nucleotide at a time. These short oligonucleotides are then assembled into a full-length gene using enzymatic methods such as ligation or Gibson Assembly, and cloned into a plasmid for verification and use. I would use this method because it is the current standard for accurate, high-fidelity DNA synthesis and allows precise control over the exact sequence being produced. It also enables incorporation of specific design elements such as codon optimization, regulatory sequences, or tags, making it great for constructing engineered therapeutic genes.
Solid-phase phosphoramidite synthesis builds DNA one nucleotide at a time on a solid support. The process occurs in repeated chemical cycles. First, the growing DNA strand is attached to a solid surface. A protected nucleotide (phosphoramidite) is then added to the chain in a coupling reaction. After coupling, a capping step inactivates any strands that did not successfully react, which helps reduce errors. The protecting group on the newly added nucleotide is then removed (deprotection) to expose a reactive site for the next cycle. Finally, an oxidation step stabilizes the newly formed bond. This cycle repeats until the full oligonucleotide sequence is synthesized. After synthesis is complete, the DNA is cleaved from the solid support, fully deprotected, and purified. Since this chemical method efficiently produces short oligonucleotides, multiple synthesized fragments are then assembled using enzymes into a longer gene construct and sequence-verified..
In solid-phase phosphoramidite synthesis, each chemical coupling step is slightly less than 100% efficient, meaning that errors accumulate as the sequence length increases. For this reason, individual oligonucleotides are typically limited to around 150–200 base pairs, and longer genes must be assembled from shorter fragments. Additionally, sequences that are highly repetitive or have extreme GC content can be more difficult to synthesize accurately.
5.3 DNA Edit
(i) What DNA would you want to edit and why? I would like to explore editing the DNA of cancerous cells to selectively induce apoptosis (programmed cell death) and prevent uncontrolled cell division as a therapeutic strategy for cancer treatment. Many cancers arise from mutations in genes that regulate cell growth and survival. By using targeted gene editing tools to either restore normal tumor suppressor function or disrupt oncogenes specifically in cancer cells, it may be possible to stop tumor progression while minimizing damage to healthy tissue. A major challenge in this approach would be ensuring precise delivery to cancer cells while avoiding unintended edits in healthy cells.
(ii) What technology or technologies would you use to perform these DNA edits and why? To perform these DNA edits, I would use a CRISPR-based editing platform, specifically base editing or prime editing, depending on the type of mutation involved. These technologies allow precise genetic modifications without introducing full double-strand breaks, which reduces the risk of unintended insertions or deletions. However, significant challenges remain, particularly achieving efficient delivery to cancer cells while minimizing off-target effects in healthy tissue.
I would use prime editing, a CRISPR-based technology that uses a Cas9 nickase fused to a reverse transcriptase and a specialized guide RNA, pegRNA (a specially designed guide RNA that identifies the target site and contains the template for the desired genetic edit). The guide RNA directs the enzyme to a specific mutation in the cancer cell DNA. Instead of creating a full double-strand break, the system makes a single-strand nick and directly writes the corrected DNA sequence into the genome. The cell’s repair machinery then stabilizes the edit, making the change permanent.
First, I would identify a mutation specific to the cancer type and design a pegRNA targeting that sequence. The editing components (Cas9 nickase–reverse transcriptase and pegRNA) would need to be packaged into a delivery system, such as targeted lipid nanoparticles that bind receptors overexpressed on cancer cells. The key inputs include the editing enzyme, the guide RNA, and a selective delivery vehicle to minimize effects on healthy cells.
Although prime editing is more precise than traditional CRISPR-Cas9, challenges remain. Editing efficiency may be incomplete, and off-target effects are still possible. The biggest limitation is safe and selective delivery to cancer cells, since unintended edits in healthy tissues could cause harm.
References
Fortier, L. A., Barker, J. U., Strauss, E. J., McCarrel, T. M., & Cole, B. J. (2011). The role of growth factors in cartilage repair.Clinical Orthopaedics and Related Research, 469(10), 2706–2715.
Week 3 HW Lab Automation
*You can find the Python Script for Opentrons Artwork part in the Week 3 Lab section!
Post-Lab Questions
1. Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
The paper I chose, “An Automated Versatile Diagnostic Workflow for Infectious Disease Detection in Low-Resource Settings” (Urrutia Iturritza et al., 2024), presents a fully automated diagnostic pipeline built using the Opentrons OT-One-S Hood robot. The authors combined open-source modular automation with molecular biology protocols to create a workflow capable of detecting Neisseria meningitidis, a pathogen responsible for meningitis. What I found especially compelling is that the system automates the entire process - from DNA isolation using magnetic beads to isothermal amplification (RPA), enzymatic digestion, and final detection on a paper-based vertical flow microarray. Instead of focusing solely on analytical novelty, the study emphasizes integration: connecting multiple biological modules into a single continuous, robot-executed workflow.
The workflow processes eight samples in parallel and completes the full diagnostic pipeline in about 110 minutes, about 18% faster than manual processing by trained personnel. Even more interesting is the cost analysis: the estimated cost per sample (~USD 16) is significantly lower (~5.8× less per sample) than traditional PCR-based diagnostic tests. The use of recombinase polymerase amplification (RPA), which operates at constant temperature, eliminates the need for expensive thermocycling infrastructure. The detection step uses gold nanoparticles on paper-based microarrays, producing a visible signal, which makes the system potentially adaptable for decentralized or low-resource settings.
It is important to note that the workflow was not completely autonomous. Before running the protocol, researchers manually prepared the samples and placed the required reagents, consumables, microarrays, and supporting equipment on the robot deck. Some user interaction was still needed during the thermal cycling steps, and imaging and quantitative analysis of the microarrays were carried out manually after the run. Overall, while the liquid-handling and main molecular processes were automated, the system still relied on human setup and post-processing - showing both the strengths and current practical limits of laboratory automation.
This work highlights automation not as a luxury but as a strategy for improving accessibility, reproducibility, and safety in molecular diagnostics. By using an open-source robot such as Opentrons, standard laboratory consumables, and modular protocols, the authors demonstrate a strong proof of concept for automating nearly an entire diagnostic workflow. Importantly, this approach minimizes human handling steps, thereby reducing the risk of contamination and operational errors, while allowing scientists to focus on experimental design, interpretation, and optimization. In this sense, the engineering contribution lies not only in the individual modules, but in the successful integration of these modules into a cohesive, largely automated system with clear translational potential.
Resources:
Miren, U. I., Mlotshwa, P., Gantelius, J., Alfvén, T., Loh, E., Karlsson, J., . . . Gaudenzi, G. (2024). An automated versatile diagnostic workflow for infectious disease detection in low-resource settings. Micromachines, 15(6), 708. doi:https://doi.org/10.3390/mi15060708
2. Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
Automation plan for BioGlue Meniscal Repair project
For my final project, I intend to use lab automation to systematically screen BioGlue formulations that deliver a cartilage-supporting growth factor (e.g., a TGF-β family protein). The goal of automation is to remove pipetting variability and enable parallel testing of many conditions (protein dose, gel composition, crosslinking conditions) in a reproducible way.
What I would automate:
A) Hydrogel formulation + dose matrix (OT-2 / Opentrons)
Prepare a concentration series of growth factor (e.g., 0×, low, medium, high, very high).
Dispense hydrogel precursor components into a 96-well plate (or small molds).
Add growth factor to each well according to a planned matrix (dose × gel composition).
Mix consistently (pipette mixing program).
Start a standardized “release study” (how much protein leaves the gel over time) by overlaying buffer and scheduling timed sampling.
B) Automated sampling for release kinetics
6. At defined timepoints (e.g., 1h, 6h, 24h, 48h), the robot removes a small aliquot of supernatant and transfers it to a readout plate (for ELISA / fluorescence / total protein assay).
Final Project Ideas
As requested, I uploaded my ideas to the slide deck, but here they are too:
BioGlue for Meniscal Repair
At-Home Androgen Biosensing for PCOS (Wearable / Microfluidic Kit)
Programmable RGB Fluorescence System
Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Answer: On average, raw meat is ~20–25% protein by weight. If we assume 20%, then in a 500 g sample we have:
500 × 0.20 = 100 g protein Since 1 g ≈ 6.022 × 10^{23} Daltons, then:
100 g = 6.022 × 10^{25} Daltons If the average amino acid has a mass of ~100 Daltons, then:
Number of AAs = (6.022 × 10^{25}) / 100 = 6.022 × 10^{23}
So there are approximately 6 × 10^23 AAs!
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Answer: When we eat beef or fish, we are not incorporating their cells into our bodies. During digestion, proteins are broken down into amino acids, and DNA is broken down into nucleotides by enzymes in the stomach. What gets absorbed into our bloodstream are these small building blocks - not intact cells, genes, or whole DNA molecules. There is no natural mechanism in our body that takes DNA from food and integrates it into our genome. For DNA to become part of our cells, it would need very specific enzymes and delivery systems (like viruses use), which do not occur through normal digestion. So biologically, we are not “becoming” cow or fish; we are breaking their biomolecules down and reusing the basic components to build and maintain our own human cells.
3. Why are there only 20 natural amino acids?
Answer: There are 20 natural amino acids, likely because evolution settled on a set that is chemically diverse enough to build all necessary protein structures, while still being manageable for the translation machinery. Each amino acid has a different side chain. With 20 options, we already get a very wide range of chemical behaviors, which is enough to create stable, functional, and highly specific proteins. The genetic code is built around triplet codons (64 possible codons), and these encode 20 amino acids plus stop signals. Expanding the number significantly would require major changes to the ribosome, tRNAs, and the entire translation system. It is possible that once this system became fixed early in evolution, it was strongly conserved.
4. Can you make other non-natural amino acids? Design some new amino acids.
Answer: Yes, we can design non-natural amino acids. Synthetic biology already does this by engineering new tRNAs and enzymes that insert artificial amino acids into proteins. To design one, I would keep the same backbone so the ribosome can still recognize it, and modify only the side chain. For example, a fluorescent amino acid that allows direct visualization of proteins.
5. Where did amino acids come from before enzymes that make them, and before life started?
Answer: Before life existed, amino acids were formed by simple chemical reactions on early Earth. The early atmosphere had small molecules like methane, ammonia, hydrogen, and water. Energy from lightning, heat, and UV radiation drove reactions between them and produced amino acids. This was shown in the Miller–Urey experiment. Amino acids were also found in meteorites, so some may have formed in space.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Answer: L-amino acids and D-amino acids are stereoisomers, or mirror images of each other. Natural proteins are made of L-amino acids, and they form right-handed α-helices. If we build an α-helix using D-amino acids instead, the geometry is mirrored. So I would expect the helix to be left-handed.
7. Can you discover additional helices in proteins?
Answer: Yes, additional helices can be discovered both experimentally and computationally. Experimentally, techniques like X-ray crystallography, NMR, and cryo-EM allow us to determine protein structures and identify helical regions. Computationally, secondary structure prediction algorithms and structure prediction tools can analyze amino acid sequences and detect potential α-helices or other helical motifs.
8. Why are most molecular helices right-handed?
Answer: Most biological helices are right-handed because life is built mainly from L-amino acids. The stereochemistry of L-amino acids restricts the backbone angles in a way that makes the right-handed α-helix energetically more favorable and sterically stable.
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
Answer: β-sheets tend to aggregate because their backbone hydrogen bond donors and acceptors are exposed and can easily form hydrogen bonds with neighboring β-strands from other molecules. When multiple β-strands align, they form extended intermolecular hydrogen-bonding networks, which are very stable. The main driving force for β-sheet aggregation is hydrogen bonding between backbones, together with the hydrophobic effect - hydrophobic side chains pack together, which further stabilizes the aggregate.
Part B: Protein Analysis and Visualization
1. Briefly describe the protein you selected and why you selected it.
Answer: Nav1.7 is a voltage-gated sodium channel located in neuronal membranes that initiates and propagates action potentials by allowing Na⁺ ions to enter the cell in response to changes in membrane voltage. It plays a crucial role in controlling the excitability of nociceptor neurons and is therefore essential for the sensory perception of pain. This protein demonstrates a clear relationship between 3D structure and electrical function and has significant physiological and clinical relevance. I was particularly drawn to it because of my interest in understanding how the brain works and because its structural properties provide an opportunity to explore the mechanisms underlying neuronal signaling.
2. Identify the amino acid sequence of your protein:
As shown in the sequence, the Nav1.7 complex includes auxiliary beta subunits that regulate channel function (chains B and C). For the purposes of this assignment, I will focus on chain A, which corresponds to the alpha subunit forming the pore and voltage-sensing domains of the channel!
How long is it? 2031 AAs!
What is the most frequent amino acid? L, Leucine, which appears 202 times.
How many protein sequence homologs are there for your protein? There are 250 protein sequence homologs identified in UniProtKB using BLAST (I took sequences with percent identity > 78%).
Does your protein belong to any protein family? Yes, according to UniProt, it belongs to the sodium channel (TC 1.A.1.10) family.
3D Protein Visualization
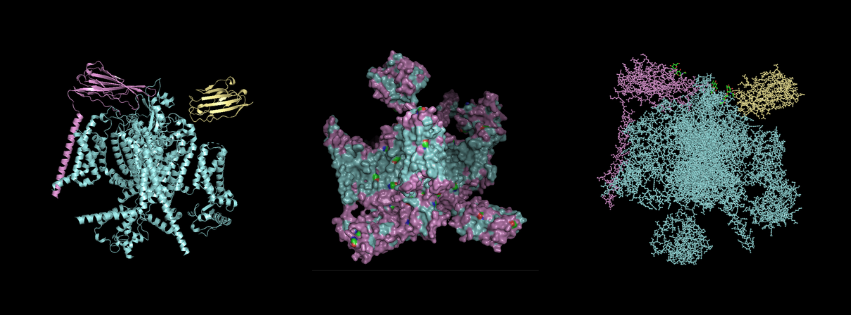
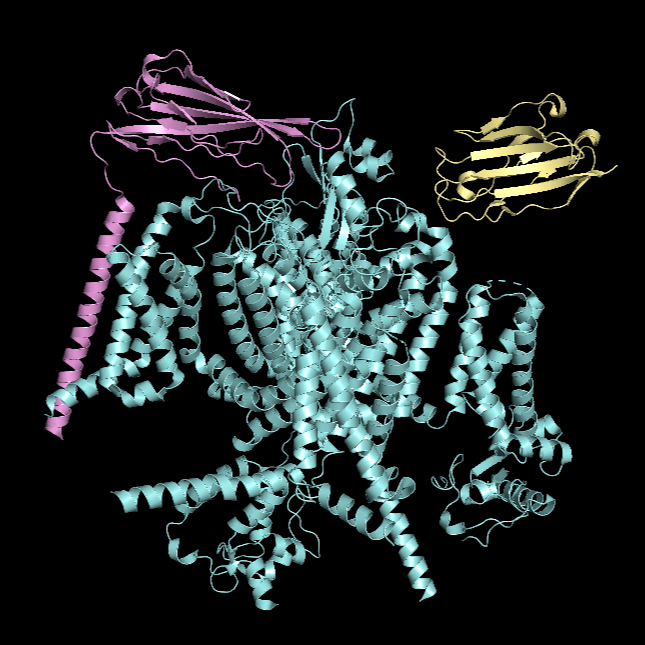
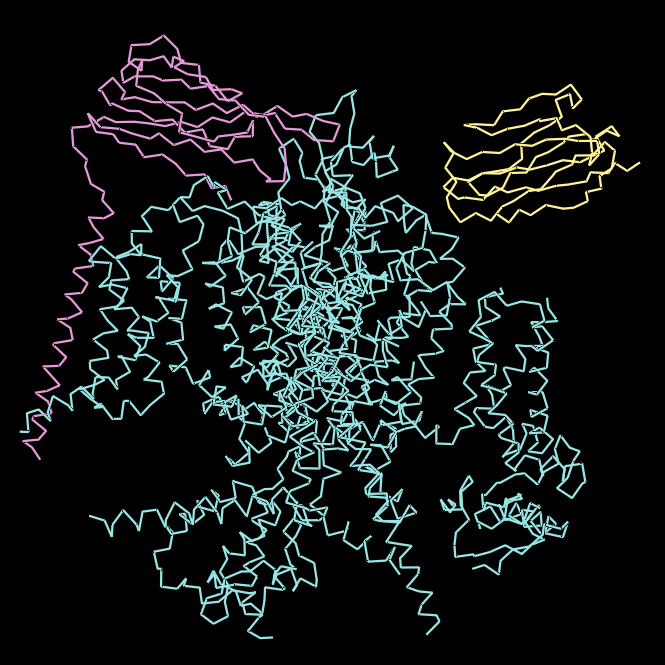
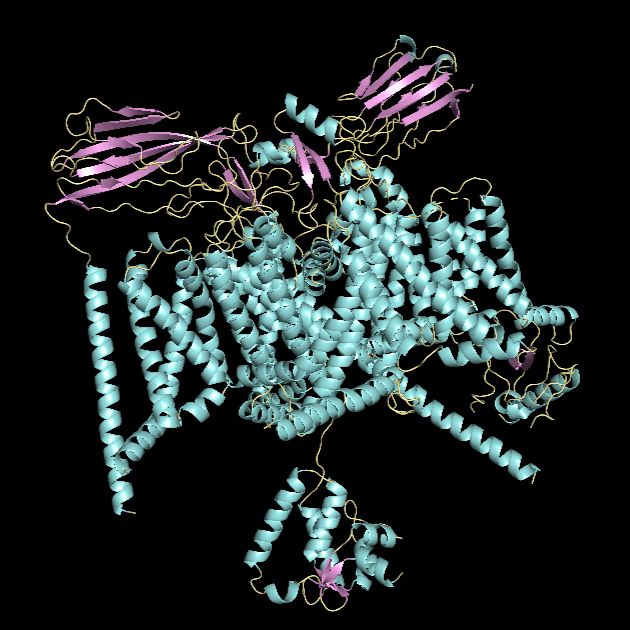
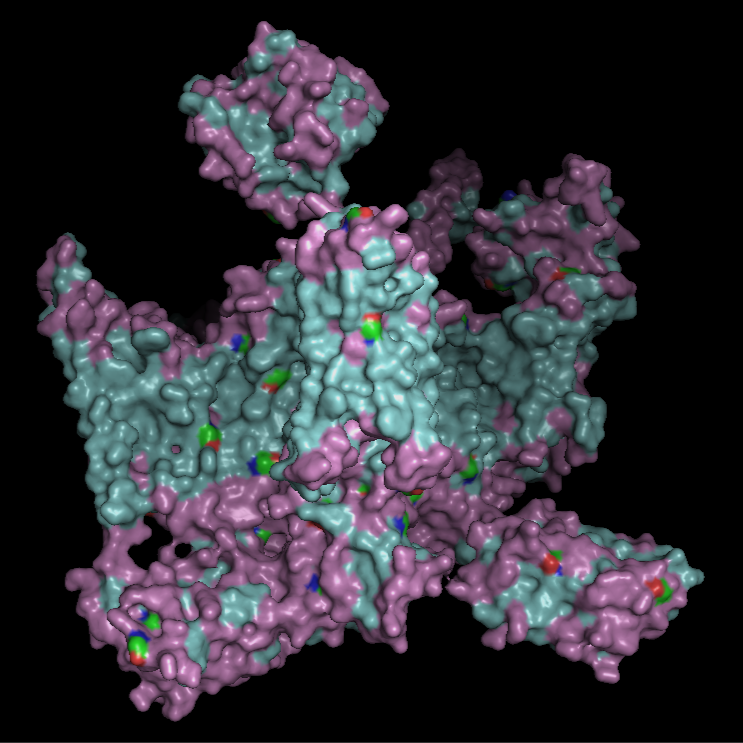
For this part, I first downloaded the .pdb file of the Nav1.7-beta1-beta2 complex from RCSB PDB. I decided to include the entire structure, not just chain A, so that we can see the protein’s main form.
After downloading the file, I went to PyMOL and loaded it there.
Cartoon Visualisation
*Chain A is colored in cyan, B in magenta, and C in yellow.
Ribbon Visualisation
*Chain A is colored in cyan, B in magenta, and C in yellow.
Ball and Stick Visualisation
*Chain A is colored in cyan, B in magenta, and C in yellow.
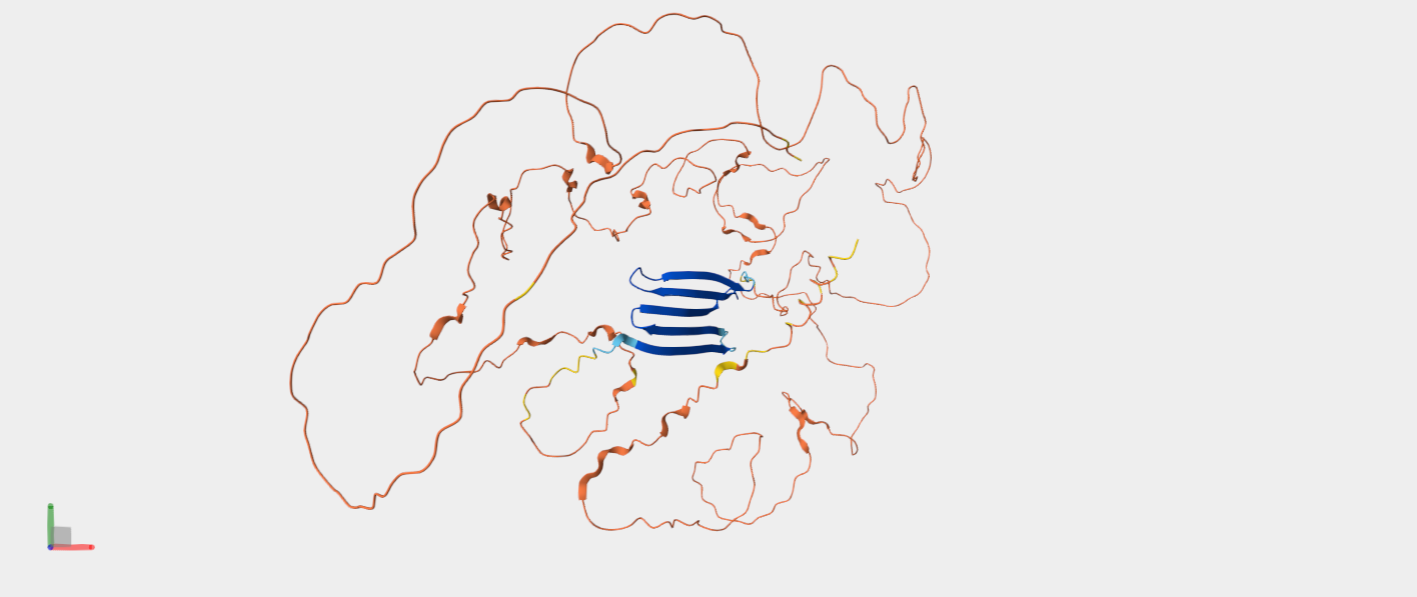
α-helices vs. β-sheets
*α-helices are colored in cyan, β-sheets in magenta, and loops in yellow.
The quaternary structure has a significantly greater number of α-helices than β-sheets. Most of the helices are concentrated in chain A, whereas the majority of the β-sheets are located in chains B and C.
Hydrophobic vs Hydrophilic Residue
*Hydrophobic residues are colored in cyan and hydrophilic in magenta.
Hydrophobic residues are predominantly located in the interior of the complex and at inter-chain interfaces, forming a stabilizing core. In contrast, hydrophilic residues are more frequently exposed on the outer surface. The central region of the protein displays a substantial hydrophobic surface, which is consistent with its role as a transmembrane protein, as this region is embedded within the lipid bilayer, whose interior is hydrophobic.
Surface Visualisation
The surface view shows that the protein has a roughly cross-like shape with visible cavities. There appear to be small holes or pockets that may help with binding to the membrane. These cavities are primarily located at the same level as the hydrophobic region embedded within the membrane, which is consistent with the protein’s transmembrane nature.
Part C. Using ML-Based Protein Design Tools
For this assignment, I wanted to choose once again Pro-Resilin, the highly elastic, rubber-like structural protein found in insects that I used in Homework 2. But unfortunatly I couldn’t find its PDB page, likely since it is mostly intrinsically disordered, so there is no experimental PDB structure (only prediction of AlphaFold).
So, I chose instead human alpha-thrombin (PDB ID: 1PPB), which is a serine protease in the blood coagulation cascade (a chain reaction of clotting proteins activated after vessel damage). It converts fibrinogen into fibrin, forming blood clots that seal damaged blood vessels following injury. Here is the link to its PDB page. The structure was solved by X-ray crystallography at 1.92 Å resolution.
Since it has two chains, I’m going to choose chain B for this exercise.
C1. Protein Language Modeling
1. Deep Mutational Scans
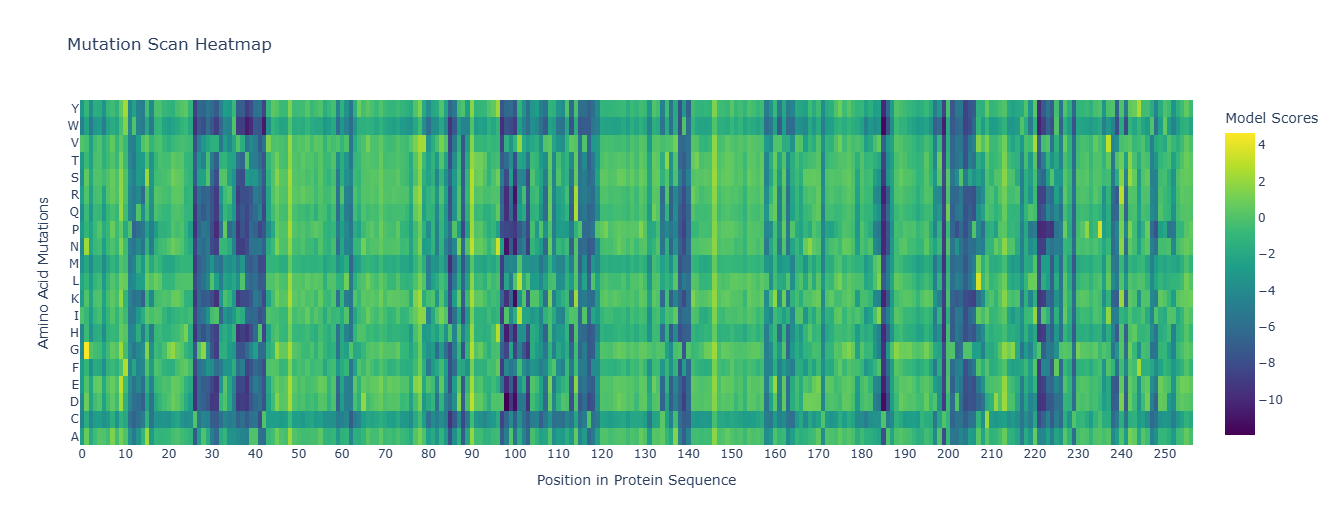
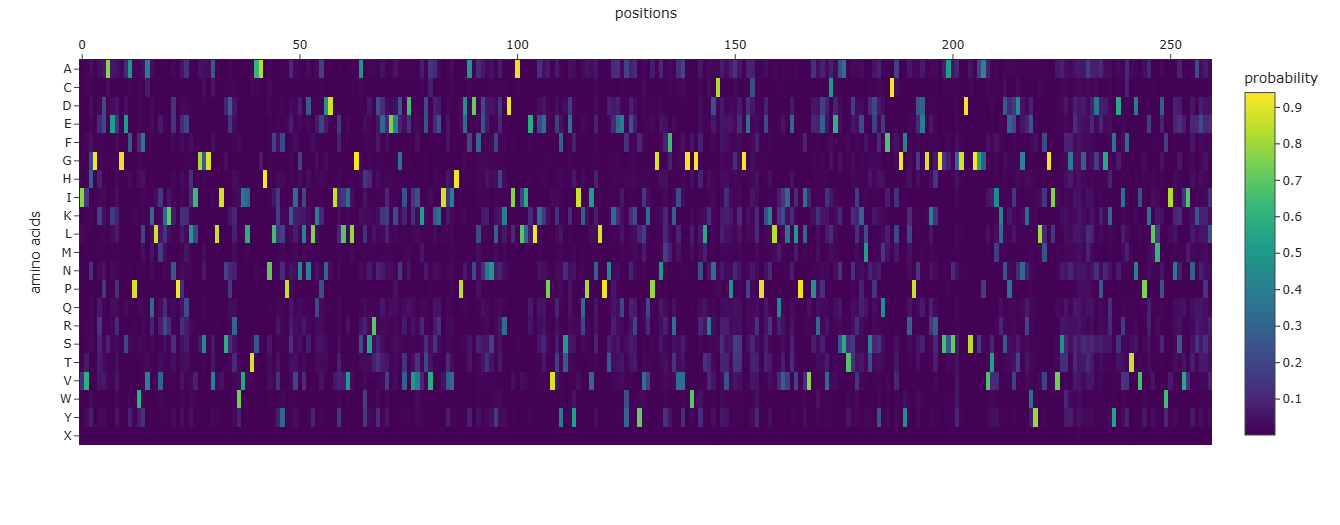
*To improve readability, I modified the x-axis to display every 10th residue instead of labeling every position.
The heatmap shows how evolutionarily “allowed” each possible mutation is at each position in thrombin, according to an LLM for proteins. One clear pattern appears at position 26, where nearly all substitutions are strongly disfavored by the model (dark colors), indicating that this residue is likely structurally important and evolutionarily constrained. Interestingly, substitution to cysteine is comparatively less unfavorable than other mutations. This may suggest that a cysteine at this position could sometimes be tolerated, potentially due to structural compatibility or possible disulfide stabilization in related proteins.
2. Latent Space Analysis
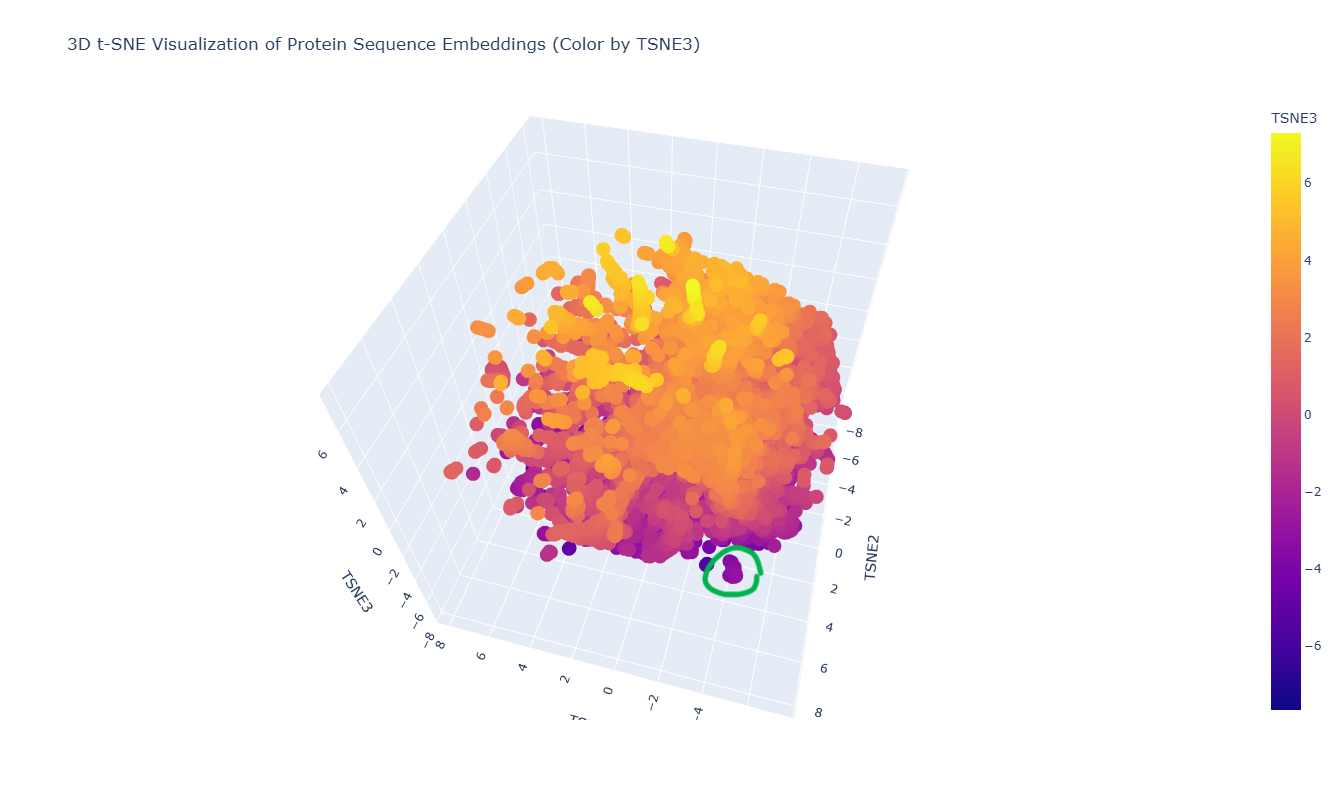
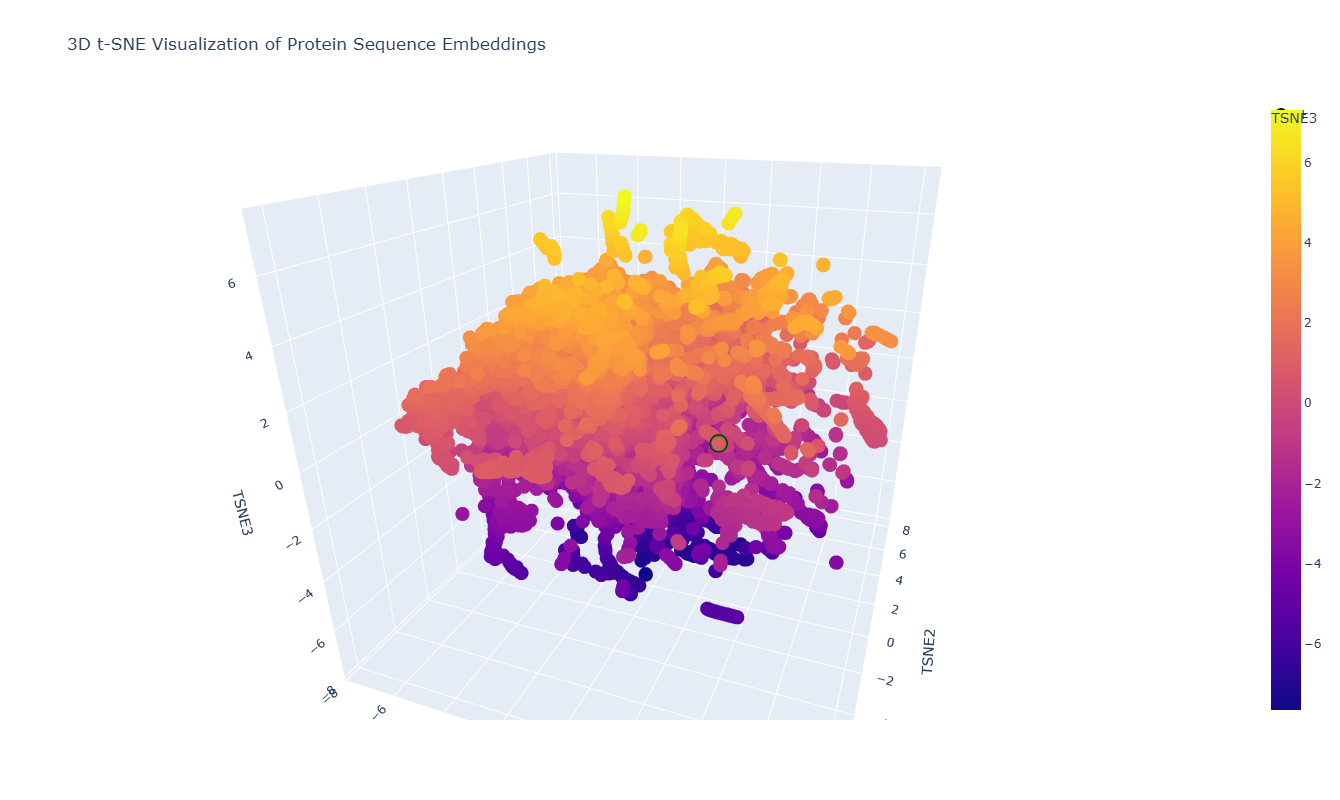
The 3D t-SNE visualization represents protein sequence embeddings generated by ESM2 and reduced to three dimensions. Each point corresponds to a protein domain from the SCOPe/ASTRAL dataset, and the spatial proximity between points reflects similarity in their learned embedding representations. Because t-SNE preserves local relationships, nearby points are expected to correspond to proteins that share sequence or structural similarities.
At first glance, the embedding space appears as one large, dense cluster rather than clearly separated groups. This suggests that many of the proteins in the dataset occupy a continuous similarity space rather than forming sharply distinct families. However, upon zooming in, smaller local neighborhoods become visible, indicating finer-grained organization within the global cluster.
For example, in the distinct local cluster highlighted in green in the figure, the neighboring proteins include:
Hypothetical protein NMA1147 (Neisseria meningitidis)
PhoU homolog TM1734 (Thermotoga maritima)
Although their annotations differ, these proteins are all bacterial in origin and belong to related structural or metabolic functional classes. Their proximity in embedding space suggests that ESM2 captures shared structural folds or evolutionary relationships beyond simple sequence identity. Even the hypothetical protein may share structural motifs with annotated metabolic proteins, explaining its placement within the same neighborhood.
Overall, the latent space organization indicates that ESM2 embeddings encode biologically meaningful information: proteins with similar structural domains or evolutionary backgrounds tend to cluster locally, even if their functional annotations are not identical. The continuous nature of the embedding space further reflects the gradual evolutionary relationships between protein families rather than strictly discrete categories.
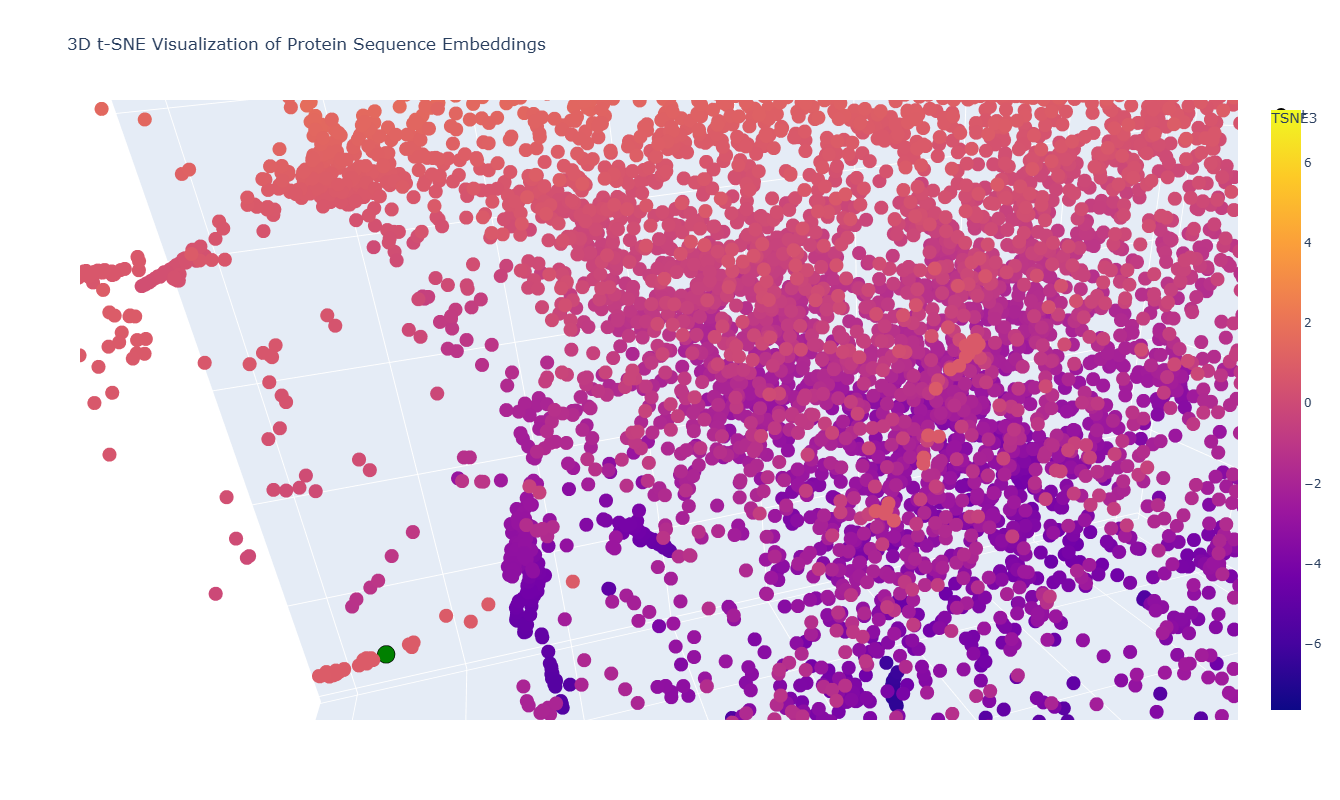
In the 3D t-SNE map, thrombin (highlighted in green in the figure) is located within a distinct local cluster rather than being isolated.
On closer inspection, this neighborhood includes proteins such as:
This makes sense because thrombin is a trypsin-like serine protease, and these neighboring proteins share the same catalytic fold and similar active-site architecture. Their proximity in the embedding space suggests that the model captures underlying structural and evolutionary similarities between these proteases.
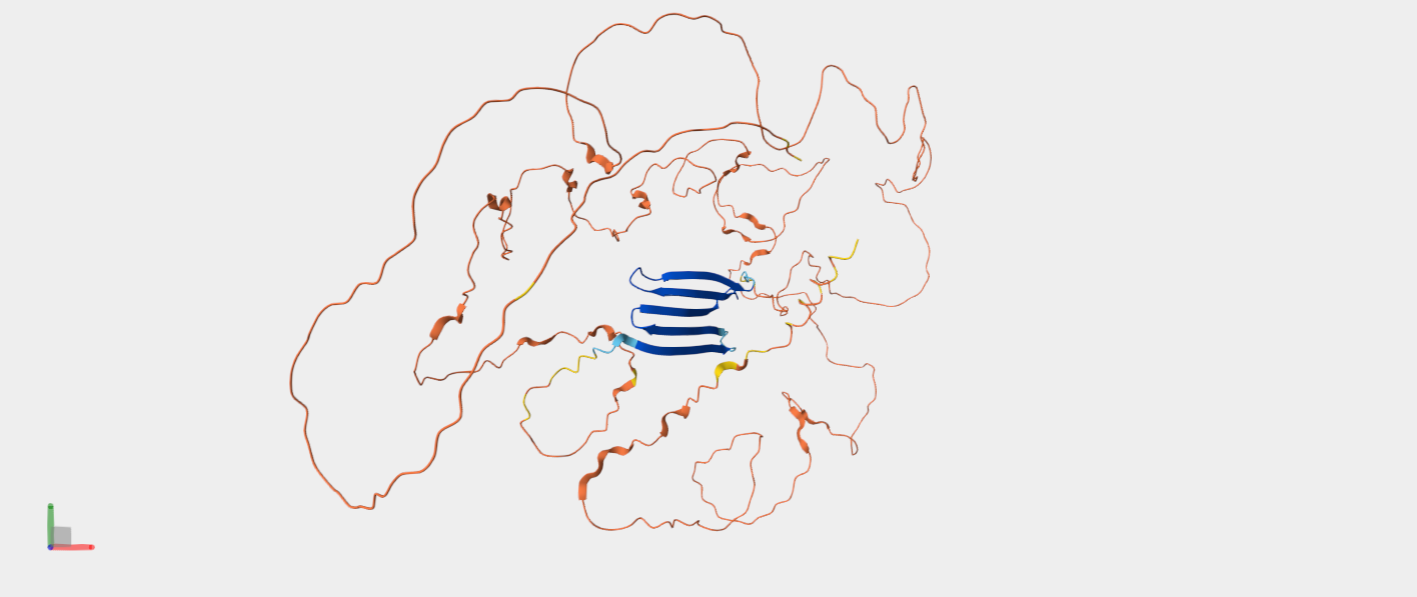
C2. Protein Folding
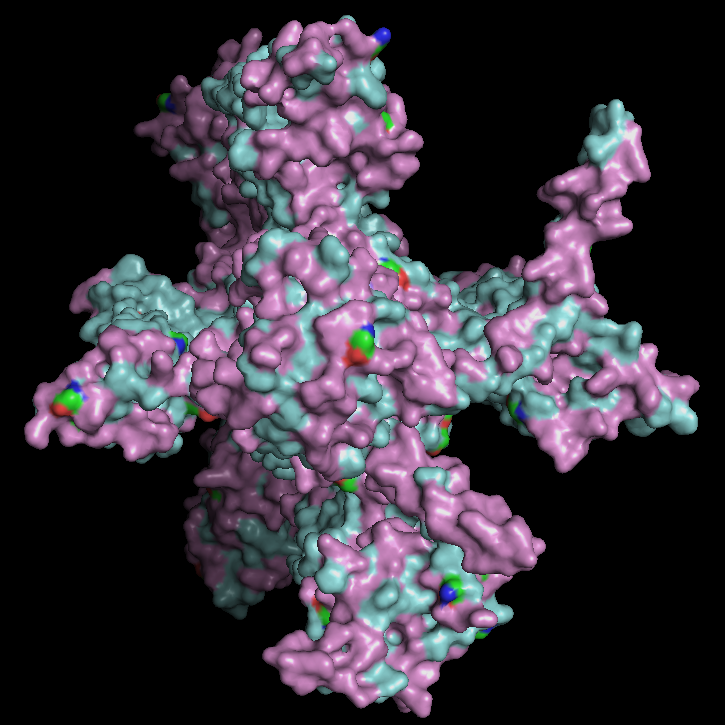
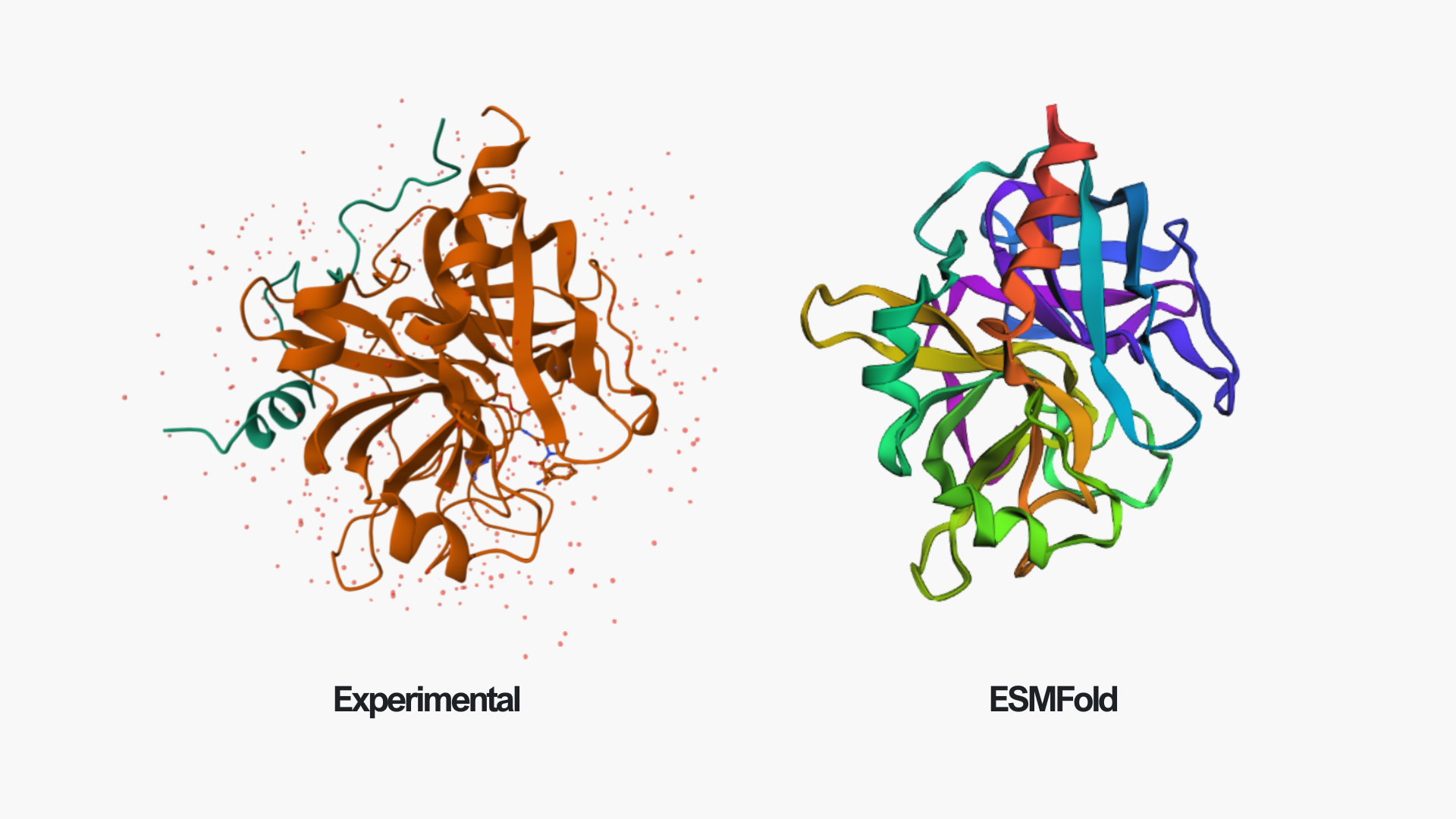
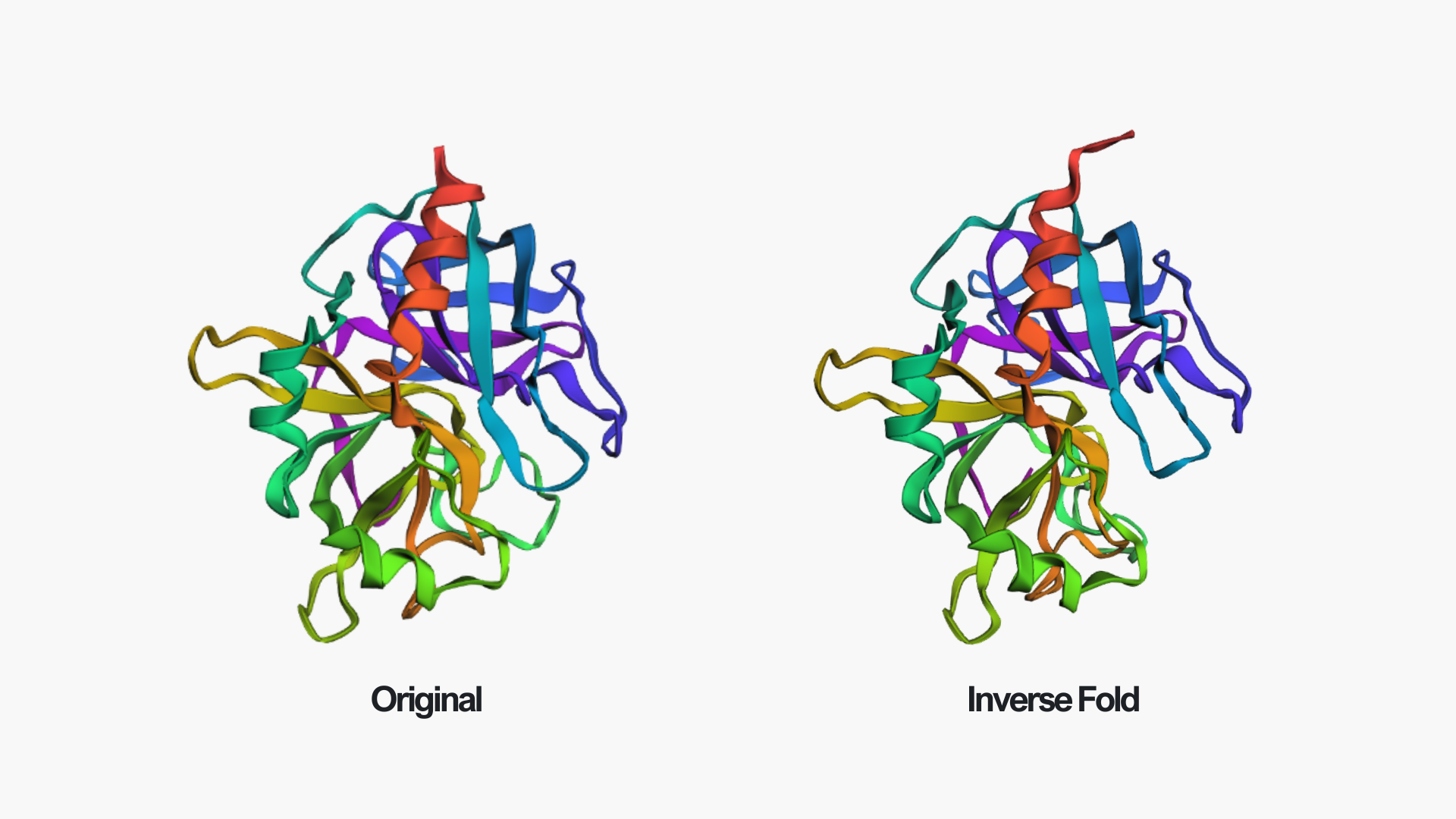
In the above picture, you can see the experimental structure on the left and the ESMFold structure on the right. They look very similar overall. The main difference is chain A (colored in green in the experimental structure), which I did not include in the ESMFold prediction. This likely also contributes to some small differences, since removing a chain can slightly affect the overall folding and interactions. However, the overall fold and structure are highly similar between the two models.
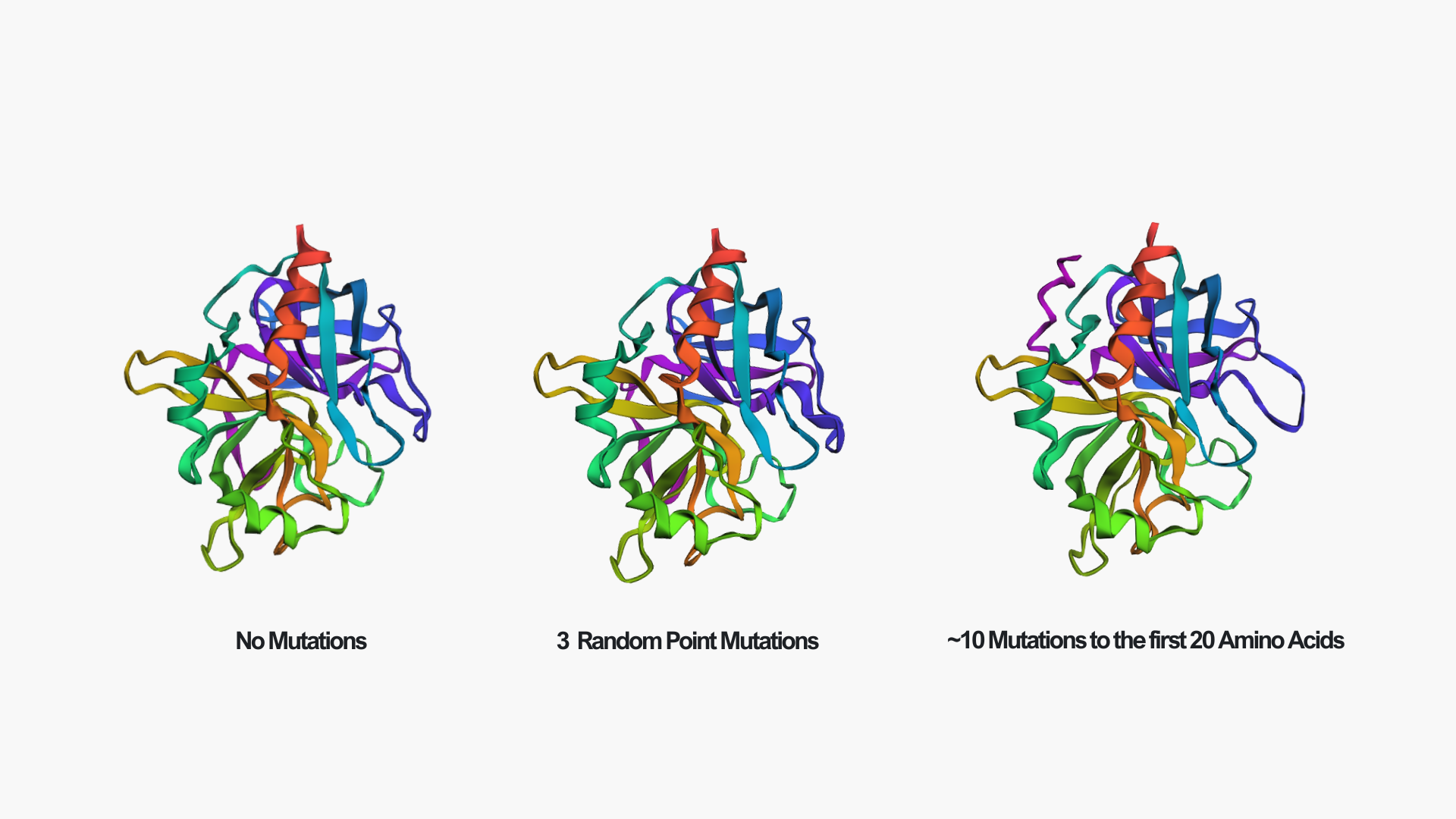
Creating three random point mutations (replacing one amino acid with another) led to the structure shown above. The mutated structure is incredibly similar to the original one. These few mutations did not have a significant effect on the overall fold of the protein, suggesting that thrombin is structurally resilient to small sequence changes.
After making ~10 mutations in just the first 20 amino acids, it seems the structure started to be affected. You can see this clearly in the mutated model by the extra pink strand/loop on the top left, and the overall shape looks a bit more distorted compared to the no-mutation structure. So thrombin seems pretty resilient to a few point mutations, but once you start mutating a larger chunk (specifically at the N-terminus), the predicted fold begins to change noticeably.
C3. Protein Generation
Here is the predicted sequence I got running the ProteinMPNN:
IVGGRDAAPGEAPWFAQLIRKNPRQLIGSGALISDTWVITAAHNLYYPKINLNLKPDDIFIRLGALSRTEVEEGVEVIKTVKEIVIHPDYDAENNLDKDIALIKLEEPVEYSEYIRPIDLPNKEIAEALLKPGNMGRVVGWGNLRNCSVPGSGPCLPTKAQVLEVPLVPREVCEASTDRKMTDNQFCAGFLPGEGKRGSACGGDSGGAFTILNPVDGRWYLMGIVSWGEXGCDDPGKFDIFTNVYKLMPWIDSVINAEPE
ProteinMPNN generated a redesigned thrombin sequence that is similar in length and retains many motifs typical of trypsin-like serine proteases (including conserved glycines/cysteines and the overall pattern of hydrophobic vs. polar residues). This makes sense because ProteinMPNN is designing a sequence to fit the same backbone coordinates. One position was returned as an “X” (unknown token) (I added this option since the code raised an error every time I ran it), which would need to be replaced with a standard amino acid before using the sequence experimentally. For the next part, I will replace X with A since it is conservative.
From the heatmap, most positions show one amino acid with a very high probability (bright yellow), while the rest are close to zero. This means ProteinMPNN is quite confident about the residue choice at many positions, suggesting the backbone strongly constrains the sequence.
There are also regions where the probabilities are more spread out (more green/blue across multiple residues). These positions are likely more tolerant to mutation and structurally less constrained.
Comparing the predicted sequence to the original thrombin sequence, many residues differ. However, the overall pattern still looks like a serine protease (conserved glycines, cysteines, and hydrophobic residues in core regions).
After folding the ProteinMPNN-designed sequence with ESMFold, the predicted structure is almost identical to the original thrombin structure. The overall fold, including the β-sheet core and surrounding helices, is preserved. Only minor local differences can be seen, for example near the edge of the red helix and in some loop regions.
This suggests that the redesigned sequence is fully compatible with the original backbone and that ProteinMPNN successfully generated a sequence that maintains the thrombin fold.
Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design
Part 1: Generate Binders with PepMLM
Searching the human SOD1 (P00441) page on UniProt, I found its amino acid sequence:
*Pay attention that the actual place of the Alanine is the 5th, maybe we should start counting at 0.
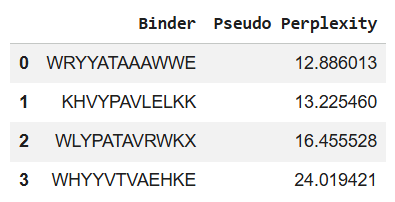
Using the PepMLM Colab, I generated the following 4 peptides of length 12 aa, which are conditioned to the mutant SOD1 sequence:
*You can see the perplexity scores that indicate PepMLM’s confidence in the binders in the table above too.
To these four peptides, we now add the known SOD1-binding peptide FLYRWLPSRRGG for comparison (control).
Part 2: Evaluate Binders with AlphaFold3
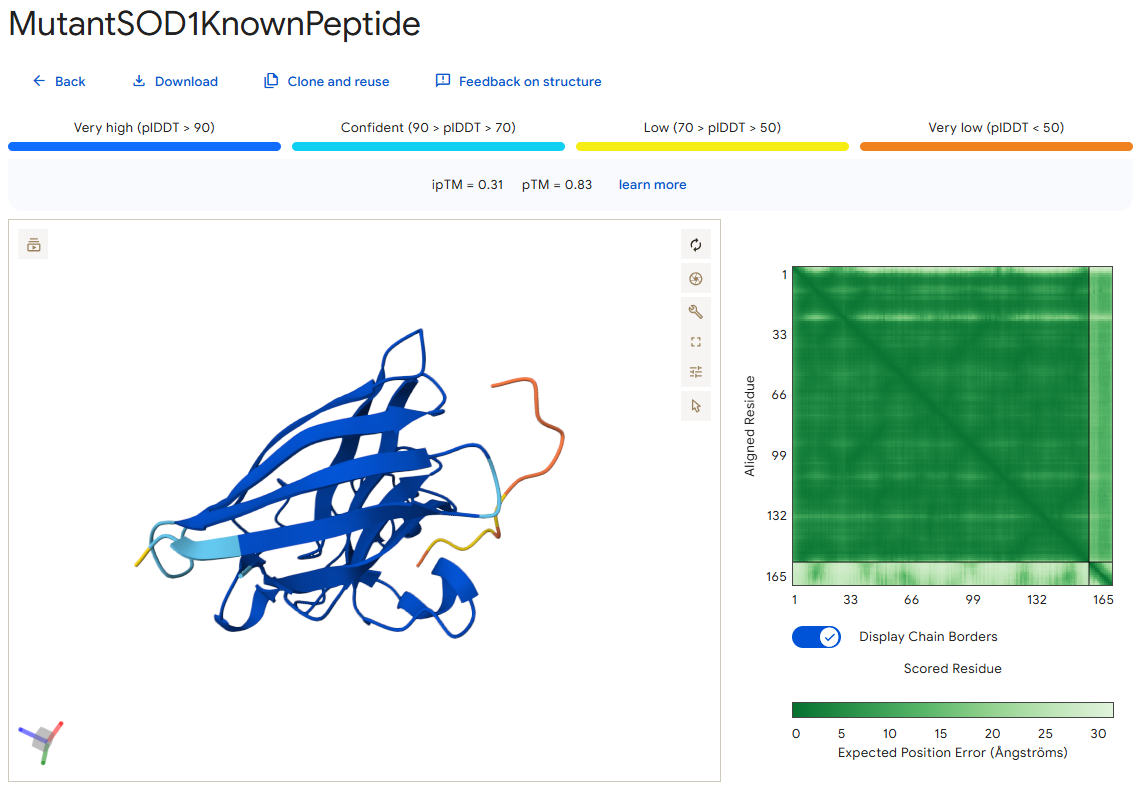
I then moved to the AlphaFold Server, where I submitted, for each peptide separately, the mutant SOD1 sequence, followed by the peptide sequence as a separate chain to model the protein-peptide complex.
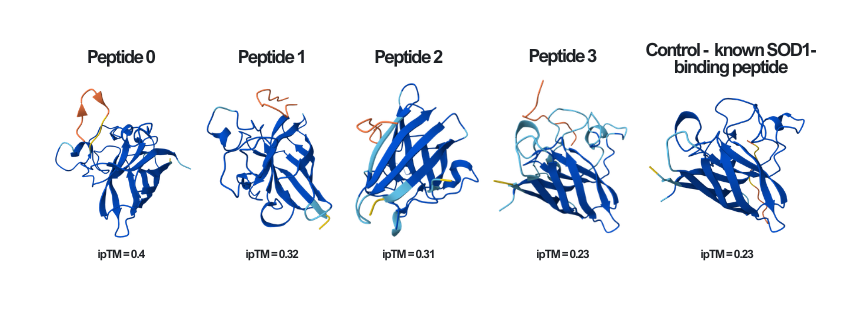
Here you can see the results of the modeling of all of the different peptides (including the known SOD1-binding peptide - the control), as well as their ipTM score, side by side:
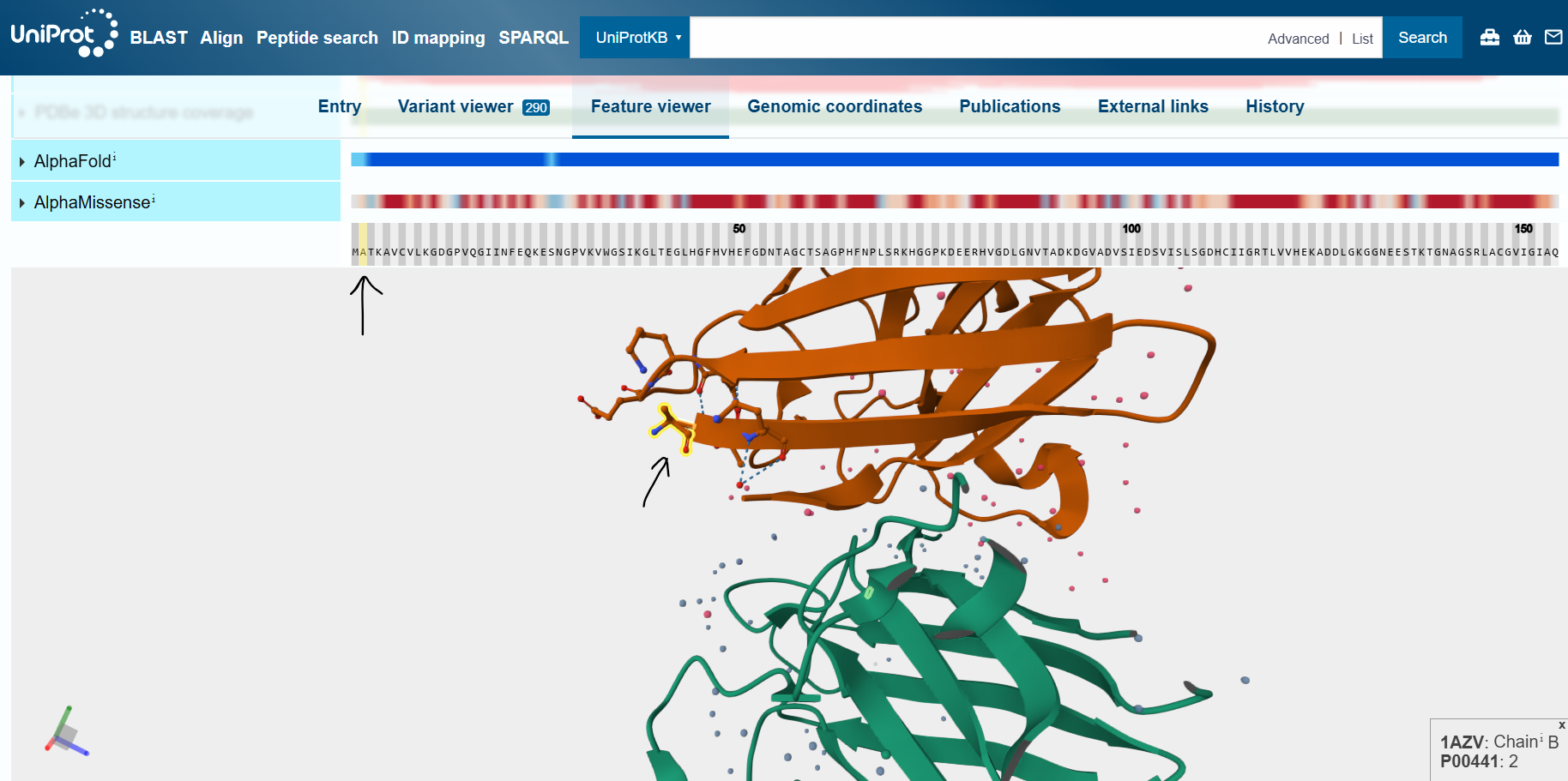
By inspecting the wild-type human SOD1 protein in UniProt’s feature viewer feature, I identified the N- and C-termini of the mutant protein.
The ipTM (Interface Predicted Template Modeling) score in AlphaFold measures the accuracy of the predicted relative positions of the subunits within the complex. Values higher than 0.8 represent confident, high-quality predictions, while values below 0.6 suggest a likely failed prediction. ipTM values between 0.6 and 0.8 are a gray zone where predictions could be correct or incorrect.
In the following pictures, the peptides are colored in orange, and the mutant human SOD1 in blue.
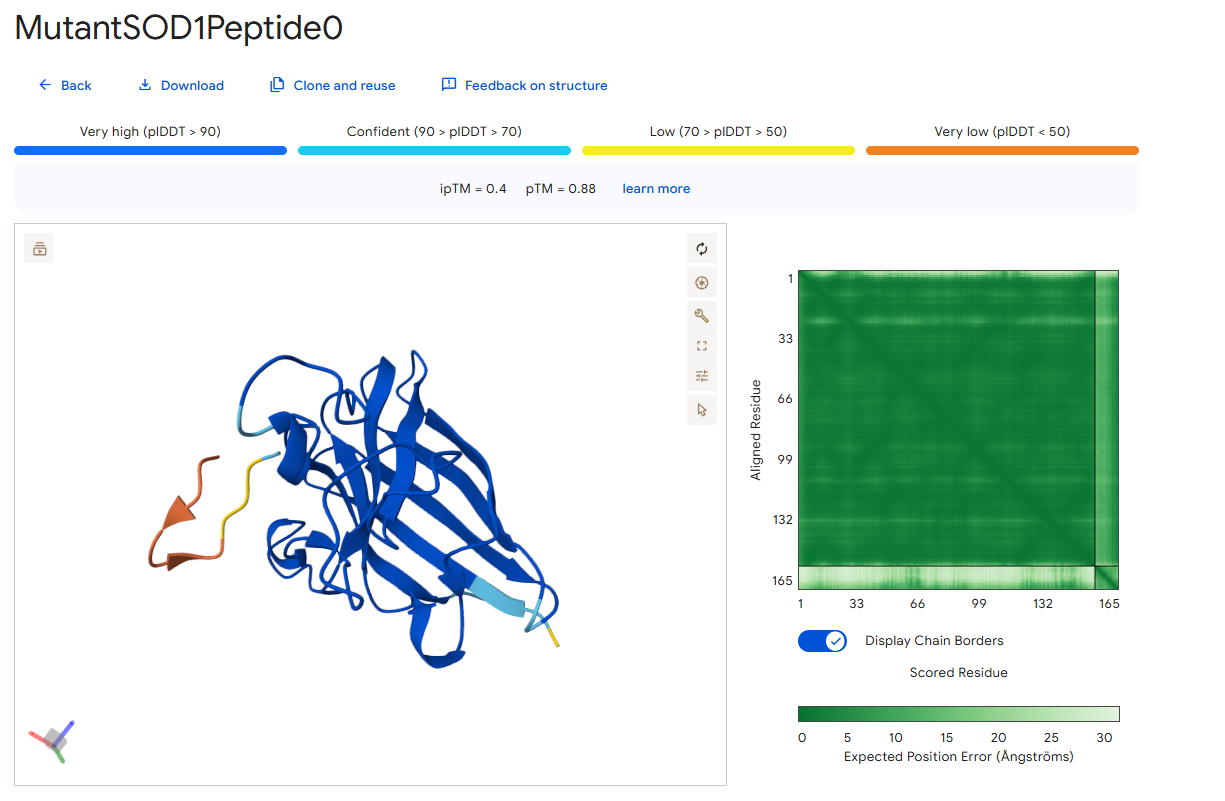
Peptide 0
According to the AlphaFold simulation, the peptide does not bind close to the N-terminus, it doesn’t engage with the β-barrel region, doesn’t approach the dimer interface, and seems to be surface-bound.
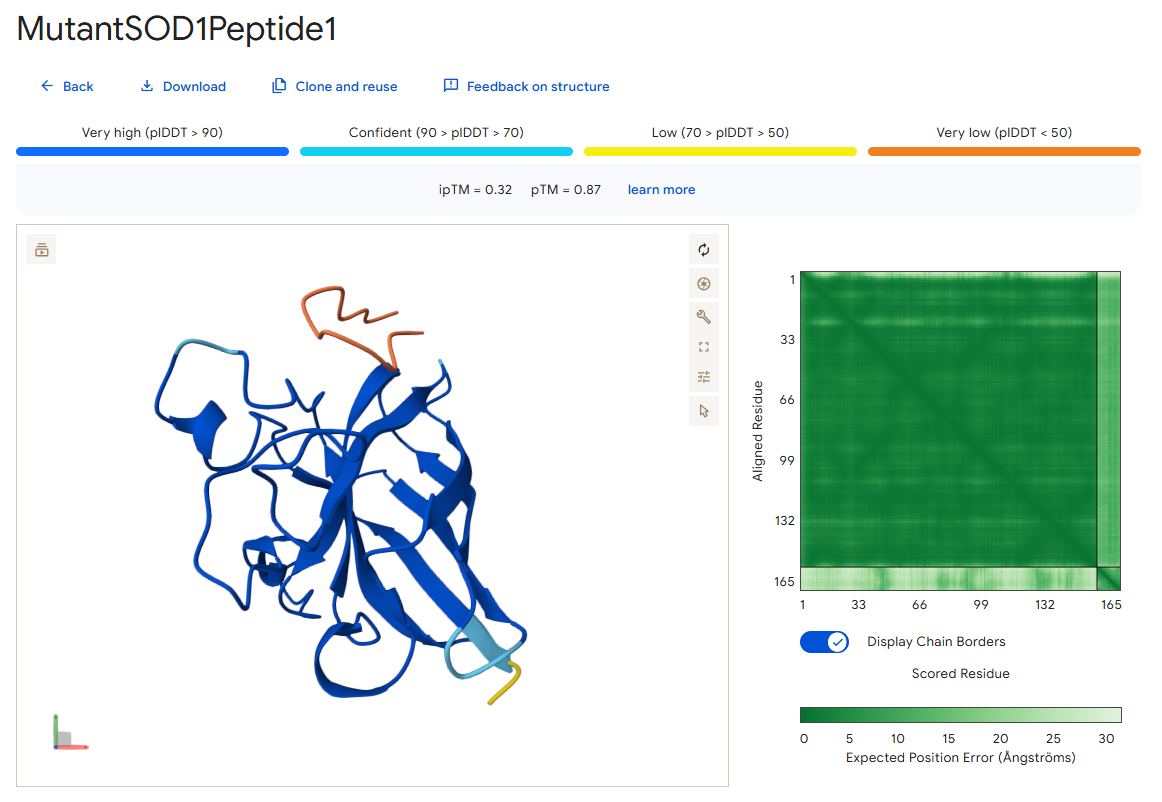
Peptide 1
According to the AlphaFold simulation, the peptide does not bind close to the N-terminus, it engages with the β-barrel region on the side, doesn’t approach the dimer interface (very far from it), and seems to be surface-bound.
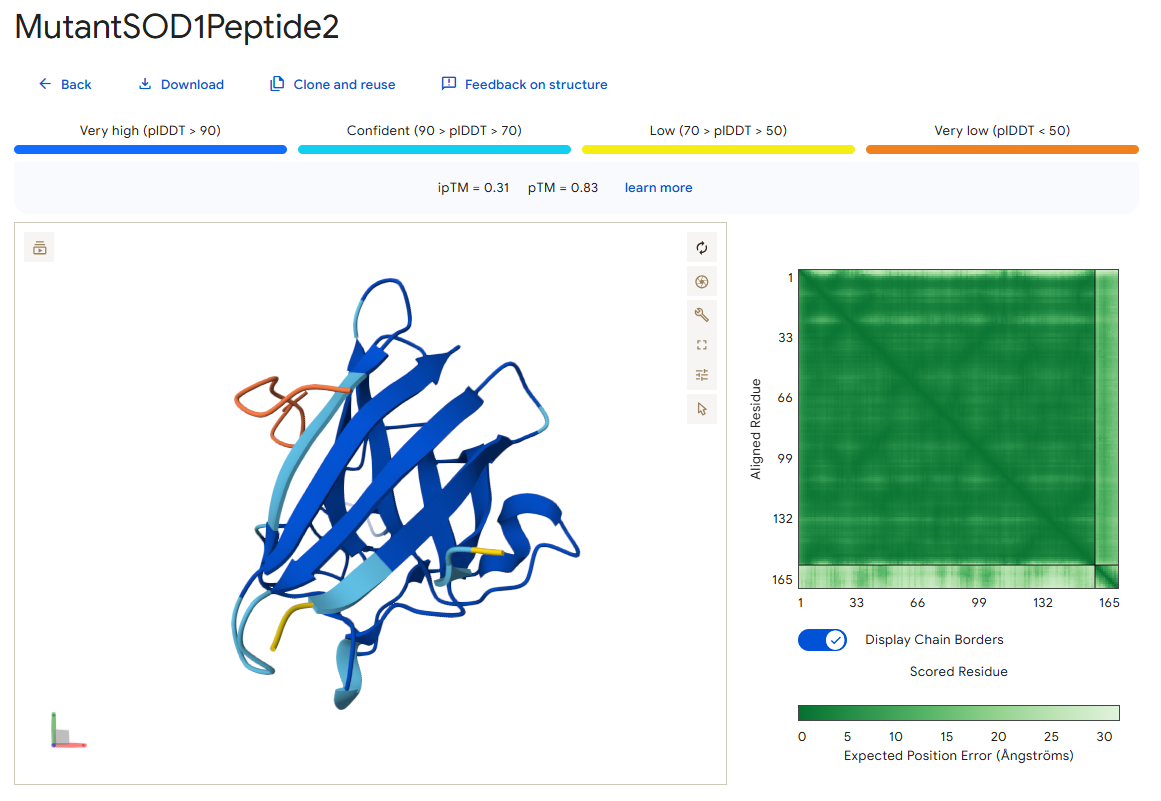
Peptide 2
According to the AlphaFold simulation, the peptide binds pretty close to the N-terminus (at least compared to the previous two peptides), it does engage with the β-barrel region, doesn’t approach the dimer interface, and seems to be very lightly buried.
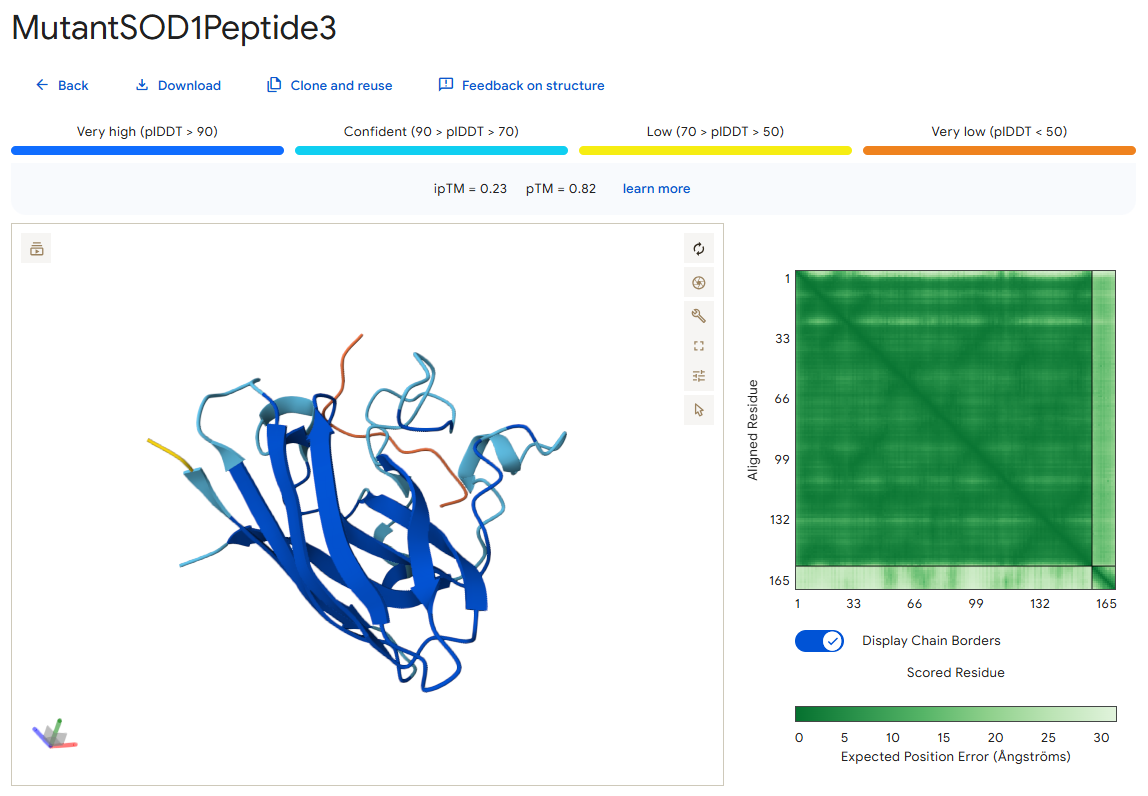
Peptide 3
According to the AlphaFold simulation, the peptide does not bind close to the N-terminus, it doesn’t engage with the β-barrel region, doesn’t approach the dimer interface, and seems to be very lightly buried.
The known SOD1-binding peptide
According to the AlphaFold simulation, the peptide does not bind close to the N-terminus, it engages with the β-barrel region from the “hole” part of it, doesn’t approach the dimer interface, and seems to be partially buried.
Surprisingly, except for peptide 3, all of the peptide sequences generated by PepMLM had higher ipTM values than the known peptide. For example, peptide0 reached an ipTM value of 0.4 compared to 0.31 for the known peptide. It is impressive that within a few minutes it was possible to identify peptides that might be better candidates for drug development than the currently known ones. However, all ipTM scores were below 0.6, meaning AlphaFold is not very confident in these predictions. This is also the case for the known SOD1-binding peptide, which we know binds experimentally. Therefore, the accuracy of these predictions cannot be confirmed without experimental testing.
Additionally, since the peptides bind far from the dimer interface, each SOD1 dimer could potentially bind two peptides, one on each chain. Moreover, because all peptides except peptide2 bind far from the N-terminus, they may not be specific to the mutant human SOD1 protein.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
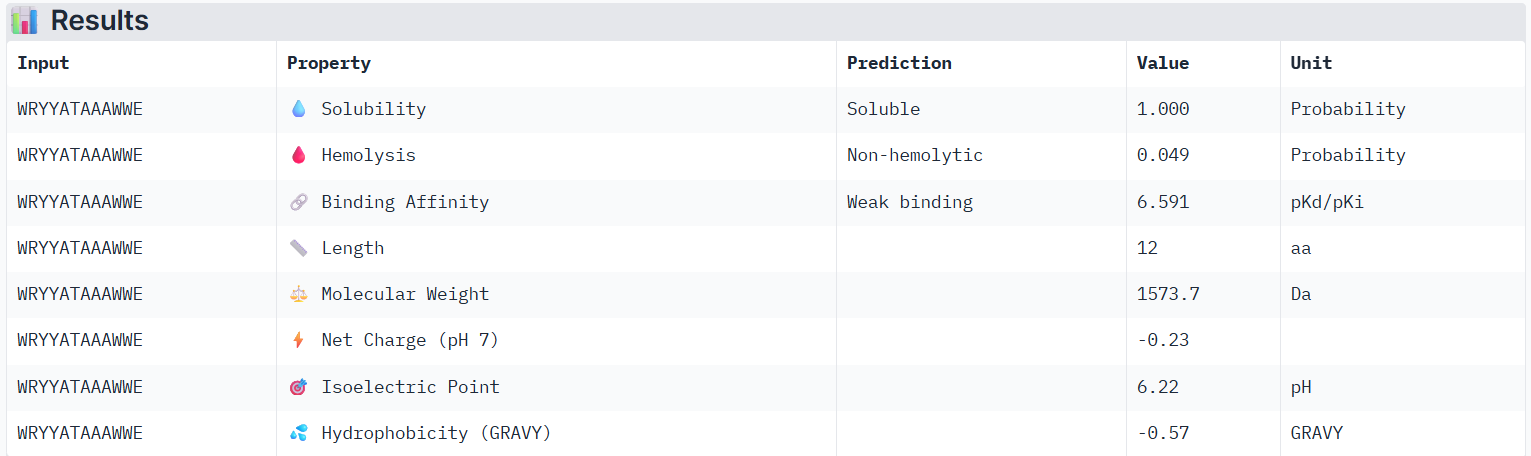
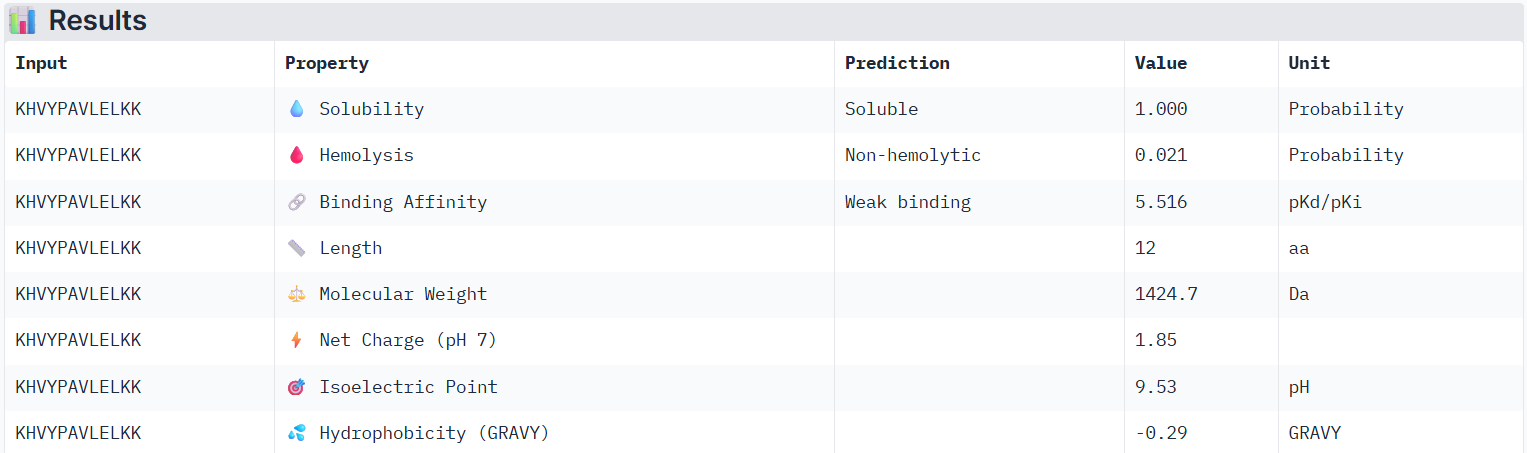
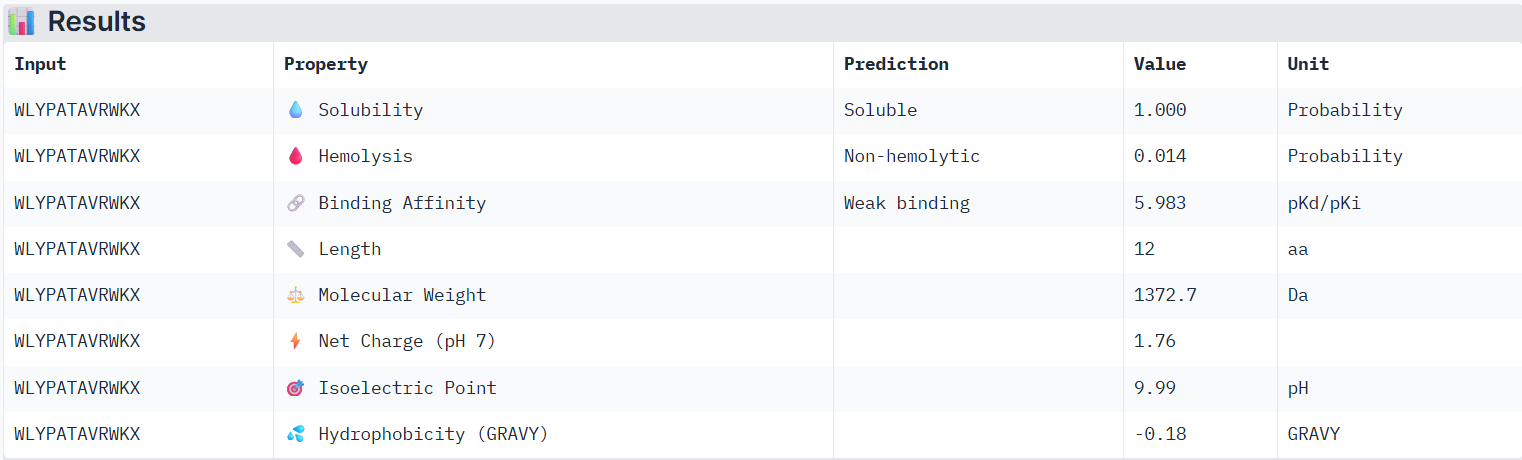
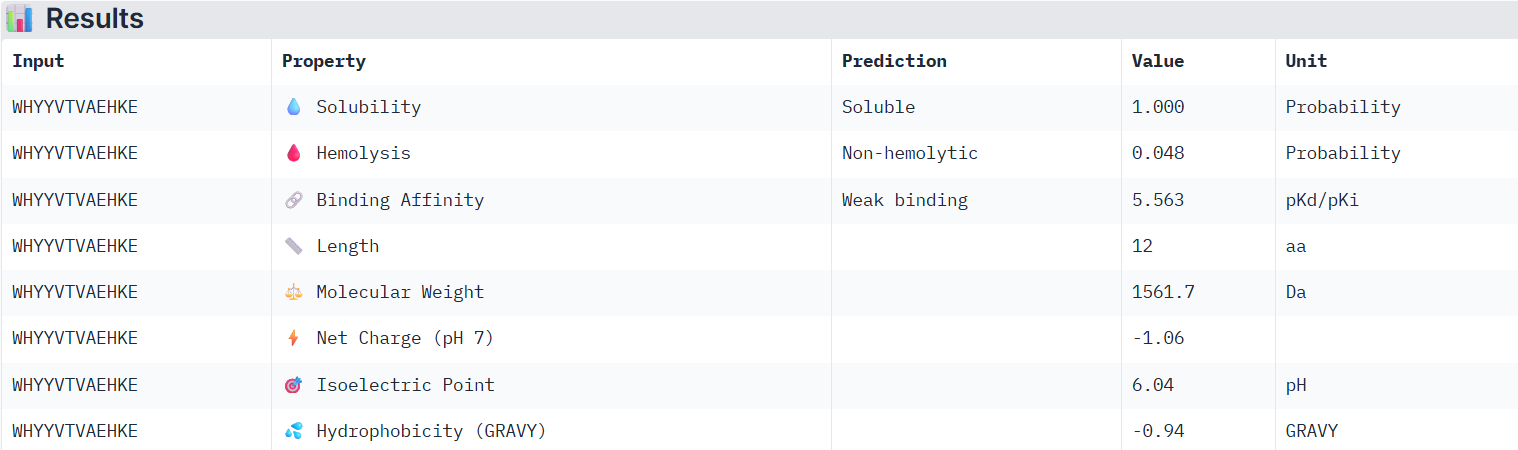
Using PeptiVerse, I evaluated the therapeutic properties of all of the peptids, and here are the results:
Peptide 0
According to PeptiVerse’s prediction, peptide0 has a weak binding affinity, even though it had the highest ipTM score (0.4) in AlphaFold. It is also soluble and is non-hemolytic. A non-hemolytic peptide is a peptide that does not cause lysis of red blood cells, indicating that it is less likely to damage human cell membranes and therefore may have lower toxicity.
Peptide 1
According to PeptiVerse’s prediction, peptide1 has a weak binding affinity, even weaker than peptide0, even though it had the second-highest ipTM score (0.32) in AlphaFold. It is also soluble and is non-hemolytic.
Peptide 2
According to PeptiVerse’s prediction, peptide2 has a weak binding affinity (it had a 0.31 ipTM score on AlphaFold). It is also soluble and is non-hemolytic.
Peptide 3
According to PeptiVerse’s prediction, peptide3 has a weak binding affinity, which matches with itslow ipTM score of 0.23 on AlphaFold. It is also soluble and is non-hemolytic.
The known SOD1-binding peptide
According to PeptiVerse’s prediction, the known peptide has a weak binding affinity (it had an ipTM score of 0.31 in AlphaFold), which is even weaker than the weak affinity predicted for PepMLM peptide0. This again suggests that we cannot fully determine whether a peptide will bind well to the human SOD1 protein until it is tested experimentally. The peptide is also predicted to be soluble and non-hemolytic.
For the next part, I will continue working with peptide0 WRYYATAAAWWE. Although it has an ipTM score below 0.6 (0.4) and is predicted to have weak binding affinity to the mutant human SOD1 protein, it still shows the best values among the generated peptides and even performs better than the known SOD1-binding peptide.
Part 4: Generate Optimized Peptides with moPPIt
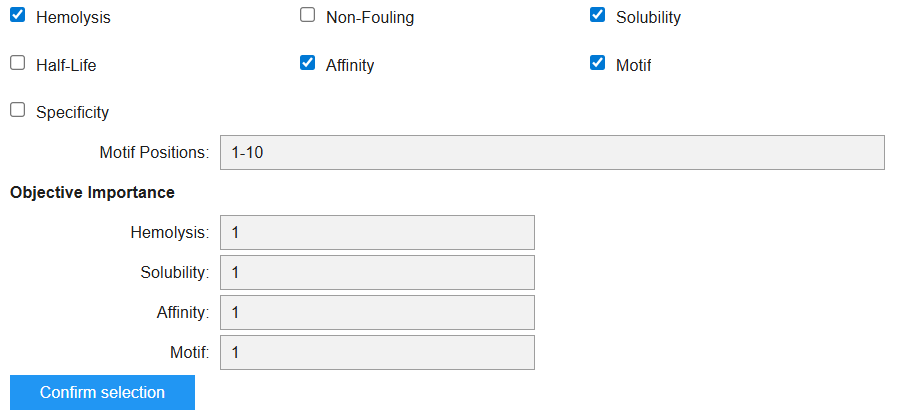
In the moPPIT Colab, I pasted the mutated SOD1 sequence, chose a peptide length of 12 amino acids, and set the objectives and weights for the following properties:
I then ran the model and it generated the following peptide:
Looking at the objectives I chose and their order, I was able to create the following table:
Value
Property
Meaning
0.9349
Hemolysis
High probability of being non-hemolytic (???)
0.75
Solubility
Predicted fairly soluble
5.72
Affinity (pKd/pKi)
Weak binding
0.6226
Motif score
Moderate match to the motif constraint
The moPPIT peptides differ from the PepMLM peptides because moPPIT generates peptides using controlled multi-objective optimization, while PepMLM mainly samples sequences that could bind the target protein. The PepMLM peptides were all predicted to be soluble, non-hemolytic, and to have weak binding affinity. In comparison, the moPPIT peptide also satisfies these constraints but shows lower predicted solubility, suggesting a trade-off between optimizing different properties such as motif matching and binding.
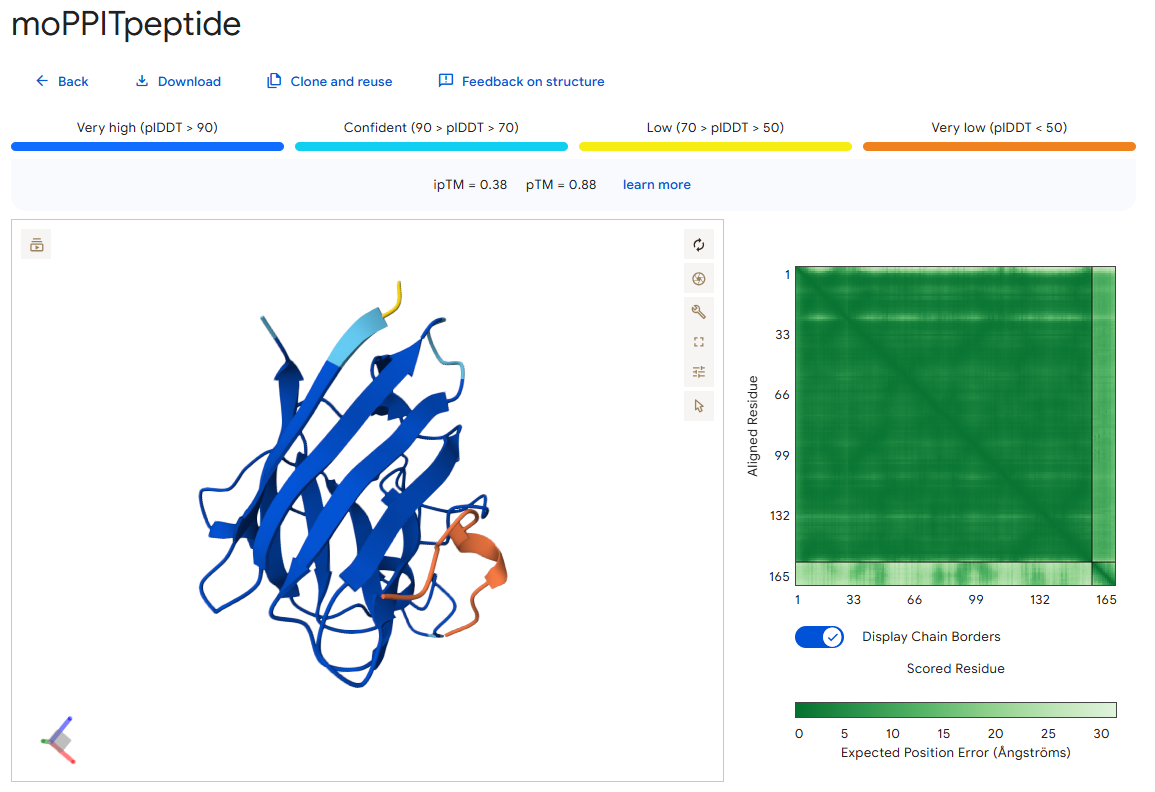
I also used AlphaFold to predict the binding of this peptide with the mutant SOD1 protein, and this is what I got:
As you can see, it has an ipTM score of 0.38 and does not appear to bind close to the N-terminus, which was one of the objectives during its generation.
Before advancing these peptides toward clinical studies, I would first evaluate them computationally using tools such as AlphaFold and PeptiVerse to assess their structure and predicted binding affinity. Next, I would test them in in vitro experiments to measure binding strength, toxicity, and stability. If the results are promising, the peptides could then be tested in in vivo experiments to evaluate safety, pharmacokinetics, and therapeutic effectiveness.
Part C: Final Project: L-Protein Mutants
For this part of the assignment, I was not sure what were the actual tasks we had to do, so I hope I did everything we needed to do:)
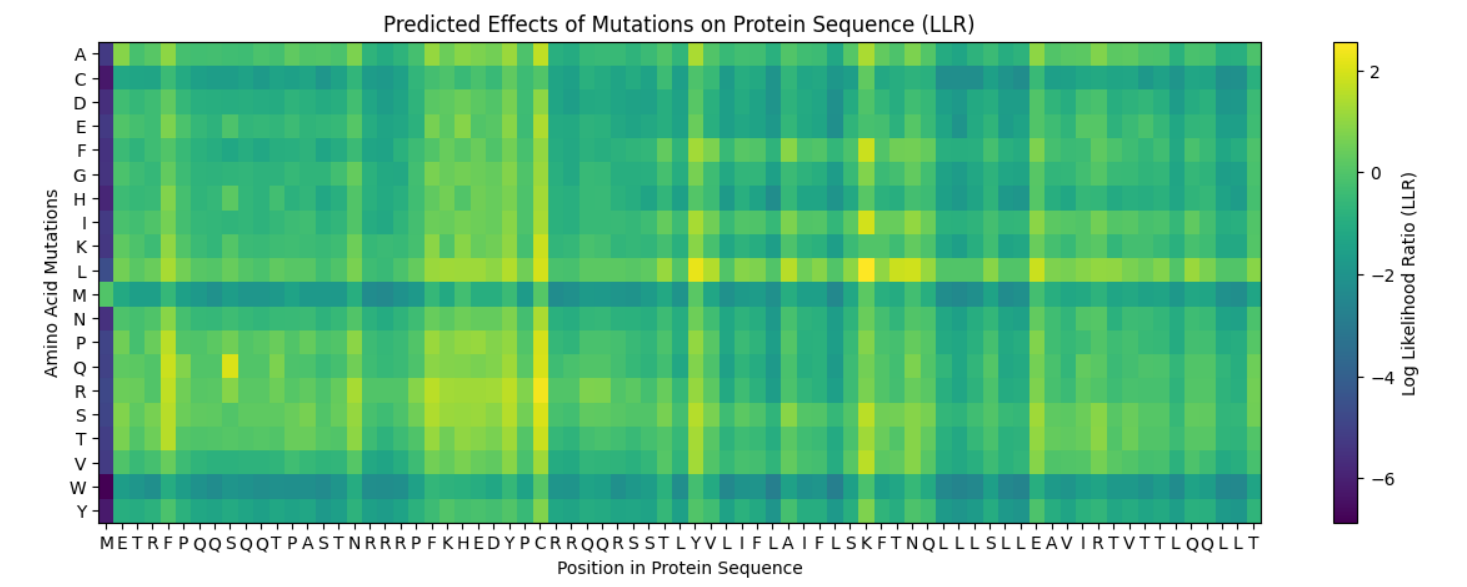
L-Protein Engineering | Option 1: Mutagenesis
I ran the given notebook and got the following heatmap, representing the predicted effects of mutations on the lysis Protein Sequence:
Week 6 HW: Genetic Circuits Part I
DNA Assembly Questions
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion High-Fidelity PCR Master Mix contains several key components that enable accurate and efficient DNA amplification. First, it includes the Phusion DNA polymerase, which is responsible for synthesizing new DNA strands. This enzyme has both 5’→3’ polymerase activity and 3’→5’ exonuclease (proofreading) activity, which allows it to correct mistakes during DNA replication and results in very high fidelity compared to standard polymerases like Taq. The mix also contains deoxynucleotides (dNTPs), which are the building blocks used by the polymerase to construct the new DNA strands. In addition, it includes an optimized reaction buffer with MgCl₂, which provides the proper chemical environment for the enzyme to function. Because all of these components are already included in the master mix, only the template DNA, primers, and water need to be added to perform the PCR.
What are some factors that determine primer annealing temperature during PCR?
The primer annealing temperature is mainly determined by the primer’s melting temperature (Tm), which depends on:
Primer length - longer primers have higher Tm
GC content - more G–C pairs increase stability and raise Tm
Salt concentration - higher ionic strength stabilizes primer–template binding and increases Tm
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Both PCR and restriction enzyme digests can be used to generate linear DNA fragments, but they differ in how they work and when they are best used.
PCR:
Uses primers and a DNA polymerase to amplify a specific region of DNA
Can introduce mutations or modifications (e.g., through primer design)
Does not require pre-existing restriction sites
Useful when you want flexibility, to create new sequences, or to amplify a certain region
Restriction enzyme digest:
Uses enzymes to cut DNA at specific recognition sites
Produces predictable ends (sticky or blunt)
Requires that the desired cut sites already exist in the DNA
Useful when you want precise, reproducible cuts without introducing mutations
In general, PCR is preferable when you need to modify or design DNA fragments and amplify them, while restriction digests are preferable when you want to cut DNA in a precise and controlled way using existing sequences.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To be appropriate for Gibson cloning, the DNA fragments need to have overlapping homologous ends. This means that the ends of the fragments to be assembled should share matching sequences, usually around 20–40 bp, so they can anneal to each other during the Gibson reaction.
You can ensure this by:
Designing PCR primers that add the correct overlap sequences to the ends of the amplified fragment
Choosing restriction sites carefully so that the digested fragments have ends compatible with each other
So overall, the main idea is to make sure the fragments have the right matching overlaps for Gibson assembly to join them correctly.
How does the plasmid DNA enter the E. coli cells during transformation?
Plasmid DNA enters E. coli during transformation when competent cells are heat shocked. The sudden temperature change causes the cell membrane to temporarily “open up,” allowing the DNA to enter the cell.
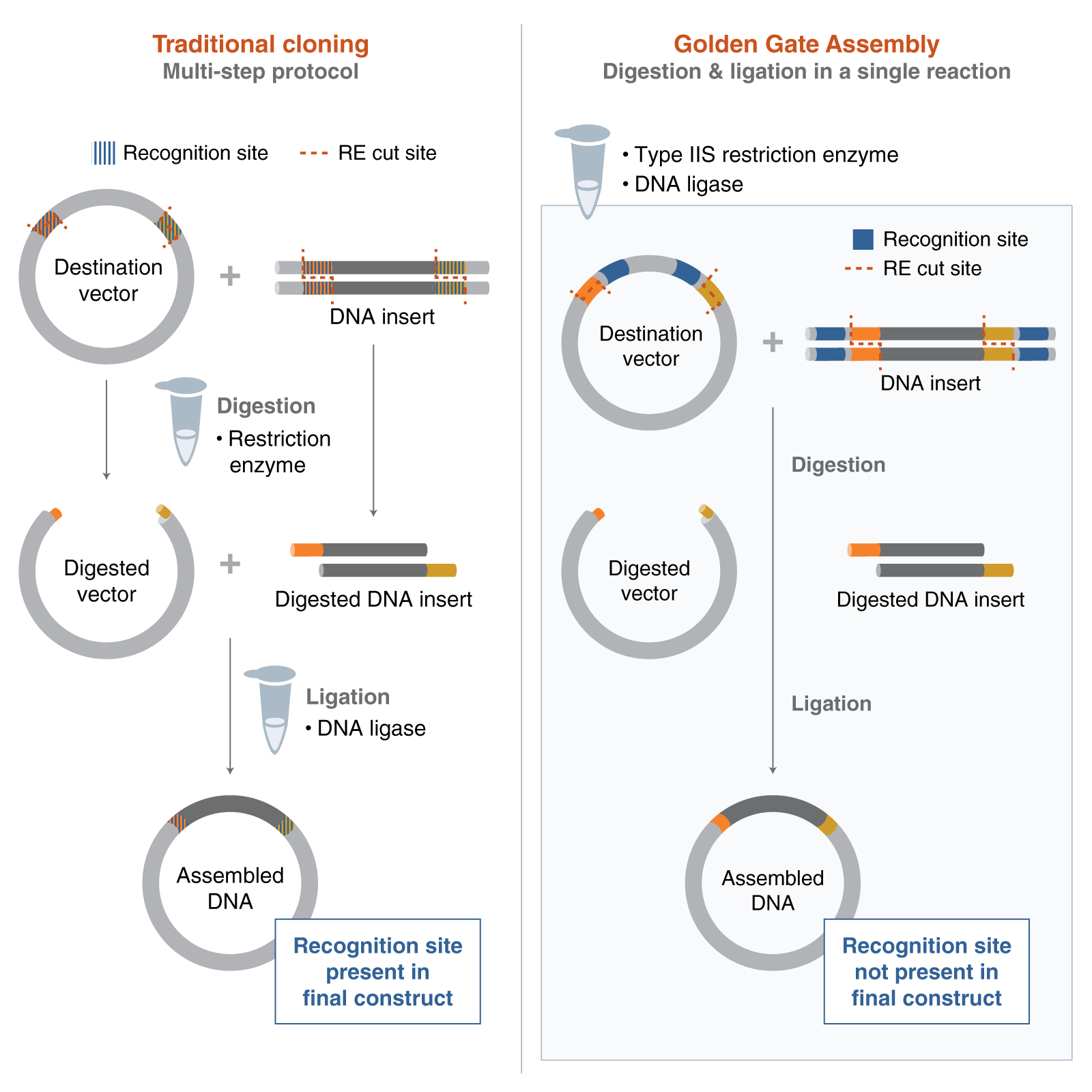
Describe another assembly method in detail (such as Golden Gate Assembly)
Golden Gate Assembly is a DNA assembly method that uses Type IIS restriction enzymes and DNA ligase to join multiple fragments in a single reaction. Unlike standard restriction enzymes, Type IIS enzymes cut outside of their recognition site, which allows the creation of custom overhangs. These overhangs can be designed so that different DNA fragments assemble in a specific order and orientation.
In the reaction, the DNA is repeatedly cut and ligated in cycles, which increases the efficiency of correct assembly. Because the recognition sites are removed during the process, the final construct does not contain unwanted extra sequences. This makes Golden Gate especially useful for assembling multiple fragments seamlessly.
Model this assembly method with Benchling or Asimov Kernel!
Asimov Kernel
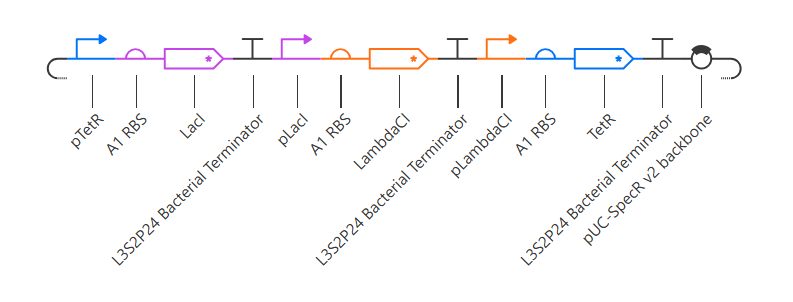
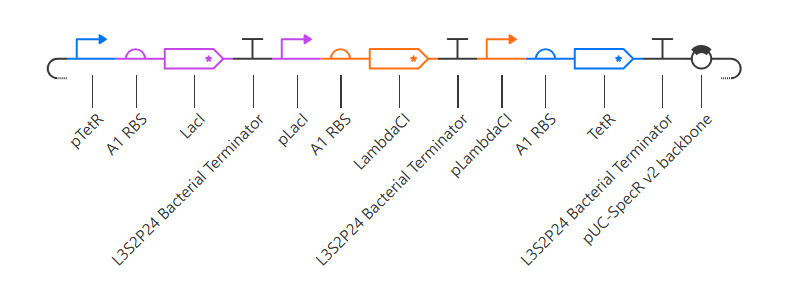
Repressilator
Here is my recreated repressilator:
Note that I used pUC-SpecR v2 backbone instead of pUC-SpecR v1 backbone, since I couldn’t find it.
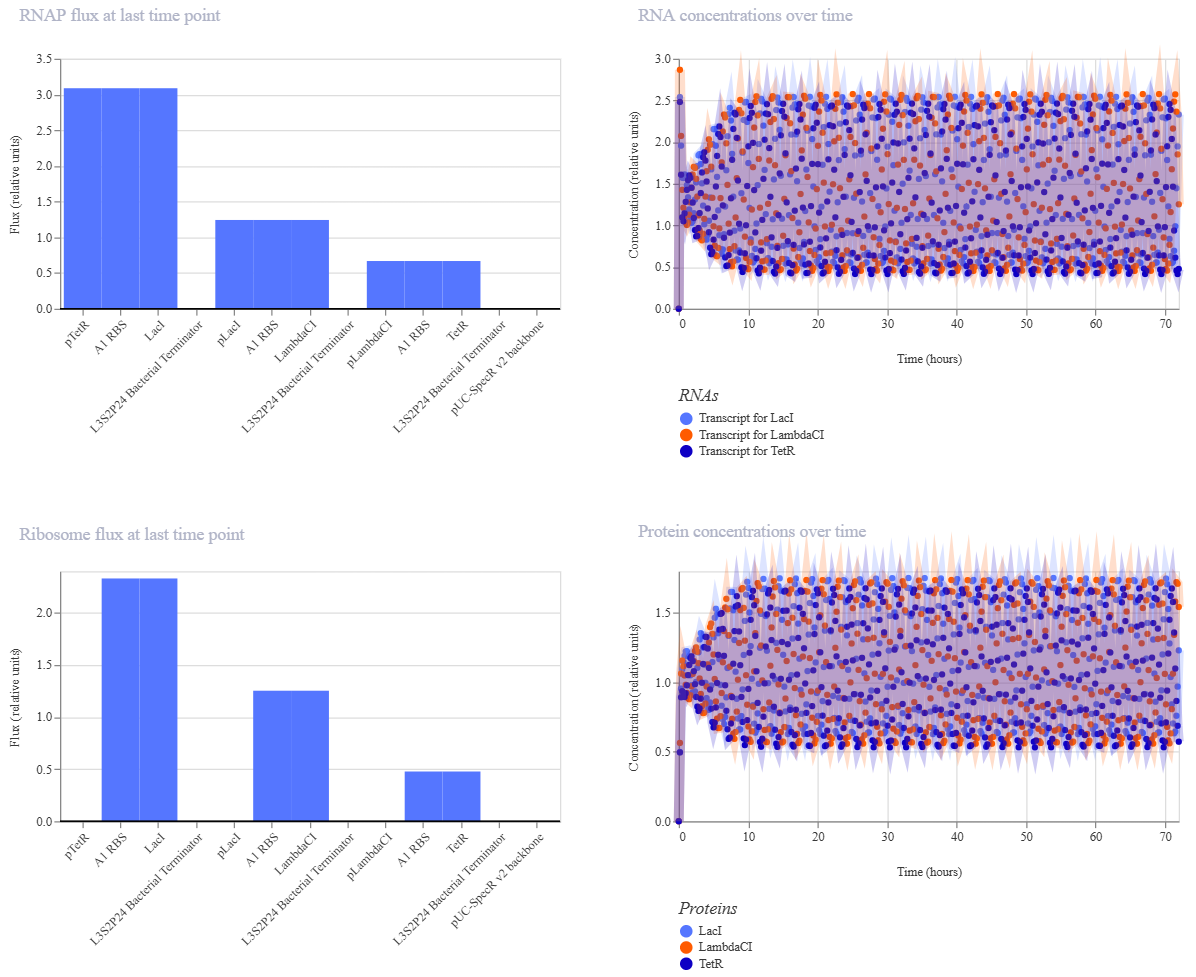
My Repressilator’s simulation:
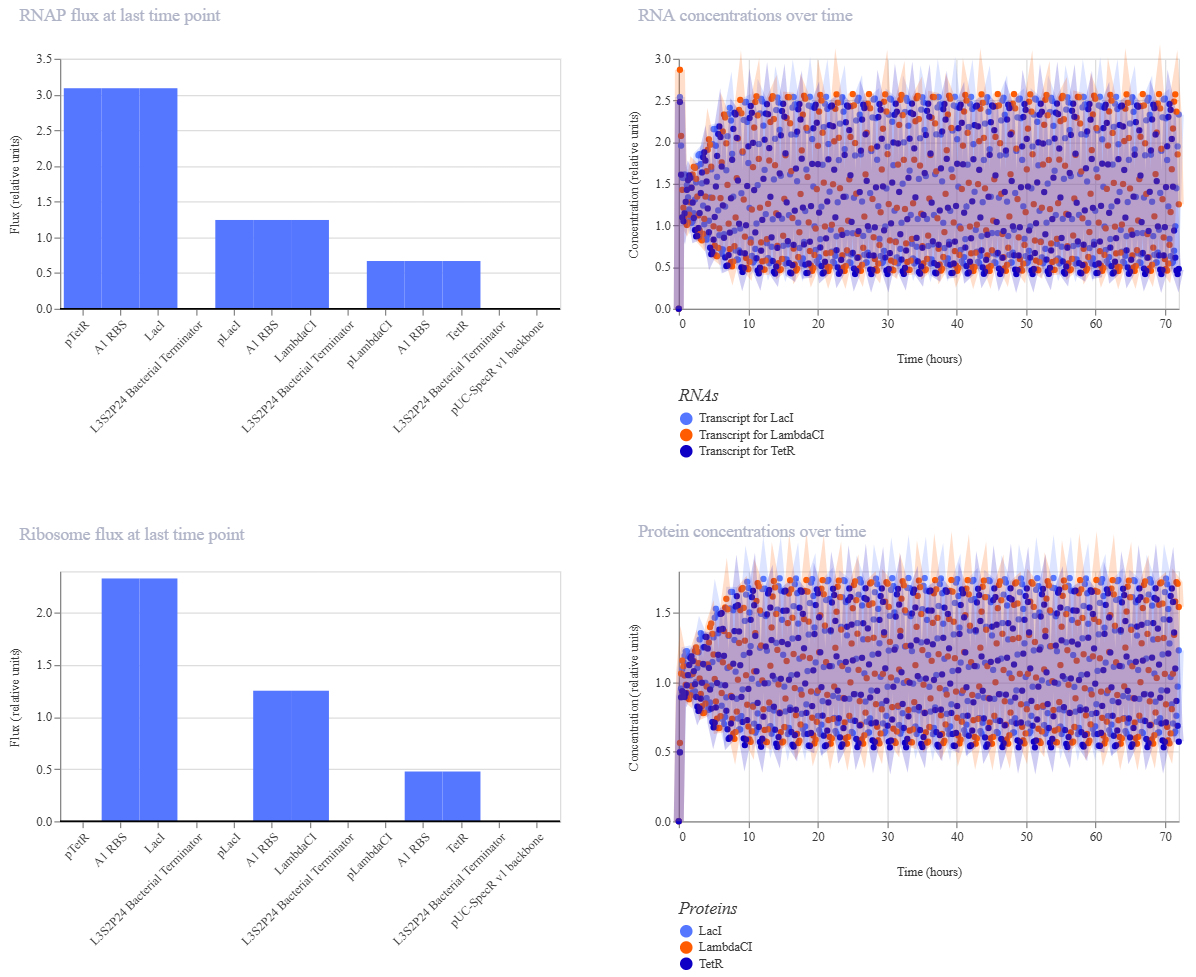
The Repressilator Construct from the Bacterial Demos repository:
The two simulations look identical to the naked eye.
Original Constructs
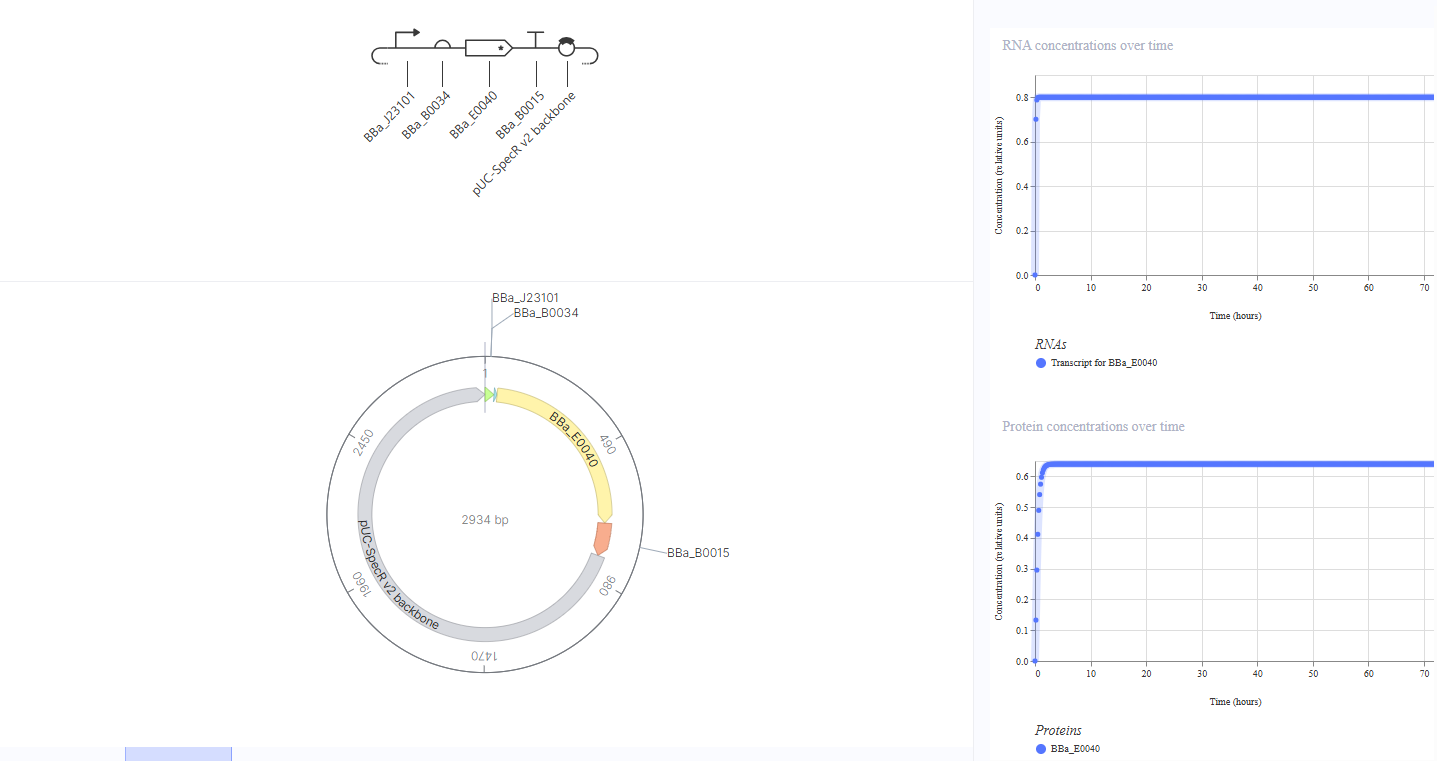
Construct 1: Constitutive GFP Expression
Idea: A simple “always on” circuit where GFP is continuously produced without any external signal or inducer. This serves as a baseline to demonstrate that the core expression machinery (promoter, RBS, CDS, terminator) is functioning correctly.
Here is the circuit and its simulation:
This construct uses a constitutive promoter (BBa_J23101) to drive constant GFP expression via RBS BBa_B0034 and GFP CDS BBa_E0040, terminated by BBa_B0015. Since the promoter is always active and not regulated by any inducer, I expect constant RNA and protein levels over time. The simulation confirms this - RNA stays stable, and GFP protein rises quickly to a steady state, showing constitutive expression.
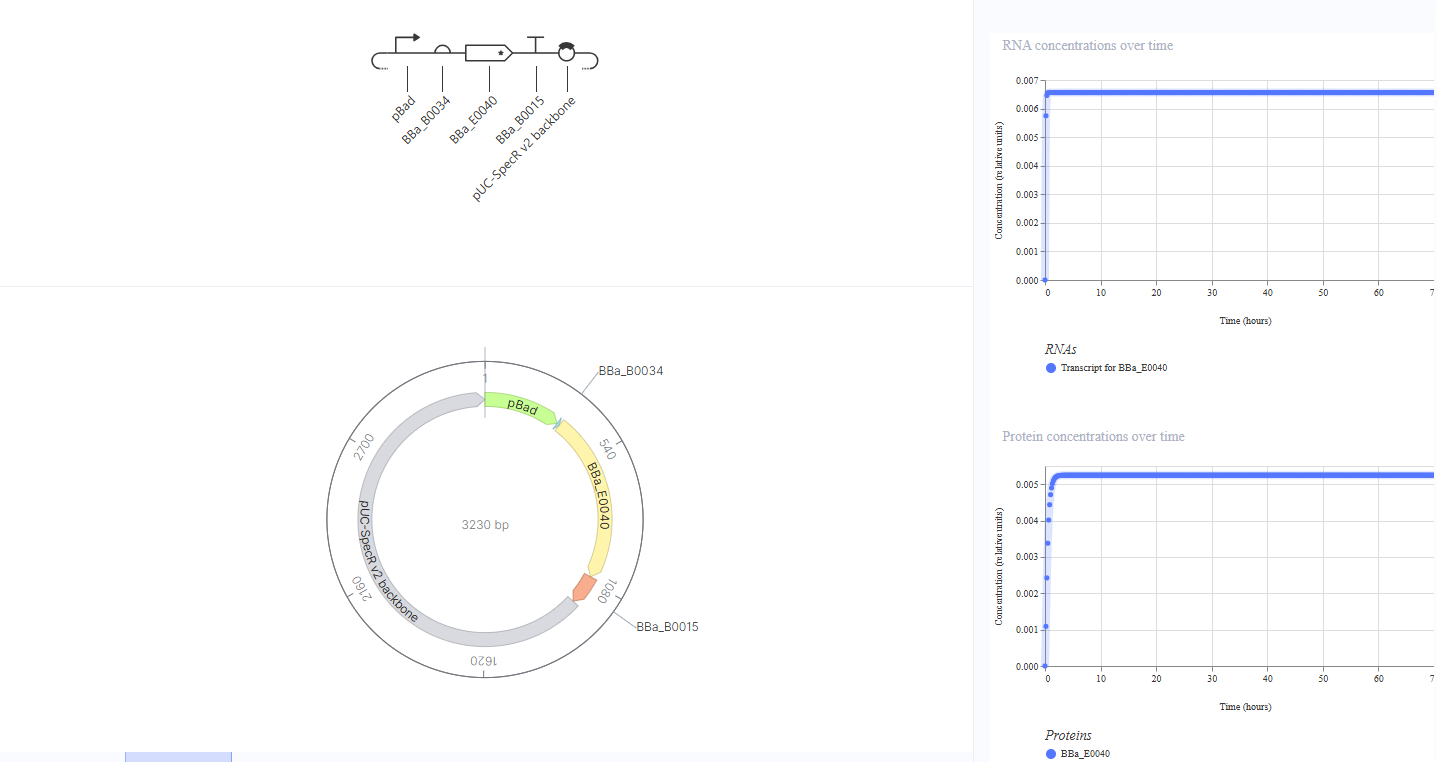
Construct 2: Arabinose-Inducible GFP Expression
Idea: An inducible “switch on” circuit where GFP production is controlled by the pBAD promoter, which activates only in the presence of arabinose. Without arabinose, GFP should be off; with arabinose, GFP should turn on.
This construct replaces the constitutive promoter with pBAD, an arabinose-sensitive promoter, upstream of RBS BBa_B0034 and GFP CDS BBa_E0040. I expected no GFP expression without arabinose and high expression with arabinose. The simulation showed low but constant GFP RNA and protein levels even in the absence of any inducer, consistent with the known leaky basal transcription of pBAD. The expression level is much lower than in Construct 1 (~0.006 vs ~0.8), suggesting the promoter is mostly repressed but not completely silent. To test the inducible response, I added L-arabinose as a ligand at hour 35, but GFP levels remained unchanged. This suggests the simulator does not model the arabinose-pBAD activation interaction, which might be a limitation of the simulation environment rather than a flaw in the biological design.
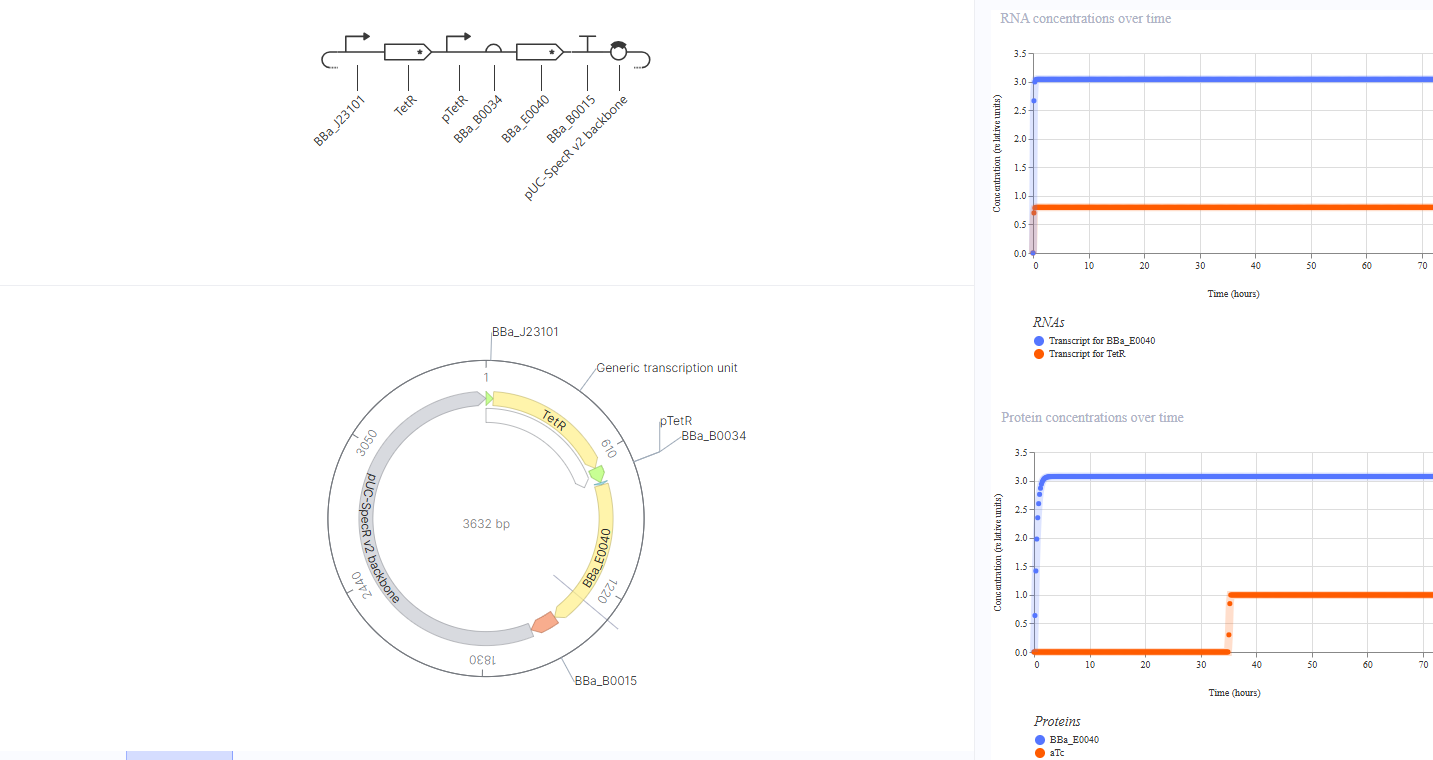
Construct 3: TetR-Repressible GFP Expression
Idea: A “switch off” circuit where TetR protein, produced constitutively, represses GFP expression by binding the pTetR promoter. Adding aTc would sequester TetR and allow GFP to turn back on, demonstrating inducible de-repression.
This construct uses BBa_J23101 to constitutively drive TetR expression, with TetR intended to repress pTetR, which controls GFP expression via RBS BBa_B0034 and CDS BBa_E0040. I expected low or no GFP expression by default, since TetR should suppress pTetR. However, the simulation showed high GFP expression (~3.0) rather than repression. To investigate, I attempted adding aTc as a ligand at hour 35, which should release TetR from pTetR and allow GFP expression - but GFP levels remained unchanged throughout. This confirms that the simulator does not model the regulatory interaction between TetR protein and the pTetR promoter automatically. The biological design remains sound in principle, but this is a limitation of the simulation environment that prevented us from observing the expected repression behavior.
Week 7 HW: Genetic Circuits Part II
Part 1: Intracellular Artificial Neural Networks
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Answer: The main advantage of IANNs over traditional genetic circuits is their ability to operate with analog (continuous) inputs and outputs, rather than being limited to Boolean (ON/OFF) behavior. This allows cells to respond more precisely to different signal levels instead of only detecting their presence or absence.
Additionally, IANNs can combine multiple inputs and balance their effects, allowing for more complex decision-making. Unlike Boolean circuits, which are limited to simple logic operations, IANNs can approximate many different types of functions, making them much more flexible for designing complex biological behaviors.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
Answer: A useful application of an IANN is a smart therapeutic cell that releases a drug only under specific biological conditions, for example, in response to disease signals.
The inputs to the system could be concentrations of molecules such as inflammatory markers or hormones. Each of these inputs would affect the system differently (some activating, some repressing), and the IANN would combine them in a weighted way. The output would be the level of expression of a therapeutic protein, such as an anti-inflammatory drug or a cancer-targeting molecule. Instead of turning fully ON or OFF, the output could be graded, meaning the cell releases more drug when the signals are stronger or more relevant.
This allows the cell to make more precise decisions compared to traditional Boolean circuits, for example, only producing a high drug dose when multiple disease signals are present at certain levels.
However, there are several limitations. Biological systems are noisy and variable, so the same inputs may not always produce the same output. It is also difficult to precisely control the “weights” (strength of interactions), since they depend on complex factors like binding affinity and gene expression levels. Additionally, responses can be slow due to transcription and translation times, and the system may be affected by changes in the cellular environment. These challenges can make it hard to achieve reliable and predictable behavior.
Another important limitation is that the circuit exists inside a living cell, which has its own regulatory systems. Native proteins or molecules in the cell may unintentionally interact with parts of the circuit, activating or repressing it in unexpected ways. In the best case, this disrupts the intended logic of the system, and in the worst case, it could lead to incorrect levels of drug production, which may be harmful.
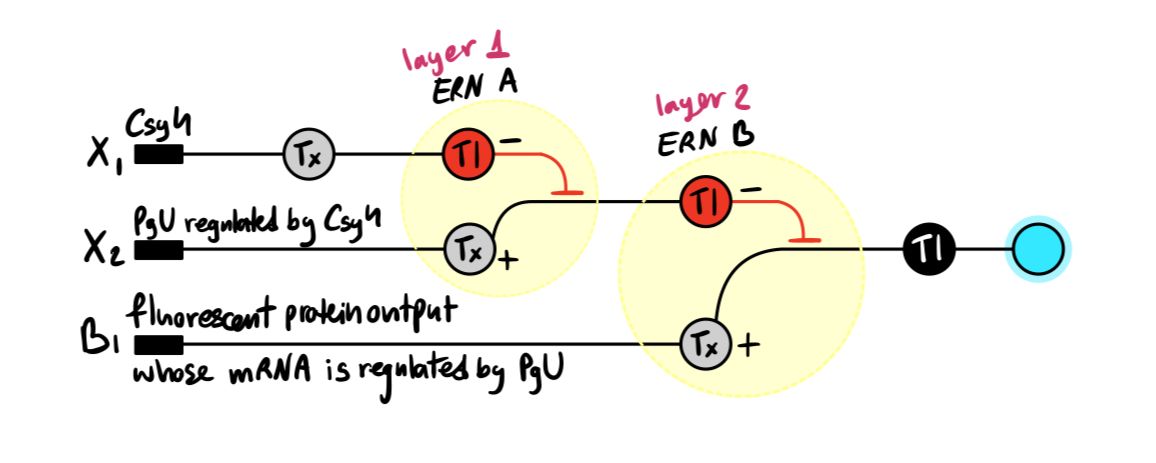
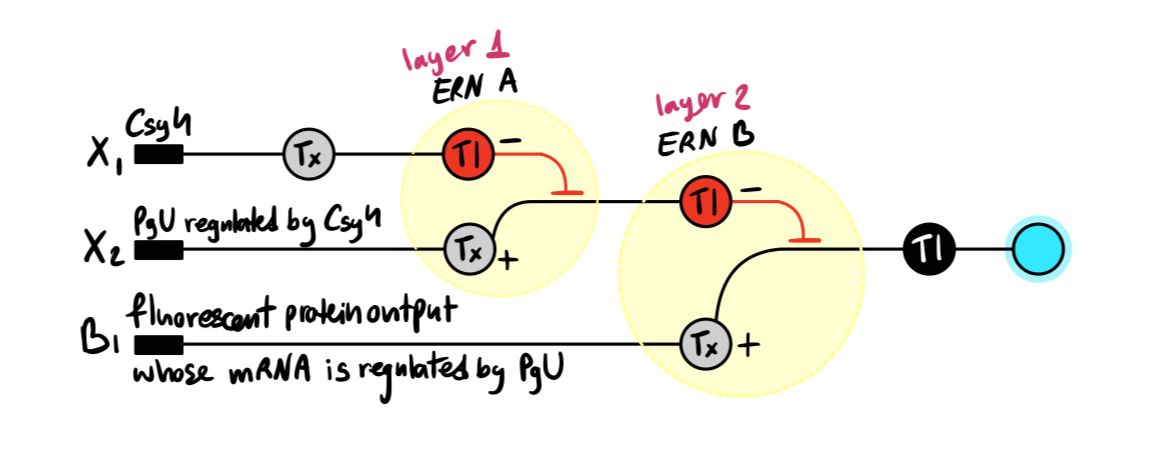
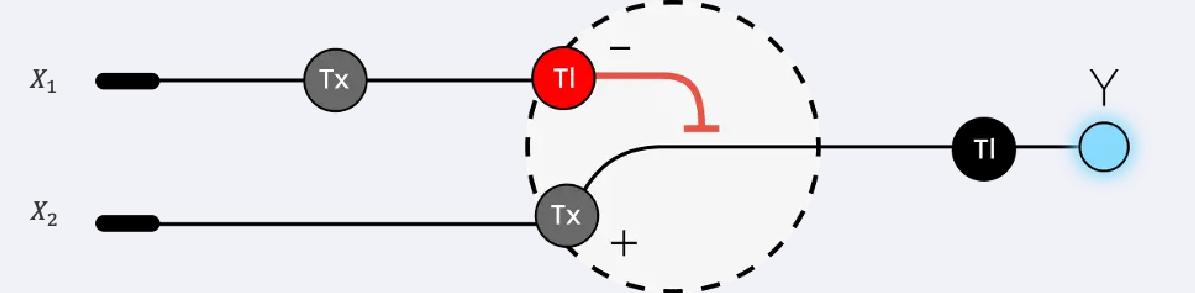
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Answer:
Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Answer: Examples of fungal materials include mycelium-based materials and biocement.
Mycelium materials are used for packaging, insulation, furniture, and building, and are even being explored by NASA for space habitats. Biocement uses microbes to bind materials like sand into solid structures.
Their advantages include being lightweight, biodegradable, moldable into many shapes, and good for thermal and acoustic insulation. They can also be grown from waste materials, making them sustainable.
However, they can be brittle and weaker than traditional materials, harder to scale, and slower to produce. Since they are biologically grown, they can also be inconsistent and sensitive to environmental conditions.
What might you want to engineer fungi to do and why genetically? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
Answer: One goal would be to genetically engineer fungi to produce more flexible and less brittle materials so that they could be used in applications like textiles, wearable materials, or more durable building components. This could be done by modifying how the fungal cell wall is built or by introducing new proteins that change the material’s mechanical properties. Making fungal materials more flexible would expand their use beyond insulation and packaging into areas that require strength and durability.
An advantage of using fungi over bacteria is that fungi naturally grow as large, interconnected networks, which makes them well-suited for forming macroscopic materials. In contrast, bacteria are typically single cells and are better at producing molecules rather than structured materials. Additionally, fungi can grow on low-cost substrates like agricultural waste and can be easily shaped in molds as they grow, which is useful for fabrication.
Part 3: First DNA Twist Order
My final project ultimately did not involve bacterial expression or plasmid insert design - but check out what I did end up doing here!
Week 9 HW: Cell Free Systems
General homework questions
1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Answer: Cell-free protein synthesis offers greater flexibility and control compared to traditional in vivo methods because the reaction occurs outside of living cells. This allows precise control over experimental variables such as temperature, pH, ion concentrations, and the addition of specific components (e.g., cofactors, inhibitors, or non-natural amino acids), without being constrained by cellular viability or stress responses. Additionally, cell-free systems are not time-dependent on cell growth, making them faster and more consistent. DNA constructs can also be prepared and stored for long periods and used on demand.
Cell-free expression is particularly beneficial in cases such as:
Toxic protein production, where the protein would harm or kill living cells.
Production of proteins with non-natural amino acids or modified components, since cell-free systems allow you to directly add these into the reaction without worrying about cellular metabolism or toxicity.
2. Describe the main components of a cell-free expression system and explain the role of each component.
Answer:The main components of a cell-free expression system:
DNA / RNA template – encodes the protein of interest; can be plasmid DNA or mRNA, and determines what protein is made
RNA polymerase – transcribes DNA into mRNA (only needed if starting from DNA)
Ribosomes – translate mRNA into protein
tRNAs – deliver the correct amino acids to the ribosome by matching codons in the mRNA
Amino acids – building blocks of the protein
Nucleotides (ATP, GTP, etc.) – used for RNA synthesis and provide energy for transcription and translation
Energy regeneration system (e.g., 3-PGA) – regenerates ATP to sustain protein synthesis over time
Cofactors and coenzymes (e.g., NAD, CoA, folic acid) – support enzymatic reactions required for transcription and translation
Buffer system (e.g., HEPES) – maintains stable pH and optimal chemical conditions for the reaction
Salts and small molecules (e.g., Mg²⁺, K⁺, spermidine) – stabilize ribosomes, nucleic acids, and improve reaction efficiency
*Cell extract (lysate) – most of these components are provided by the cell extract; it supplies the essential cellular machinery, including ribosomes, enzymes, and translation factors needed for protein synthesis.
**3. Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Answer: Energy provision and regeneration are critical in cell-free systems because transcription and translation require a large amount of ATP and GTP. Since there is no living cell to continuously produce new energy molecules, in the absence of an energy source, the reaction would quickly run out of energy, and protein synthesis would stop. In addition, ATP is needed not only as an energy source but also for many enzymatic steps in the system.
One way to ensure a continuous ATP supply is to include an energy regeneration system, such as 3-phosphoglycerate (3-PGA). In this method, 3-PGA is added to the reaction mixture and is metabolized by enzymes present in the cell extract to regenerate ATP over time. This helps maintain the energy level in the reaction and allows protein synthesis to continue for longer.
4. Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Answer:Prokaryotic vs. Eukaryotic Cell-Free Systems
Prokaryotic systems (e.g., E. coli lysate): Faster, cheaper, and higher yield, but lack post-translational modifications (PTMs) such as glycosylation and proper folding for complex proteins.
Eukaryotic systems (e.g., wheat germ, rabbit reticulocyte, or mammalian lysates): Slower and more expensive, but support proper folding and PTMs, making them suitable for complex proteins.
Examples:
1. Prokaryotic system → Green Fluorescent Protein (GFP) - GFP is a relatively simple protein that does not require complex post-translational modifications, so it can be efficiently produced in an E. coli-based cell-free system with high yield and speed.
2. Eukaryotic system → Human insulin (or a glycosylated antibody) - Proteins like antibodies or hormones often require proper folding and post-translational modifications (e.g., disulfide bonds, glycosylation). An eukaryotic cell-free system is better suited to produce these functional proteins.
5. How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Answer: I would design the experiment using a cell-free system supplemented with membrane mimics such as liposomes, nanodiscs, or mild detergents, so that the hydrophobic regions of the membrane protein have an environment to insert into as it is being synthesized. I would then optimize conditions such as temperature, magnesium concentration, and the type/amount of membrane mimic.
The main challenge is that membrane proteins are hydrophobic, so they tend to aggregate or misfold without a membrane-like environment. This can be addressed by adding membrane mimics and evaluating both protein yield and functionality to ensure proper folding and insertion.
6. Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Answer:
Not enough ribosomes – limits translation efficiency → Troubleshooting: increase the amount or quality of cell extract (which provides ribosomes)
Insufficient energy supply (ATP/GTP depletion) – reaction stops early → Troubleshooting: add or optimize an energy regeneration system (e.g., 3-PGA, creatine phosphate)
Poor DNA template quality or concentration – low transcription/translation → Troubleshooting: increase DNA concentration, check purity, or use a stronger promoter
Homework question from Kate Adamala
1. Synthetic minimal cell: glucose-sensing cell
Function: A synthetic minimal cell detects high glucose levels and produces insulin or an insulin-like signal.
Input and output:
Input: glucose
Output: insulin (and optional reporter signal)
Cell-free Tx/Tl only? Partly. It can sense and produce protein, but lacks stability and containment without encapsulation. Additionally, it is time-constrained.
Genetically modified natural cell? Yes. Natural cells can do this, but are more complex and less controllable since they have other processes happening in the background.
Desired outcome: Inactive at normal glucose levels; activates at high glucose to help regulate sugar levels in the blood.
2. Design all components of the synthetic glucose-sensing cell
a. What would the membrane be made of? A simple lipid vesicle membrane, made from phospholipids, so it can act like a small artificial cell and keep the internal components contained.
b. What would you encapsulate inside? Enzymes, small molecules. Inside, I would encapsulate the Tx/Tl machinery, a DNA circuit for glucose sensing and insulin production, energy molecules, ribosomes, enzymes, amino acids, and a reporter protein for signaling.
c. Which organism will your Tx/Tl system come from? Is bacterial OK, or do you need a mammalian system for some reason? A bacterial Tx/Tl system would likely be enough if the goal is just to sense glucose and produce a simple protein output.
d. How will your synthetic cell communicate with the environment? The membrane would need to allow glucose to enter, either through natural permeability if sufficient, or by including a membrane channel or pore protein. The produced output could be released by diffusion or membrane leakage.
Experimental details
a. List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel,” pick the actual gene.) Answer:
b. How will you measure the function of your system? Answer:
3. Experimental details
a. List all lipids and genes.
Lipids: POPC and cholesterol for the vesicle membrane.
Tx/Tl system: bacterial cell-free system, for example from E. coli.
Genes: a glucose-sensing genetic circuit, a reporter gene such as gfp, and a membrane pore gene such as α-hemolysin (ahl) to allow communication with the environment.
Output gene: an insulin or insulin-like peptide gene.
b. How will you measure the function of your system? Measure the output signal as glucose concentration changes. This could be done by tracking GFP fluorescence and by measuring insulin production with an assay such as ELISA.
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material.
Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept. Answer: A wearable skin patch that changes color in response to electrolyte loss in sweat (primarily sodium, potassium, and magnesium), enabling athletes and their teams to monitor hydration levels during competition (and outside of it) and optimize electrolyte intake in real time.
How will the idea work, in more detail? Write 3–4 sentences or more. Answer:
The idea is a skin sticker with a freeze-dried cell-free sensing layer in the center and an adhesive border around it so it can stay attached during physical activity. The sensing region would be transparent from the outside, and when sweat reaches it, the water in the sweat would rehydrate and activate the cell-free reaction. Inside this layer, a DNA circuit would respond to sodium concentration: at low sodium levels, the patch would stay mostly transparent, while higher sodium levels would produce a stronger visible color change (the color would be due to a chromoprotein). If expanded to multiple inputs, the same concept could be adapted into a neuromorphic-style circuit that senses sodium, potassium, and magnesium together, assigns each ion a different weight based on its importance, and uses the combined signal to determine the final patch color.
What societal challenge or market need will this address? Answer: This addresses the challenge of monitoring hydration and electrolyte balance in athletes in a simple, real-time, and non-invasive way. Currently, assessing electrolyte loss often requires lab tests or indirect measures, which are not practical during training or competition. This patch would provide immediate visual feedback, helping prevent dehydration, fatigue, and heat-related illnesses. The market need is especially strong in sports, military, and outdoor work, where performance and safety depend on maintaining proper electrolyte levels.
How do you envision addressing the limitations of cell-free reactions (e.g., activation with water, stability, one-time use)? Answer: Most sports activities and competitions are shorter than the typical working time of a cell-free reaction, so the limited lifetime of the system should still be sufficient for a single session of use. A key design goal would be to optimize the freeze-dried reaction so it can be activated directly by sweat, rather than requiring pure water. In this application, one-time use is not a major limitation, since the patch is meant to be disposable and replaced with a new one for each practice or competition. Stability during storage could be addressed by keeping the sticker sealed and dry until use.
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/.
My Idea: Effect of Microgravity on the Time Required for Functional GFP Expression in Cell-Free Systems
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words) Answer: Cell-free protein expression systems could become an important tool for long-duration space missions because they allow astronauts to produce proteins on demand without maintaining living cells. But, how does microgravity affect the speed or efficiency of protein production in these systems? In this project, I propose to compare the production of a fluorescent protein in freeze-dried cell-free reactions performed in space and on Earth. Understanding these effects is important for the future development of space-based biotechnology, including diagnostics and medicine production. While studying one protein cannot represent all proteins, any observed differences could be important in future experiments.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words) Answer: The expression and maturation time of green fluorescent protein (GFP) produced from plasmid DNA in the BioBits cell-free expression system.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words) Answer: GFP serves as a measurable reporter for protein production in the BioBits cell-free expression system. By comparing the time required for functional fluorescent GFP to appear in reactions performed in microgravity (or the space environment in general) versus on Earth, I can evaluate whether space conditions affect protein expression or protein maturation kinetics. Because GFP fluorescence only appears after the protein is properly folded and matured, differences in fluorescence timing or intensity may indicate that spaceflight conditions influence important biochemical processes involved in protein production. Although this experiment focuses on one protein, any observed effects could suggest that future space biotechnology systems require additional testing and optimization.
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words) Answer: I hypothesize that the time required for functional GFP fluorescence to appear in a freeze-dried cell-free expression system will differ between reactions performed in space and identical reactions performed on Earth. This difference could result from microgravity or other spaceflight-related conditions affecting biochemical processes such as transcription, translation, protein folding, or GFP maturation. Because future long-duration missions may rely on cell-free systems for on-demand production of medicines, diagnostics, and biomaterials, it is important to understand whether these systems behave differently in space environments. While this experiment investigates only one protein, observing altered expression or maturation kinetics could indicate that space conditions influence broader aspects of cell-free biotechnology and should therefore be considered in future system design and testing.
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words) Answer: I will test identical BioBits cell-free reactions containing plasmid DNA encoding GFP. One set will be activated and monitored in space, and the control set will be activated and monitored on Earth under similar conditions. A negative control without GFP DNA will be included (both on Earth and in space) to confirm that fluorescence comes from GFP expression. The main measurements will be time until visible fluorescence appears, fluorescence intensity over time, and final fluorescence intensity. These data will be used to compare functional GFP production kinetics in space versus on Earth.
Week 10 HW: Imaging and Measurement
Final Project
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
My final project is inherently related to measurement, as it is going to be a measurement instrument. The main aspect that I will measure for my final project is therefore its core purpose — measuring testosterone levels in sweat. To do that, I will use either cell-free systems, bacterial expression, or an aptamer-based method to create a fluorescent or electrical signal that correlates with the levels of testosterone in the sample.
Moreover, I would measure the following:
The length of the different DNA fragments I designed for the project, using gel electrophoresis, to make sure they are consistent with my design.
The time it takes for the sensor I create to detect the level of testosterone, or in earlier versions, the time it takes to produce a reliable signal that indicates that there is testosterone in the sample.
The sensitivity and accuracy of the sensor - specifically, the range of testosterone concentrations it can reliably detect, its limit of detection, and how consistent the signal is across repeated measurements. This will help me evaluate how precise and usable the system is in practice.
The specificity of the sensor - testing whether the system responds uniquely to testosterone versus similar molecules (e.g., other hormones in sweat), to ensure that the signal is not due to interference.
Additionally, this is a measurement that would be done further down the line, but is very much needed: I will measure both the levels of testosterone in sweat using my sensor, and the levels of testosterone in the blood using traditional blood tests, to create a correlation between the two (and verify that it exists). This will allow me to deduce what certain levels of testosterone in sweat, detected by my sensor, actually mean in terms of blood levels, and thereby validate the method.
Waters Part I — Molecular Weight
Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
Answer: Using the full predicted eGFP sequence provided, including the LE linker and C-terminal His-purification tag, the calculated molecular weight is 28,006.60 Da, or approximately 28.01 kDa. The theoretical pI is 5.90.
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation.
Answer: I selected two adjacent peaks from the intact LC-MS spectrum: m/z = 875.4421 and m/z = 903.7148. Using the adjacent charge state approach, these correspond approximately to charge states z = 32 and z = 31, respectively.
Using the relationship between m/z, MW, and z, the calculated molecular weight is approximately 27,982 Da, or 27.98 kDa.
Compared to the theoretical molecular weight from Question 1, 28,006.60 Da, the percent error is:
So, the experimental molecular weight is very close to the theoretical molecular weight.
Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
Answer: No, the charge state cannot be directly observed from the zoomed-in peak alone. The zoomed-in peak shows the isotope pattern of one charge state, but the charge state is determined by comparing the spacing between adjacent charge-state peaks in the full spectrum. Therefore, the zoomed-in peak helps show isotopic resolution, but we need the full charge envelope to assign the charge state confidently.
Waters Part II — Secondary/Tertiary structure
Based on learnings in the lab, please explain the difference between native and denatured protein conformations. For example, what happens when a protein unfolds? How is that determined with a mass spectrometer? What changes do you see in the mass spectrum between the native and denatured protein analyses (Figure 2)?
Answer: In the native state, the protein stays folded in its natural 3D structure. In the denatured state, the protein unfolds due to conditions such as solvent composition and pH. When the protein unfolds, more amino acids become exposed to the solvent, which allows the protein to pick up more charges during electrospray ionization.
This can be seen in the mass spectrum by the different charge state distributions. The denatured eGFP spectrum shows many peaks across a wider m/z range, corresponding to higher charge states. In contrast, the native eGFP spectrum has fewer peaks at higher m/z values, corresponding to lower charge states, because the folded protein has fewer exposed sites.
The molecular weight of the protein stays approximately the same, but the charge state distribution changes significantly between the native and denatured conditions.
Zooming into the native mass spectrum of eGFP from the Waters Xevo G3 QTof MS (see Figure 3), can you discern the charge state of the peak at ~2800 m/z? What is the charge state? How can you tell?
Answer: Yes. The charge state can be determined from the spacing between the isotope peaks in the zoomed-in spectrum. For a protein ion, the spacing between isotope peaks is approximately 1/z.
For the peak around 2800 m/z, the charge state is approximately z = 10, because the molecular weight of eGFP is around 28,000 Da, and:
28,000 / 10 ≈ 2800 m/z
So the peak at ~2800 m/z corresponds to the 10+ charge state.
Waters Part III — Peptide Mapping - primary structure
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above.
Answer: There are 20 Lysines (K) and 6 Arginines (R) in the eGFP sequence.
How many peptides will be generated from tryptic digestion of eGFP?
Answer: Using the ExPasy PeptideMass tool with trypsin selected and allowing 0 missed cleavages, the eGFP sequence is predicted to generate 27 tryptic peptides total.
Out of these, 19 peptides have masses greater than 500 Da and are therefore more likely to be detected in the LC-MS analysis under the selected settings. The remaining peptides are very small and may fall below the practical detection range.
The predicted sequence coverage for the detected peptides is 90.7%.
Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
Answer: Between 0.5 and 6 minutes, there are approximately 18 chromatographic peaks with greater than 10% relative abundance visible in the eGFP peptide map TIC chromatogram.
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
Answer: The numbers are close, but not exactly the same. In question 2, 19 peptides were predicted to be above 500 Da, and in the chromatogram, I counted about 18 peaks above 10% relative abundance between 0.5 and 6 minutes.
This is slightly fewer than the predicted number, but it still makes sense. Some peptides may not be detected clearly, some peptides may co-elute and appear as one chromatographic peak, and some smaller or lower-abundance peptides may fall below the cutoff. Also, if counting smaller peaks below 10%, there would be more peaks in the chromatogram.
Overall, the observed number of peaks is reasonably consistent with the predicted number of detectable tryptic peptides.
Identify the mass-to-charge (m/z) of the peptide shown in Figure 5b. What is the charge (z) of the most abundant charge state of the peptide? Calculate the mass of the singly charged form of the peptide ([M+H]+) based on its m/z and z.
Answer: The most abundant peptide peak is at m/z = 525.76712.
The isotope peaks are separated by about 0.5 m/z, which means the charge state is z = 2 because isotope spacing is approximately 1/z.
Assuming the relationship for electrospray ionization:
:contentReference[oaicite:0]{index=0}
and using z = 2, the singly charged peptide mass is::
[M+H]+ = (525.76712 × 2) - 1.0073 = 1050.53 Da
So, the singly charged mass of the peptide is approximately 1050.53 Da.
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm.
Answer: Based on the PeptideMass results, the peptide with a predicted mass closest to the measured value is FEGDTLVNR, with a theoretical [M+H]+ mass of 1050.5214 Da.
From Figure 5b, the calculated experimental singly charged mass was approximately 1050.53 Da.
So, the peptide is most likely FEGDTLVNR, and the mass accuracy is approximately 8 ppm.
What is the percentage of the sequence that is confirmed by peptide mapping? (see Figure 6)
Answer: Based on Figure 6, the peptide mapping analysis confirmed 88% sequence coverage of the eGFP protein.
Waters Part IV — Oligomers
Using the known masses of the polypeptide subunits for KLH, identify where the following oligomeric species are on the spectrum shown from the CDMS: 7FU Decamer, 8FU Didecamer, 8FU 3-Decamer, and 8FU 4-Decamer.
Answer: To identify the oligomeric species, I calculated the expected mass by multiplying the mass of each subunit by the number of subunits in the oligomer, and then compared it to the peaks in the CDMS spectrum.
Oligomeric Species
Calculation
Expected Mass
Approximate Peak in Spectrum
7FU Decamer
10 × 340 kDa
3.4 MDa
~3.4 MDa
8FU Didecamer
20 × 400 kDa
8.0 MDa
~8.33 MDa
8FU 3-Decamer
30 × 400 kDa
12.0 MDa
~12.67 MDa
8FU 4-Decamer
40 × 400 kDa
16.0 MDa
weak peak/feature around ~16–17 MDa
Overall, the expected masses match reasonably well with the main peaks in the CDMS spectrum, although the experimental peaks are slightly shifted from the simple calculated masses.
Waters Part V — Did I make GFP?
Property
Theoretical
Measured from LC-MS
PPM Error
eGFP molecular weight (kDa)
28.0066
~27.984
~880 ppm
Sequence coverage from peptide mapping
90.7%
88%
—
Peptide FEGDTLVNR [M+H]+ (Da)
1050.5214
1050.53
~8 ppm
Week 11 HW: Bioproduction and Cloud Labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
My Contribution
My involvement in the community bioart project started right at the beginning. I found myself in a “digital battle” with other users over whether the top row on the left plate would be blue or yellow - I was on Team Yellow! No particular reason, just a gut feeling:) After a few minutes, others joined me:
It did indeed end up yellow in the final design!
What I liked
I really enjoyed the global aspect of the project - the fact that hundreds of people from all around the world contributed to a single piece of art is remarkable. I also loved the “evolutionary” dimension of it: being able to trace how the design changed and developed over time gave it a unique, living quality.
My favourite snapshot from the evolution:
What could be improved
For next year, I’d suggest two things:
More colors - a broader palette would give participants more creative flexibility and make the process even more fun.
More surface area - a larger canvas would allow for higher-resolution details and give more people the space to leave their mark on the final piece.
Overall, it was a wonderful idea and a genuinely fun experience to be part of!
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
1. Cell-Free Protein Synthesis Reaction Components
E. coli Lysate
Component
Role
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase)
Provides the core cellular machinery needed for transcription and translation, including ribosomes, enzymes, and co-factors. The T7 RNA Polymerase specifically transcribes DNA into mRNA using the T7 promoter system.
Salts / Buffer
Component
Role
Potassium Glutamate
Maintains ionic strength and mimics the intracellular environment, supporting enzyme activity and ribosome stability.
HEPES-KOH pH 7.5
A buffering agent that maintains a stable, physiological pH (7.5), which is optimal for transcription and translation enzymes.
Magnesium Glutamate
Provides Mg²⁺ ions, which are essential cofactors for ribosomes, RNA polymerase, and many enzymes involved in protein synthesis.
Potassium Phosphate Monobasic
Works together with the dibasic form to maintain pH and provide phosphate ions, which are important for energy metabolism and nucleotide synthesis.
Potassium Phosphate Dibasic
Works together with the monobasic form to maintain pH and provide phosphate ions, which are important for energy metabolism and nucleotide synthesis.
Energy / Nucleotide System
Component
Role
Ribose
A simple sugar that serves as a carbon and energy source; can be metabolized to regenerate nucleotides and sustain the reaction over longer incubation periods.
Glucose
Another energy source that feeds into central metabolic pathways to regenerate ATP and other nucleotides needed for sustained transcription and translation.
AMP
A nucleotide monophosphate precursor that is phosphorylated to ATP, the primary energy currency that drives protein synthesis.
CMP
Precursor to CTP, which is required for RNA synthesis during transcription.
GMP
Precursor to GTP, which is required for both transcription and translation (e.g., ribosome translocation).
UMP
Precursor to UTP, which is required for RNA synthesis during transcription.
Guanine
A nucleobase that can be salvaged and converted into GMP/GTP, supplementing the guanosine nucleotide pool for transcription and translation.
Translation Mix (Amino Acids)
Component
Role
17 Amino Acid Mix
Provides the bulk of the amino acid building blocks needed for the ribosome to synthesize polypeptide chains.
Tyrosine
Supplied separately because of its low solubility at neutral pH; it is an essential amino acid for building complete proteins.
Cysteine
Supplied separately due to its reactivity; it is essential for forming disulfide bonds and maintaining the structural integrity of many proteins.
Additives
Component
Role
Nicotinamide
A precursor to NAD⁺, which is a critical cofactor in metabolic redox reactions that regenerate energy to sustain the cell-free reaction.
Backfill
Component
Role
Nuclease-Free Water
Used to bring the reaction to its final volume without introducing RNases (ribonucleases) and DNases (deoxyribonucleases), enzymes that degrade RNA and DNA respectively, that could degrade the nucleic acids in the reaction.
2. 1-Hour PEP/NTP vs. 20-Hour NMP-Ribose Master Mix: Key Differences
The most fundamental difference between the two master mixes is their energy and nucleotide system: the 1-hour PEP/NTP mix supplies energy directly via pre-made NTPs (ATP, GTP, CTP, UTP) and PEP-Mono (phosphoenolpyruvate monopotassium salt; regenerates NTPs by donating phosphate groups to NDPs produced during transcription) as an immediate energy source, while the 20-hour NMP-Ribose mix supplies energy indirectly via NMPs and simple sugars (ribose and glucose) that the lysate enzymes must first convert into usable NTPs. This makes the PEP/NTP system fast but short-lived, while the NMP-Ribose system is slower to get going but more sustainable over longer incubation periods. The 1-hour mix also contains more additives (Spermidine, DMSO, cAMP, NAD, Folinic Acid) to boost immediate transcription/translation activity, whereas the 20-hour mix keeps additives minimal (only Nicotinamide) since the reaction has more time to proceed on its own.
3. Bonus question
Although GMP is listed as 0.00 uM in the 20-hour mix, Guanine (the nucleobase) is still provided and can be converted into GMP through a process called nucleotide salvage, where enzymes in the lysate attach a ribose-phosphate group to the free Guanine base. The resulting GMP is then phosphorylated up to GTP, which can be used for transcription. This is a more cost-effective way to supply guanosine nucleotides compared to adding pre-made GMP directly.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
A. Biophysical and Functional Properties of the 6 Fluorescent Proteins
1. sfGFP (Superfolder GFP)
sfGFP is engineered to fold extremely rapidly and robustly, even when fused to poorly folding proteins, making it one of the most reliable reporters in cell-free systems. Like all GFP-based proteins, its chromophore maturation requires molecular oxygen, meaning fluorescence will be delayed or absent in low-oxygen conditions.
2. mRFP1 (Monomeric Red Fluorescent Protein 1)
mRFP1 is a true monomer, which is important in cell-free systems because oligomerizing proteins can cause aggregation artifacts. However, it has relatively low brightness and photostability compared to newer red fluorescent proteins, which can limit signal readout in cell-free reactions where protein yields may already be low.
3. mKO2 (Monomeric Kusabira Orange 2)
mKO2 is an orange fluorescent protein with moderate acid sensitivity, meaning its fluorescence can be partially quenched if the pH of the cell-free reaction drifts below neutral. It also has a relatively slow and complex maturation process (requiring both a folding and a separate maturation step), which can delay fluorescence readout in cell-free systems.
4. mTurquoise2
mTurquoise2 is a rapidly maturing cyan fluorescent protein with very low acid sensitivity, making it a stable and reliable reporter across different reaction conditions. Its fast maturation is particularly advantageous in cell-free systems, where getting a quick fluorescence readout is important.
5. mScarlet-I
mScarlet-I is a synthetically derived, rapidly maturing monomeric red fluorescent protein with high brightness, making it one of the best-performing red reporters in cell-free systems. Like other GFP-family proteins, it requires oxygen for chromophore maturation, so adequate oxygenation of the reaction is important for good signal.
6. Electra2
Electra2 is a blue fluorescent protein (excitation ~405 nm, emission ~456 nm) derived from Entacmaea quadricolor, engineered for high intracellular brightness. Its blue emission provides good spectral separation from green and red fluorescent proteins, making it useful in multicolor cell-free experiments — though blue fluorescent proteins generally have lower brightness than their green and red counterparts, which can affect detection sensitivity.
B. Cell-Free Optimization Experiment: mRFP1 vs mScarlet-I over 36 Hours
Hypothesis
mRFP1’s main limitations are slow maturation and low brightness. I hypothesize that
supplementing the 36-hour master mix with additional Cysteine (critical for mRFP1’s disulfide
bonds and folding) and GMP (to boost GTP availability for sustained transcription) will improve
folding efficiency and protein yield, thereby maximizing fluorescence over 36 hours. I will use
mScarlet-I as a positive control reference since it is a fast-maturing red protein, to see
whether my optimization can close the brightness gap.
Experimental Design
mRFP1 Wells (4 wells — varying Cysteine + GMP)
Well
Protein
GMP added
Cysteine added
Notes
A1
mRFP1
0 mM
0 mM
Baseline control
A2
mRFP1
0.250 mM
0 mM
GMP only
A3
mRFP1
0 mM
0.500 mM
Cysteine only
A4
mRFP1
0.250 mM
0.500 mM
Combined
mScarlet-I Wells (4 wells — same design, for comparison)
Well
Protein
GMP added
Cysteine added
Notes
B1
mScarlet-I
0 mM
0 mM
Baseline control
B2
mScarlet-I
0.250 mM
0 mM
GMP only
B3
mScarlet-I
0 mM
0.500 mM
Cysteine only
B4
mScarlet-I
0.250 mM
0.500 mM
Combined
All wells contain: 6 µL Lysate + 10 µL 2X Master Mix + 2 µL DNA template + 0–2 µL custom supplements (backfilled with nuclease-free water to 2 µL)
I input these concentrations on the website
Reagent Rationale
GMP → GTP: The baseline 36-hour mix has GMP = 0, relying entirely on Guanine salvage.
Adding a small GMP boost gives the system more immediate GTP for transcription, which should
increase mRNA production early in the reaction and improve total protein yield.
Cysteine: mRFP1 underwent 33 mutations during its engineering and contains cysteines important
for proper folding. The baseline Cysteine concentration is 4.00 mM — adding 0.25 mM is a modest,
safe increase that could improve folding efficiency without disrupting the reaction balance.
A1 and B1 as controls: Essential baselines to compare against — if A4 is brighter than A1, the supplementation worked.
Expected Outcome
I expect fluorescence in the mRFP1 wells to follow the order A4 > A3 > A2 > A1,
with the combined GMP + Cysteine well giving the best result. The mScarlet-I wells
(B1–B4) are expected to be brighter than their mRFP1 counterparts overall, given
mScarlet-I’s faster maturation and higher intrinsic brightness. However, the supplements
may also boost mScarlet-I fluorescence - comparing B1 vs B4 will reveal whether these
additions benefit fast-maturing proteins as much as slow-maturing ones like mRFP1.