Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Answer: On average, raw meat is ~20–25% protein by weight. If we assume 20%, then in a 500 g sample we have:
500 × 0.20 = 100 g protein
Since 1 g ≈ 6.022 × 10^{23} Daltons, then: 100 g = 6.022 × 10^{25} Daltons
If the average amino acid has a mass of ~100 Daltons, then: Number of AAs = (6.022 × 10^{25}) / 100 = 6.022 × 10^{23}
So there are approximately 6 × 10^23 AAs!
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Answer: When we eat beef or fish, we are not incorporating their cells into our bodies. During digestion, proteins are broken down into amino acids, and DNA is broken down into nucleotides by enzymes in the stomach. What gets absorbed into our bloodstream are these small building blocks - not intact cells, genes, or whole DNA molecules. There is no natural mechanism in our body that takes DNA from food and integrates it into our genome. For DNA to become part of our cells, it would need very specific enzymes and delivery systems (like viruses use), which do not occur through normal digestion. So biologically, we are not “becoming” cow or fish; we are breaking their biomolecules down and reusing the basic components to build and maintain our own human cells.
3. Why are there only 20 natural amino acids?
Answer: There are 20 natural amino acids, likely because evolution settled on a set that is chemically diverse enough to build all necessary protein structures, while still being manageable for the translation machinery. Each amino acid has a different side chain. With 20 options, we already get a very wide range of chemical behaviors, which is enough to create stable, functional, and highly specific proteins.
The genetic code is built around triplet codons (64 possible codons), and these encode 20 amino acids plus stop signals. Expanding the number significantly would require major changes to the ribosome, tRNAs, and the entire translation system. It is possible that once this system became fixed early in evolution, it was strongly conserved.
4. Can you make other non-natural amino acids? Design some new amino acids.
Answer: Yes, we can design non-natural amino acids. Synthetic biology already does this by engineering new tRNAs and enzymes that insert artificial amino acids into proteins. To design one, I would keep the same backbone so the ribosome can still recognize it, and modify only the side chain. For example, a fluorescent amino acid that allows direct visualization of proteins.
5. Where did amino acids come from before enzymes that make them, and before life started?
Answer: Before life existed, amino acids were formed by simple chemical reactions on early Earth. The early atmosphere had small molecules like methane, ammonia, hydrogen, and water. Energy from lightning, heat, and UV radiation drove reactions between them and produced amino acids. This was shown in the Miller–Urey experiment. Amino acids were also found in meteorites, so some may have formed in space.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Answer: L-amino acids and D-amino acids are stereoisomers, or mirror images of each other. Natural proteins are made of L-amino acids, and they form right-handed α-helices. If we build an α-helix using D-amino acids instead, the geometry is mirrored. So I would expect the helix to be left-handed.
7. Can you discover additional helices in proteins?
Answer: Yes, additional helices can be discovered both experimentally and computationally. Experimentally, techniques like X-ray crystallography, NMR, and cryo-EM allow us to determine protein structures and identify helical regions. Computationally, secondary structure prediction algorithms and structure prediction tools can analyze amino acid sequences and detect potential α-helices or other helical motifs.
8. Why are most molecular helices right-handed?
Answer: Most biological helices are right-handed because life is built mainly from L-amino acids. The stereochemistry of L-amino acids restricts the backbone angles in a way that makes the right-handed α-helix energetically more favorable and sterically stable.
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
Answer: β-sheets tend to aggregate because their backbone hydrogen bond donors and acceptors are exposed and can easily form hydrogen bonds with neighboring β-strands from other molecules. When multiple β-strands align, they form extended intermolecular hydrogen-bonding networks, which are very stable. The main driving force for β-sheet aggregation is hydrogen bonding between backbones, together with the hydrophobic effect - hydrophobic side chains pack together, which further stabilizes the aggregate.
Part B: Protein Analysis and Visualization
1. Briefly describe the protein you selected and why you selected it.
Answer: Nav1.7 is a voltage-gated sodium channel located in neuronal membranes that initiates and propagates action potentials by allowing Na⁺ ions to enter the cell in response to changes in membrane voltage. It plays a crucial role in controlling the excitability of nociceptor neurons and is therefore essential for the sensory perception of pain. This protein demonstrates a clear relationship between 3D structure and electrical function and has significant physiological and clinical relevance. I was particularly drawn to it because of my interest in understanding how the brain works and because its structural properties provide an opportunity to explore the mechanisms underlying neuronal signaling.
2. Identify the amino acid sequence of your protein:
Here is the AA sequence of Nav1.7:
As shown in the sequence, the Nav1.7 complex includes auxiliary beta subunits that regulate channel function (chains B and C). For the purposes of this assignment, I will focus on chain A, which corresponds to the alpha subunit forming the pore and voltage-sensing domains of the channel!
- How long is it? 2031 AAs!
- What is the most frequent amino acid? L, Leucine, which appears 202 times.
- How many protein sequence homologs are there for your protein? There are 250 protein sequence homologs identified in UniProtKB using BLAST (I took sequences with percent identity > 78%).
- Does your protein belong to any protein family? Yes, according to UniProt, it belongs to the sodium channel (TC 1.A.1.10) family.
3D Protein Visualization
For this part, I first downloaded the .pdb file of the Nav1.7-beta1-beta2 complex from RCSB PDB. I decided to include the entire structure, not just chain A, so that we can see the protein’s main form. After downloading the file, I went to PyMOL and loaded it there.
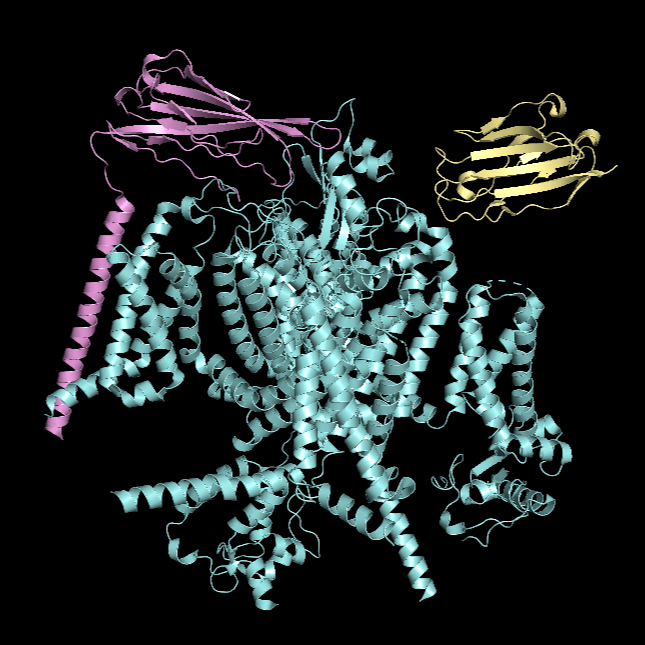
Cartoon Visualisation
*Chain A is colored in cyan, B in magenta, and C in yellow.
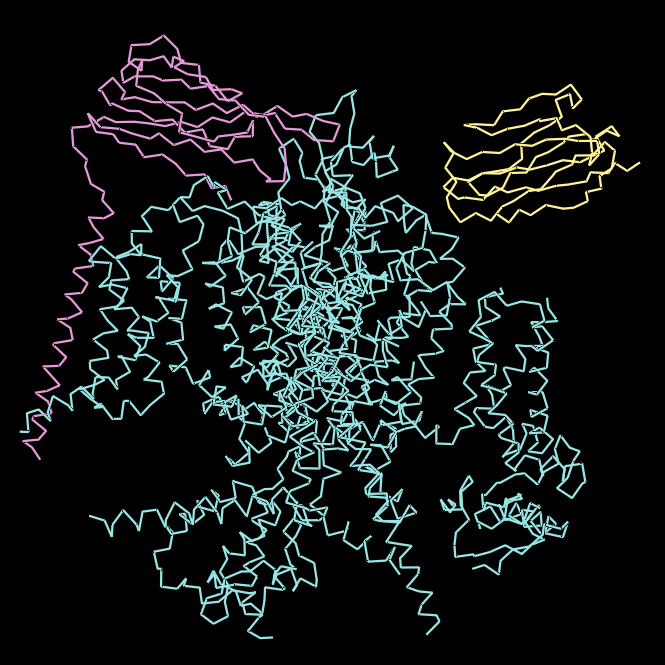
Ribbon Visualisation
*Chain A is colored in cyan, B in magenta, and C in yellow.
Ball and Stick Visualisation
*Chain A is colored in cyan, B in magenta, and C in yellow.

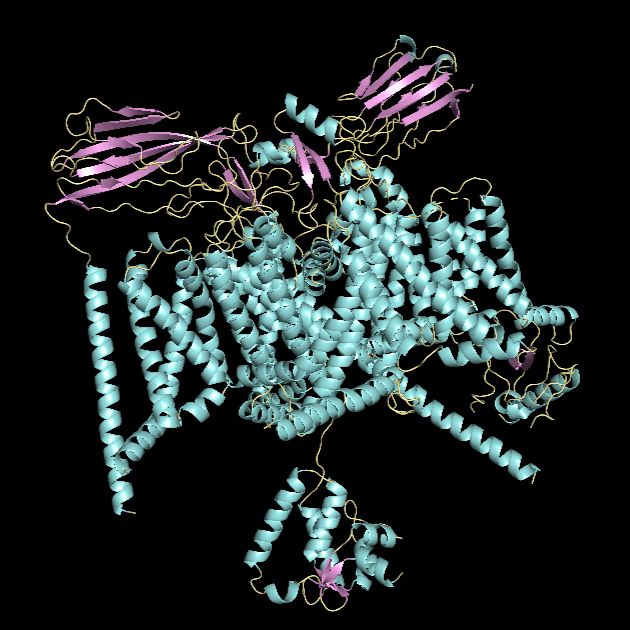
α-helices vs. β-sheets
*α-helices are colored in cyan, β-sheets in magenta, and loops in yellow.
The quaternary structure has a significantly greater number of α-helices than β-sheets. Most of the helices are concentrated in chain A, whereas the majority of the β-sheets are located in chains B and C.
Hydrophobic vs Hydrophilic Residue
*Hydrophobic residues are colored in cyan and hydrophilic in magenta.
Hydrophobic residues are predominantly located in the interior of the complex and at inter-chain interfaces, forming a stabilizing core. In contrast, hydrophilic residues are more frequently exposed on the outer surface. The central region of the protein displays a substantial hydrophobic surface, which is consistent with its role as a transmembrane protein, as this region is embedded within the lipid bilayer, whose interior is hydrophobic.
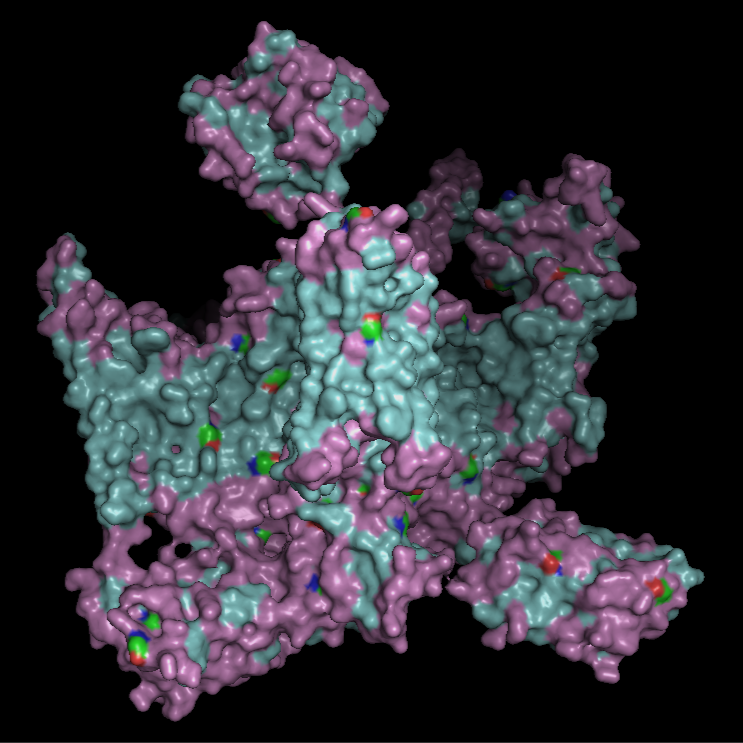
Surface Visualisation
The surface view shows that the protein has a roughly cross-like shape with visible cavities. There appear to be small holes or pockets that may help with binding to the membrane. These cavities are primarily located at the same level as the hydrophobic region embedded within the membrane, which is consistent with the protein’s transmembrane nature.
Part C. Using ML-Based Protein Design Tools
For this assignment, I wanted to choose once again Pro-Resilin, the highly elastic, rubber-like structural protein found in insects that I used in Homework 2. But unfortunatly I couldn’t find its PDB page, likely since it is mostly intrinsically disordered, so there is no experimental PDB structure (only prediction of AlphaFold).
So, I chose instead human alpha-thrombin (PDB ID: 1PPB), which is a serine protease in the blood coagulation cascade (a chain reaction of clotting proteins activated after vessel damage). It converts fibrinogen into fibrin, forming blood clots that seal damaged blood vessels following injury. Here is the link to its PDB page. The structure was solved by X-ray crystallography at 1.92 Å resolution.
Here is the protein sequence:
1PPB_2|Chain B[auth H]|ALPHA-THROMBIN (LARGE SUBUNIT)|Homo sapiens (9606) IVEGSDAEIGMSPWQVMLFRKSPQELLCGASLISDRWVLTAAHCLLYPPWDKNFTENDLLVRIGKHSRTRYERNIEKISMLEKIYIHPRYNWRENLDRDIALMKLKKPVAFSDYIHPVCLPDRETAASLLQAGYKGRVTGWGNLKETWTANVGKGQPSVLQVVNLPIVERPVCKDSTRIRITDNMFCAGYKPDEGKRGDACEGDSGGPFVMKSPFNNRWYQMGIVSWGEGCDRDGKYGFYTHVFRLKKWIQKVIDQFGE 1PPB_1|Chain A[auth L]|ALPHA-THROMBIN (SMALL SUBUNIT)|Homo sapiens (9606) TFGSGEADCGLRPLFEKKSLEDKTERELLESYIDGR
Since it has two chains, I’m going to choose chain B for this exercise.
C1. Protein Language Modeling
1. Deep Mutational Scans *To improve readability, I modified the x-axis to display every 10th residue instead of labeling every position.
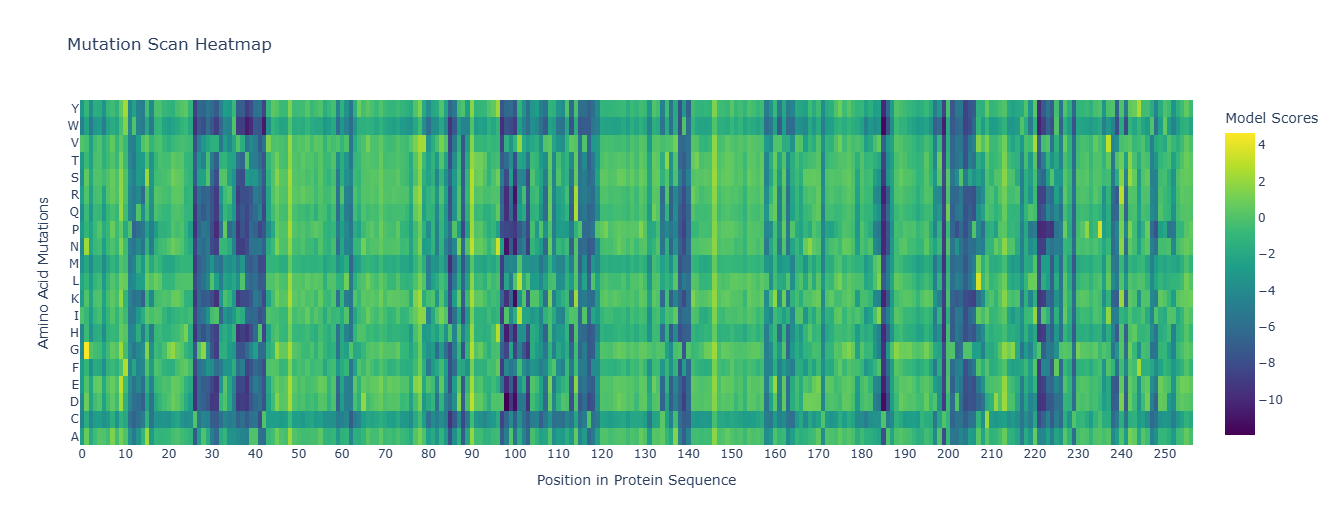
The heatmap shows how evolutionarily “allowed” each possible mutation is at each position in thrombin, according to an LLM for proteins. One clear pattern appears at position 26, where nearly all substitutions are strongly disfavored by the model (dark colors), indicating that this residue is likely structurally important and evolutionarily constrained. Interestingly, substitution to cysteine is comparatively less unfavorable than other mutations. This may suggest that a cysteine at this position could sometimes be tolerated, potentially due to structural compatibility or possible disulfide stabilization in related proteins.
2. Latent Space Analysis
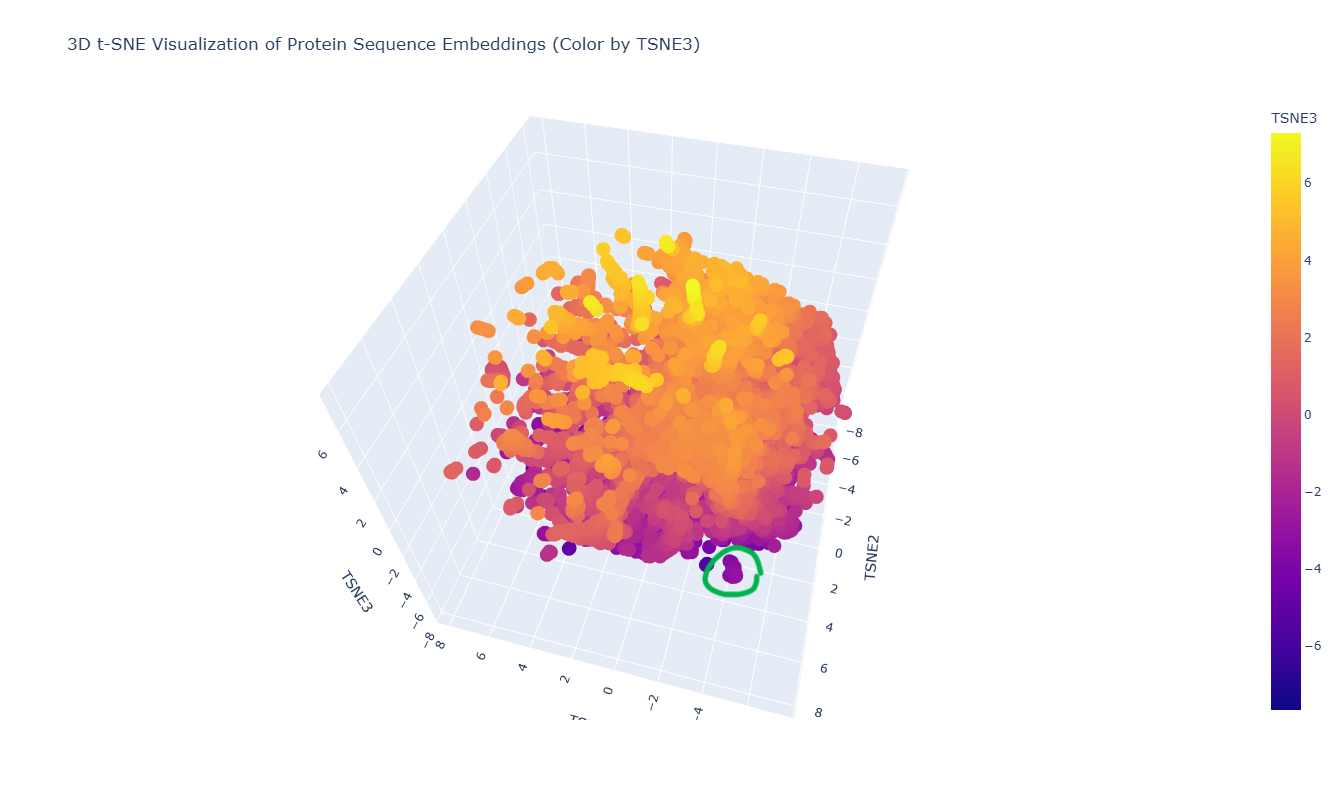
The 3D t-SNE visualization represents protein sequence embeddings generated by ESM2 and reduced to three dimensions. Each point corresponds to a protein domain from the SCOPe/ASTRAL dataset, and the spatial proximity between points reflects similarity in their learned embedding representations. Because t-SNE preserves local relationships, nearby points are expected to correspond to proteins that share sequence or structural similarities.
At first glance, the embedding space appears as one large, dense cluster rather than clearly separated groups. This suggests that many of the proteins in the dataset occupy a continuous similarity space rather than forming sharply distinct families. However, upon zooming in, smaller local neighborhoods become visible, indicating finer-grained organization within the global cluster.
For example, in the distinct local cluster highlighted in green in the figure, the neighboring proteins include:
- Mciothiol-dependent maleylpyruvate isomerase (Corynebacterium glutamicum)
- Hypothetical protein NMA1147 (Neisseria meningitidis)
- PhoU homolog TM1734 (Thermotoga maritima)
Although their annotations differ, these proteins are all bacterial in origin and belong to related structural or metabolic functional classes. Their proximity in embedding space suggests that ESM2 captures shared structural folds or evolutionary relationships beyond simple sequence identity. Even the hypothetical protein may share structural motifs with annotated metabolic proteins, explaining its placement within the same neighborhood.
Overall, the latent space organization indicates that ESM2 embeddings encode biologically meaningful information: proteins with similar structural domains or evolutionary backgrounds tend to cluster locally, even if their functional annotations are not identical. The continuous nature of the embedding space further reflects the gradual evolutionary relationships between protein families rather than strictly discrete categories.
In the 3D t-SNE map, thrombin (highlighted in green in the figure) is located within a distinct local cluster rather than being isolated.
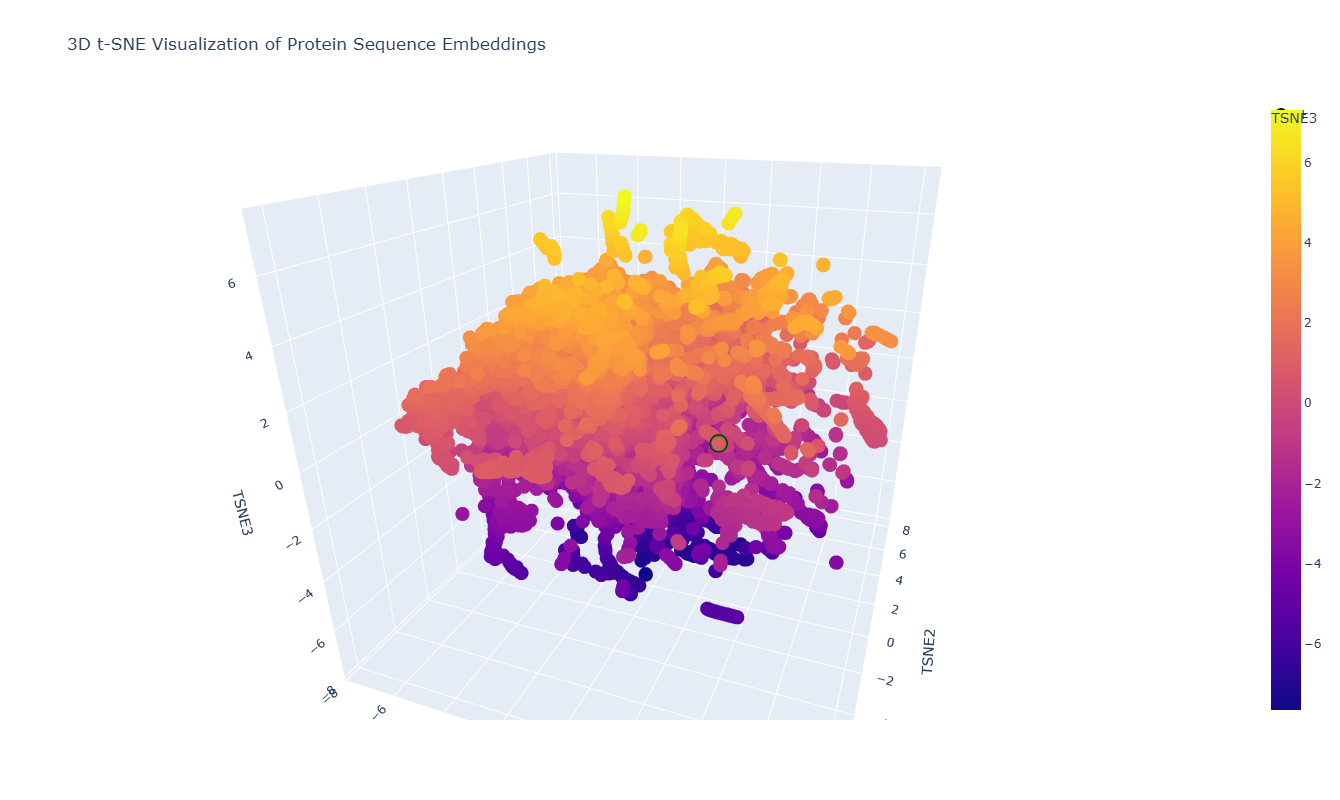
On closer inspection, this neighborhood includes proteins such as:
- Mannan-binding lectin serine protease 2 (MASP-2), catalytic domain (human)
- Complement C1s protease, catalytic domain (human)
- Other human serine protease domains
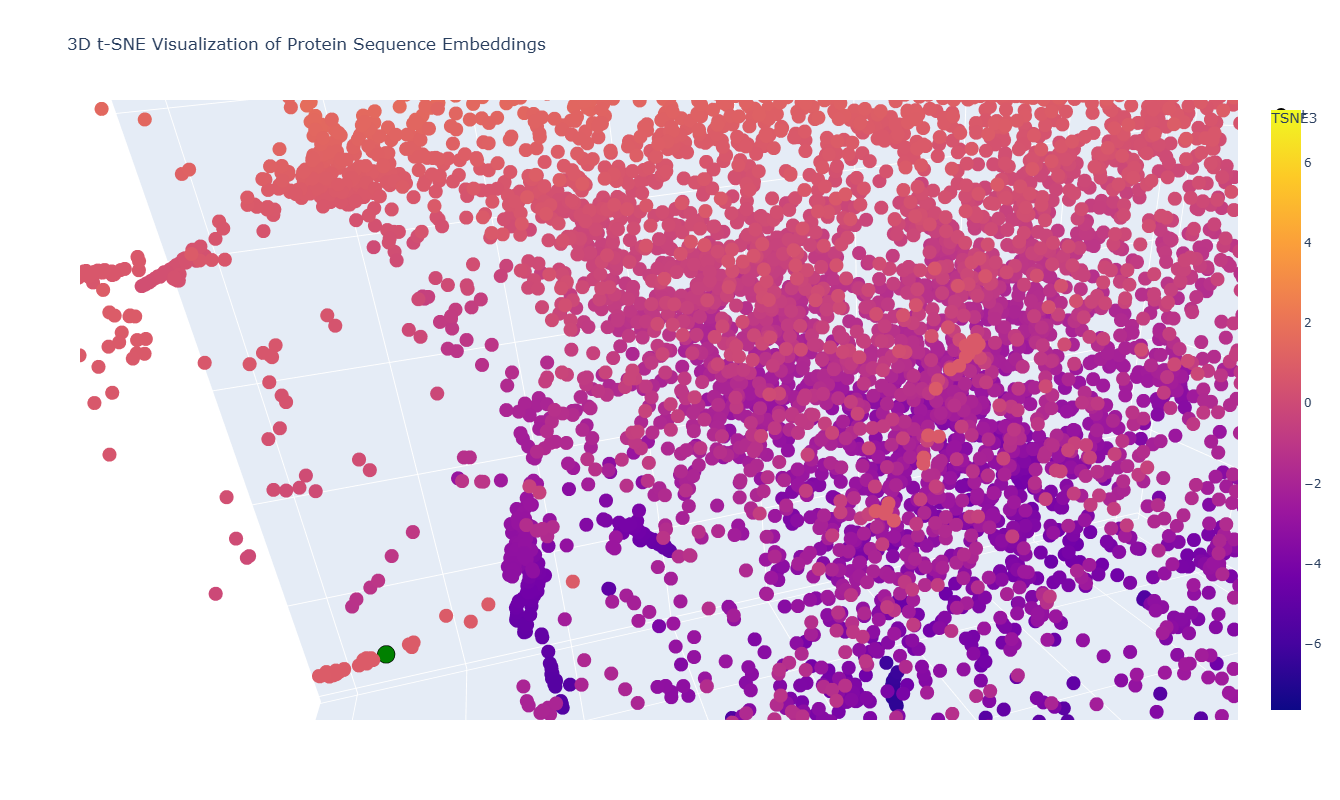
This makes sense because thrombin is a trypsin-like serine protease, and these neighboring proteins share the same catalytic fold and similar active-site architecture. Their proximity in the embedding space suggests that the model captures underlying structural and evolutionary similarities between these proteases.
C2. Protein Folding
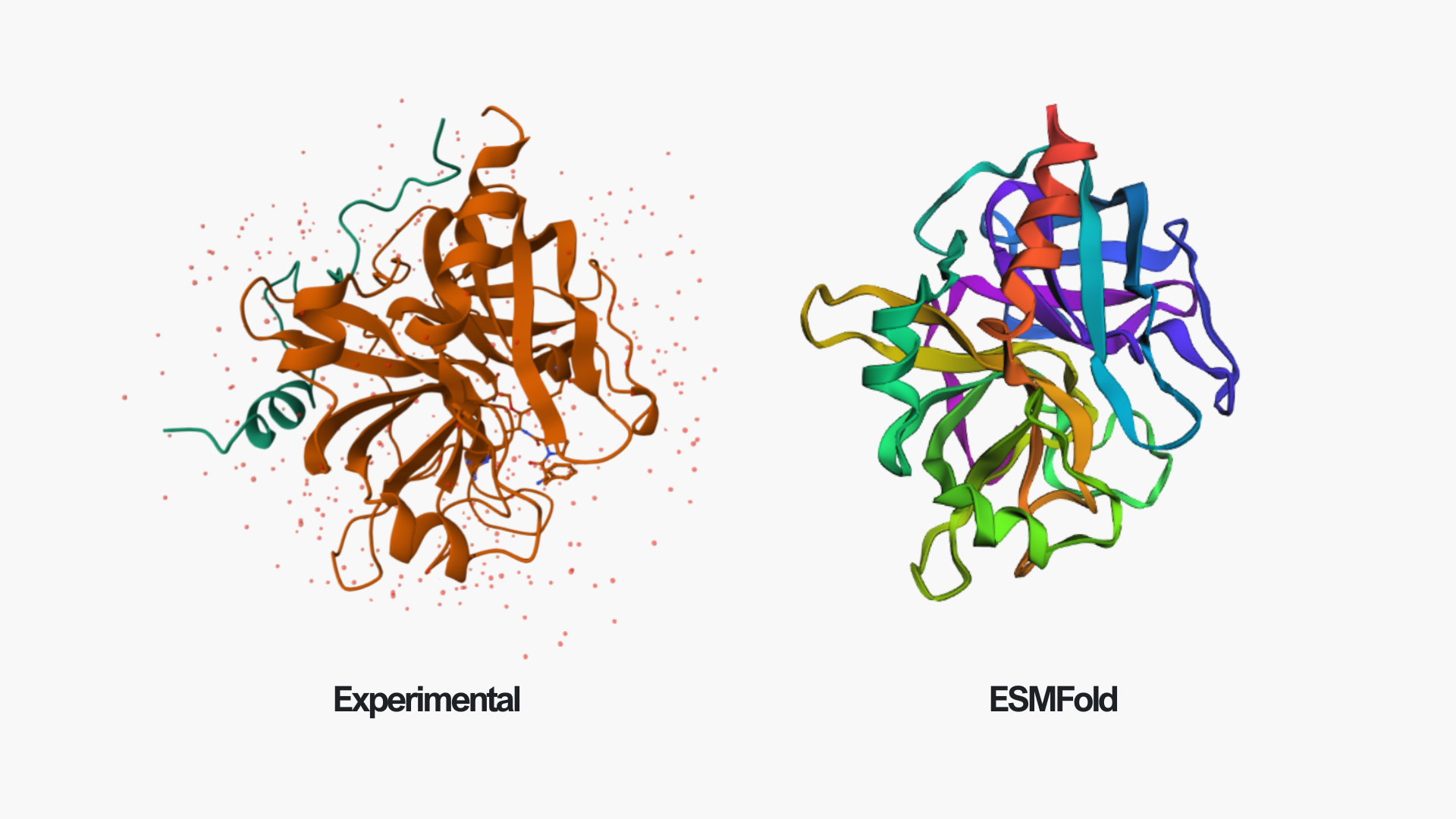
In the above picture, you can see the experimental structure on the left and the ESMFold structure on the right. They look very similar overall. The main difference is chain A (colored in green in the experimental structure), which I did not include in the ESMFold prediction. This likely also contributes to some small differences, since removing a chain can slightly affect the overall folding and interactions. However, the overall fold and structure are highly similar between the two models.
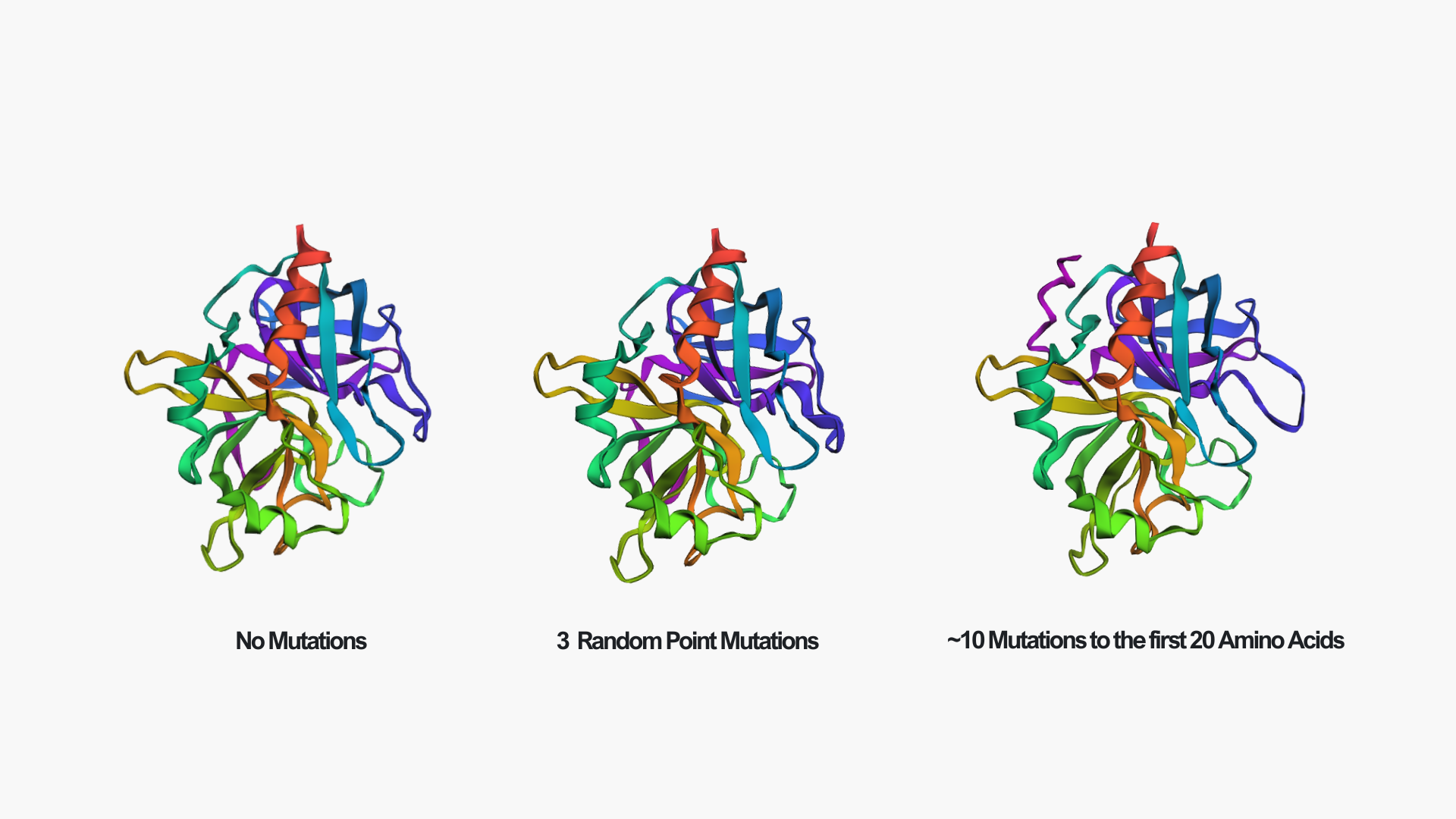
Creating three random point mutations (replacing one amino acid with another) led to the structure shown above. The mutated structure is incredibly similar to the original one. These few mutations did not have a significant effect on the overall fold of the protein, suggesting that thrombin is structurally resilient to small sequence changes.
After making ~10 mutations in just the first 20 amino acids, it seems the structure started to be affected. You can see this clearly in the mutated model by the extra pink strand/loop on the top left, and the overall shape looks a bit more distorted compared to the no-mutation structure. So thrombin seems pretty resilient to a few point mutations, but once you start mutating a larger chunk (specifically at the N-terminus), the predicted fold begins to change noticeably.
C3. Protein Generation
Here is the predicted sequence I got running the ProteinMPNN: IVGGRDAAPGEAPWFAQLIRKNPRQLIGSGALISDTWVITAAHNLYYPKINLNLKPDDIFIRLGALSRTEVEEGVEVIKTVKEIVIHPDYDAENNLDKDIALIKLEEPVEYSEYIRPIDLPNKEIAEALLKPGNMGRVVGWGNLRNCSVPGSGPCLPTKAQVLEVPLVPREVCEASTDRKMTDNQFCAGFLPGEGKRGSACGGDSGGAFTILNPVDGRWYLMGIVSWGEXGCDDPGKFDIFTNVYKLMPWIDSVINAEPE
ProteinMPNN generated a redesigned thrombin sequence that is similar in length and retains many motifs typical of trypsin-like serine proteases (including conserved glycines/cysteines and the overall pattern of hydrophobic vs. polar residues). This makes sense because ProteinMPNN is designing a sequence to fit the same backbone coordinates. One position was returned as an “X” (unknown token) (I added this option since the code raised an error every time I ran it), which would need to be replaced with a standard amino acid before using the sequence experimentally. For the next part, I will replace X with A since it is conservative.
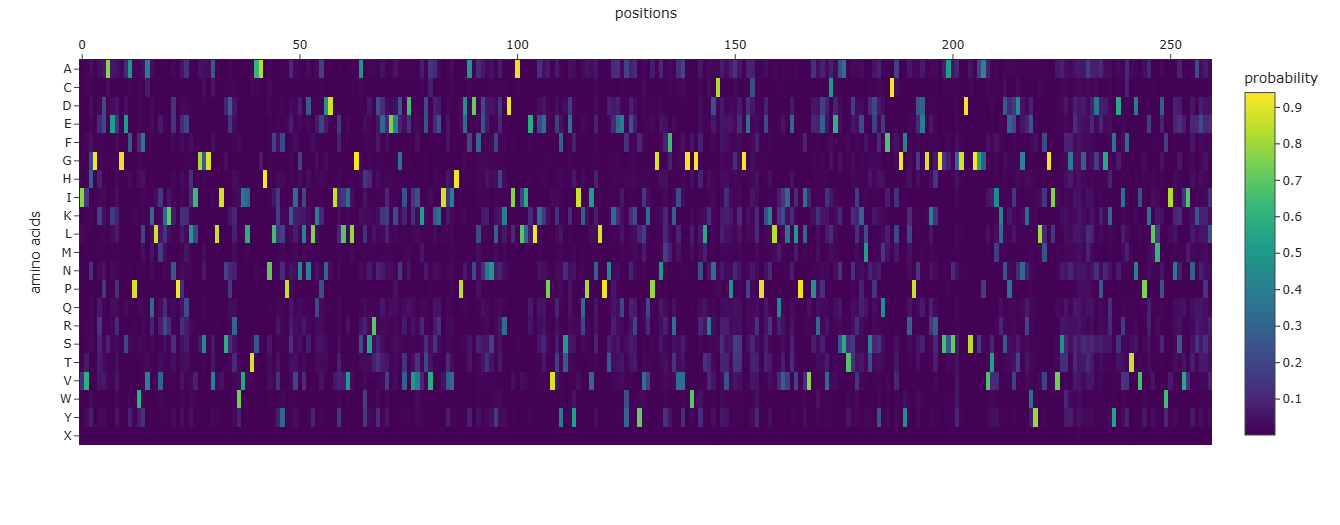
From the heatmap, most positions show one amino acid with a very high probability (bright yellow), while the rest are close to zero. This means ProteinMPNN is quite confident about the residue choice at many positions, suggesting the backbone strongly constrains the sequence.
There are also regions where the probabilities are more spread out (more green/blue across multiple residues). These positions are likely more tolerant to mutation and structurally less constrained.
Comparing the predicted sequence to the original thrombin sequence, many residues differ. However, the overall pattern still looks like a serine protease (conserved glycines, cysteines, and hydrophobic residues in core regions).
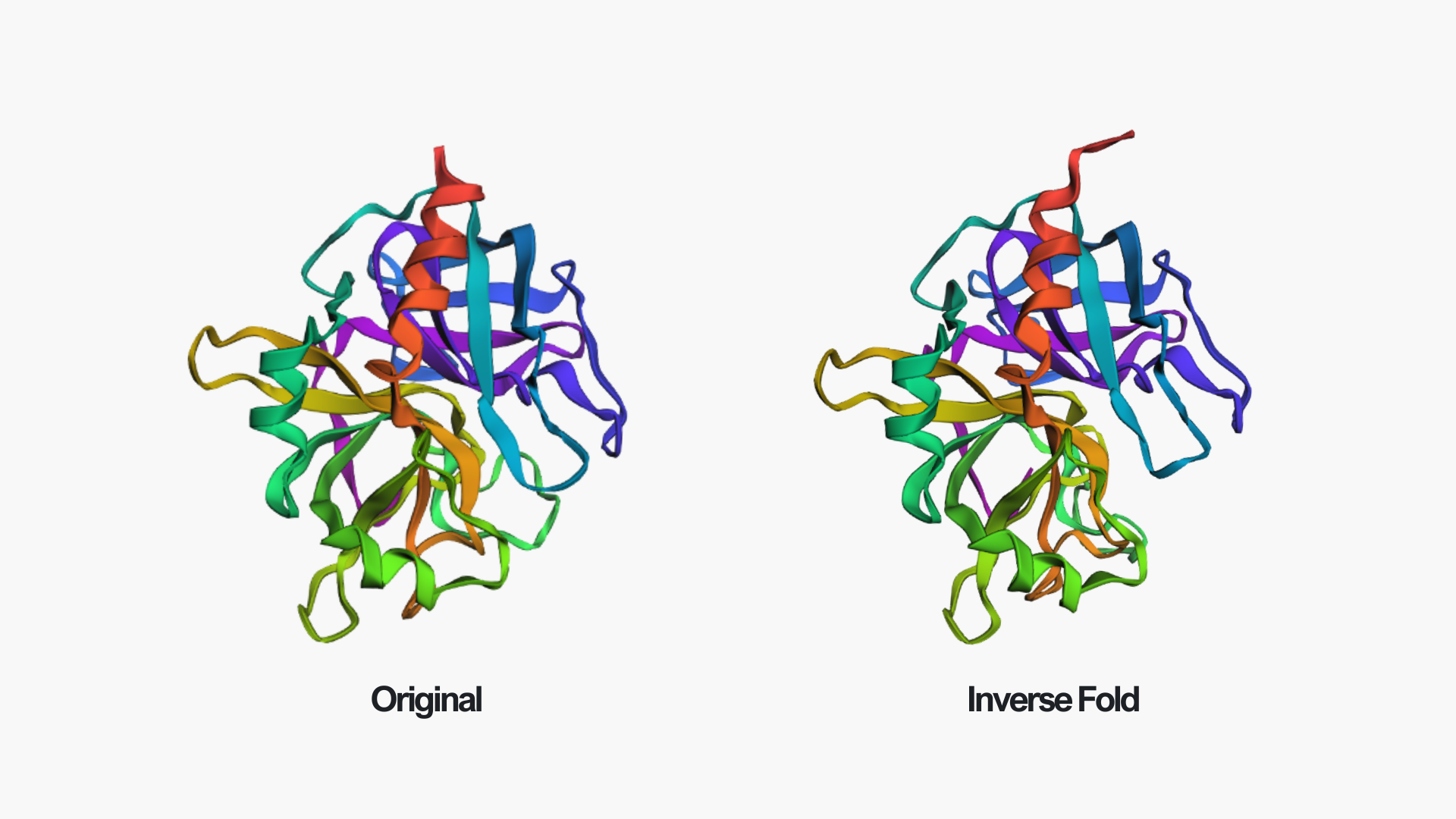
After folding the ProteinMPNN-designed sequence with ESMFold, the predicted structure is almost identical to the original thrombin structure. The overall fold, including the β-sheet core and surrounding helices, is preserved. Only minor local differences can be seen, for example near the edge of the red helix and in some loop regions.
This suggests that the redesigned sequence is fully compatible with the original backbone and that ProteinMPNN successfully generated a sequence that maintains the thrombin fold.