Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design
Part 1: Generate Binders with PepMLM
Searching the human SOD1 (P00441) page on UniProt, I found its amino acid sequence:
sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2 MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Introducing the A4V mutation to this sequence, we get:
sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2 MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
*Pay attention that the actual place of the Alanine is the 5th, maybe we should start counting at 0.
Using the PepMLM Colab, I generated the following 4 peptides of length 12 aa, which are conditioned to the mutant SOD1 sequence:
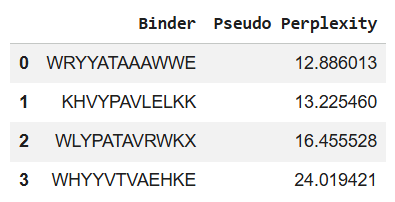
*You can see the perplexity scores that indicate PepMLM’s confidence in the binders in the table above too.
To these four peptides, we now add the known SOD1-binding peptide FLYRWLPSRRGG for comparison (control).
Part 2: Evaluate Binders with AlphaFold3
I then moved to the AlphaFold Server, where I submitted, for each peptide separately, the mutant SOD1 sequence, followed by the peptide sequence as a separate chain to model the protein-peptide complex.
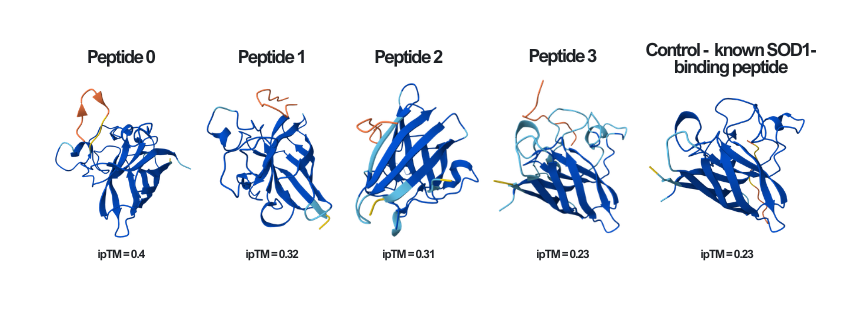
Here you can see the results of the modeling of all of the different peptides (including the known SOD1-binding peptide - the control), as well as their ipTM score, side by side:
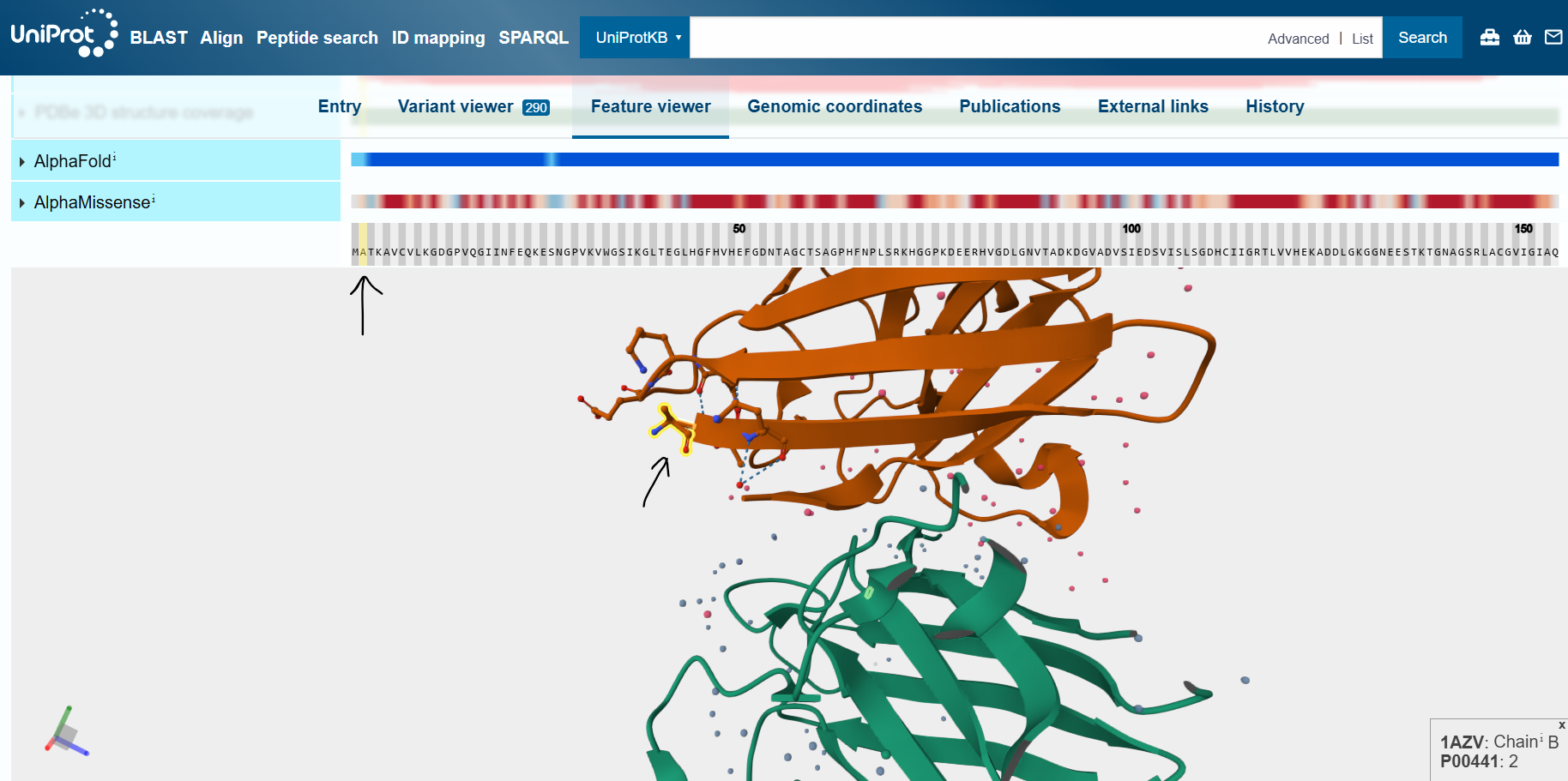
By inspecting the wild-type human SOD1 protein in UniProt’s feature viewer feature, I identified the N- and C-termini of the mutant protein.
The ipTM (Interface Predicted Template Modeling) score in AlphaFold measures the accuracy of the predicted relative positions of the subunits within the complex. Values higher than 0.8 represent confident, high-quality predictions, while values below 0.6 suggest a likely failed prediction. ipTM values between 0.6 and 0.8 are a gray zone where predictions could be correct or incorrect. In the following pictures, the peptides are colored in orange, and the mutant human SOD1 in blue.
Peptide 0
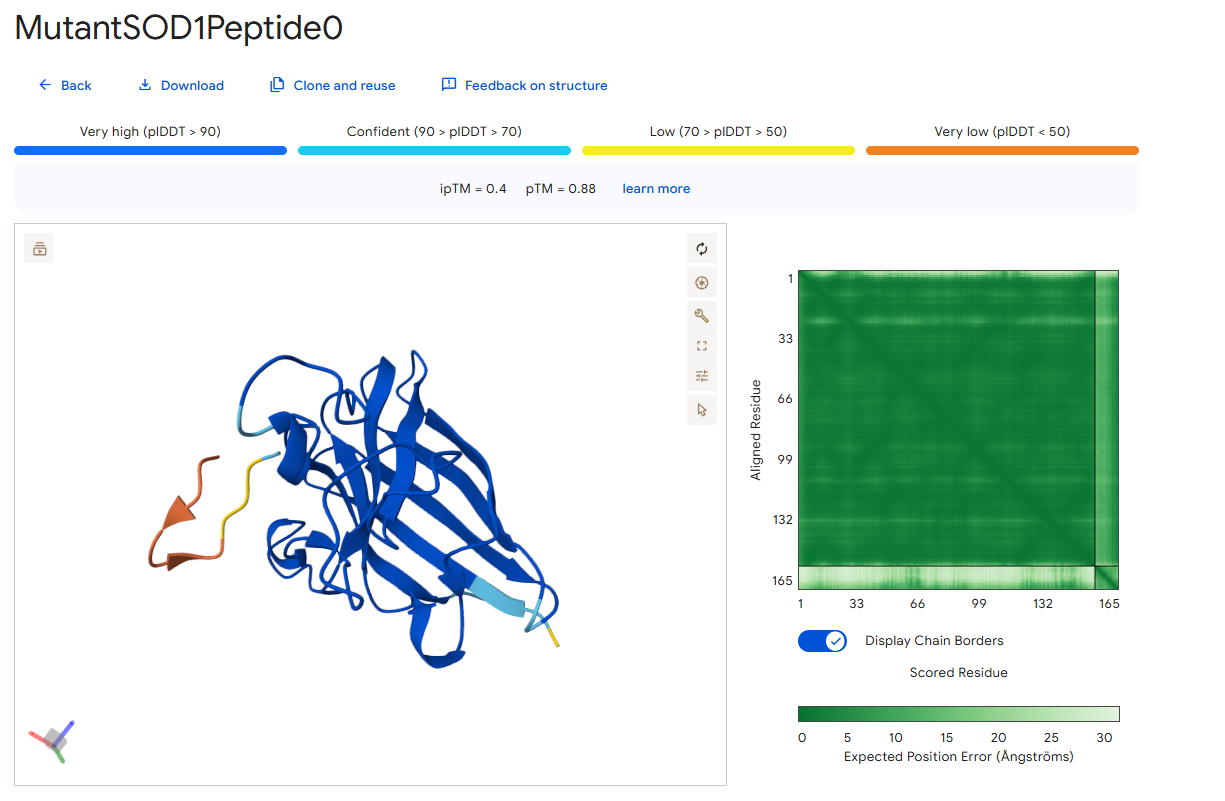
According to the AlphaFold simulation, the peptide does not bind close to the N-terminus, it doesn’t engage with the β-barrel region, doesn’t approach the dimer interface, and seems to be surface-bound.
Peptide 1
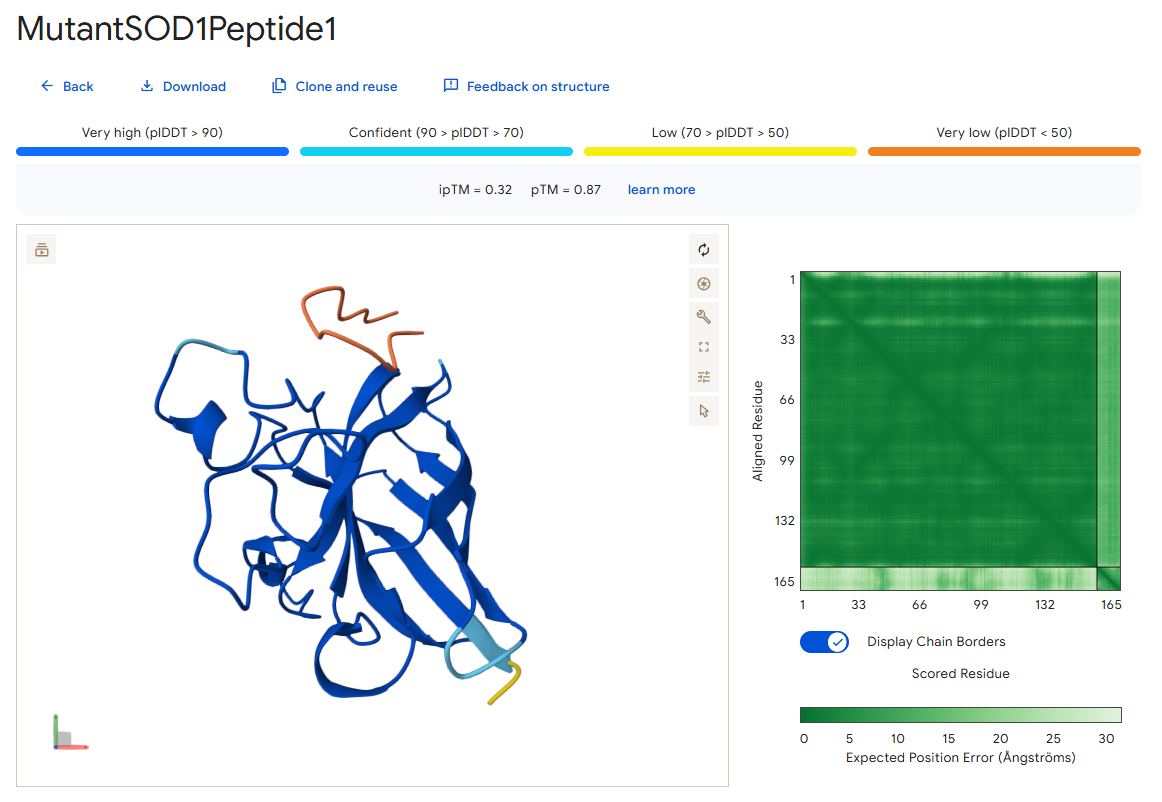
According to the AlphaFold simulation, the peptide does not bind close to the N-terminus, it engages with the β-barrel region on the side, doesn’t approach the dimer interface (very far from it), and seems to be surface-bound.
Peptide 2
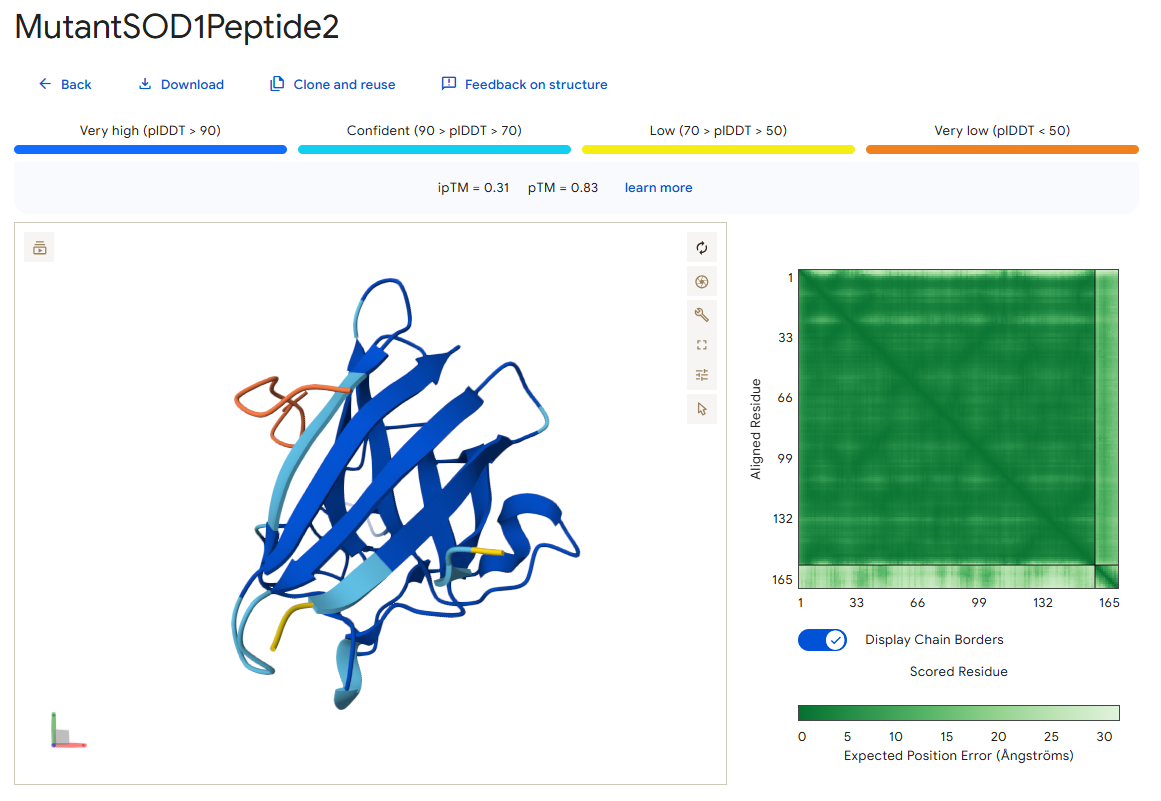
According to the AlphaFold simulation, the peptide binds pretty close to the N-terminus (at least compared to the previous two peptides), it does engage with the β-barrel region, doesn’t approach the dimer interface, and seems to be very lightly buried.
Peptide 3
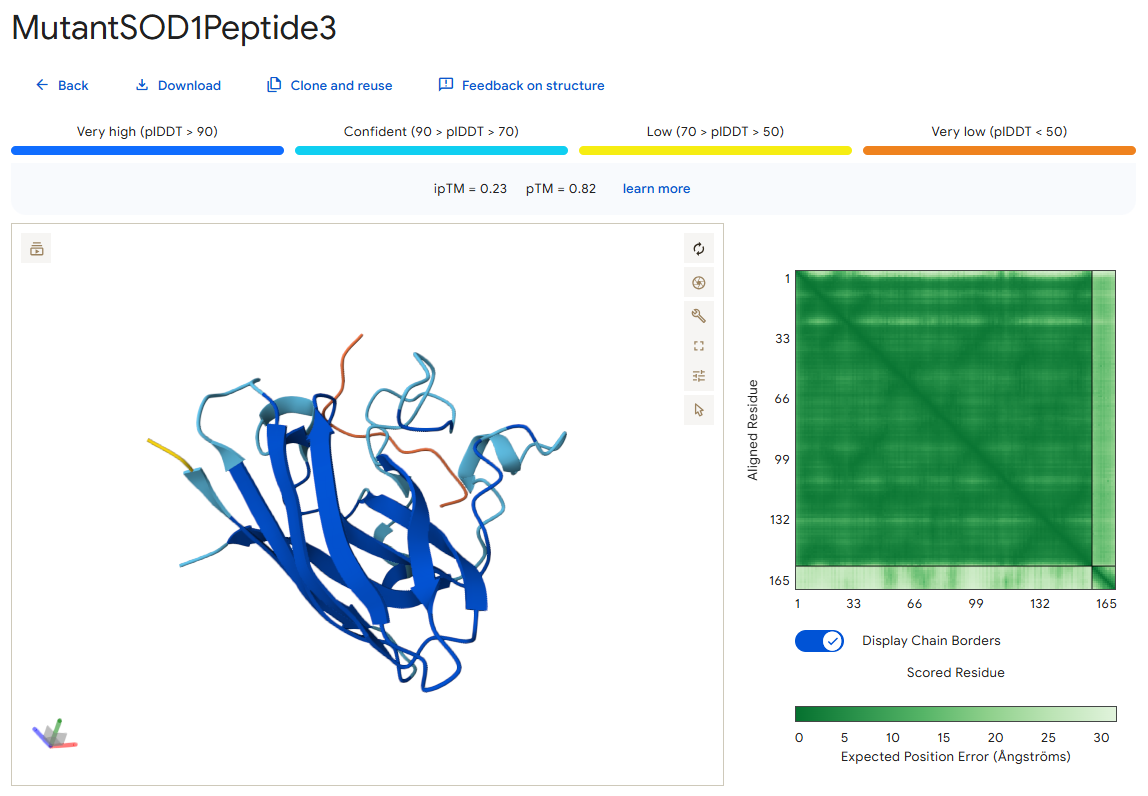
According to the AlphaFold simulation, the peptide does not bind close to the N-terminus, it doesn’t engage with the β-barrel region, doesn’t approach the dimer interface, and seems to be very lightly buried.
The known SOD1-binding peptide
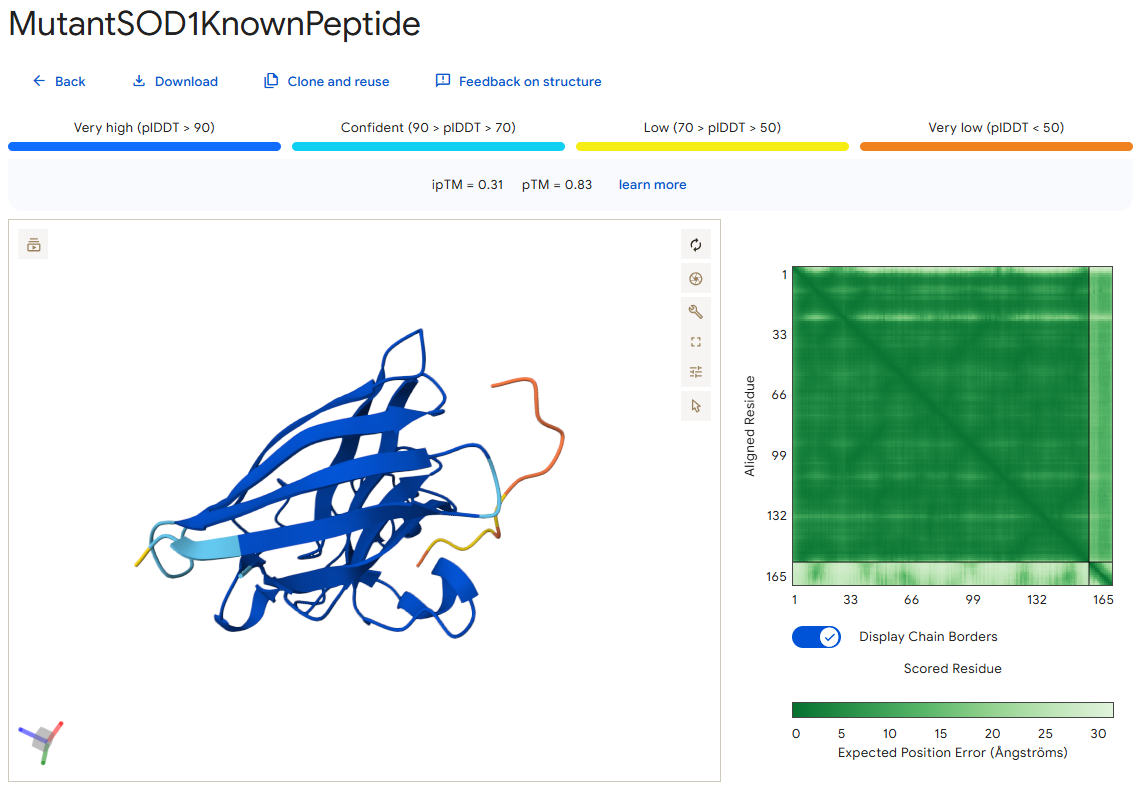
According to the AlphaFold simulation, the peptide does not bind close to the N-terminus, it engages with the β-barrel region from the “hole” part of it, doesn’t approach the dimer interface, and seems to be partially buried.
Surprisingly, except for peptide 3, all of the peptide sequences generated by PepMLM had higher ipTM values than the known peptide. For example, peptide0 reached an ipTM value of 0.4 compared to 0.31 for the known peptide. It is impressive that within a few minutes it was possible to identify peptides that might be better candidates for drug development than the currently known ones. However, all ipTM scores were below 0.6, meaning AlphaFold is not very confident in these predictions. This is also the case for the known SOD1-binding peptide, which we know binds experimentally. Therefore, the accuracy of these predictions cannot be confirmed without experimental testing.
Additionally, since the peptides bind far from the dimer interface, each SOD1 dimer could potentially bind two peptides, one on each chain. Moreover, because all peptides except peptide2 bind far from the N-terminus, they may not be specific to the mutant human SOD1 protein.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Using PeptiVerse, I evaluated the therapeutic properties of all of the peptids, and here are the results:
Peptide 0
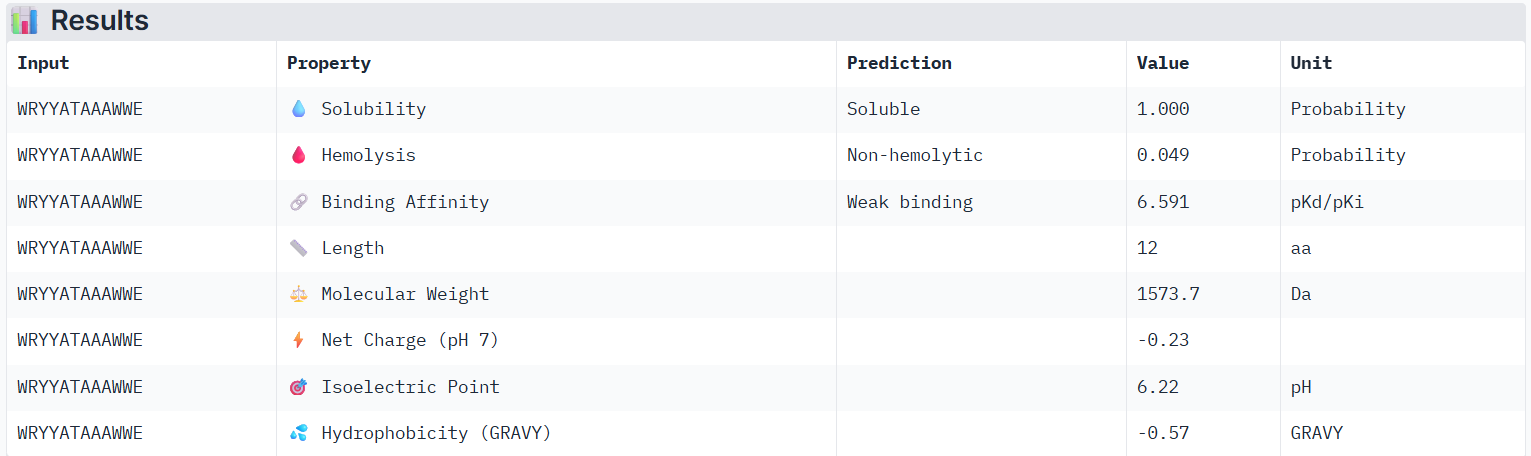
According to PeptiVerse’s prediction, peptide0 has a weak binding affinity, even though it had the highest ipTM score (0.4) in AlphaFold. It is also soluble and is non-hemolytic. A non-hemolytic peptide is a peptide that does not cause lysis of red blood cells, indicating that it is less likely to damage human cell membranes and therefore may have lower toxicity.
Peptide 1
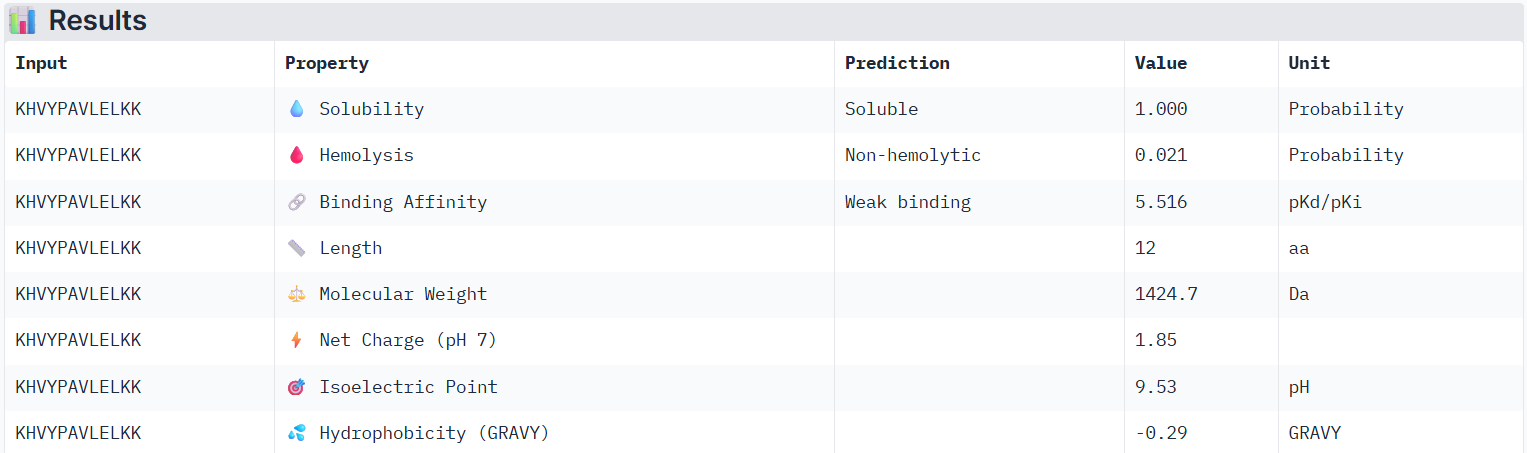
According to PeptiVerse’s prediction, peptide1 has a weak binding affinity, even weaker than peptide0, even though it had the second-highest ipTM score (0.32) in AlphaFold. It is also soluble and is non-hemolytic.
Peptide 2
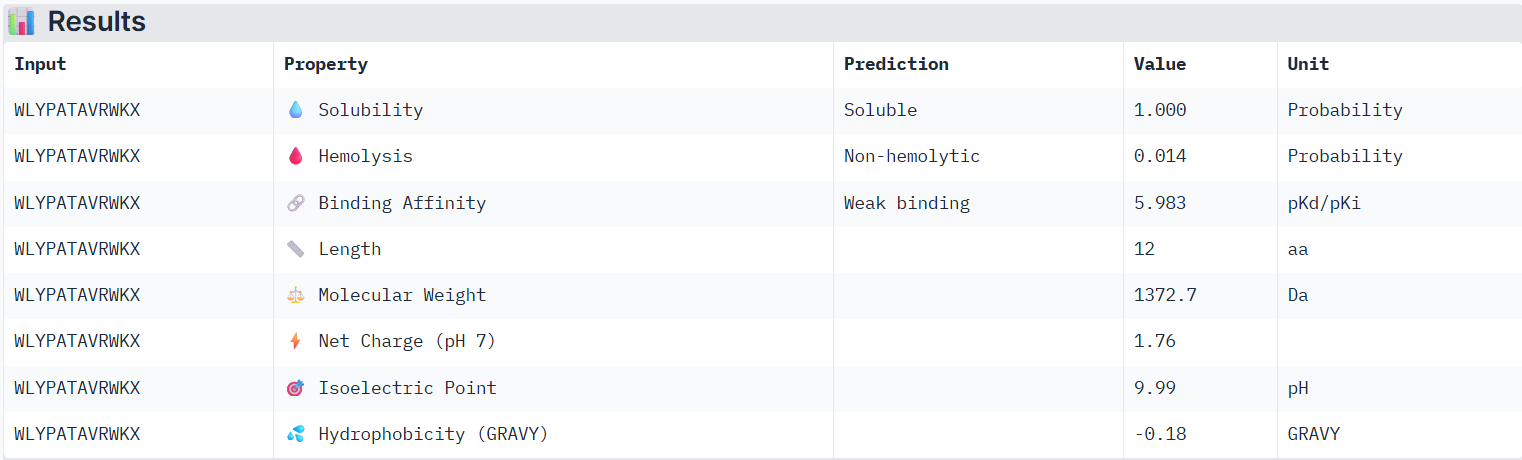
According to PeptiVerse’s prediction, peptide2 has a weak binding affinity (it had a 0.31 ipTM score on AlphaFold). It is also soluble and is non-hemolytic.
Peptide 3
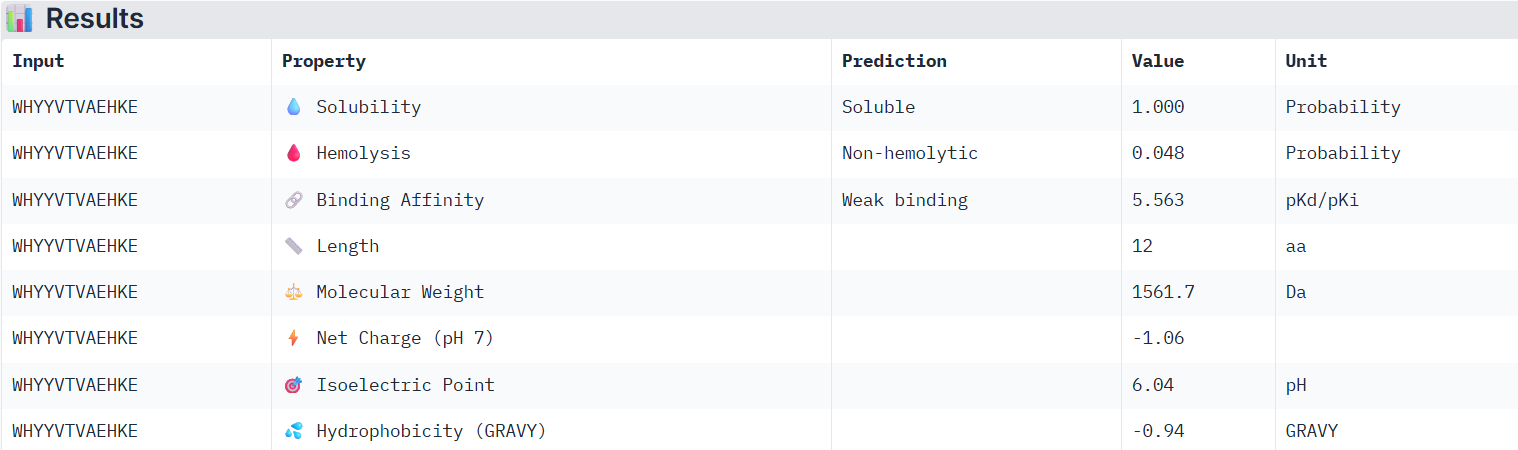
According to PeptiVerse’s prediction, peptide3 has a weak binding affinity, which matches with itslow ipTM score of 0.23 on AlphaFold. It is also soluble and is non-hemolytic.
The known SOD1-binding peptide
According to PeptiVerse’s prediction, the known peptide has a weak binding affinity (it had an ipTM score of 0.31 in AlphaFold), which is even weaker than the weak affinity predicted for PepMLM peptide0. This again suggests that we cannot fully determine whether a peptide will bind well to the human SOD1 protein until it is tested experimentally. The peptide is also predicted to be soluble and non-hemolytic.
For the next part, I will continue working with peptide0 WRYYATAAAWWE. Although it has an ipTM score below 0.6 (0.4) and is predicted to have weak binding affinity to the mutant human SOD1 protein, it still shows the best values among the generated peptides and even performs better than the known SOD1-binding peptide.
Part 4: Generate Optimized Peptides with moPPIt
In the moPPIT Colab, I pasted the mutated SOD1 sequence, chose a peptide length of 12 amino acids, and set the objectives and weights for the following properties:
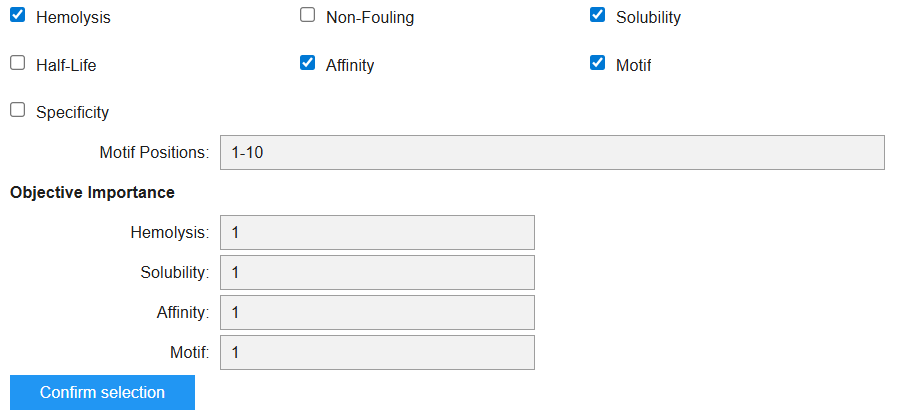
I then ran the model and it generated the following peptide:
Looking at the objectives I chose and their order, I was able to create the following table:
| Value | Property | Meaning |
|---|---|---|
| 0.9349 | Hemolysis | High probability of being non-hemolytic (???) |
| 0.75 | Solubility | Predicted fairly soluble |
| 5.72 | Affinity (pKd/pKi) | Weak binding |
| 0.6226 | Motif score | Moderate match to the motif constraint |
The moPPIT peptides differ from the PepMLM peptides because moPPIT generates peptides using controlled multi-objective optimization, while PepMLM mainly samples sequences that could bind the target protein. The PepMLM peptides were all predicted to be soluble, non-hemolytic, and to have weak binding affinity. In comparison, the moPPIT peptide also satisfies these constraints but shows lower predicted solubility, suggesting a trade-off between optimizing different properties such as motif matching and binding.
I also used AlphaFold to predict the binding of this peptide with the mutant SOD1 protein, and this is what I got:
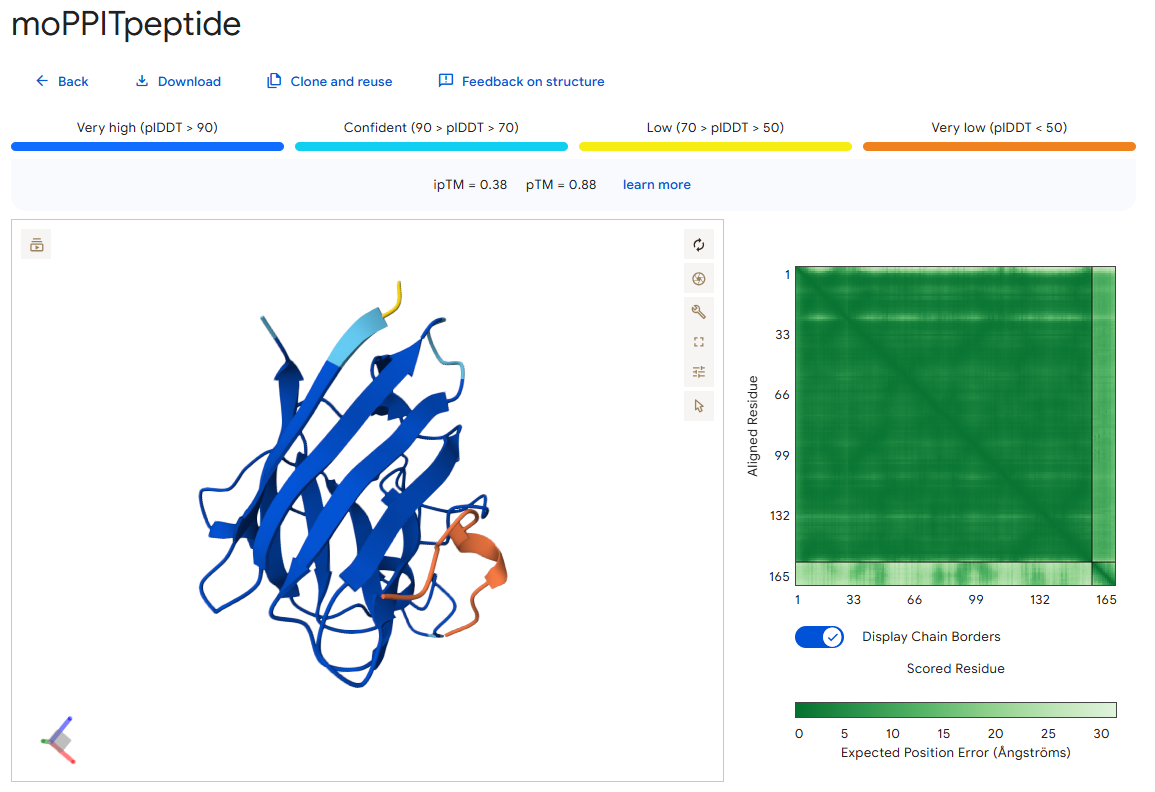
As you can see, it has an ipTM score of 0.38 and does not appear to bind close to the N-terminus, which was one of the objectives during its generation.
Before advancing these peptides toward clinical studies, I would first evaluate them computationally using tools such as AlphaFold and PeptiVerse to assess their structure and predicted binding affinity. Next, I would test them in in vitro experiments to measure binding strength, toxicity, and stability. If the results are promising, the peptides could then be tested in in vivo experiments to evaluate safety, pharmacokinetics, and therapeutic effectiveness.
Part C: Final Project: L-Protein Mutants
For this part of the assignment, I was not sure what were the actual tasks we had to do, so I hope I did everything we needed to do:)
L-Protein Engineering | Option 1: Mutagenesis
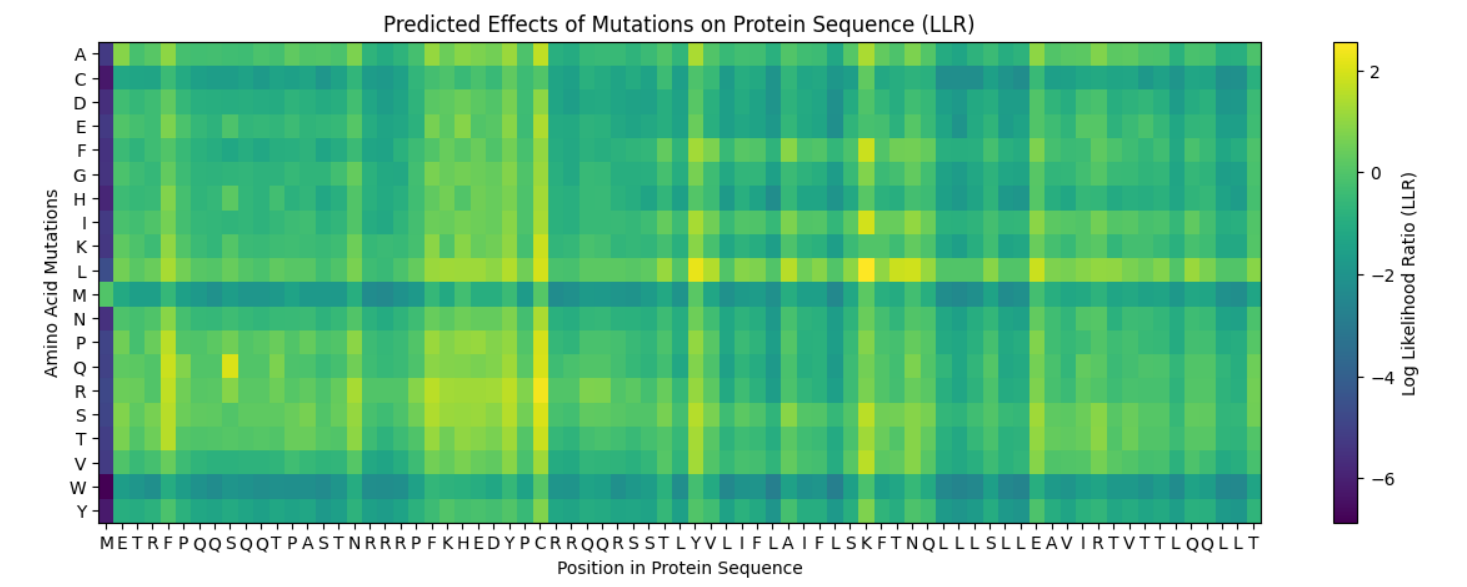
I ran the given notebook and got the following heatmap, representing the predicted effects of mutations on the lysis Protein Sequence: