Week 6 HW: Genetic Circuits Part I - Assembly Technologies
Assignment: DNA Assembly
Answer these questions about the protocol in this week’s lab:
- What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Ans:
| Component | Purpose |
|---|---|
| Phusion High-Fidelity DNA Polymerase | Synthesizes new DNA strands during PCR. It has proofreading activity, giving very low error rates and high fidelity. |
| dNTPs (deoxynucleotide triphosphates) | Building blocks (A, T, G, C) used to create new DNA strands. |
| HF or GC Buffer | Maintains the correct chemical environment (pH and salt conditions) for efficient enzyme activity. GC buffer helps amplify GC-rich templates. |
| MgCl₂ (Magnesium chloride) | Essential cofactor required for DNA polymerase activity. Helps the enzyme function properly. |
| DMSO (in some formulations) | Helps denature GC-rich DNA and reduces secondary structures, improving amplification of difficult templates. |
| Water | Used as the reaction medium to dissolve and mix all components. |
- What are some factors that determine primer annealing temperature during PCR?
Ans: Some important factors that determine the primer annealing temperature during PCR are:
| Factor | Effect on Annealing Temperature |
|---|---|
| Primer Length | Longer primers generally require higher annealing temperatures because they bind more strongly to the template DNA. |
| GC Content | Primers with higher GC content have higher melting temperatures since G-C pairs form three hydrogen bonds. |
| Melting Temperature (Tm) | Annealing temperature is usually set 3–5°C below the primer melting temperature. |
| Primer Specificity | Higher temperatures improve specificity by reducing non-specific binding. |
| Salt Concentration | Higher salt concentrations stabilize primer-template binding and can increase effective annealing temperature. |
| DNA Template Complexity | Complex or GC-rich templates may require higher annealing temperatures. |
| Presence of Additives | Compounds like DMSO can alter primer binding behavior and affect annealing temperature. |
| Primer Mismatches | Mismatches between primer and template may require lower annealing temperatures for binding. |
- There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Ans: Both PCR and restriction enzyme digestion can be used to generate linear DNA fragments, but they differ in mechanism, protocol, flexibility, and applications.
| Feature | PCR (Polymerase Chain Reaction) | Restriction Enzyme Digest |
|---|---|---|
| Basic Principle | Amplifies a specific DNA region using primers and DNA polymerase | Cuts DNA at specific recognition sequences using restriction enzymes |
| Main Components | Template DNA, primers, DNA polymerase, dNTPs, buffer | DNA sample, restriction enzyme(s), buffer |
| Procedure | Requires thermal cycling: denaturation, annealing, and extension | Usually a simple incubation at optimal enzyme temperature (commonly 37°C) |
| Specificity | Determined by primer design | Determined by restriction enzyme recognition sites |
| Flexibility | Very flexible; primers can target almost any region | Limited to naturally occurring or engineered restriction sites |
| Output | Produces amplified linear DNA fragments | Produces DNA fragments by cutting existing DNA |
| Time Required | Usually longer (~1–2 hours) | Typically shorter (~1 hour) |
| Accuracy | High-fidelity polymerases reduce mutations | Does not copy DNA, so no amplification errors |
| DNA Quantity | Can amplify tiny amounts of DNA into large quantities | Requires sufficient starting DNA |
| End Types | Usually blunt ends (with Phusion polymerase) | Can produce sticky ends or blunt ends depending on enzyme |
| Common Applications | DNA amplification, mutagenesis, cloning, diagnostics | Plasmid linearization, cloning, restriction mapping, fragment analysis |
PCR is preferable when a specific DNA fragment must be amplified, when only a small amount of template DNA is available, or when custom modifications need to be introduced using primers. Restriction enzyme digestion is preferable when precise cutting at known DNA sequences is needed, especially for cloning and plasmid manipulation.
In summary, PCR is best for amplification and flexibility, while restriction digestion is best for precise sequence-specific cutting of existing DNA molecules.
- How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Ans: To ensure that DNA fragments generated by PCR and restriction enzyme digestion are appropriate for Gibson cloning, the fragments must contain overlapping sequences that allow them to anneal and be joined together during the Gibson Assembly reaction.
| Requirement | Purpose |
|---|---|
| Overlapping DNA Ends | Adjacent DNA fragments should share 20–40 base pair overlapping sequences so they can anneal during Gibson Assembly. |
| Primer Design | PCR primers should be designed to add overlaps that match the neighboring DNA fragment or vector. |
| Correct Restriction Digest | Restriction enzymes should linearize the vector or generate fragments without removing required overlap regions. |
| High-Fidelity PCR | Using high-fidelity polymerase minimizes mutations in amplified DNA fragments. |
| Clean DNA Fragments | PCR products and digested DNA should be purified to remove enzymes, primers, and unwanted fragments. |
| Compatible Fragment Orientation | Overlaps must be designed in the correct orientation so fragments assemble properly. |
| Verify Fragment Sizes | Gel electrophoresis can confirm that DNA fragments are the expected size before assembly. |
| Avoid Secondary Structures | Overlap regions should avoid strong secondary structures or repetitive sequences that interfere with assembly. |
In Gibson cloning, the overlaps are the most important feature because they guide the assembly of multiple DNA fragments into a single continuous DNA molecule.
- How does the plasmid DNA enter the E. coli cells during transformation?
Ans:
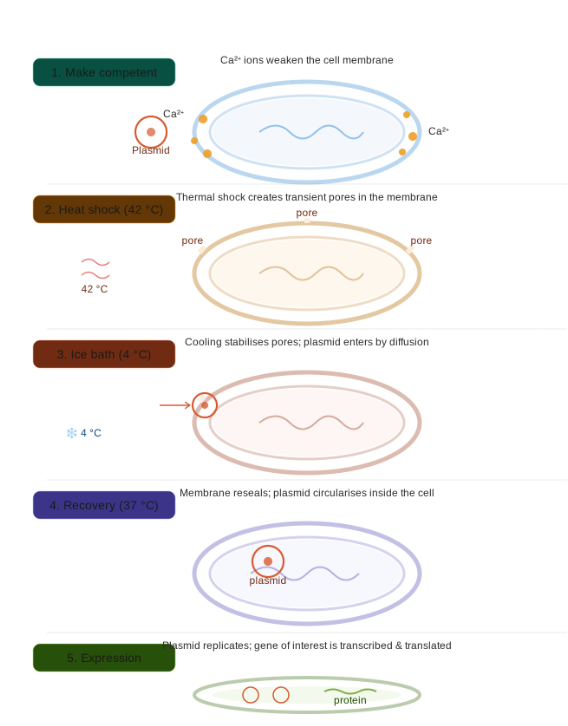
Walkthrough of the five-stage process shown in the diagram:
Making cells competent — E. coli is treated with cold calcium chloride (CaCl₂). Ca²⁺ ions associate with the negatively charged phosphate groups on both the cell membrane and the plasmid DNA, partially neutralising the repulsion between them and making the membrane more permeable.
Heat shock (42 °C, ~90 seconds) — A sudden temperature spike creates transient pores across the lipid bilayer. The thermal stress disrupts membrane packing, opening gaps large enough for DNA to pass through.
Ice bath (4 °C) — Rapid chilling halts further membrane disruption and stabilises the pores while they are still open. This temperature gradient is thought to drive a fluid “pressure differential” that helps pull DNA inward by diffusion.
Recovery (37 °C, ~45 min in rich broth) — The membrane reseals around any plasmid that entered. The plasmid circularises and begins to replicate using the cell’s own machinery. Antibiotic resistance genes on the plasmid are already being transcribed at this stage.
Expression — After plating on selective media, only cells that successfully took up the plasmid survive. The gene of interest is now transcribed into mRNA and translated into protein, completing the transformation.
The overall efficiency is low typically only 1 in 10,000–1,000,000 cells actually takes up a plasmid, which is why selection (usually antibiotic resistance) is essential to identify successful transformants.
- Describe another assembly method in detail (such as Golden Gate Assembly)
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Model this assembly method with Benchling or Asimov Kernel!
Ans: Golden Gate Assembly is a molecular cloning method used to join multiple DNA fragments together in a single reaction. It uses Type IIS restriction enzymes, such as BsaI, which cut DNA outside of their recognition sequence and generate custom overhangs. These overhangs allow DNA fragments to assemble in a specific order with high accuracy. During the reaction, the restriction enzyme cuts the DNA while DNA ligase simultaneously joins compatible fragments together. Because the recognition sites are removed after assembly, the final construct is seamless and cannot be cut again by the same enzyme. Golden Gate Assembly is highly efficient for assembling multiple genes or pathways in synthetic biology applications. It is commonly used in modular cloning systems and genetic circuit design.
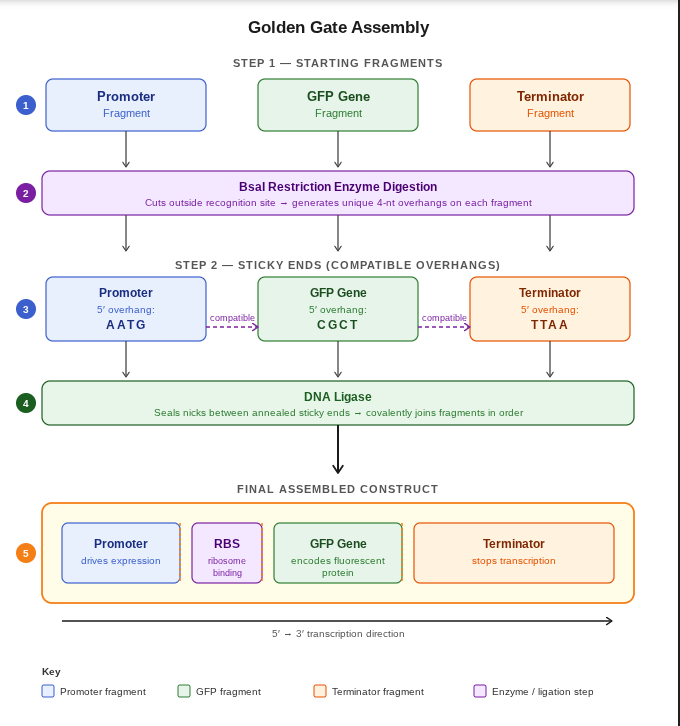 sketch by claudeai(sonnet 4.6)
sketch by claudeai(sonnet 4.6)
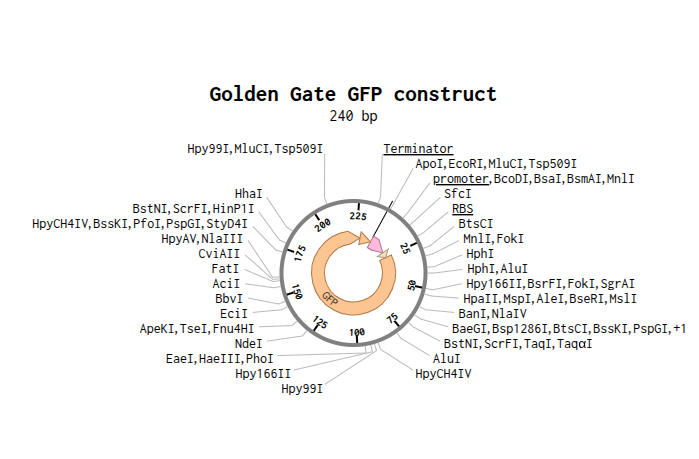
- This construct was modeled in Benchling to demonstrate Golden Gate Assembly. The sequence contains a promoter, ribosome binding site (RBS), GFP coding sequence, and terminator. Type IIS restriction enzymes such as BsaI cut outside their recognition site, creating sticky ends that allow the fragments to assemble in the correct order. DNA ligase then joins the fragments together into one complete construct. This method is efficient because digestion and ligation happen in the same reaction and multiple fragments can be assembled at once.
Assignment: Asimov Kernel
- Create a Repository for your work
Ans: Successfully created a repository for my work.
Create a blank Notebook entry to document the homework and save it to that Repository
Explore the devices in the Bacterial Demos Repo to understand how the parts work together by running the Simulator on various examples, following the instructions for the simulator found in the “Info” panel (click the “i” icon on the right to open the Info panel)
Create a blank Construct and save it to your Repository
Recreate the Repressilator in that empty Construct by using parts from the Characterized Bacterial Parts repository
Search the parts using the Search function in the right menu
Drag and drop the parts into the Construct
Confirm it works as expected by running the Simulator (“play” button) and compare your results with the Repressilator Construct found in the Bacterial Demos repository
Document all of this work in your Notebook entry - you can copy the glyph image and the simulator graphs, and paste them into your Noteboo
Ans: Created a blank construct and recreate the Repressilator using parts from the Characterized Bacterial Parts repository. Then I ran the simulator and compare the behavior with the Repressilator example from the Bacterial Demos repository.
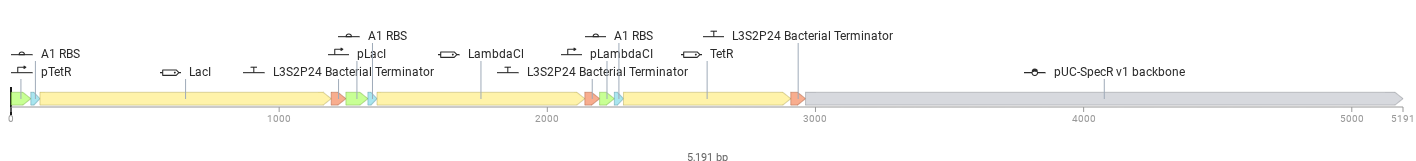
Parts Used
| Part | Function |
|---|---|
| pTetR | Promoter controlling LacI |
| A1 RBS | Ribosome binding site |
| LacI | Repressor protein |
| L3S2P24 Bacterial Terminator | Stops transcription |
| pLacI | Promoter controlling LambdaCI |
| LambdaCI | Repressor protein |
| pLambdaCI | Promoter controlling TetR |
| TetR | Repressor protein |
| pUC-SpecR v1 backbone | Plasmid backbone |
Below is the designed construct
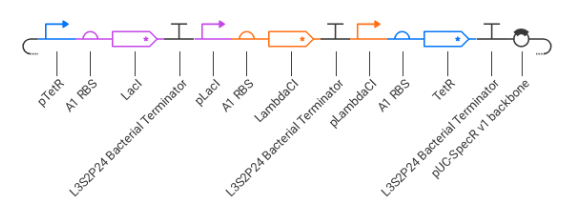
The repressilator was assembled as a cyclic repression circuit:
pTetR → LacI pLacI → LambdaCI pLambdaCI → TetR
Each protein represses another promoter:
TetR represses pTetR LacI represses pLacI LambdaCI represses pLambdaCI
This creates a feedback loop that regulates gene expression.
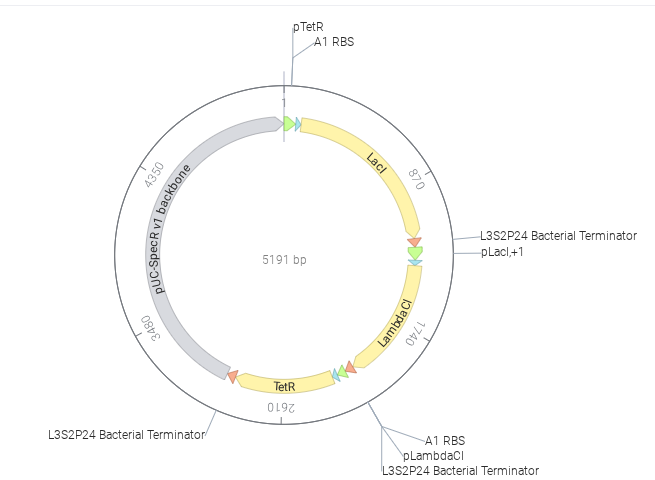
The simulator ran successfully with no errors. But the recreated repressilator did not show oscillatory behavior and instead reached a steady state. This may be due to incorrect regulatory connections, imbalance in promoter or repressor strengths, or insufficient delay in the feedback loop. As a result, one protein dominates and suppresses the others, preventing oscillation.
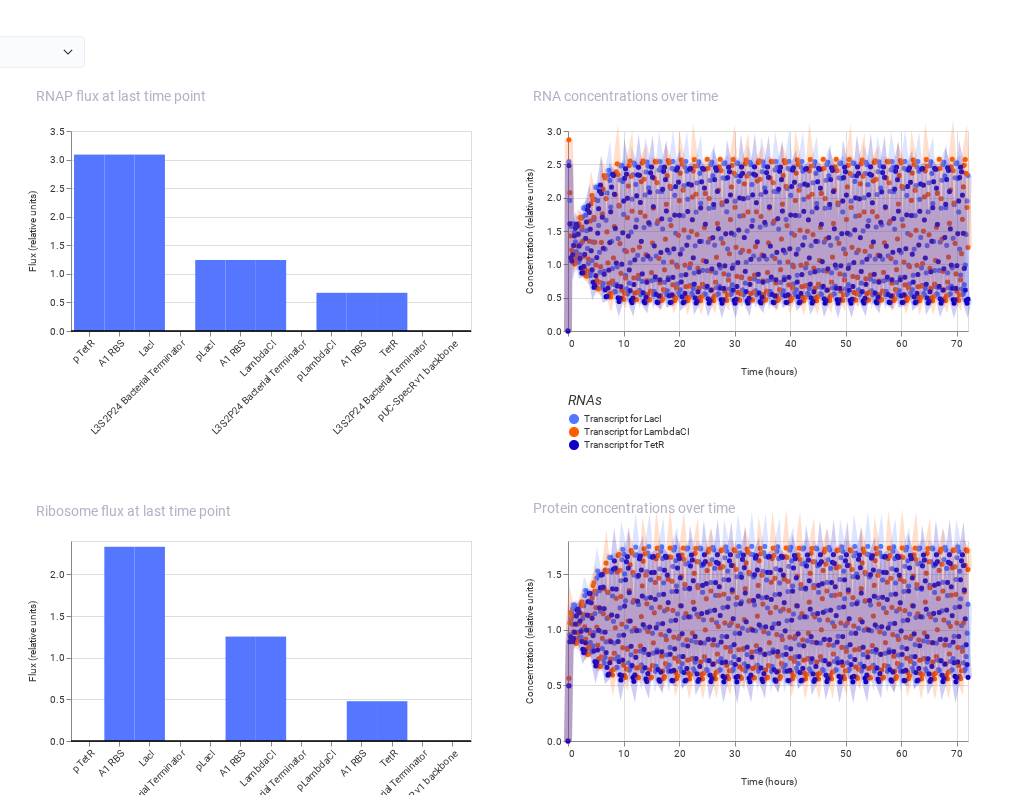
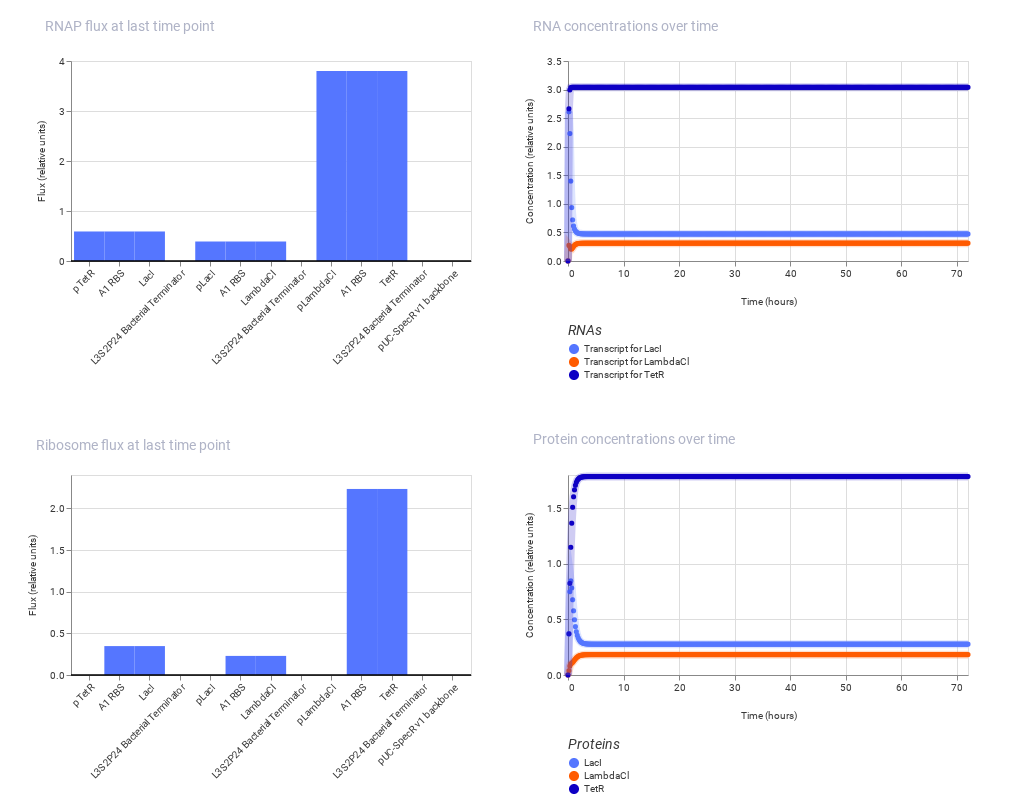
Note: The one on the above shows the result of Bacterial Demos repository and the below one shows the result of recreated repressilator.
Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo
Explain in the Notebook Entry how you think each of the Constructs should function
Run the simulator and share your results in the Notebook Entry
If the results don’t match your expectations, speculate on why and see if you can adjust the simulator settings to get the expected outcome
Ans: Construct 1:
This construct is designed for continuous gene expression using a promoter driving GFP. Since there is no regulatory element, the gene is expressed at a constant level.
Expected: Continuous, stable GFP expression.
Observed: The simulation shows a steady level of GFP over time, confirming constant expression.
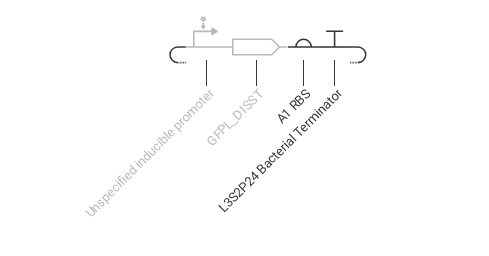
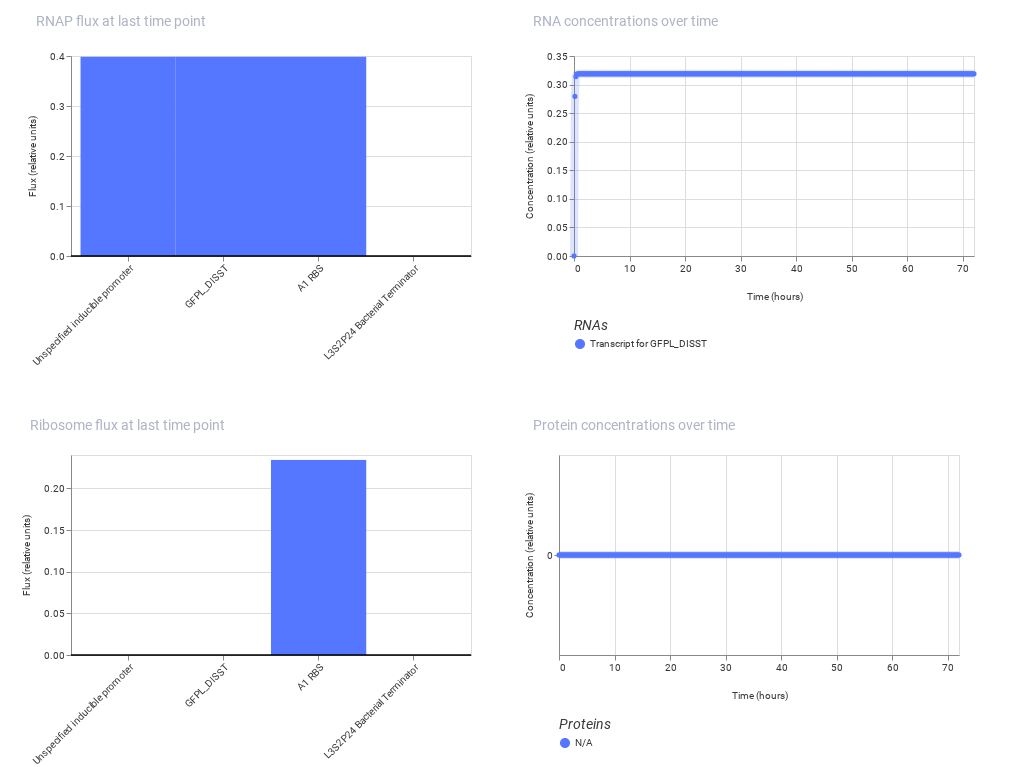
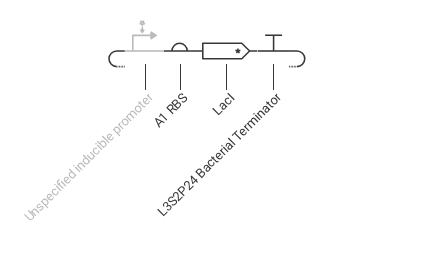
Construct 2: This construct demonstrates inducible gene expression, where a promoter responds to an external signal to control the production of a regulatory protein (LacI). The A1 ribosome binding site supports efficient translation of the protein.
Expected: LacI is produced only when the inducible promoter is activated.
Observed: The simulation shows increased production of LacI when the system is activated, indicating successful inducible expression.
This construct highlights how gene expression can be controlled by external signals.
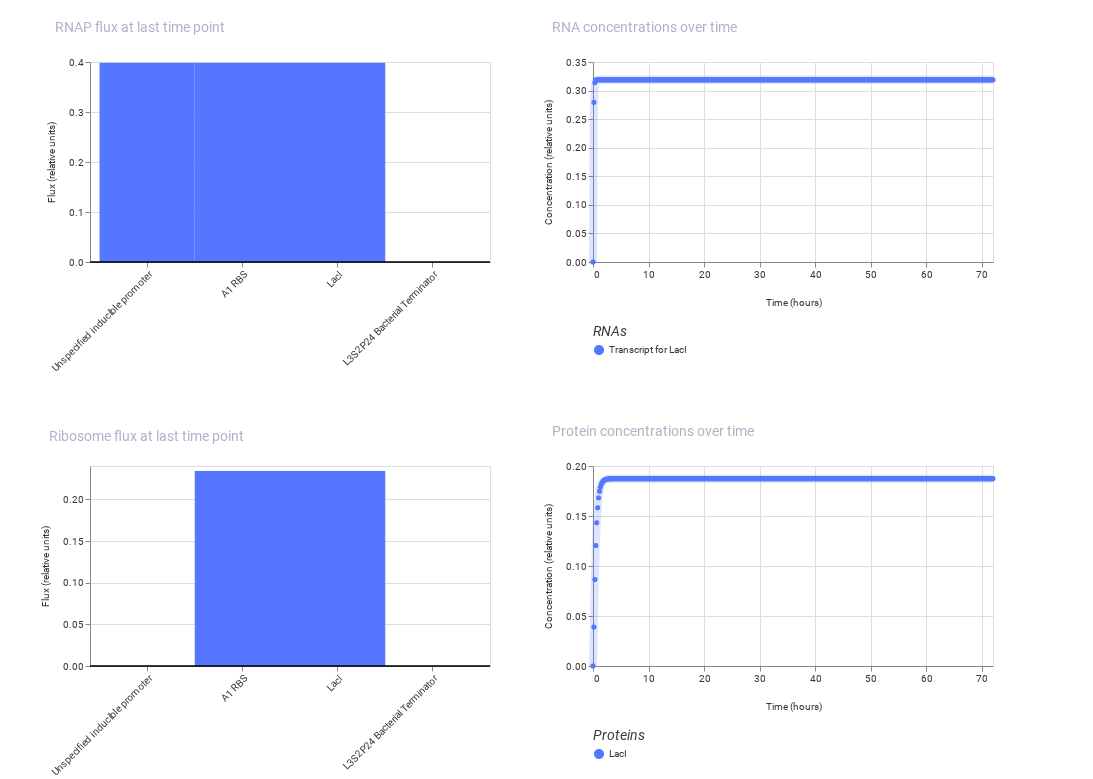
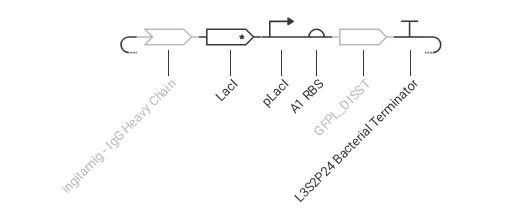
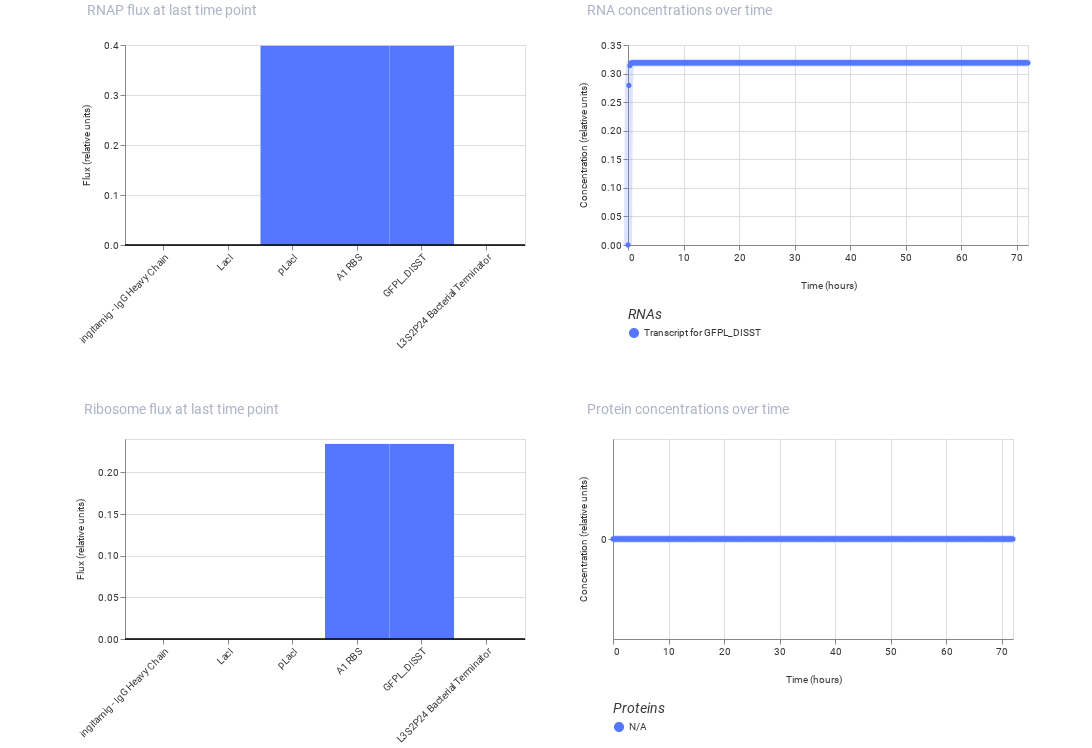
Construct 3: This construct models a molecular detection system using an IgG molecule as the input signal. The IgG activates a binding protein (modeled using LacI), which regulates a promoter controlling GFP expression.
Expected: The presence of IgG alters the activity of the regulatory protein, leading to a change in GFP expression.
Observed: The simulation shows variation in gene expression based on regulatory interactions, demonstrating a response to the molecular signal.