Week 4 HW: Protein Design Part 1
1
Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
If we assume the 500 g of meat is entirely protein, and take the average mass of one amino acid to be about 100 Da ≈ 100 g/mol, then:
Then multiply by Avogadro’s number:
So the answer is approximately 3 × 10^24 amino acid molecules.
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Humans do not become cows or fish after eating them because food is broken down during digestion. Proteins in beef or fish are digested into amino acids, fats into fatty acids, and carbohydrates into simple sugars. Your body then uses these small molecules as building blocks to make human proteins, cells, and tissues according to human DNA.
So, we do not directly turn cow tissue into human tissue. We first deconstruct the food, then rebuild it in our own biological way. In other words, we eat the raw materials, not the identity of the animal.
3. Why are there only 20 natural amino acids?
There are only 20 natural amino acids because this set is enough to make a wide variety of proteins while keeping the genetic system relatively simple and stable. They provide different chemical properties, such as size, charge, polarity, and shape, which allow proteins to fold and function in many ways. Evolution likely selected this set early because it worked well, and once the genetic code was established, it became highly conserved.
4. Can you make other non-natural amino acids? Design some new amino acids.
Yes, it is possible to make non-natural amino acids. Scientists can keep the standard amino acid backbone and change only the side chain to give it new properties. For example, they can design a fluorinated alanine by adding a fluorine atom to the side chain of alanine. This can change its electronic properties and hydrophobicity, and may also increase the molecule’s stability. Non-natural amino acids like this can help expand the functions of proteins.
5. Where did amino acids come from before enzymes that make them, and before life started?
Before life existed, amino acids were likely formed by prebiotic chemistry, meaning ordinary chemical reactions that do not require enzymes or living cells. Experiments such as the Miller–Urey type studies showed that amino acids can form from simple gases and energy sources like lightning. Scientists have also found amino acids and their precursors in meteorites, suggesting that some may have formed in space and later arrived on early Earth. Researchers have also demonstrated plausible amino-acid formation through hydrothermal, impact-driven, and other geochemical processes on the early Earth. So the best answer is that amino acids probably came from a combination of natural chemical reactions on Earth and delivery from space, before enzymes and life evolved.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
A helix made from D-amino acids would be expected to form a left-handed α-helix. This is because the common right-handed α-helix in proteins comes from L-amino acids. If you switch to the mirror-image D-amino acids, the preferred helix also becomes the mirror image, which is left-handed.
7. Can you discover additional helices in proteins?
Yes. Scientists can find extra helices in natural proteins using structure methods, and they can also design new helices by changing the amino acid sequence. But a new helix will only work if it fits the protein’s overall 3D structure — it may stabilize the protein, or it may disrupt it.
8. Why are most molecular helices right-handed?
Most molecular helices are right-handed because the shape of their building blocks makes that twist more stable. In biology, proteins are usually made from L-amino acids. When many L-amino acids fold into an α-helix, the right-handed helix avoids steric clashes better and has lower energy than a left-handed one. So it is simply the more stable shape.
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheets tend to aggregate because their extended strands can line up easily and form many backbone hydrogen bonds with each other. Hydrophobic side chains can also pack together and avoid water. So, the main driving force is intermolecular hydrogen bonding, helped by hydrophobic interactions, which makes the aggregated β-sheet structure more stable.
2
Protein Analysis and Visualization
- Briefly describe the protein you selected and why you selected it.
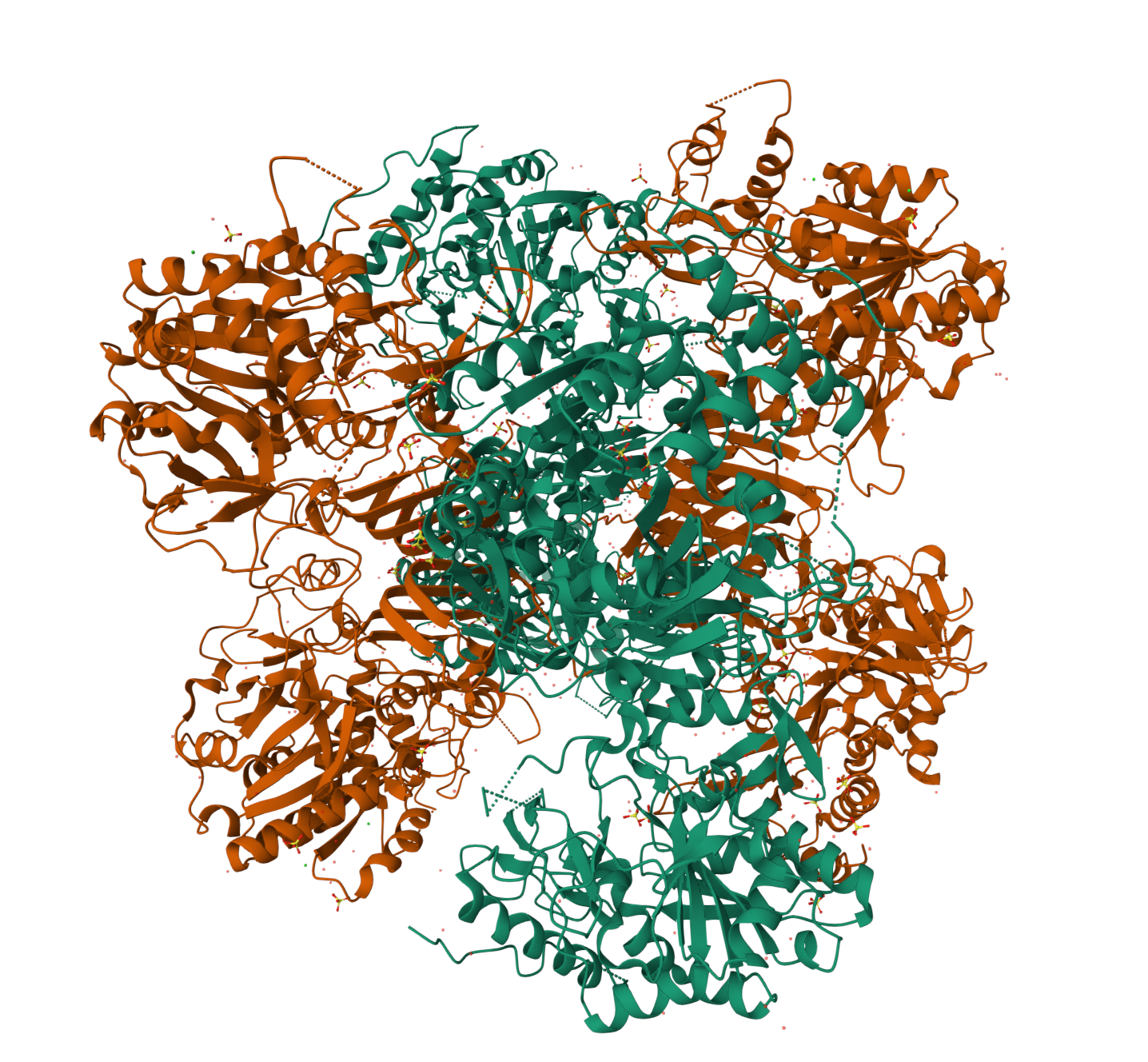
I chose UDP-glucose pyrophosphorylase (UGPase) from Komagataeibacter xylinus.
I selected this protein because it plays a central role in bacterial cellulose biosynthesis. UDP-glucose pyrophosphorylase catalyzes the conversion of glucose-1-phosphate and UTP into UDP-glucose, which is the immediate activated precursor used by cellulose synthase to polymerize β-1,4-glucan chains.
Since my final project focuses on improving bacterial cellulose production for biofilm-based food packaging materials, understanding and potentially enhancing the precursor supply through UGPase is directly relevant to my research goals.
- Identify the amino acid sequence of your protein.
- How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
The total sequence length is 336, and the most frequent amino acid is G.
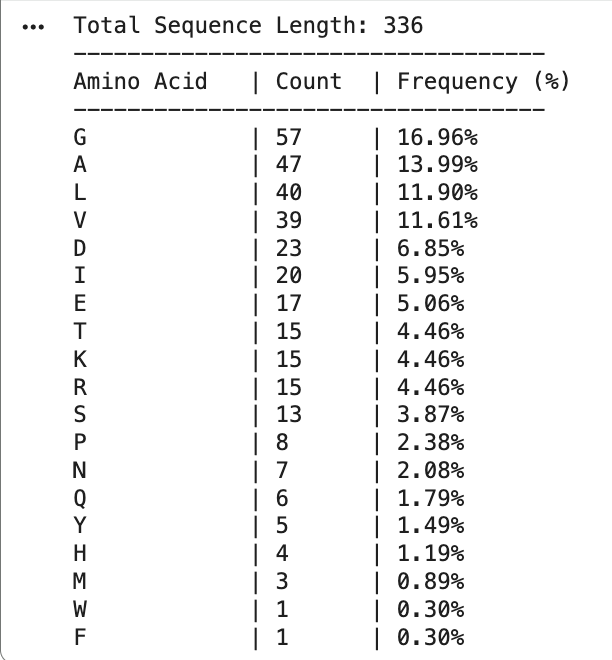
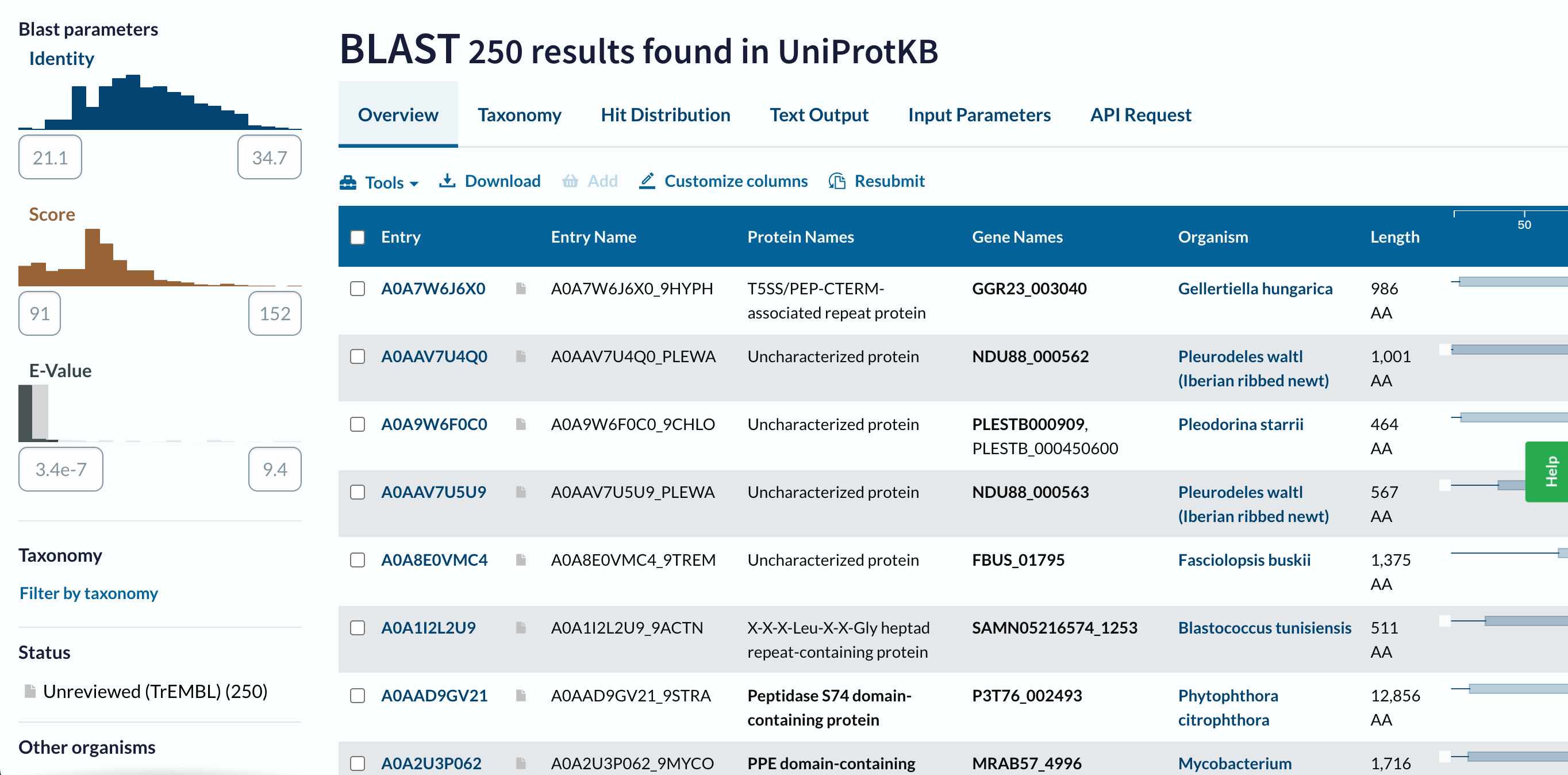
- How many protein sequence homologs are there for your protein? Hint: Use UniProt’s BLAST tool to search for homologs.
Using UniProt’s BLAST tool, I found 250 sequence homolog candidates for my protein in UniProtKB. The taxonomy results show that these homologs are distributed across a wide range of organisms. Most hits are found in Bacteria (179 results), followed by Eukaryota (70 results), and Archaea (1 result). Within bacteria, the largest groups are Pseudomonadati (91 results) and Bacillati (87 results). In eukaryotes, the hits are distributed among Metazoa (34 results), SAR (18 results), Viridiplantae (8 results), Fungi (8 results), Amoebozoa (1 result), and Haptista (1 result). This suggests that my protein, or related proteins, may be broadly distributed across different branches of life.

- Does your protein belong to any protein family?
Based on the current BLAST results, I cannot confidently assign my protein to a specific protein family yet, because the matched proteins have diverse annotations and many are uncharacterized.
- Identify the structure page of your protein in RCSB.
- When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å).
The structure was deposited on 2021-09-14 and released on 2022-09-21. So it was solved by 2021 and became publicly available in 2022. This is a reasonably good-quality structure. Its resolution is 2.57 Å, which is better than 2.70 Å. Since a smaller resolution value indicates higher structural detail, this structure can be considered good quality.
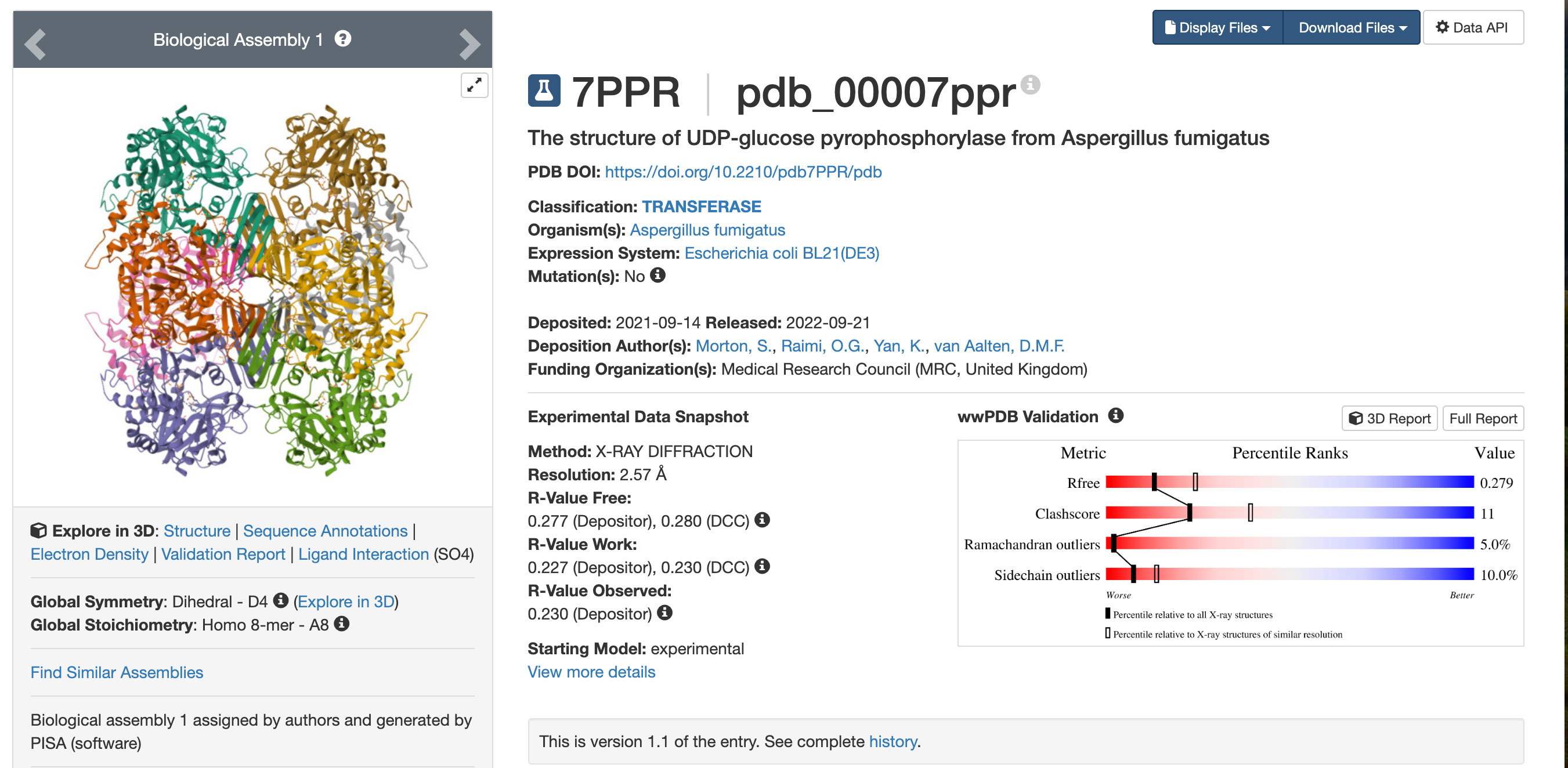
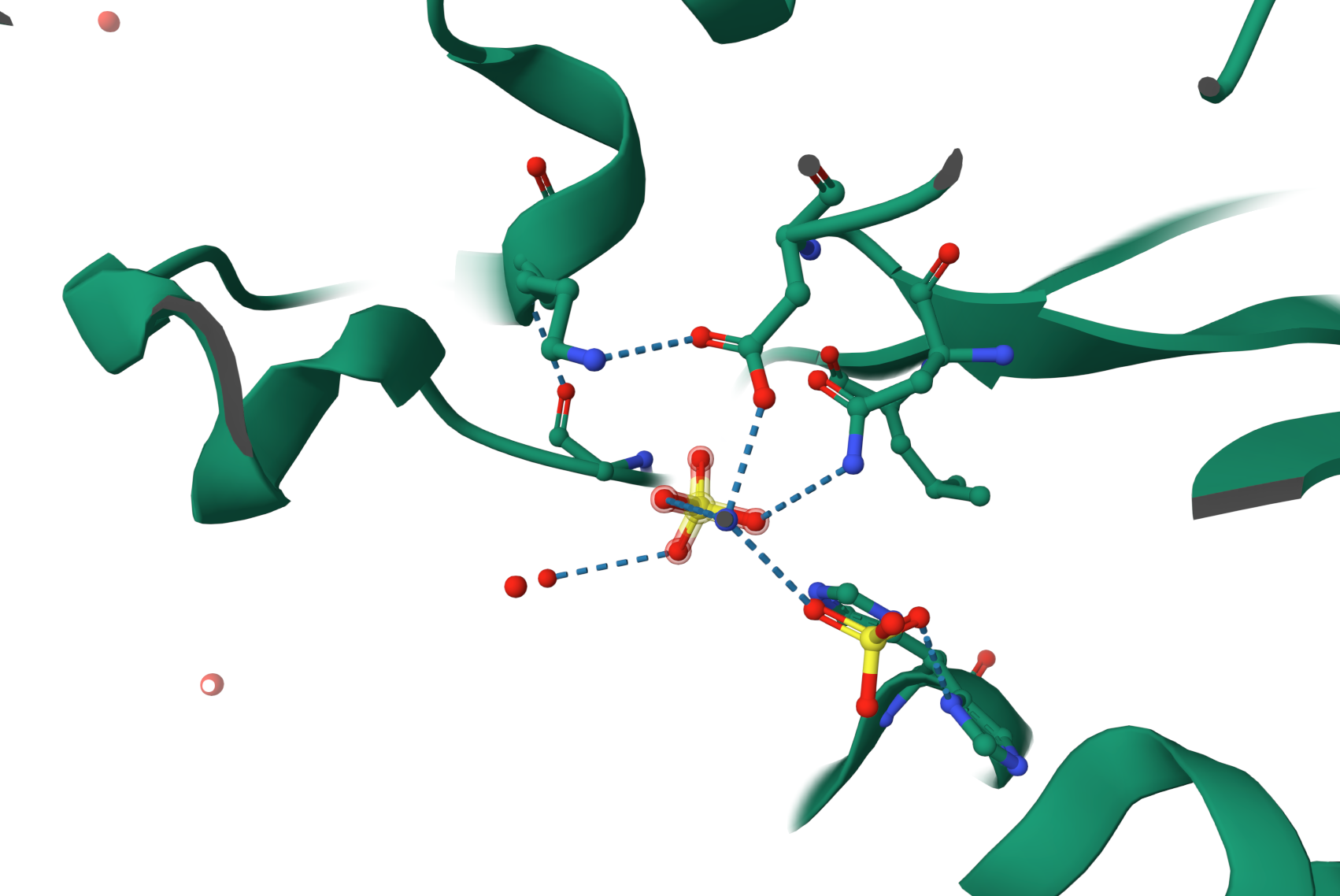
- Are there any other molecules in the solved structure apart from protein?
Yes. Besides the protein, the structure also contains SO4 (sulfate ions), as shown by the Ligand Interactions (SO4) information.
- Does your protein belong to any structure classification family?
Yes. According to the PDB entry, this protein is classified as a TRANSFERASE.
- Open the structure of your protein in any 3D molecule visualization software.
PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
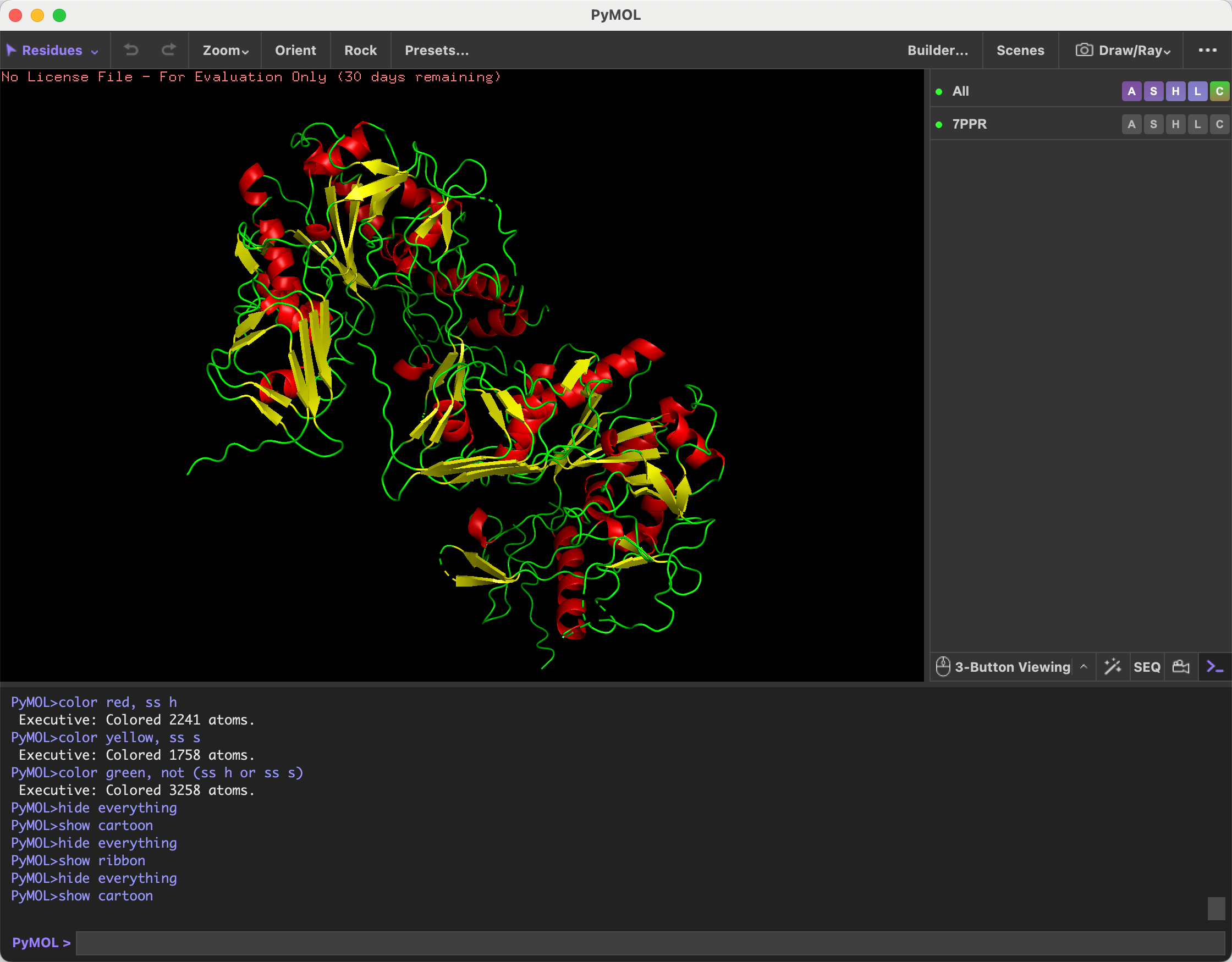
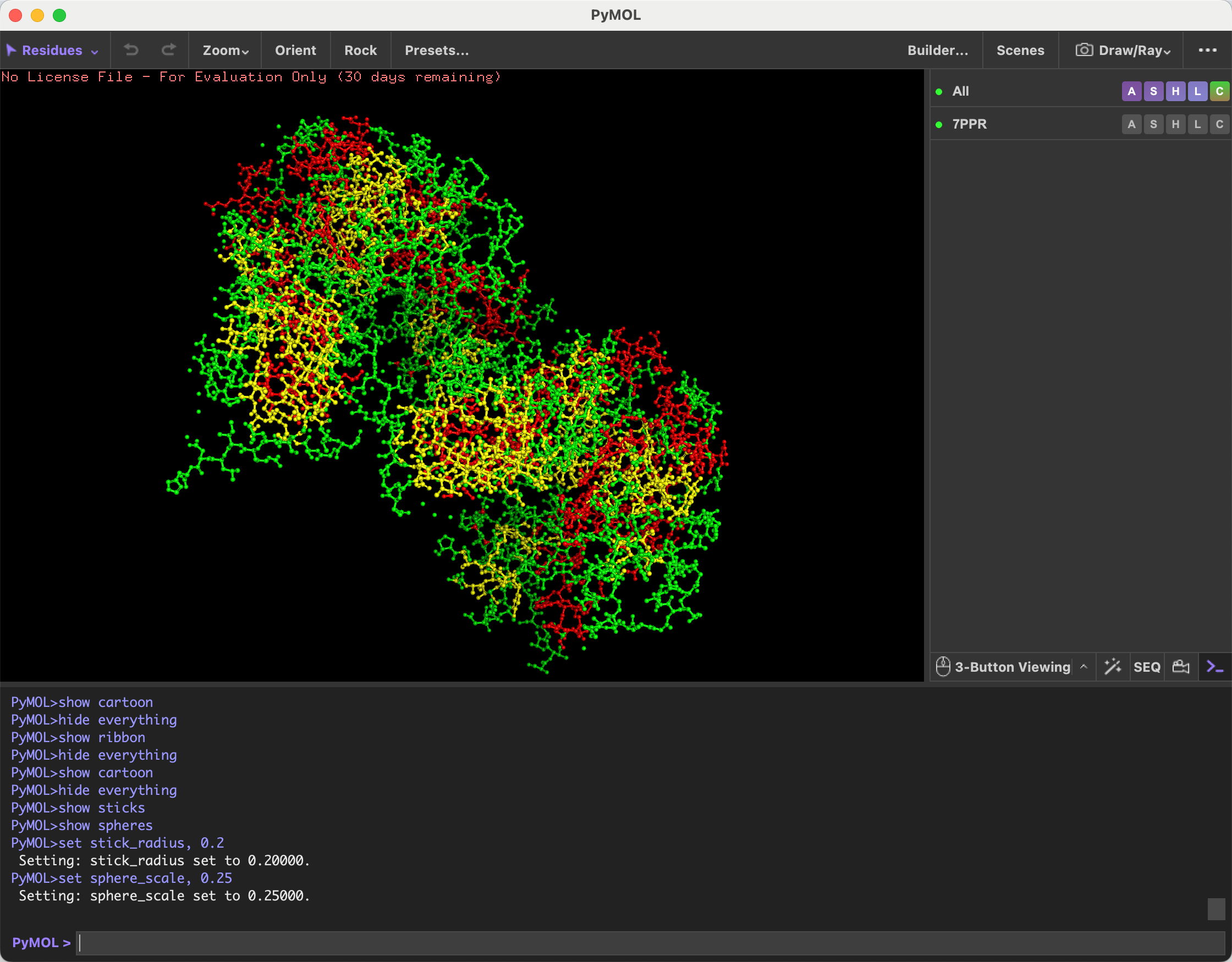
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
Screenshots attached below.
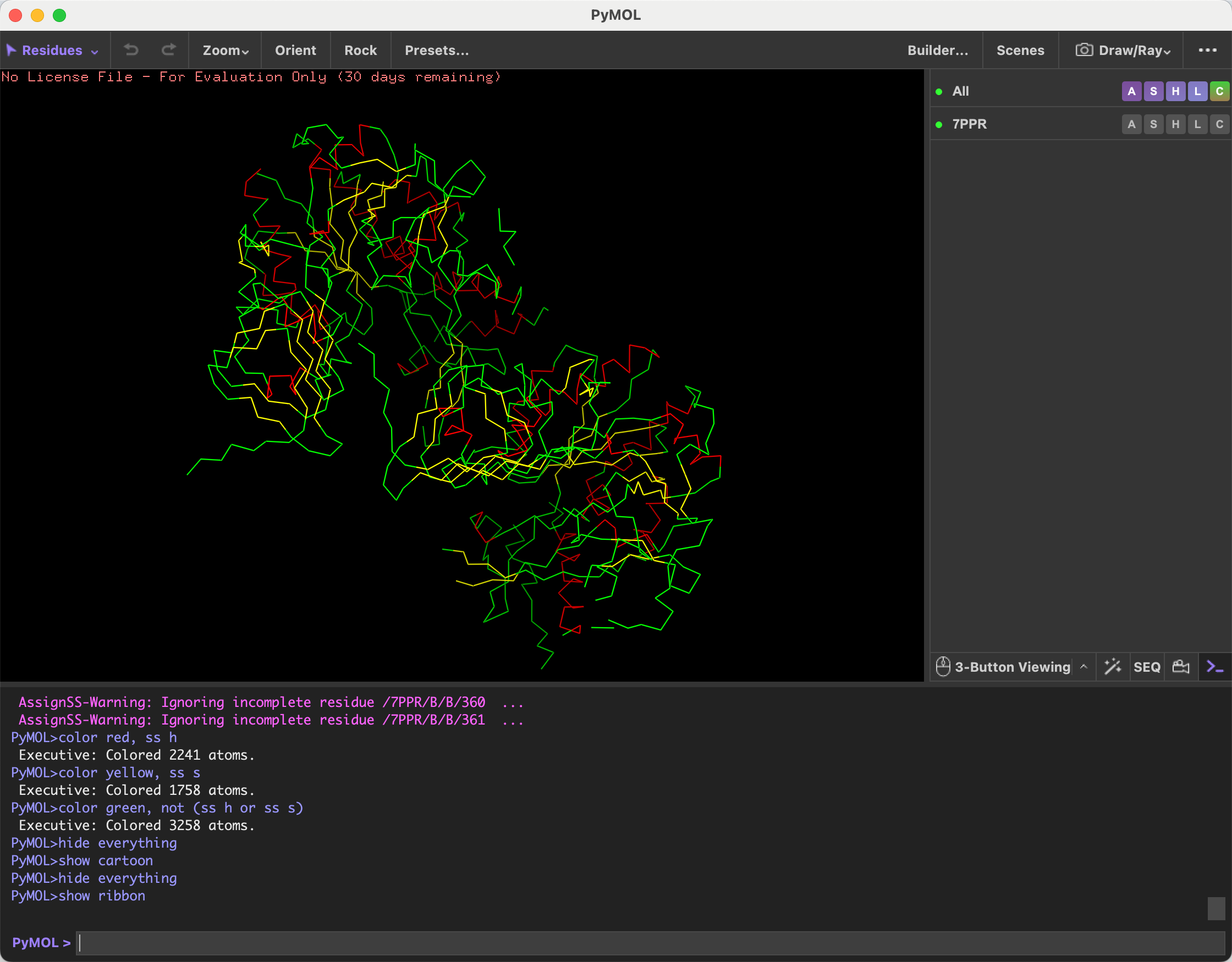
- Color the protein by secondary structure. Does it have more helices or sheets?
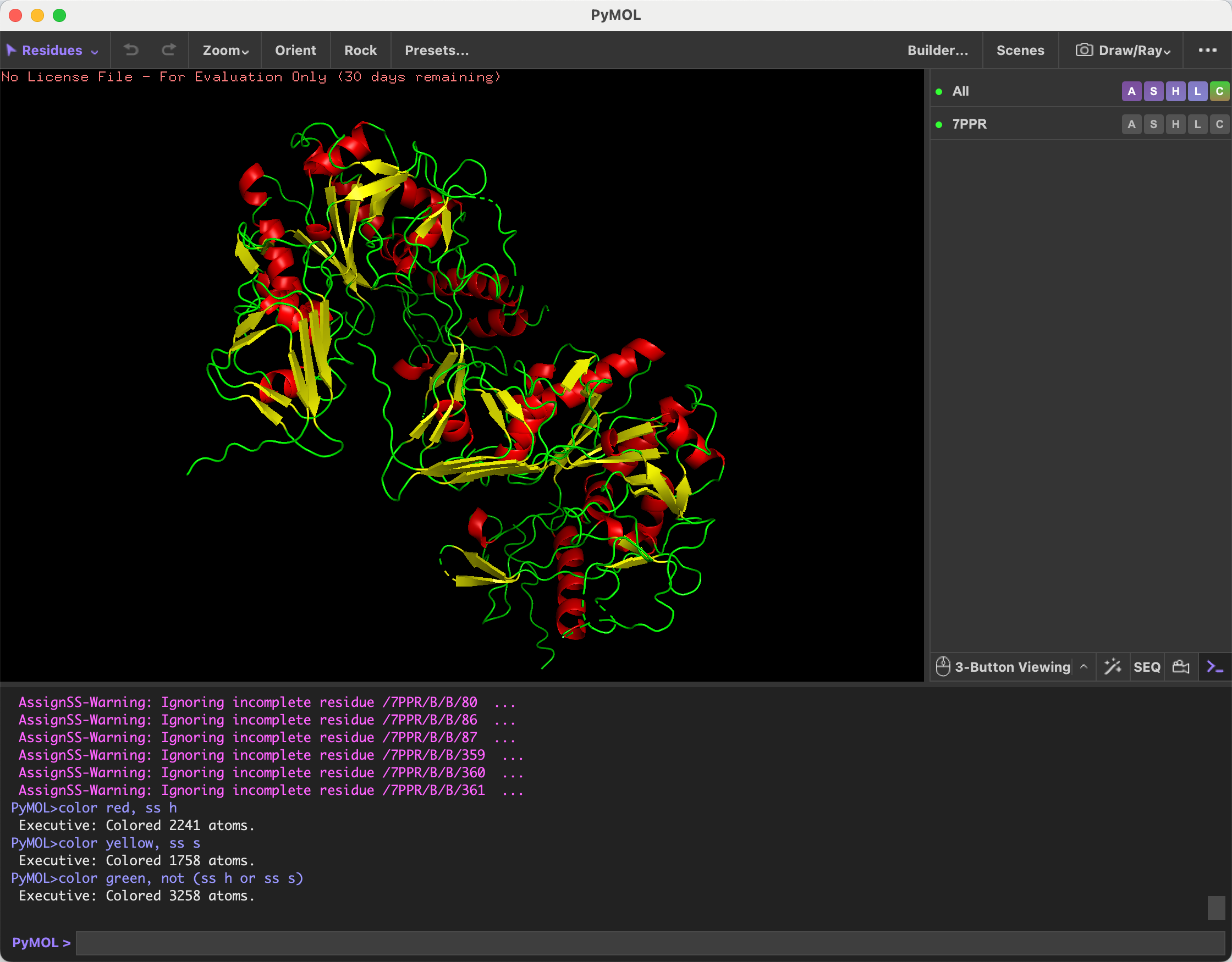
After coloring the protein by secondary structure, I observed that it contains more alpha helices than beta sheets.
- Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
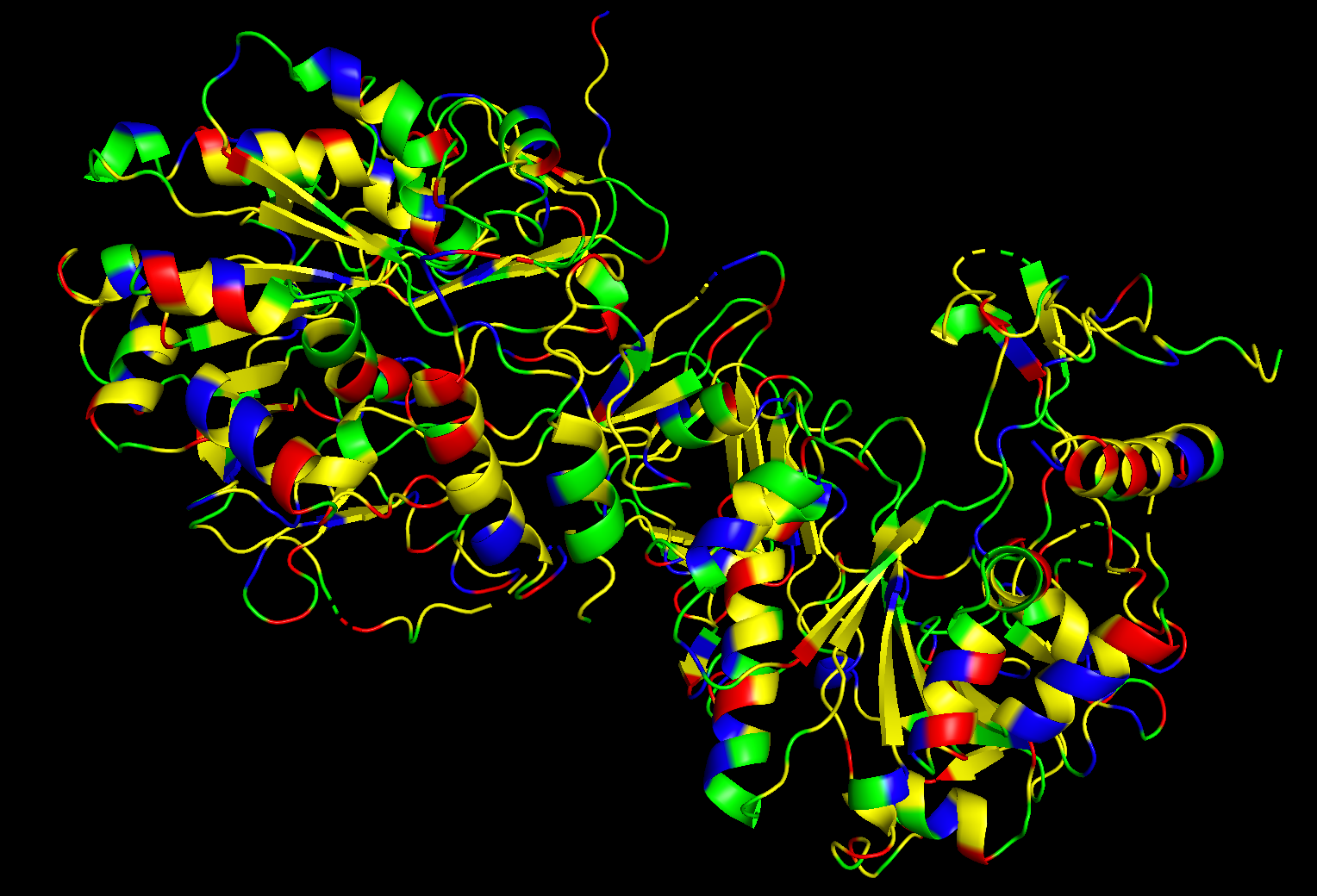
After coloring the protein by residue type, I observed that hydrophobic residues are distributed mainly throughout the interior/core regions of the protein, while hydrophilic residues, including polar and charged residues, are more exposed on the surface. This suggests a typical soluble protein organization, where hydrophobic residues help stabilize the folded core and hydrophilic residues interact with the surrounding aqueous environment.
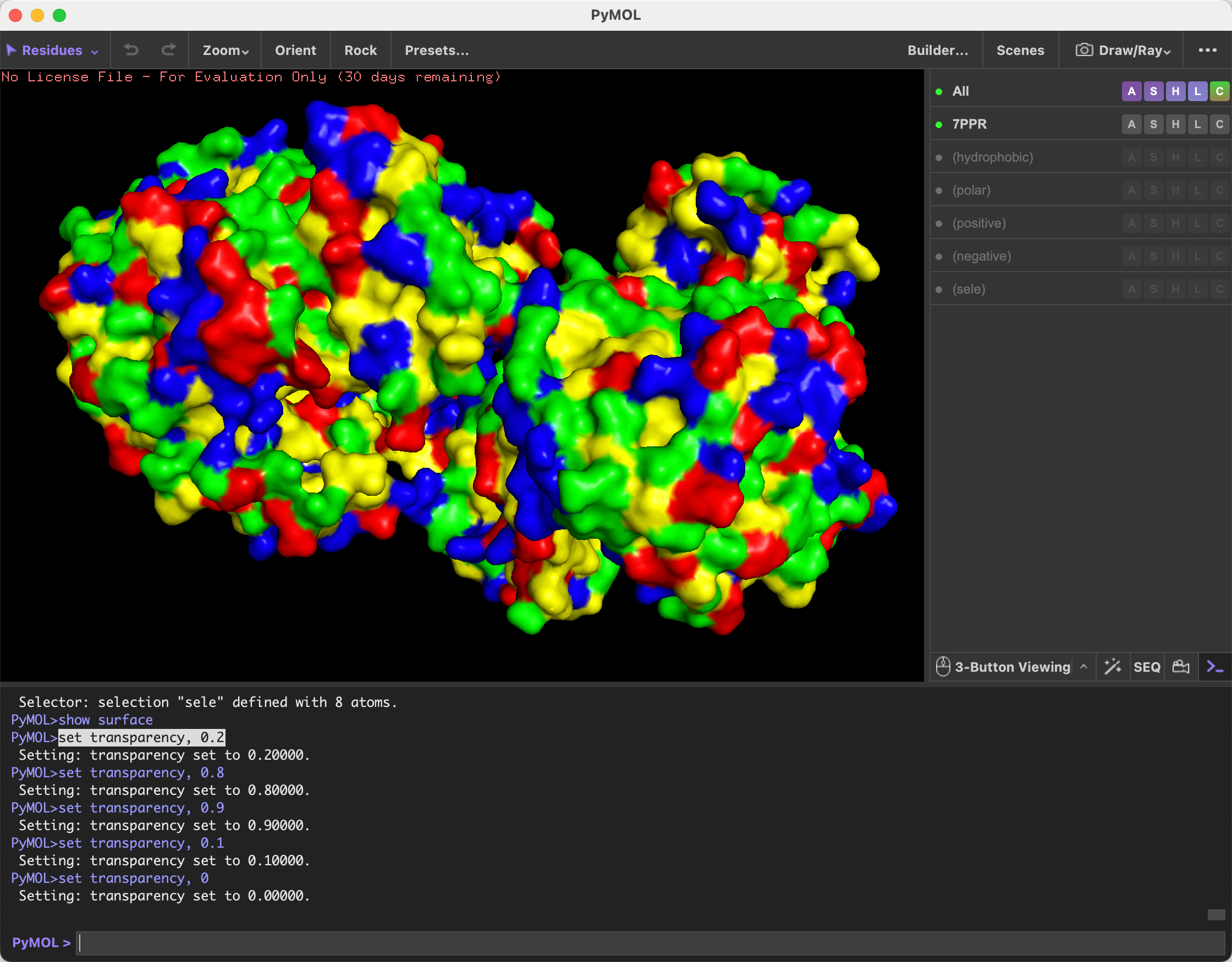
- Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
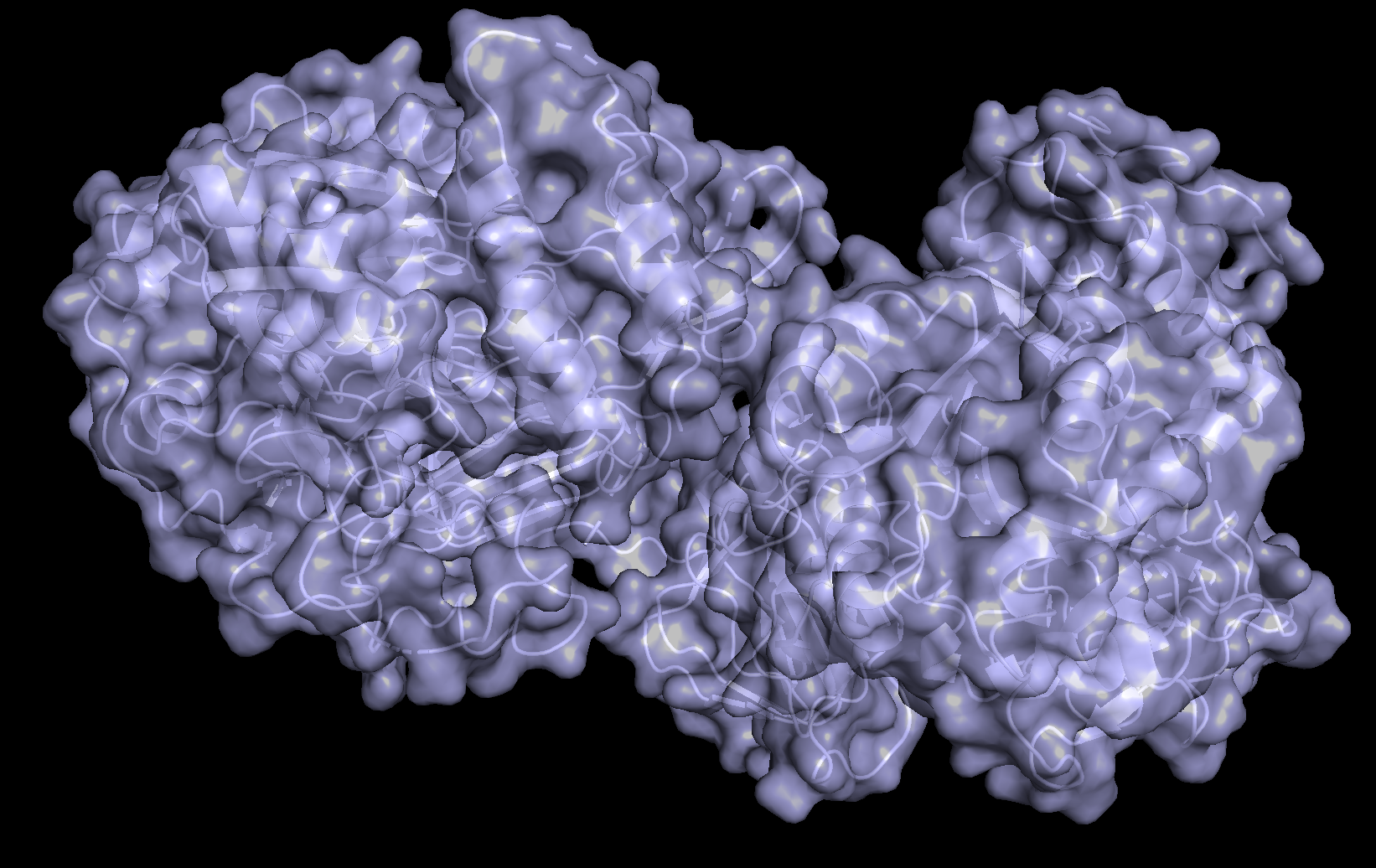
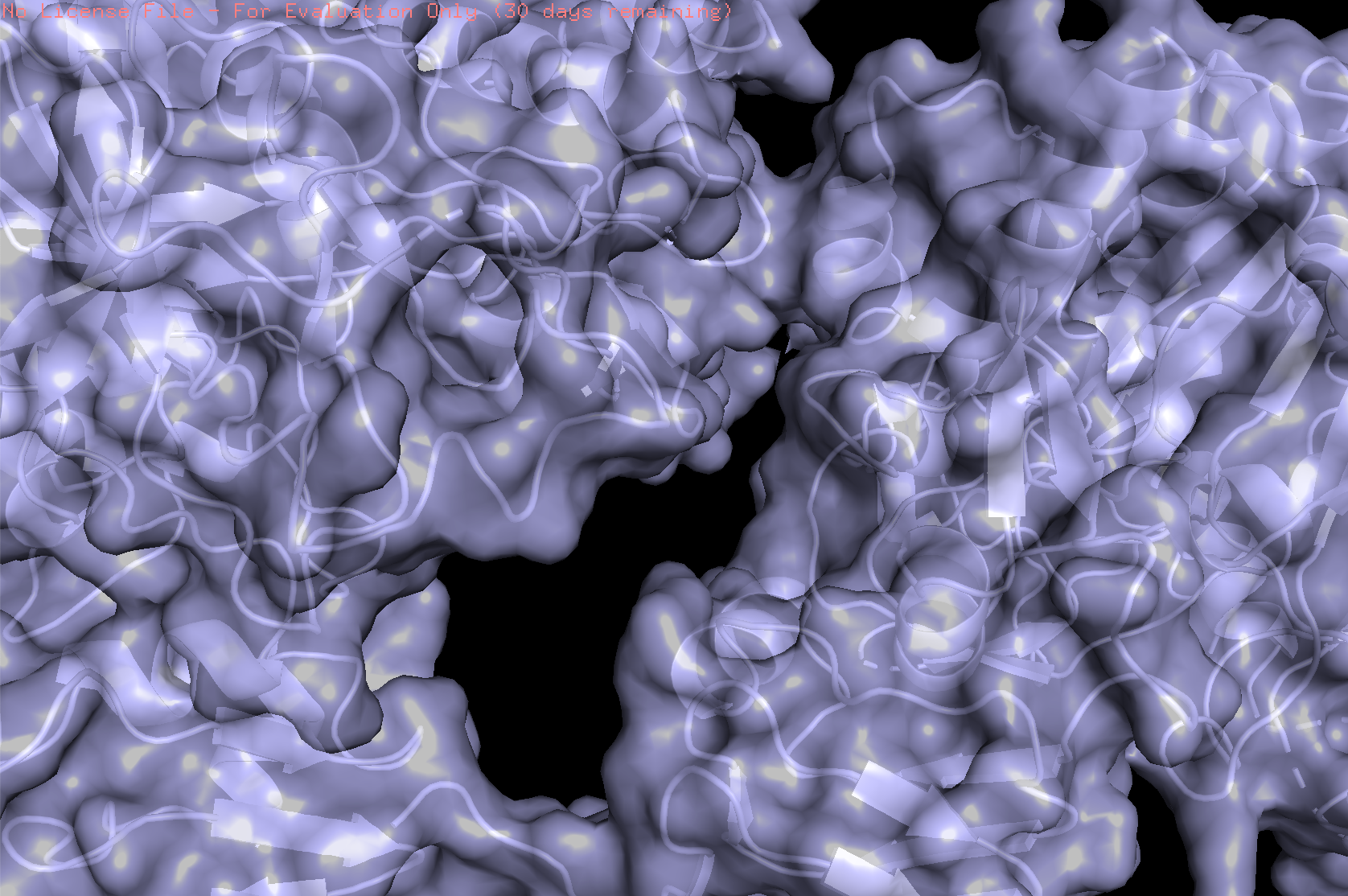

The protein surface is not smooth; it contains several pockets and cavities of different sizes. These surface depressions could serve as potential binding sites.
3
Using ML-Based Protein Design Tools
- Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
- Choose your favorite protein from the PDB.
I chose vanillin synthase because it produces vanillin, the molecule responsible for the characteristic aroma of vanilla. Vanilla is one of the most recognizable and pleasant food aromas, commonly found in desserts such as ice cream, cakes, and pastries.
- We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
C1. Protein Language Modeling
Deep Mutational Scans
a. Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
b. Can you explain any particular pattern? (choose a residue and a mutation that stands out)
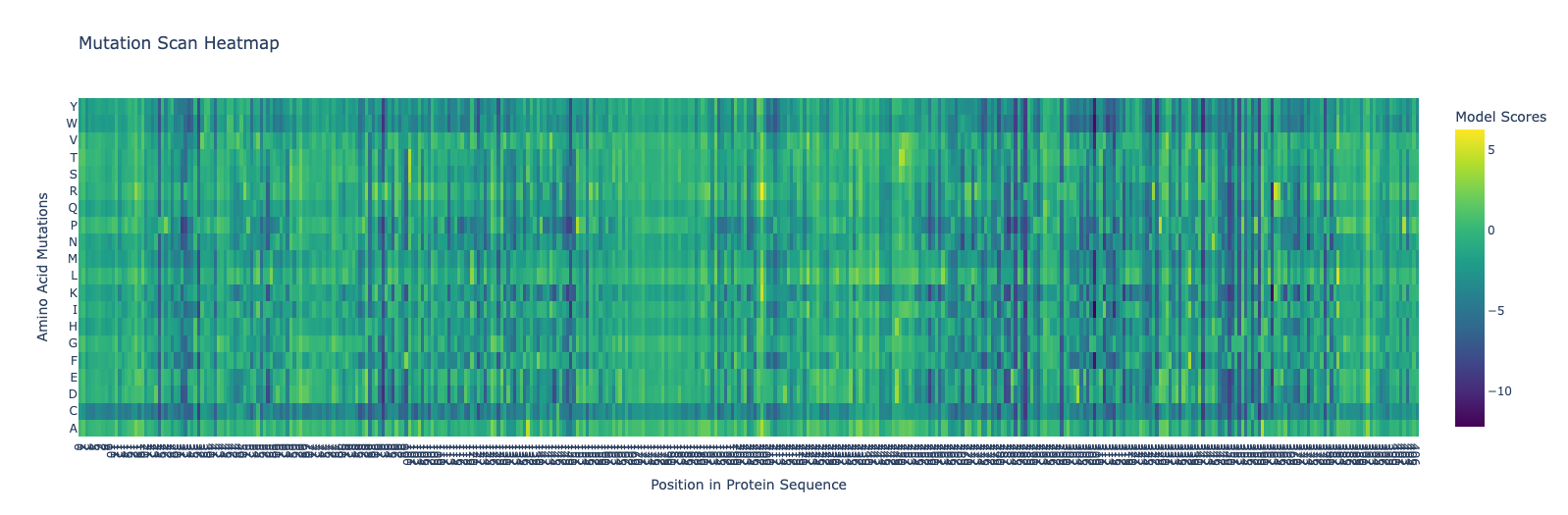
A pattern that stands out in the heatmap is that mutations to cysteine (C) are frequently more negative than many other substitutions. This may be because cysteine is chemically special: its thiol group is reactive and can introduce constraints or unwanted interactions, so the model tends to consider cysteine substitutions less favorable in many positions.
C2. Protein Folding
a. Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
b. Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
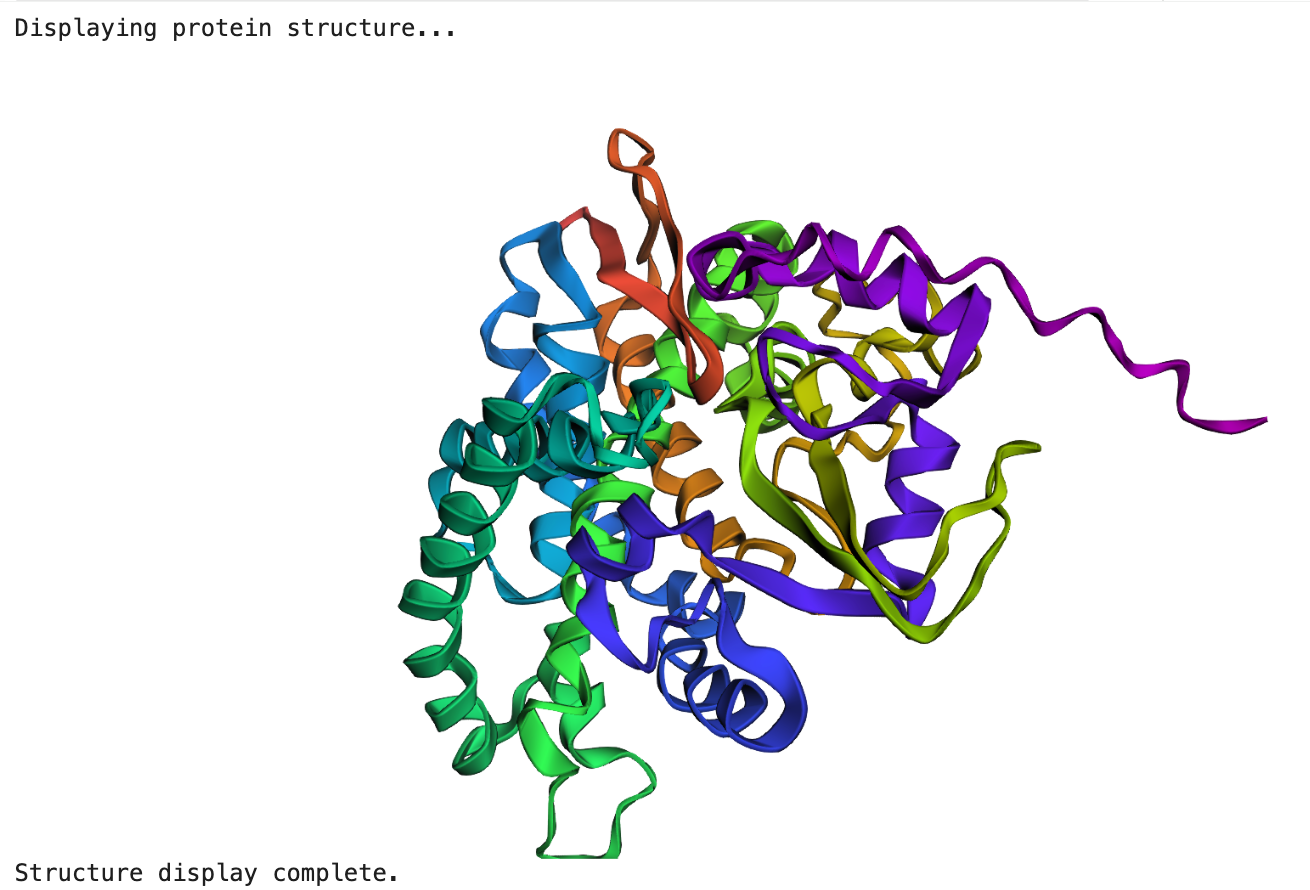
The structure predicted by ESMFold is broadly consistent with the experimentally determined structure. Both structures show a compact globular fold composed of several α-helices and β-sheets. While some local differences are visible in loop regions and the orientation of certain helices, the overall topology of the protein appears similar. This suggests that the model successfully captured the general folding pattern of the protein.
unchanged compared to the original structure. Only minor differences appeared in flexible loop regions. This suggests that the protein fold is robust to small conservative mutations.
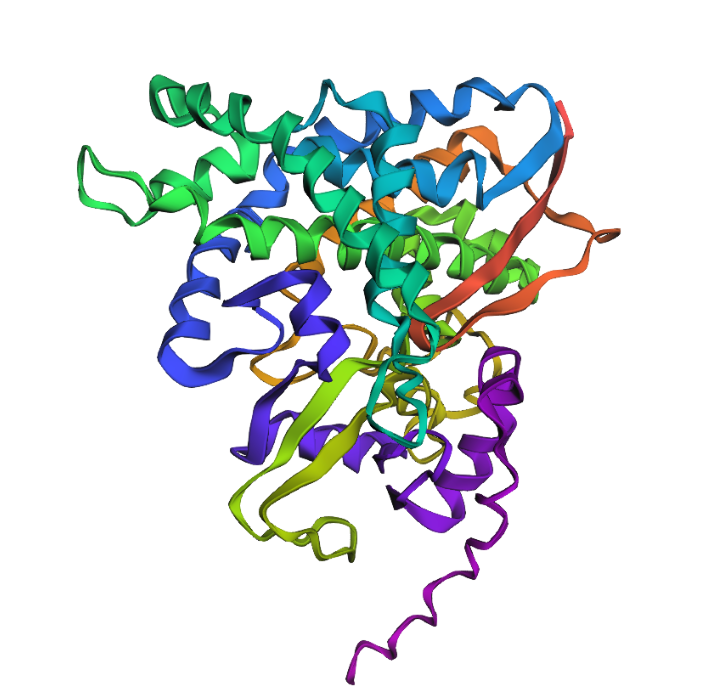 (L11I)
(L11I)
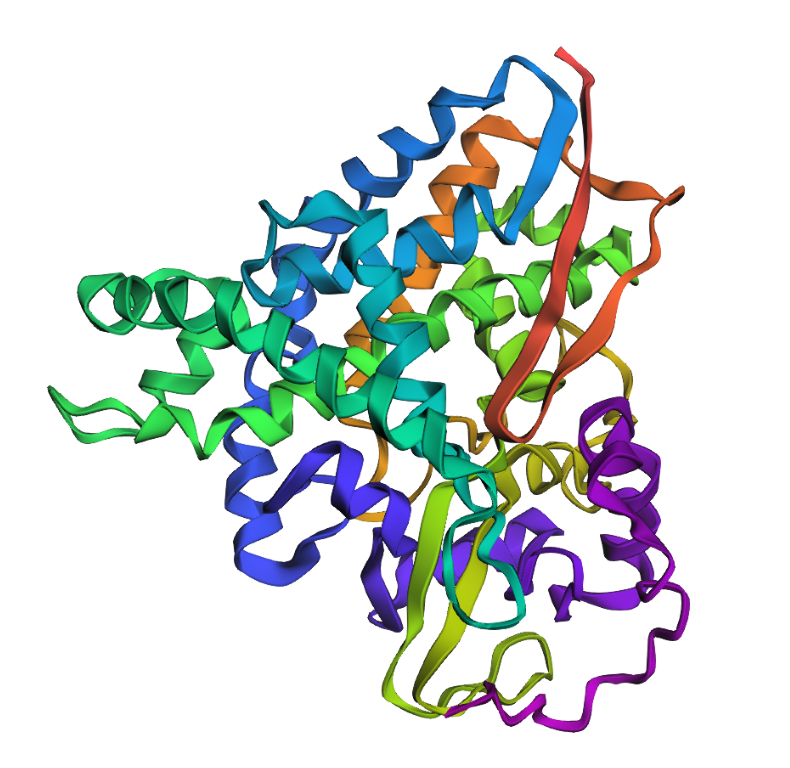 (L11P)
(L11P)I introduced both conservative (L11I) and more disruptive (L11P) mutations into the protein sequence and predicted the structures using ESMFold. In both cases, the overall protein fold remained very similar to the original structure. The positions of α-helices and β-sheets were largely preserved, and only small local differences appeared in flexible regions. This suggests that the protein structure is relatively resilient to single-point mutations. Even mutations that might disrupt local secondary structure do not necessarily change the global fold of the protein.

I replaced residues 184–191 (TNAAVDEN) with GGGGSGGG. After running the structure prediction again, I observed that the overall fold of the protein remained very similar to the original structure. Most secondary structure elements such as α-helices and β-sheets were preserved. However, some local changes appeared in flexible loop regions, especially near the tail of the protein. This suggests that the protein structure is relatively resilient to mutations, and small or moderate sequence changes mainly affect local conformations rather than the global fold.
C3. Protein Generation
a. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
b. Input this sequence into ESMFold and compare the predicted structure to your original.