Week 4 HW: Protein Design Part I

Part 1: Conceptual Questions
- Assuming protein mass of meat is 20%, 0.20 x 500=100g of actual protein in this meat. If average residue is roughly 100 Da, I can probably assume that’s roughly 100g/mol, meaning I now have 1 mol of residue. In one mole, there’s roughly 6 x 1023 molecules. Thus, there are roughly 6 x 1023 molecules of amino acids in 500 grams of meat
- We digest these proteins, not incorporate them into our bodies. We rebuild our own proteins using molecular tools in our body, that may or may not come from what we eat.
- There are only 20 “natural” amino acids because that’s what biology standardized our amino acids as. These amino acids have good chemical diversity, synthetic accessibility and different constraints. Now that we have evolutionarily reached this place, it is difficult to incorporate more without rewiring our whole system. As to why there’s only 20 versus like 100, this could be because if there were too many, it would be rather costly and inefficient, so it’s better to keep our biological systems simple.
- Yes, noncanonical amino acids are frequently made, and they can also probably be genetically encoded as well. For example, fluoroleucine or p-iodo-phenylalanine.
- Before enzymes & life, amino acids came from multiple plausible sources. For example, strecker synthsis in watery environments, delivery by meteorites (they were found in chondrites), or even atomsphere/UV activity.
- They are left handed
- Yes, proteins contain other helical motiefs like π-helix, collagen triple helix, β-helix / solenoid-like helices. New helical patterns can be found by analyzing high resolution structures.
- Most molecular helices are right hanaded because of chirality bias in L-amino acids. Another reason could be that right-handed packing avoids clashes and are better for L residues in general (so as a result of sterics).
- They have sticky edges where backbone hydrogen bond donors/acceptors are exposed at the sheet edges, and adding on another strand would satisfy H-bonds. This allows β-sheets to extend into larger assemblies easily.
- Drivers include bacbkone hydrogen bonding, hydrophobic effect and shape complentarity. Growth is probably also kinetically favorable.
- Amyloid diseases tend to form β-sheets because the cross-β amyyloid architecture is very stable and can form many sequences once misfolded. This ultimately templates further misfolding and thus createspersistent aggregates that disrupt cells.
- Yes, they are often found in nanofibers, hydrogels, templates for mineralization, etc.
- A simple, reliable design is a β-hairpin that self-assembles with controlled registry:
Design rules:
- Use alternating hydrophobic / polar residues to create amphipathic strands
- Use a tight turn motif
- Put charged residues at ends to control solubility and alignment
- Keep strands ~6–10 residues each for clean hairpins
Example motif:
- Strand 1: (Val/Lys alternating) → amphipathic
- Turn: D-Pro–Gly
- Strand 2: complementary alternating pattern
One concrete style:
X₁ X₂ X₃ X₄ X₅ X₆ – (DPro–Gly) – X₆ X₅ X₄ X₃ X₂ X₁
with hydrophobes on one face and charged/polar on the other.
Part 2: Protein Analysis and Visualizations
- I’m picking the Sonic hedgehog protein. I recently found out it exists and has a silly name. It is an important signaling molecule that plays a role of embryonic development in animals, so it’s pretty cool that such a silly protein name has quite an important role in our lives.
- The amino acid sequence is as follows: MLLLARCLLLVLVSSLLVCSGLACGPGRGFGKRRHPKKLTPLAYKQFIPNVAEKTLGASGRYEGKISRNS ERFKELTPNYNPDIIFKDEENTGADRLMTQRCKDKLNALAISVMNQWPGVKLRVTEGWDEDGHHSEESLH YEGRAVDITTSDRDRSKYGMLARLAVEAGFDWVYYESKAHIHCSVKAENSVAAKSGGCFPGSATVHLEQG GTKLVKDLSPGDRVLAADDQGRLLYSDFLTFLDRDDGAKKVFYVIETREPRERLLLTAAHLLFVAPHNDS ATGEPEASSGSGPPSGGALGPRALFASRVRPGQRVYVVAERDGDRRLLPAAVHSVTLSEEAAGAYAPLTA QGTILINRVLASCYAVIEEHSWAHRAFAPFRLAHALLAALAPARTDRGGDSGGGDRGGGGGRVALTAPGA ADAPGAGATAGIHWYSQLLYQIGTWLLDSEALHPLGMAVKSS
According to the Google Colab notebook:
The length of the protein is: 462 aminoacids.
The most common amino acid is: A, which appears 57 times.
Humans have three Hedgehog homologs: SHH, IHH and DHH. Across species, SHH is conserved.
This protein belongs to the Hedgehog signaling protein family.
- The structure page of my protein can be found here: https://www.rcsb.org/structure/6PJV?utm
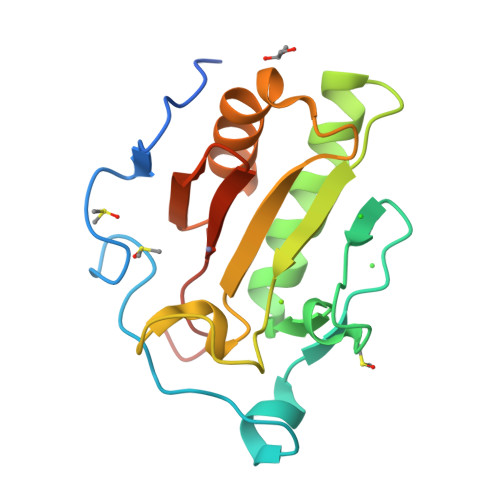
This particular structure was Deposited: 2019-06-28 and Released: 2019-11-13. It can be considered a good quality structure as the experiment type is XRD with a resolution of 1.43 Å. This is a very small resolution value, and means atomic positions are pretty well defined.
In this particular entry, there are other ions such as Zinc, Magnesium, as well as solvent molecules. This was found by clicking “Ligand Interactions”
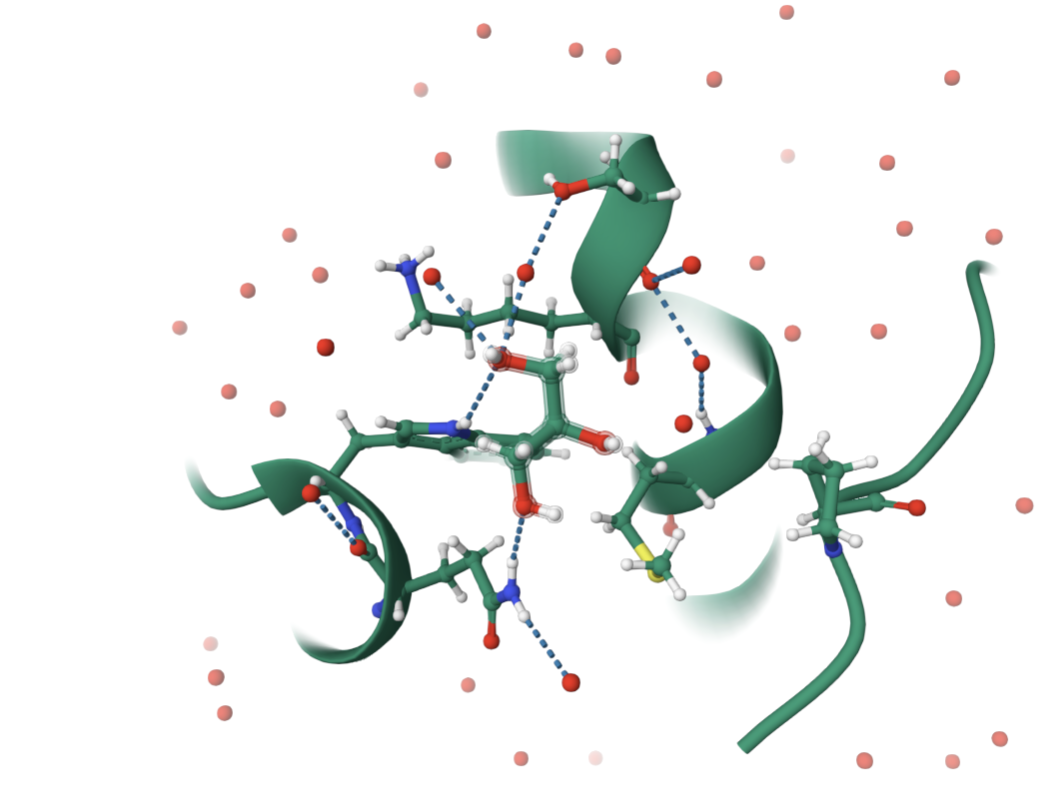
This protein belongs to the Hedgehog family of signaling proteins. SHH proteins have a conserved N-terminal signaling domain (Shh-N) found across animals. In structural classification systems (like SCOP, CATH), this domain is placed in a signaling/ligand family with a unique fold that binds Zn²⁺ and interacts with receptors such as Patched. So in structural classification terms, this is not a generic enzyme fold but rather a specific morphogen fold conserved among Hedgehog homologs.
I then used PyMol to visualize my structure.
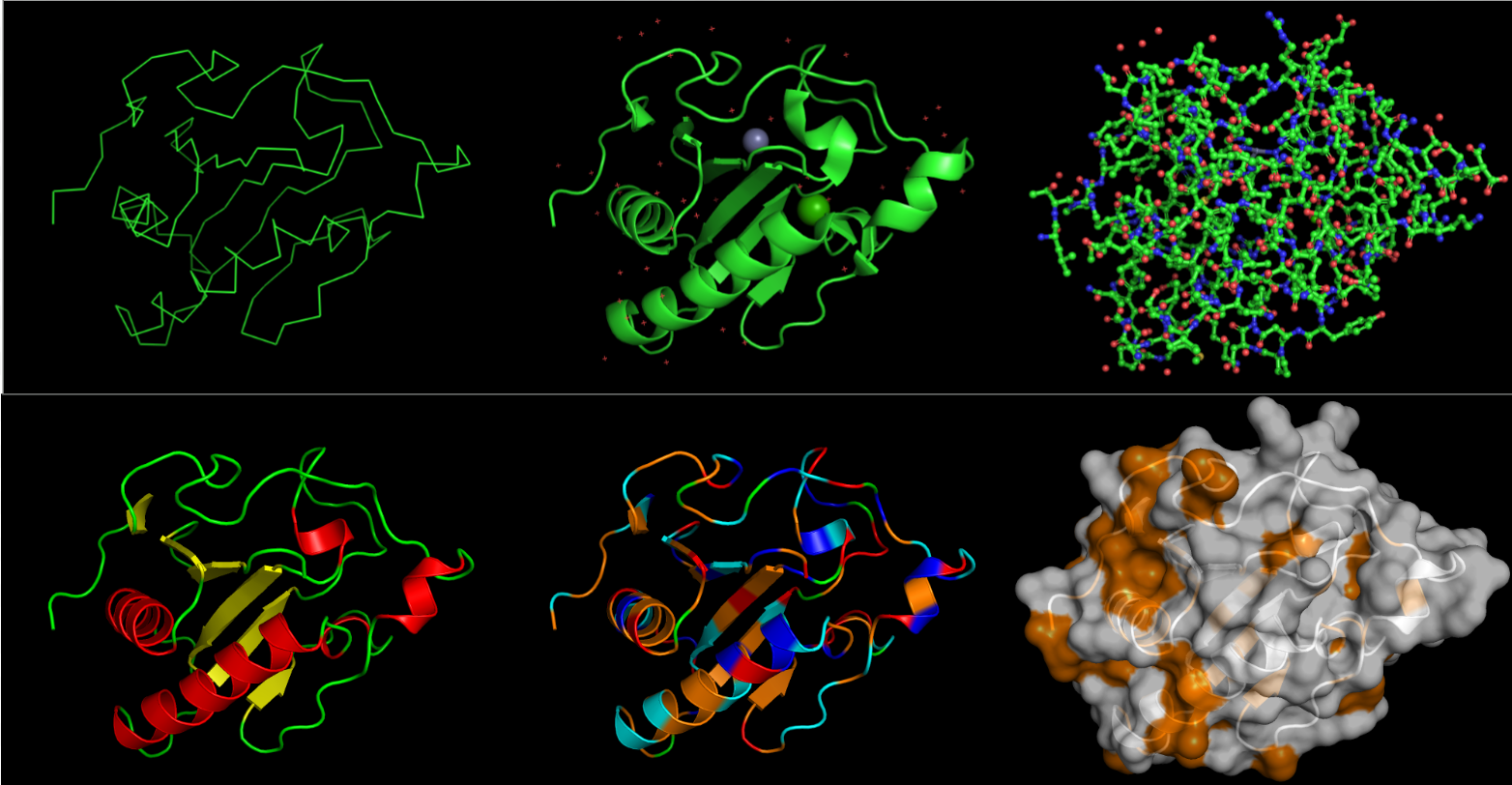 On the top, from left to right, the images are: ribbon, cartoon and ball-and-stick visualizations.
On the top, from left to right, the images are: ribbon, cartoon and ball-and-stick visualizations.
On the bottom, the protein is colored by secondary structure, residue type, and the surface.
From what I can see, it definitely has more helices than sheets. I want to say all the residues are equally represented. I represented hydrophobic residues in orange, and everythign else (varying in charge) in other colors: blue for + charge, red for - charge, cyan for no charge. But other than that, thee are less hydrophobic residues than hydrophilic. Overall, there doesn’t seem to be a very deep binding pocket from any angle.
Part 3: Using ML-Based Protein Design Tools
Deep Mutational Scans
I chose to use model esm2_t6_8M_UR50D
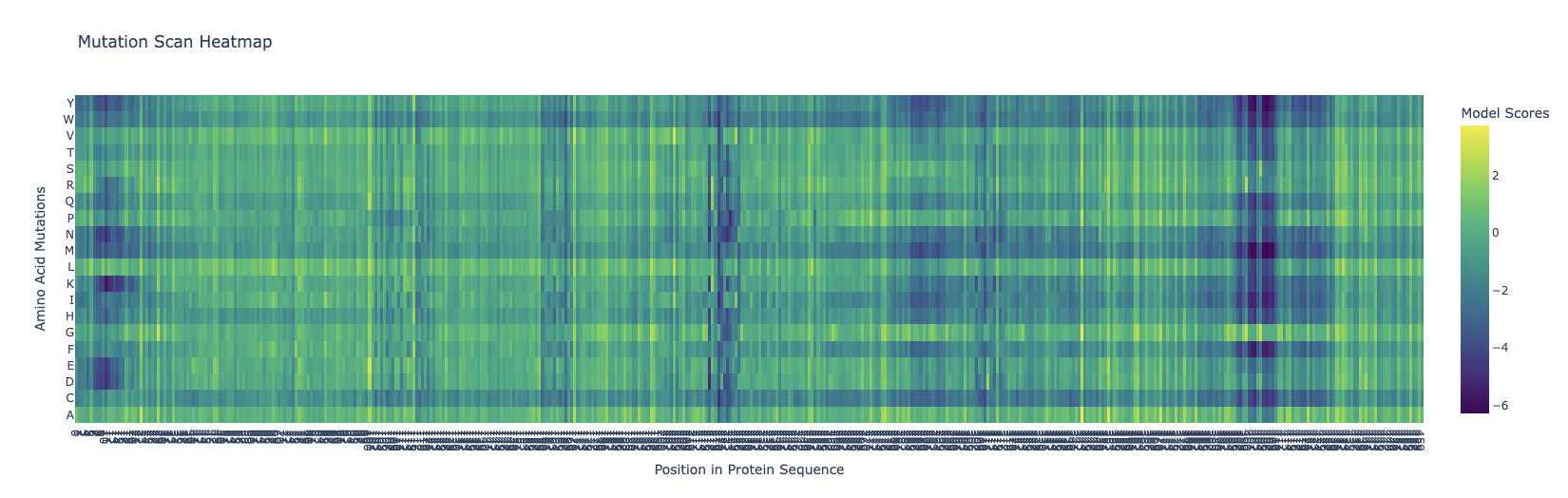 The dark blue means that mutations are not very likely to happen. Around the 400-410 mark, there is a dark blue column that is similar across all the different variations, meaning that this location is probably not extremely likely to mutate. I’m kind of intrigued as to why this could be, and frankly I don’t know that I can come with any good explanation for this.
The dark blue means that mutations are not very likely to happen. Around the 400-410 mark, there is a dark blue column that is similar across all the different variations, meaning that this location is probably not extremely likely to mutate. I’m kind of intrigued as to why this could be, and frankly I don’t know that I can come with any good explanation for this.
Latent Space Analysis
This is what the code produced.
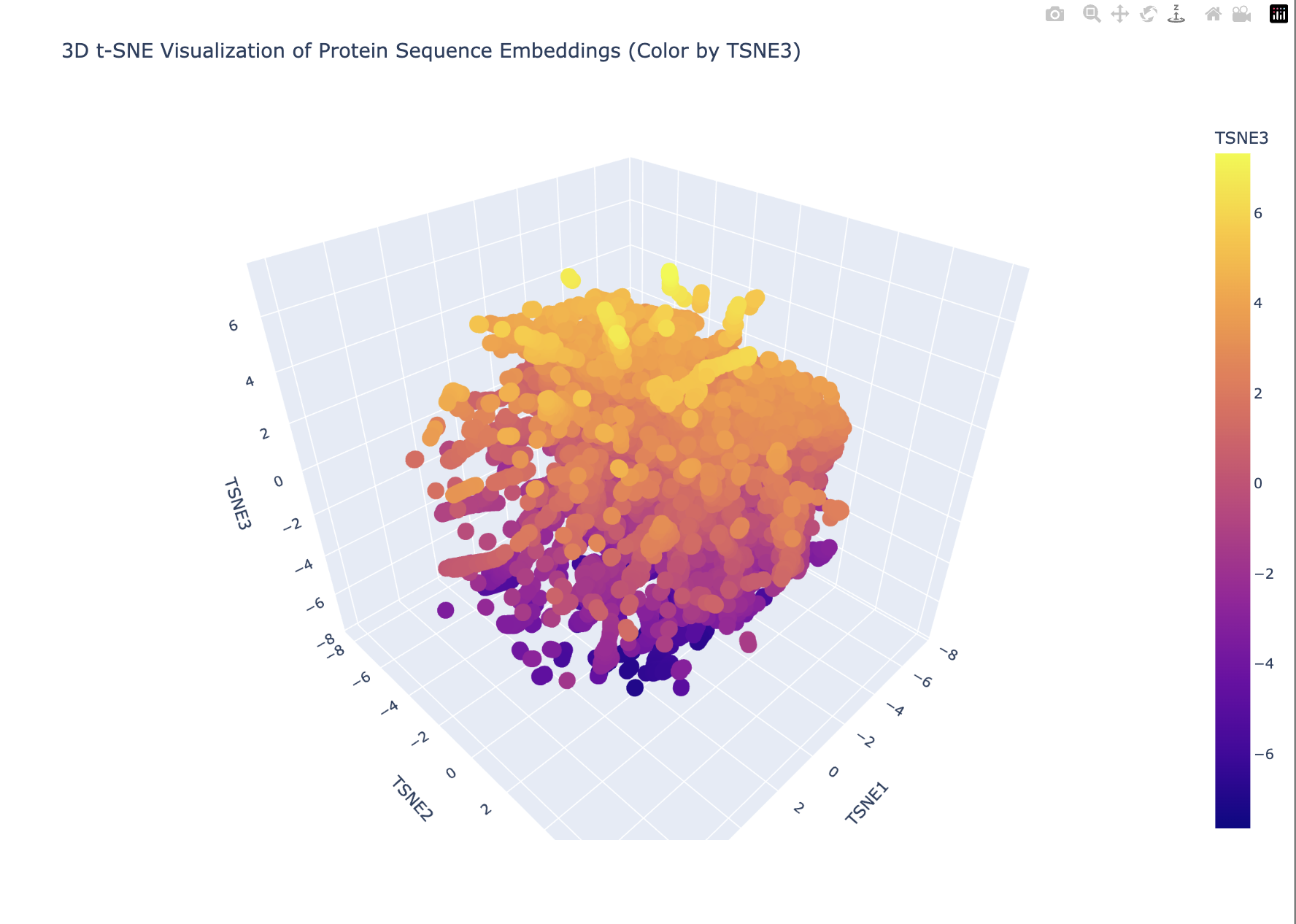 My plot looks like a single dense blob with smooth gradients (especially along TSNE3). This suggests that there are no strongly separated protein families. Furthermore, TSNE3 captures a continuous variation in possibly protein length, compositional differences or evolutionary divergence. This looks more like a continuum than discrete groups.
My plot looks like a single dense blob with smooth gradients (especially along TSNE3). This suggests that there are no strongly separated protein families. Furthermore, TSNE3 captures a continuous variation in possibly protein length, compositional differences or evolutionary divergence. This looks more like a continuum than discrete groups.
The embedding does not show sharply separated clusters, suggesting that the proteins form a continuous similarity space rather than distinct families. My protein lies within a dense region of the embedding, indicating that it shares sequence-level similarity with many neighboring proteins. The local neighborhood likely contains proteins with related sequence motifs or structural features.
Protein Folding
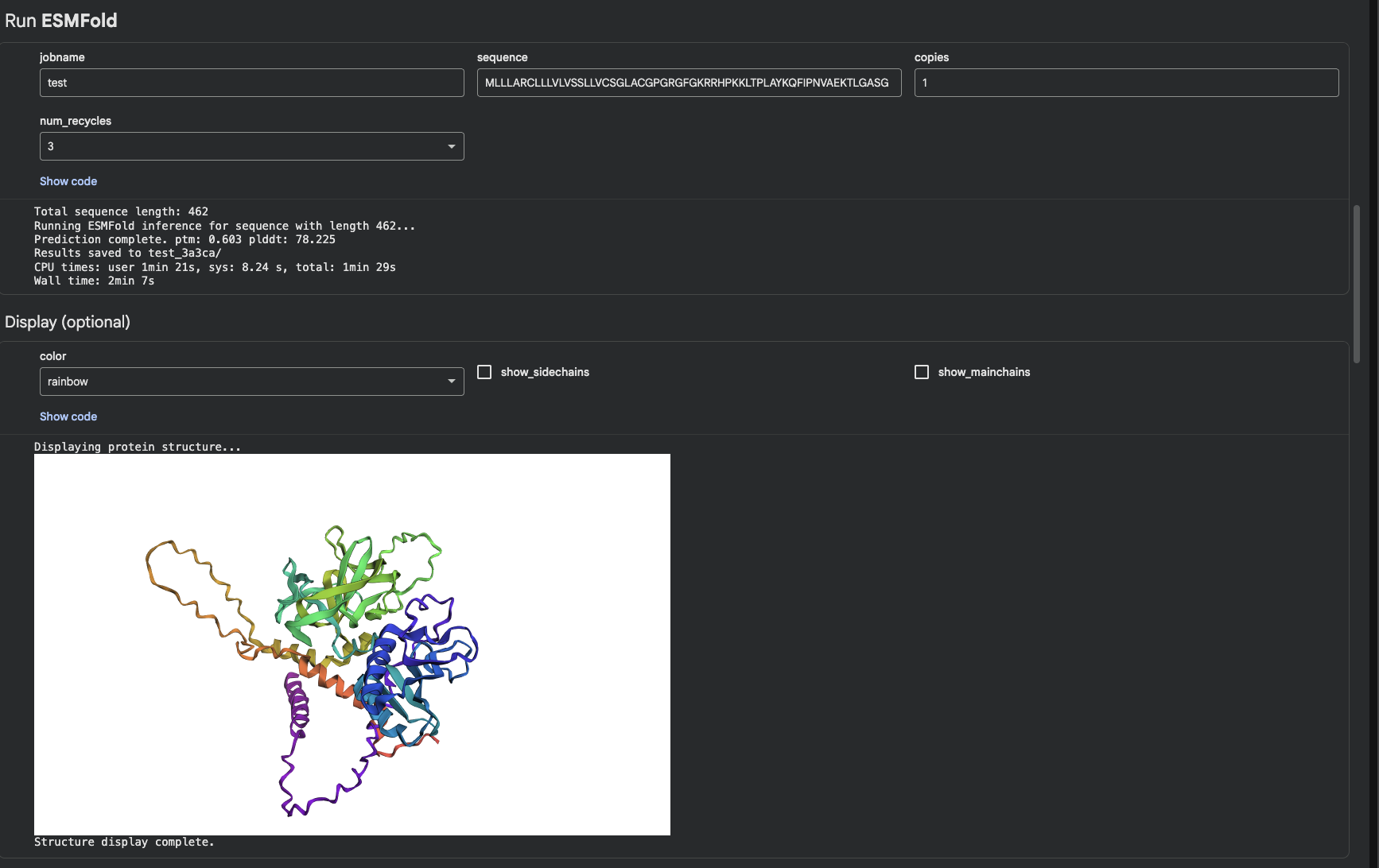
I was able to find my protein. The first image shows the complete sequence, which seems to differ from the original structure. However, it is difficult to tell because they are oriented differently.
When adding mutations (the second image), it seems that the protein is quite susceptible to mutational changes. I deleted a very large chunk, however, and it’s possible that 20 amino acids affects it much less than 50 amino acids deleted.
Protein Generation
Inverse:
Length of chain A is 150
Generated Sequences:
Generating sequences...
>tmp, score=1.5198, fixed_chains=[], designed_chains=['A'], model_name=v_48_020
LTPLAYKQFIPNVAEKTLGASGRYEGKITRNSERFKELTPNYNPDIIFKDEENTGADRLMTQRCKDKLNALAISVMNQWPGVKLRVTEGWDEDGHHSEESLHYEGRAVDITTSDRDRSKYGMLARLAVEAGFDWVYYESKAHIHCSVKAE
>T=0.1, sample=0, score=0.8737, seq_recovery=0.5067
MTPLRPGERSPPVPEHSPEAAGPYLGAITPDSPRFKLLKPNTNPRIIFEDKDGTGWDKLFTPRMHEVLDRLADLVEAAWPGLRLRVLEGYDREGNHPPGSYHYEGRAADLTNSNRDRSLLPELARLAVEAGADYVLLESPDHVYVAVRHE
Generated Sequences:
Generating sequences...
>tmp, score=1.5049, fixed_chains=[], designed_chains=['A'], model_name=v_48_020
LTPLAYKQFIPNVAEKTLGASGRYEGKITRNSERFKELTPNYNPDIIFKDEENTGADRLMTQRCKDKLNALAISVMNQWPGVKLRVTEGWDEDGHHSEESLHYEGRAVDITTSDRDRSKYGMLARLAVEAGFDWVYYESKAHIHCSVKAE
>T=0.1, sample=0, score=0.8509, seq_recovery=0.4733 MKPLKLGERSPPVPEHSPEAVGPYRGAITPDSPEFKLLKPNNNPNIIFVDKDGKGNDKLHTPKLHEVLNKAAELVEKAWPGLKLEVLRGYDFEGNHPPGSYHYEGRAVDLTFSNHDKSLLPELARLMVEAGADYVYLESENGVHAAVKWE
New Sequence: MKPLKLGERSPPVPEHSPEAVGPYRGAITPDSPEFKLLKPNNNPNIIFVDKDGKGNDKLHTPKLHEVLNKAAELVEKAWPGLKLEVLRGYDFEGNHPPGSYHYEGRAVDLTFSNHDKSLLPELARLMVEAGADYVYLESENGVHAAVKWE
Folding the final predicted sequence:
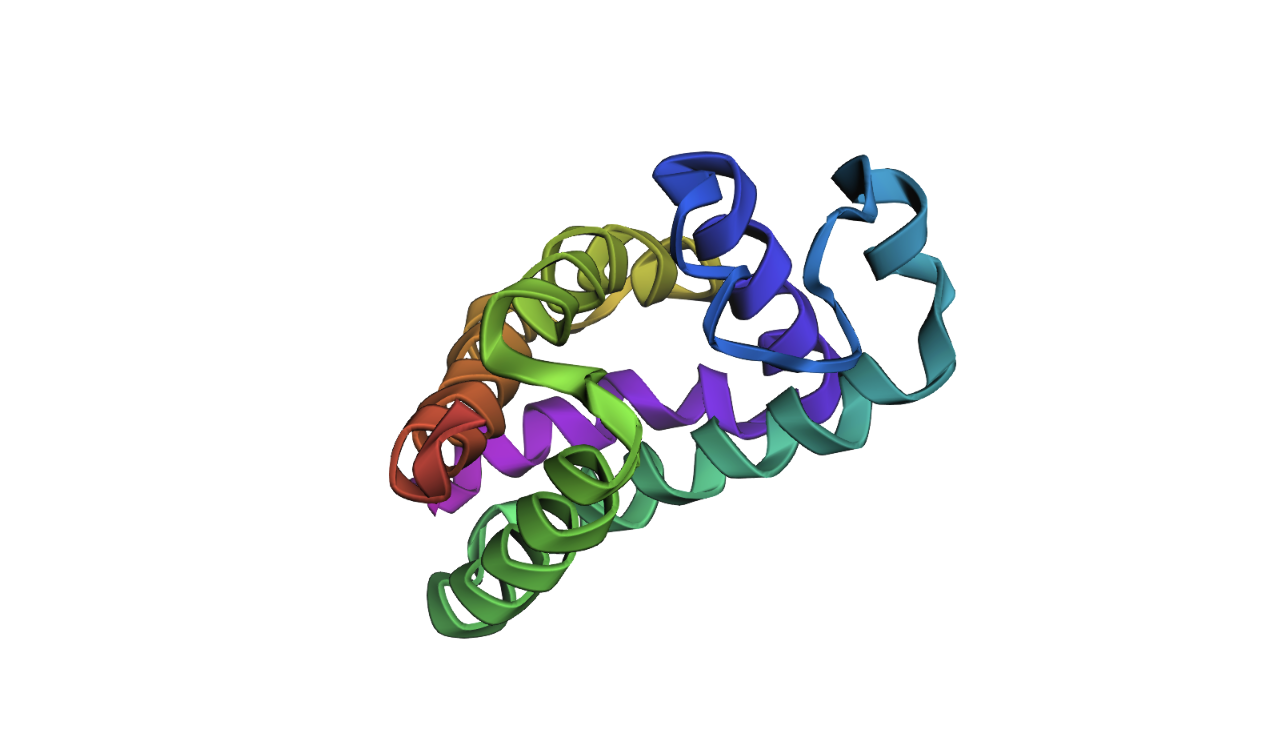
As you can tell, it is very off.