Week 5 HW: Protein Design Part II
Part 1: SOD1 Binder Peptide Design
Part A:
The retrieved SOD1 sequence is:
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Upon introducing the A4V Mutation, we get:
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
The following amino acids were generated with their subsequent perplexity scores:
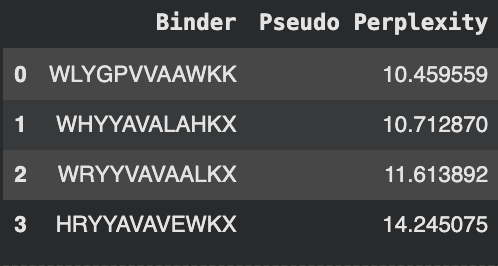
The known binder FLYRWLPSRRGG was added for comparison.
Part B:
There was an apparent issue with the predictions, as the final letter was X, though AlphaFold does not accept this. To solve this issue, any X was replaced with an A:
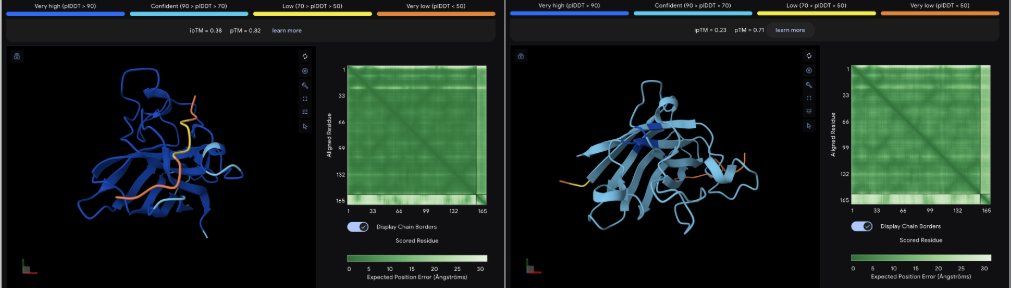
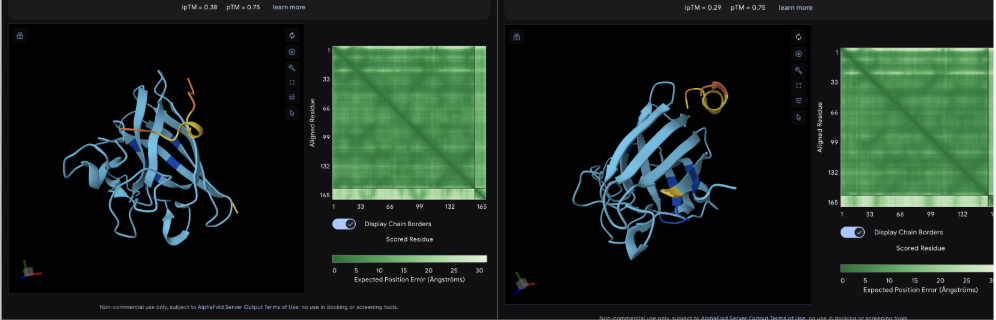
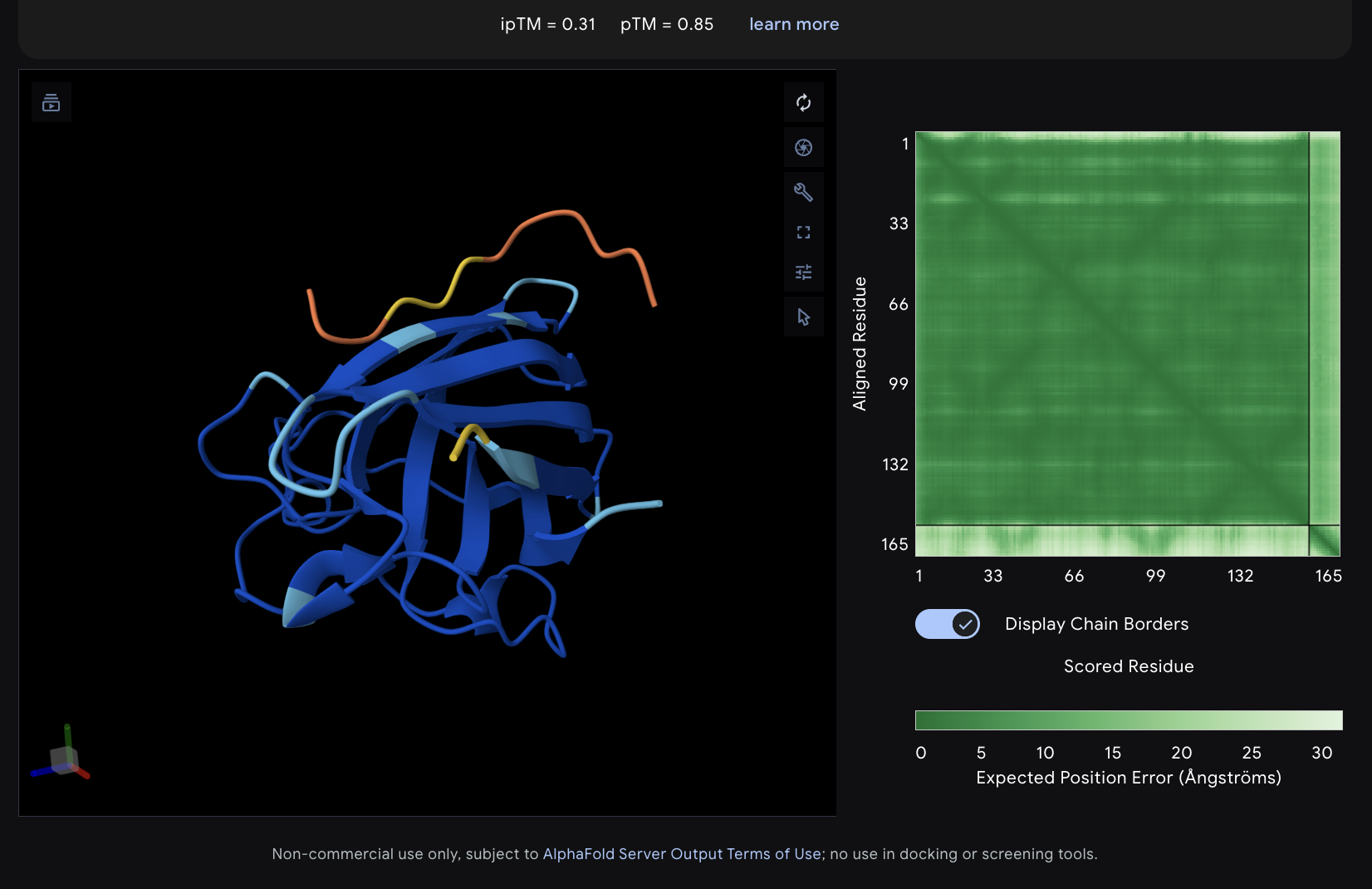
The images are somewhat blurry, but it just seems like the peptides tend to be very surface bound, typically near the β-barrel region. Interestingly enough, even the known peptide did not have a very high ipTM score, as there were still orange and yellow parts, which made me very curious.
iPTM scores were: 0.38, 0.23, 0.38, 0.29, 0.31, for known and index 0-3, respectively.
The AlphaFold ipTM score is a key metric for evaluating the accuracy of protein-protein, protein-nucleic acid, or protein-ligand interaction models. It specifically measures the confidence in the relative positions and orientation of the two interacting chains. Values above 0.8 are generally considered high-confidence predictions, while values below 0.6 usually indicate a failed prediction. Evidently, everything I had was a failed prediction, though interestingly enough, index 2 had the same score.
Part C:
Using PeptiVerse, I got the following images:
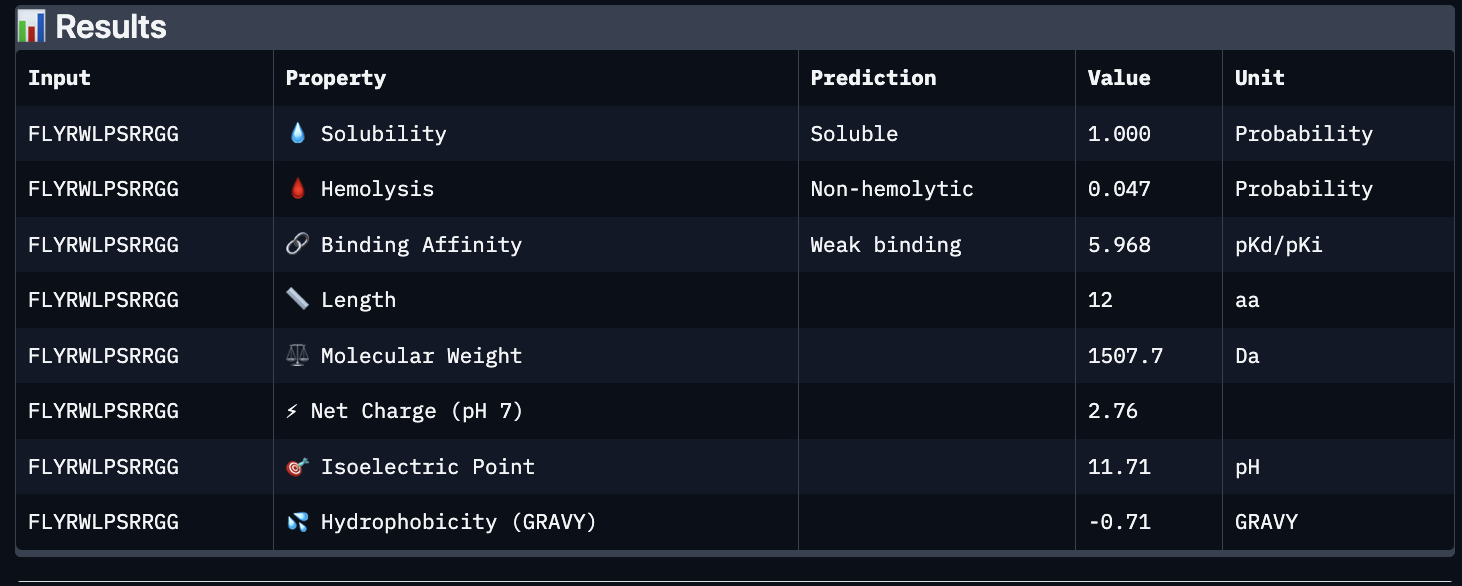
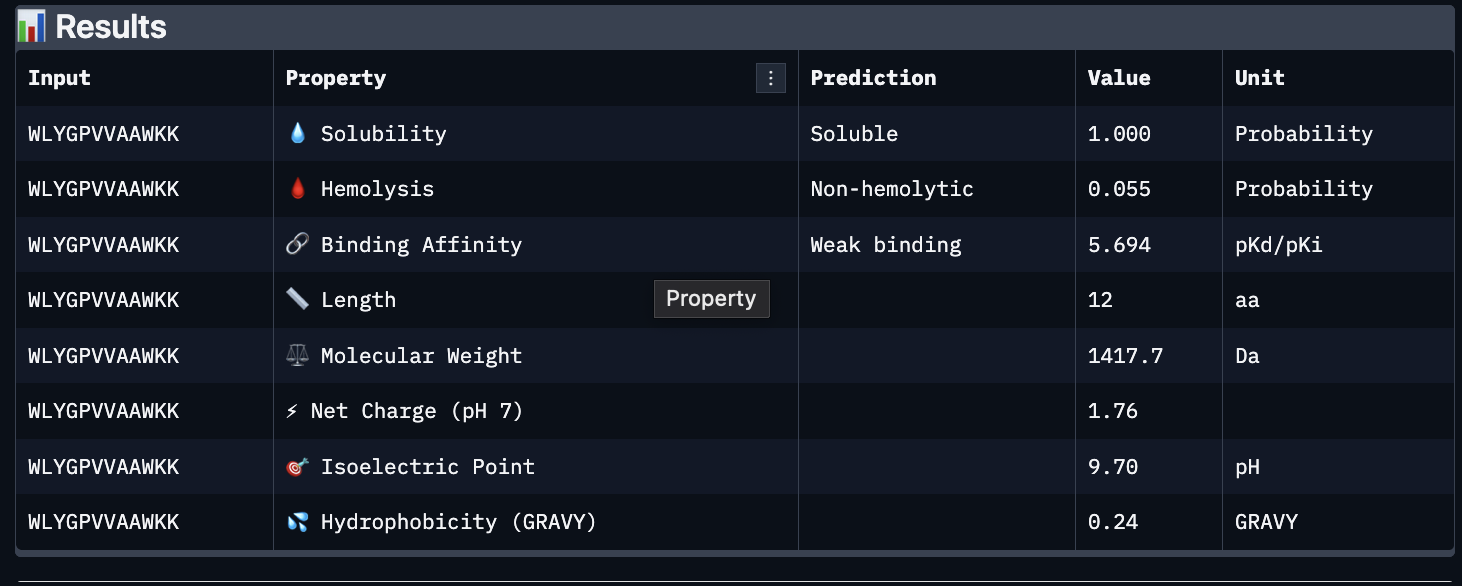
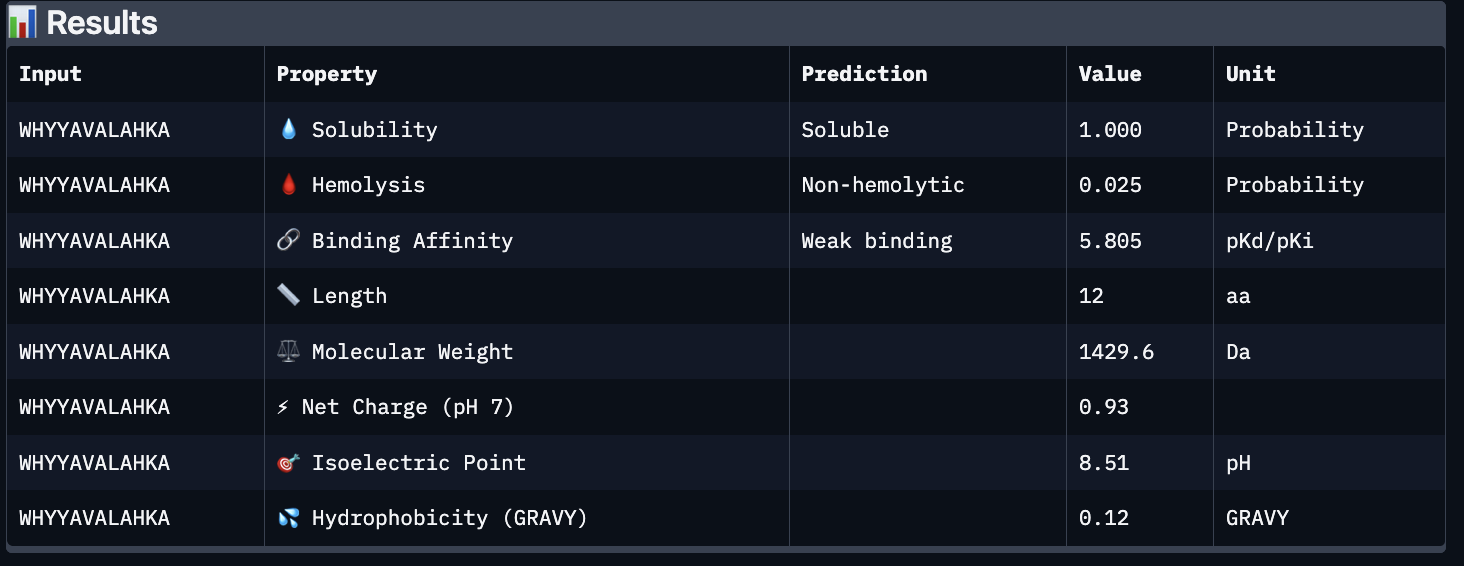
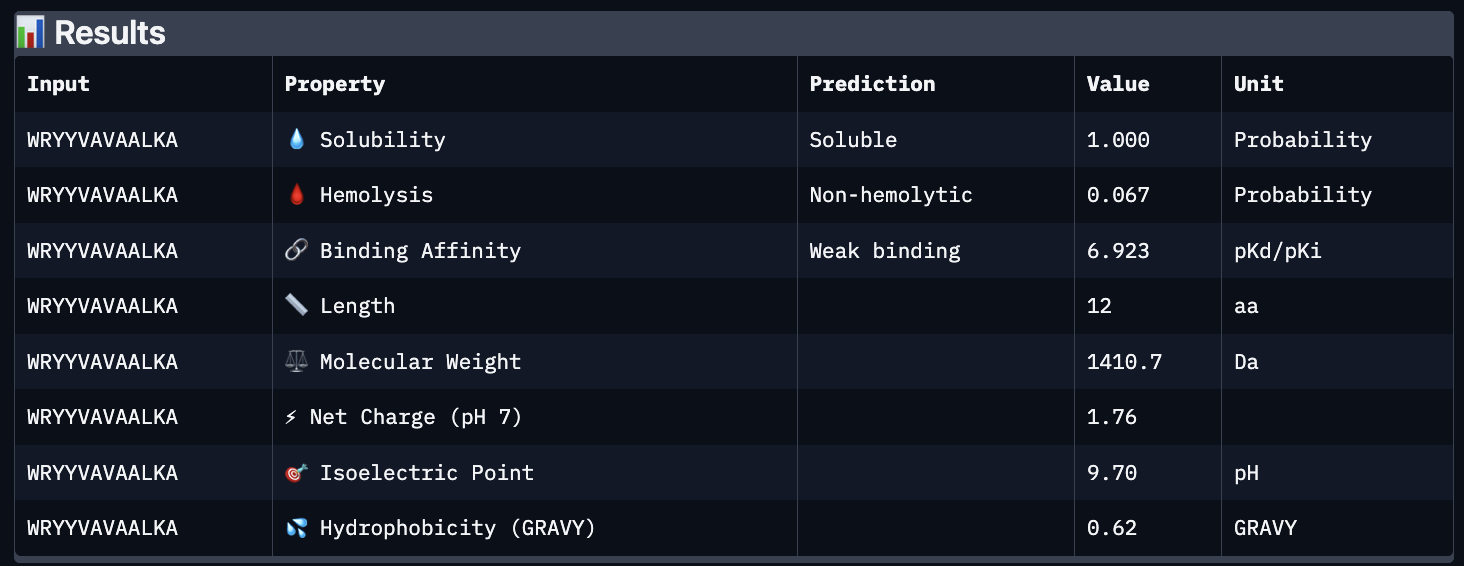
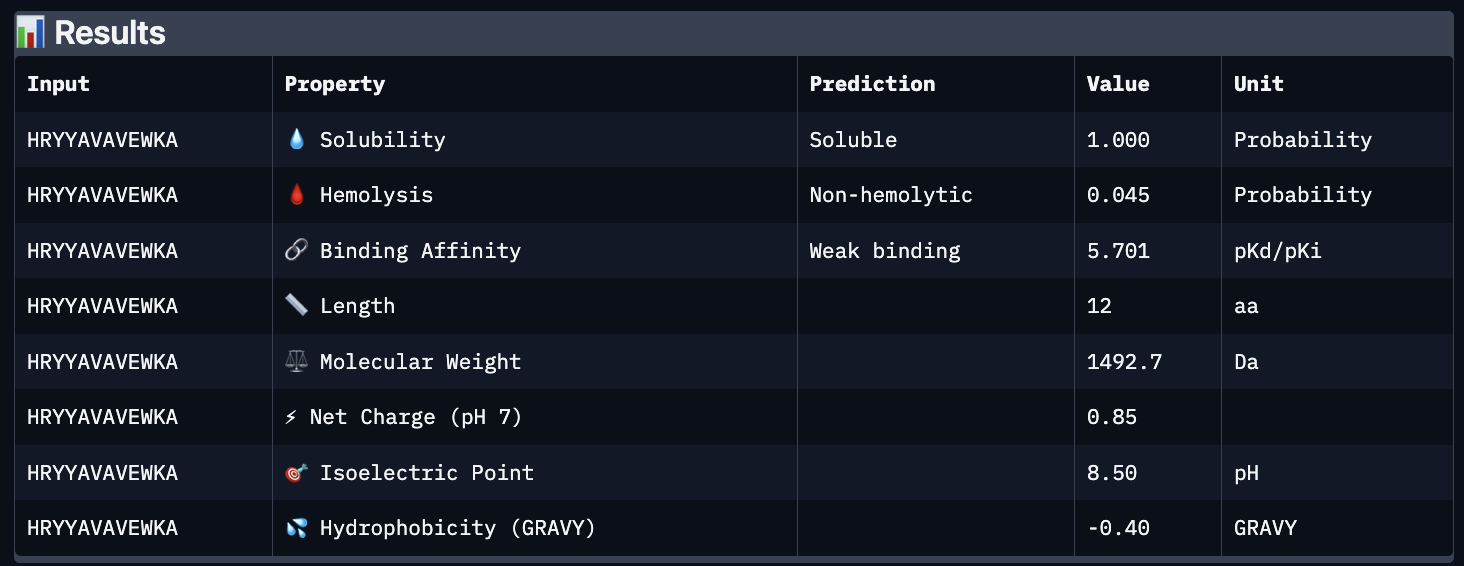
These predictions pretty much align with what I actually saw in AlphaFold, particularly with the weak binding. Between the known, index 0, 1 and 4, the binding predictions were marginally different, which is suprising because the known had a higher score, and the same score as index 2. However, index 2 has a relatively large jump in predicted affinity. Other than that, they were all predicted to be soluble and non-hemolytic.
If I had to choose a peptide to advance, I would pick index 2, only because on paper it looks slightly stronger than the other options with a slightly higher iPTM and binding affinity score. But to be honest, if I were tasked with this, I would try to generate other peptides first because these are all very poor options.
Part D:
To follow the homework, I had my target protein with the A4V mutation, and I chose 3 samples with a binder length of 12. I also checked off the boxes: Hemolysis, Affinity, Solubility and Motif. For the motif positions, I chose 2-7 because I wanted it to bind near the site 4 mutation.
The following peptides were generated:
- ‘EKLQCKKTFENQ’
- ‘KVKQCGFTQGDE’
- ‘STESGDTSYGTA’
Unsurprisingly to me, these optimized peptides had weak predicted binding affinities according to peptiverse. It’s possible that I just did not pick a good site to bind to, but part of me feels that the tools for predicting proteins are not advanced enough to make significant predictions. They differ from my PepMLM peptides very greatly though, as there seem to be a different general composition and more variety in amino acids.
In theory, if they were worth advancing, I would make sure to observe their binding and activity first. Then, I would try to assess stability and pharmacokinetic propertie,s and stability and half-life would be very useful measurements here. I think it would be worth evaluating almost everything, as in order to be used in clinical applications, peptides should be seriously screened as to not interfere with the human body.
Part 3: Lab
To be honest this lab was super confusing. I generated all of these things from the Google Colab that was generated up until the “stop here” point. This is what was generated:
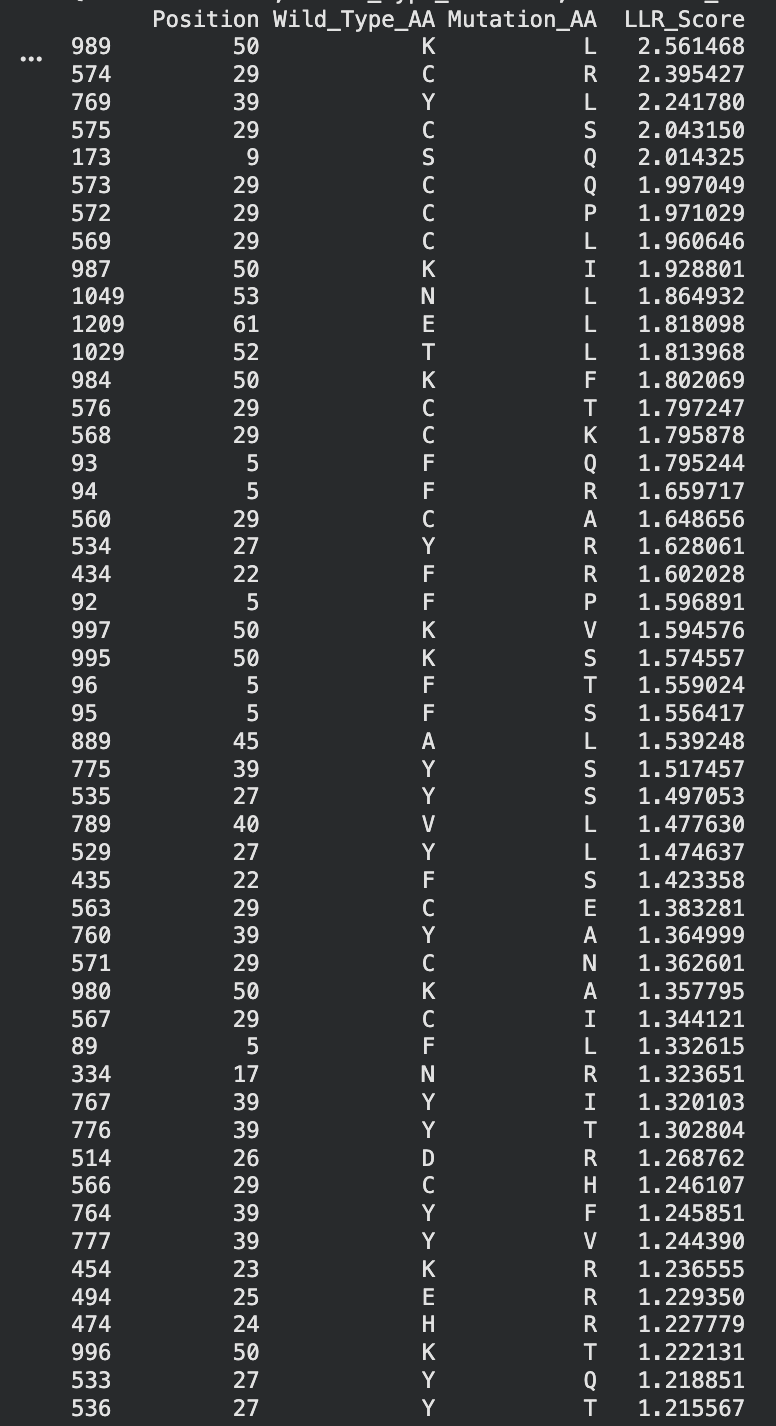
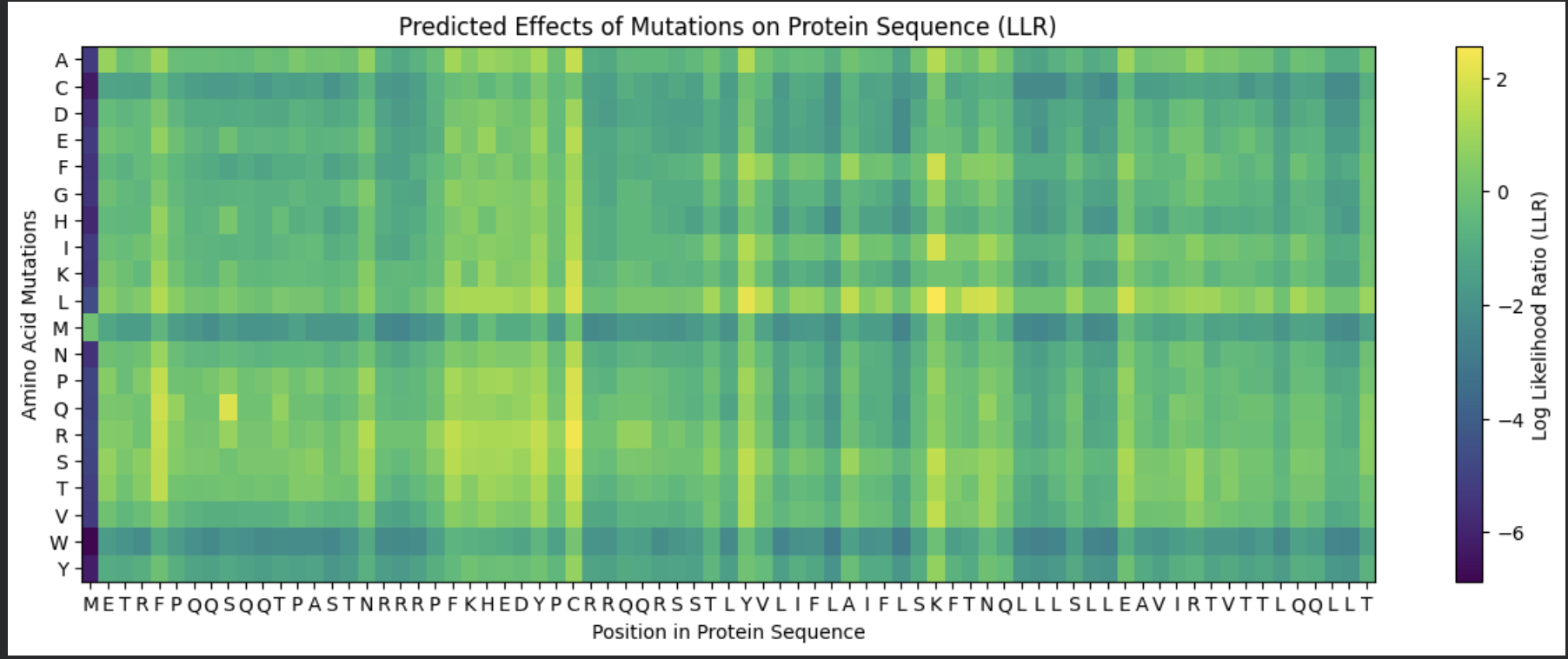
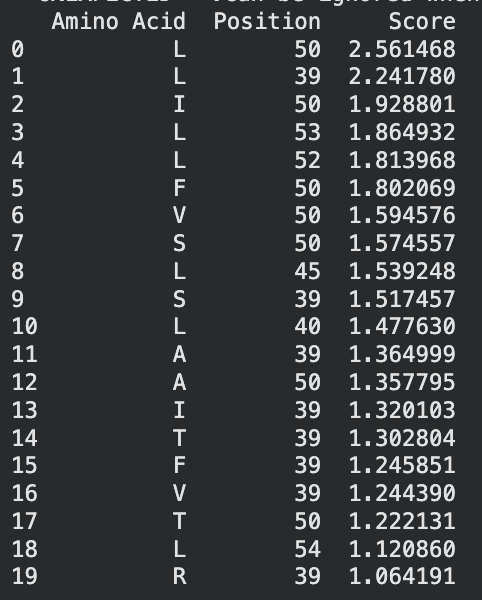
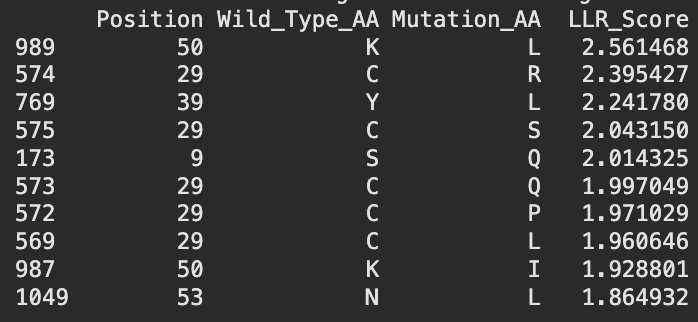
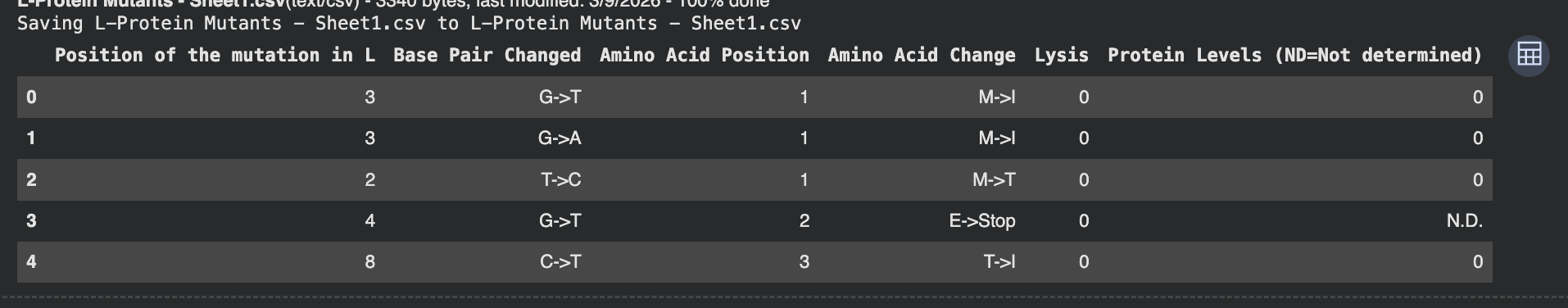
I identified the probable transmembrane helix by locating the longest hydrophobic stretch in the sequence, which spans approximately residues 40–62. Residues before this region were treated as soluble. Based on this topology estimate, I selected at least 2 mutations from the transmembrane region and at least 2 from the soluble region, prioritizing substitutions with high mutation scores and avoiding changes likely to disrupt conserved or experimentally sensitive residues.
A good set of five mutations would be:
S9Q (soluble region) – Position 9 is in the N-terminal soluble region. The mutation S→Q had a high computational score and similar polarity, so it likely preserves structure while allowing variation.
C29R (soluble region) – Position 29 is also in the soluble domain and appears multiple times in the mutation-score ranking. Substituting arginine introduces a charged residue that may improve solubility while remaining tolerated experimentally.
Y39L (soluble / boundary region) – Position 39 sits right before the transmembrane helix. The mutation to leucine had a strong score and may stabilize the transition into the hydrophobic helix because leucine is hydrophobic.
K50L (transmembrane region) – Position 50 lies inside the predicted transmembrane helix. Replacing lysine (charged) with leucine (hydrophobic) should better match the membrane environment. It also had the highest LLR score, suggesting it is strongly favored.
N53L (transmembrane region) – Position 53 is also inside the hydrophobic helix. Substituting leucine increases hydrophobicity and is consistent with residues typically found in membrane-spanning helices.
I selected mutations with high predicted scores, avoided stop mutations or positions that eliminated protein expression in the experimental dataset, and ensured the substitutions were biophysically reasonable for their regions. For soluble regions I chose substitutions that maintain polarity or introduce tolerated charges, while for the transmembrane helix I favored hydrophobic substitutions that stabilize membrane insertion