Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
1. Briefly describe the protein you selected and why you selected it.
Vibrio fischeri LuxA
2. Identify the amino acid sequence of your protein.
sp|P19907|LUXA_ALIFS Alkanal monooxygenase alpha chain OS=Aliivibrio fischeri OX=668 GN=luxA PE=3 SV=1 MKFGNICFSYQPPGETHKLSNGSLCSAWYRLRRVGFDTYWTLEHHFTEFGLTGNLFVAAA NLLGRTKTLNVGTMGVVIPTAHPVRQLEDVLLLDQMSKGRFNFGTVRGLYHKDFRVFGVD MEESRAITQNFYQMIMESLQTGTISSDSDYIQFPKVDVYPKVYSKNVPTCMTAESASTTE WLAIQGLPMVLSWIIGTNEKKAQMELYNEIATEYGHDISKIDHCMTYICSVDDDAQKAQD VCREFLKNWYDSYVNATNIFNDSNQTRGYDYHKGQWRDFVLQGHTNTNRRVDYSNGINPV GTPEQCIEIIQRDIDATGITNITCGFEANGTEDEIIASMRRFMTQVAPFLKEPK
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
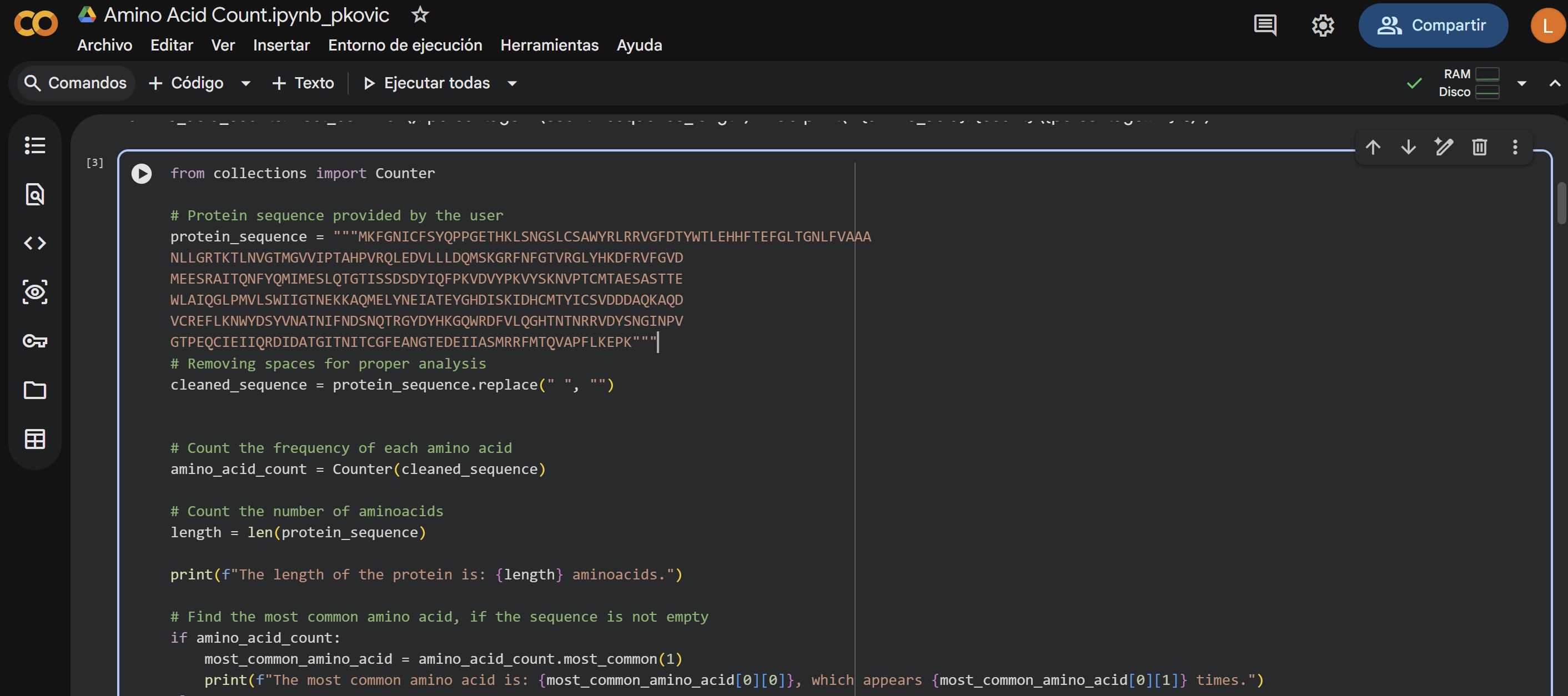
The length of the protein is: 359 aminoacids. The most common amino acid is: T, which appears 30 times.
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
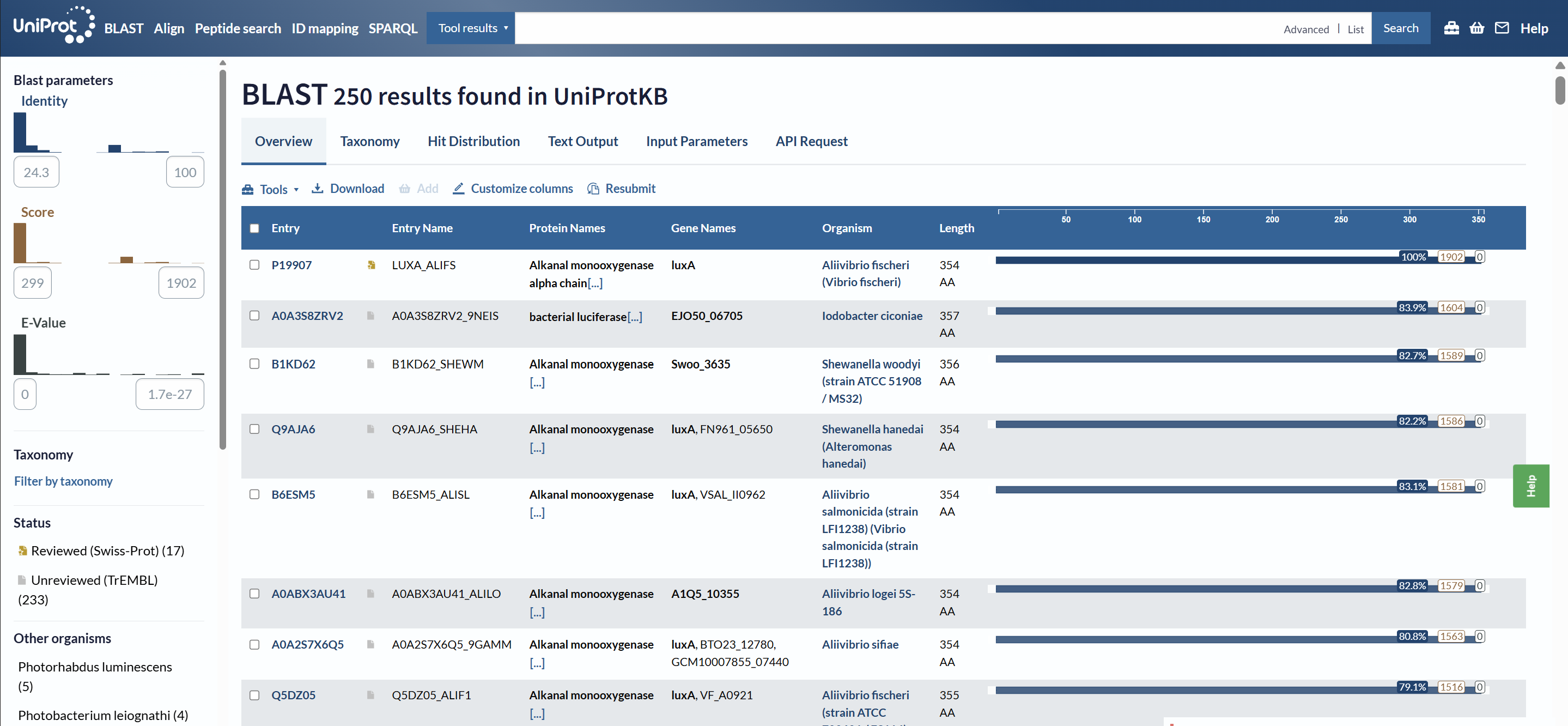
Using UniProt BLAST, 250 sequence homologs were found in UniProtKB (17 reviewed in Swiss-Prot and 233 unreviewed in TrEMBL).
3.Identify the structure page of your protein in RCSB
A direct crystal structure for LuxA from Aliivibrio fischeri was not found in the RCSB PDB database. Therefore, a highly homologous bacterial luciferase structure (PDB ID: 1LUC) from Vibrio harveyi was used as a structural reference.
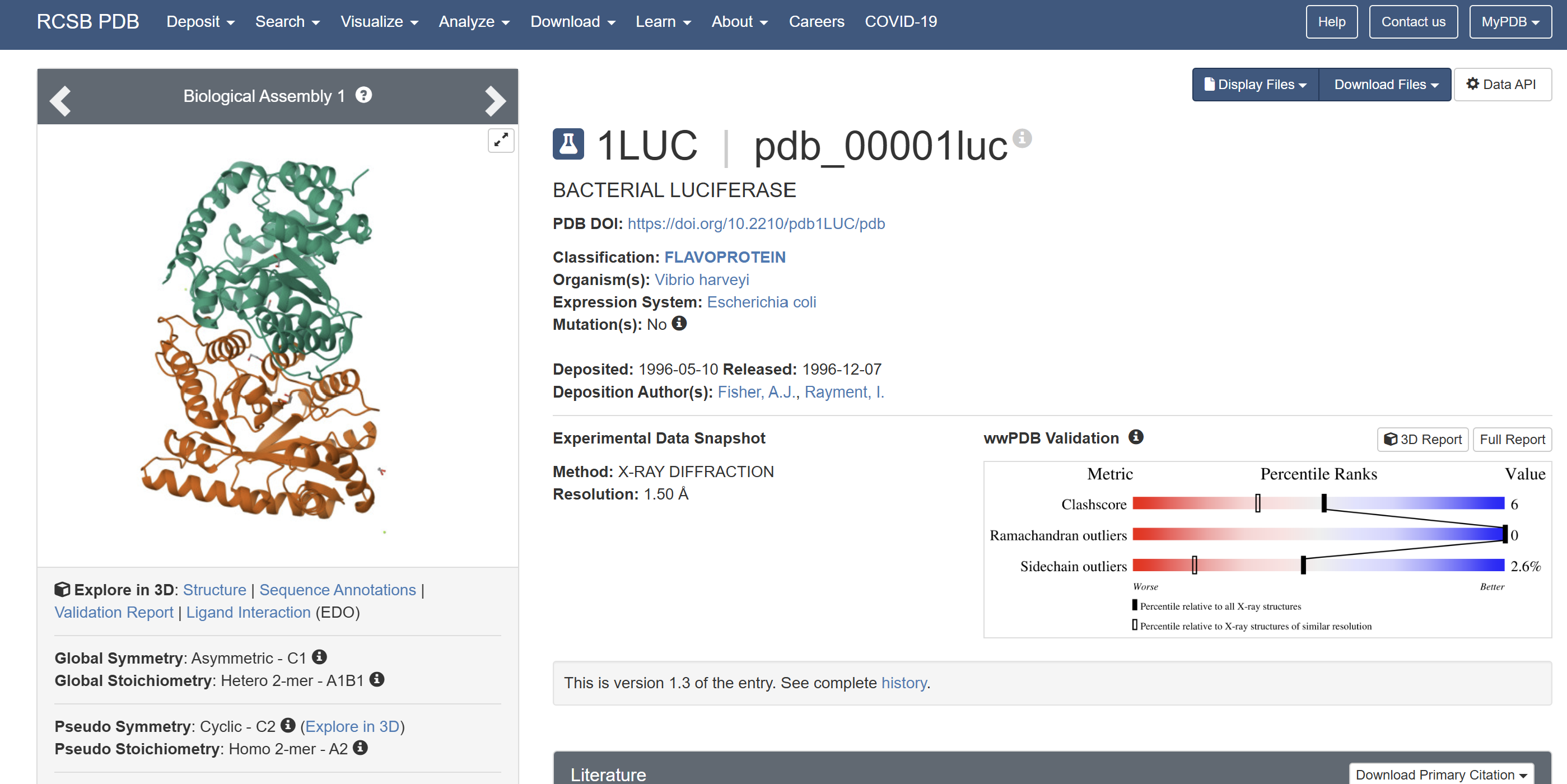
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
The structure was solved in 1996 with a resolution of 1.50 Å, which is considered high quality because lower resolution values (<2.0 Å) indicate more detailed atomic information.
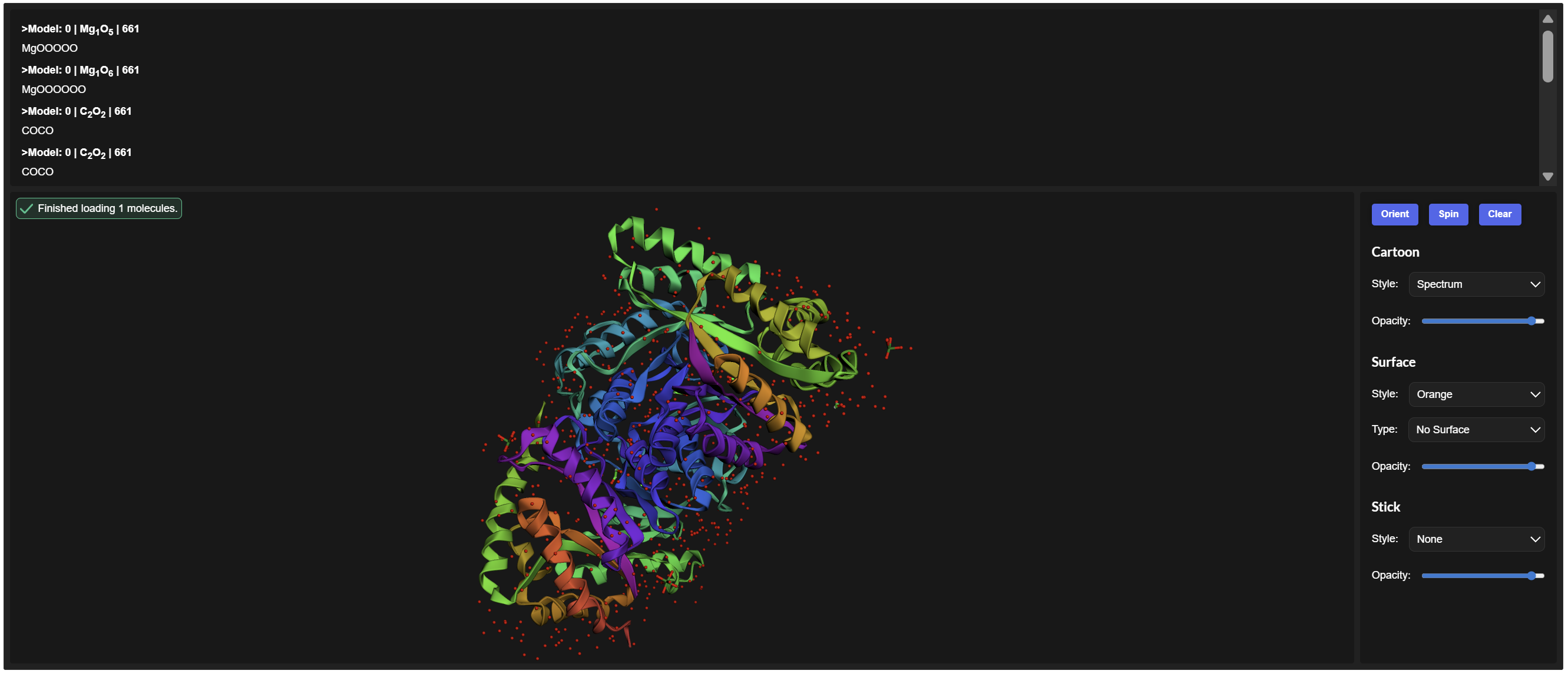
Are there any other molecules in the solved structure apart from protein?
Yes. In addition to the protein chains, the structure contains 1,2-ethanediol (EDO) and a magnesium ion (Mg²⁺).
Does your protein belong to any protein family?
Yes. LUXA belongs to the bacterial luciferase family and is a flavin-dependent monooxygenase involved in bioluminescence.
4. Open the structure of your protein in any 3D molecule visualization software
- PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
I encountered technical difficulties while attempting to install PyMOL on my local system. To complete the assignment, I opted to use online molecular visualization software (such as the RCSB PDB 3D Viewers). These web-based tools allowed me to accurately generate and analyze the required protein structures, including the Cartoon, Spacefill, and Surface representations shown in my results
- Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
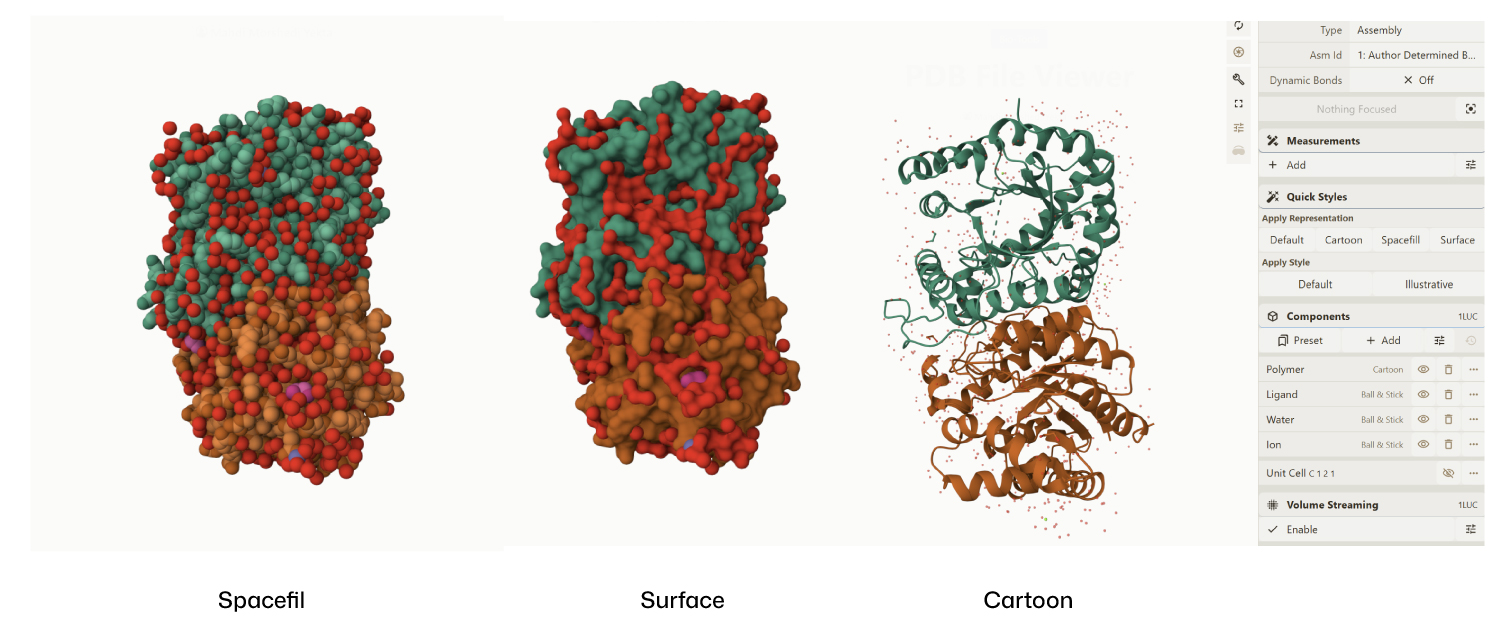
- Color the protein by secondary structure. Does it have more helices or sheets?
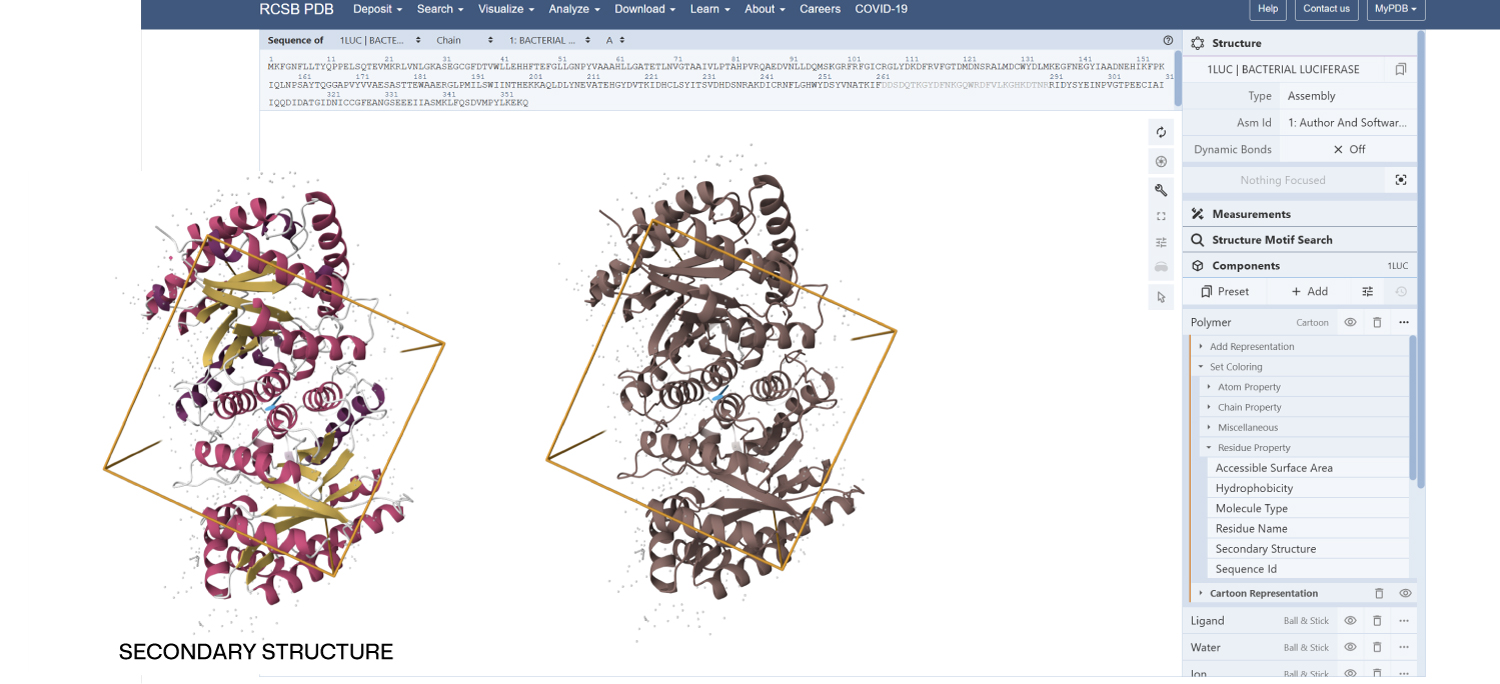
Secondary Structure: According to the previsualization,the protein has more helices than sheets. The alpha-helices are prominent and surround the internal beta-strands
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
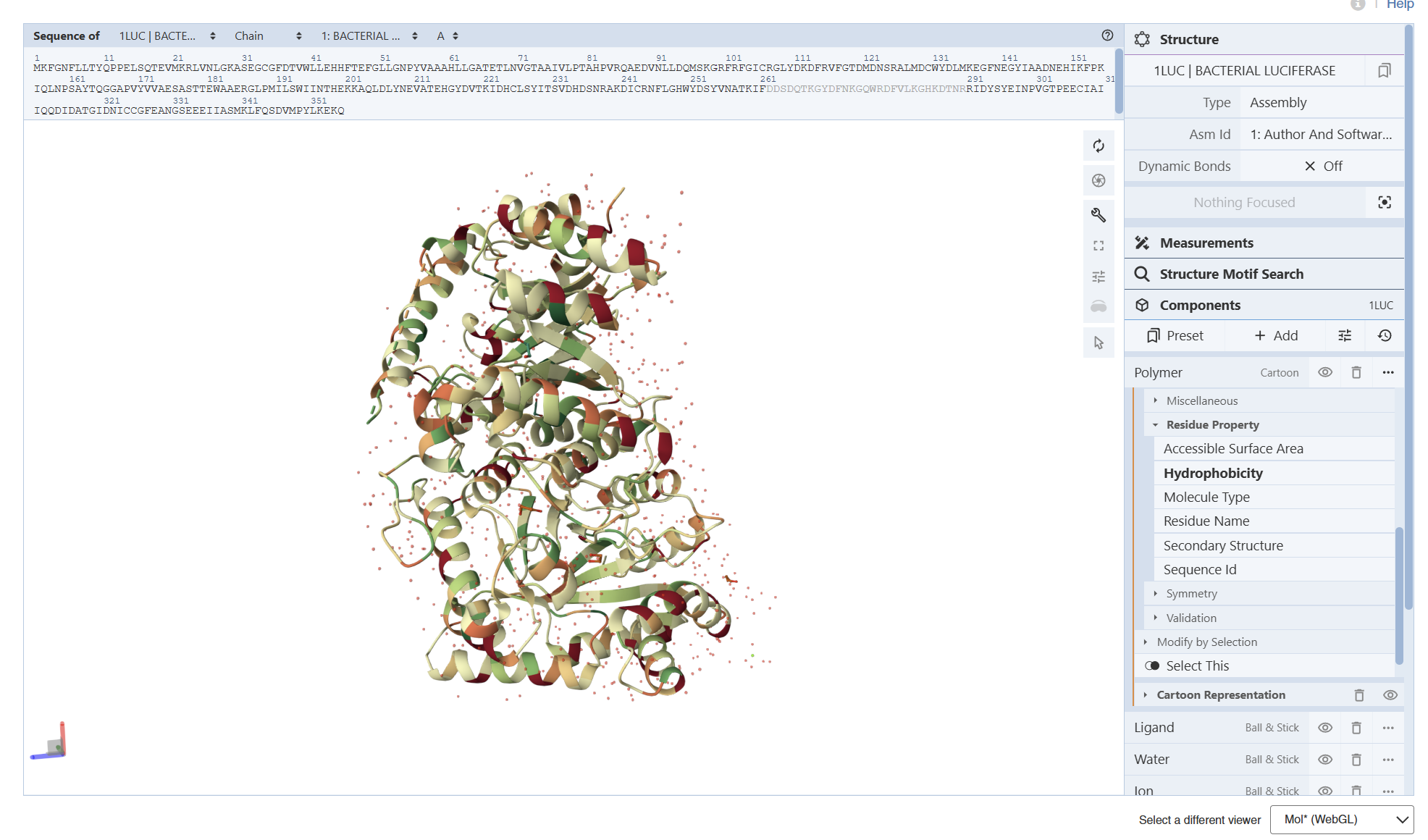
I am not entirely sure, but based on what I can observe in the visualization, the hydrophobic residues (represented by the darker/reddish tones) appear to be predominantly located in the interior of the protein. From what I understand, these residues ‘hide’ in the core, and as shown in the model, they are mostly tucked away from the surface
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
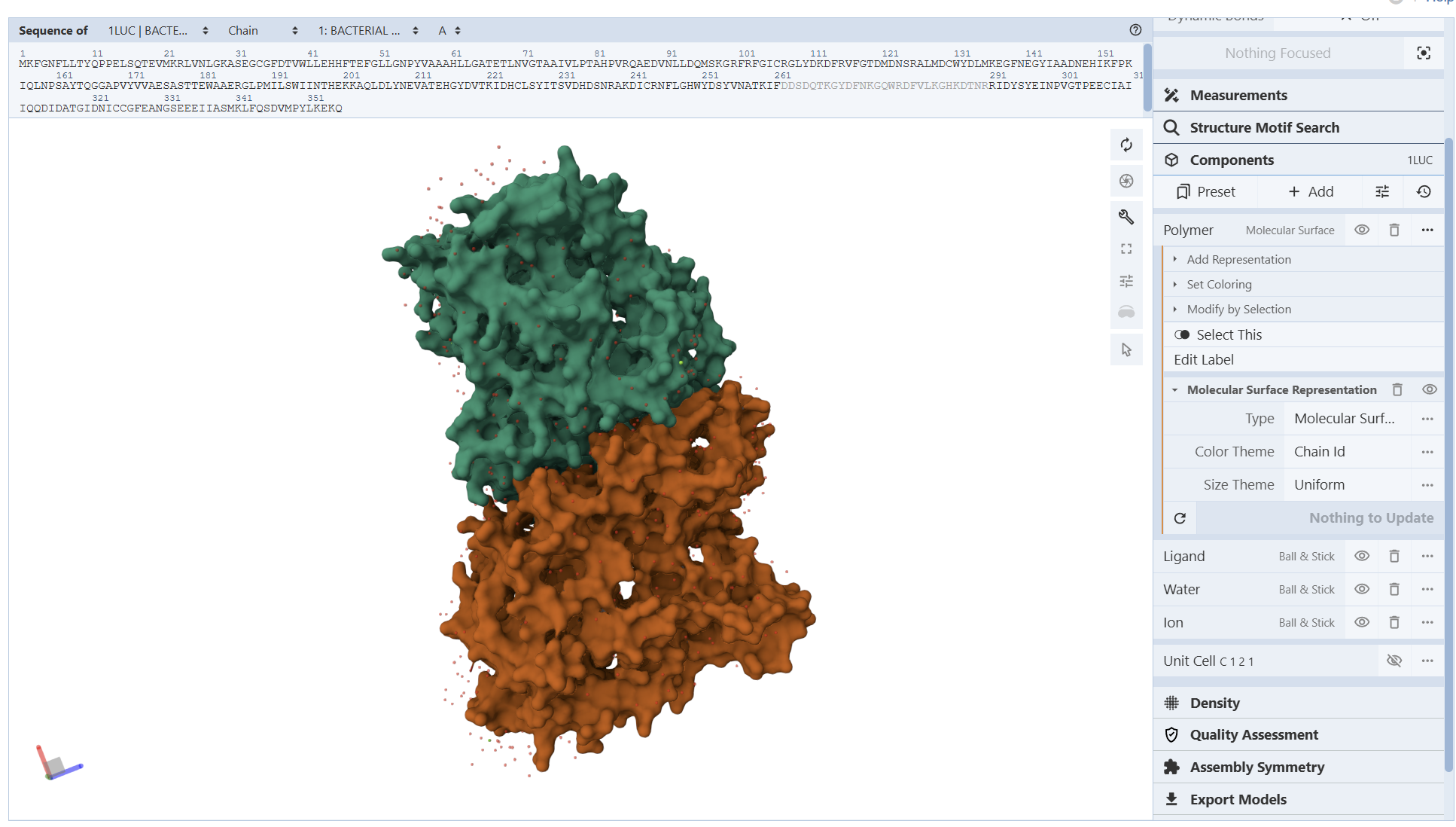
From these visualization results, it can be observed that the protein structure features various holes and cavities, which demonstrates the presence of several prominent binding pockets. As I understand it, these spaces are necessary for molecules to enter and fit into the protein, allowing the chemical reaction that produces light to occur.
Part C. Using ML-Based Protein Design Tools
- Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
- Choose your favorite protein from the PDB.
- We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
C1. Protein Language Modeling
- Deep Mutational Scans
- Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
PDB ID: 1LUC
sp|P07740|LUXA_VIBHA Alkanal monooxygenase alpha chain OS=Vibrio harveyi OX=669 GN=luxA PE=1 SV=1 MKFGNFLLTYQPPELSQTEVMKRLVNLGKASEGCGFDTVWLLEHHFTEFGLLGNPYVAAA HLLGATETLNVGTAAIVLPTAHPVRQAEDVNLLDQMSKGRFRFGICRGLYDKDFRVFGTD MDNSRALMDCWYDLMKEGFNEGYIAADNEHIKFPKIQLNPSAYTQGGAPVYVVAESASTT EWAAERGLPMILSWIINTHEKKAQLDLYNEVATEHGYDVTKIDHCLSYITSVDHDSNRAK DICRNFLGHWYDSYVNATKIFDDSDQTKGYDFNKGQWRDFVLKGHKDTNRRIDYSYEINP VGTPEECIAIIQQDIDATGIDNICCGFEANGSEEEIIASMKLFQSDVMPYLKEKQ
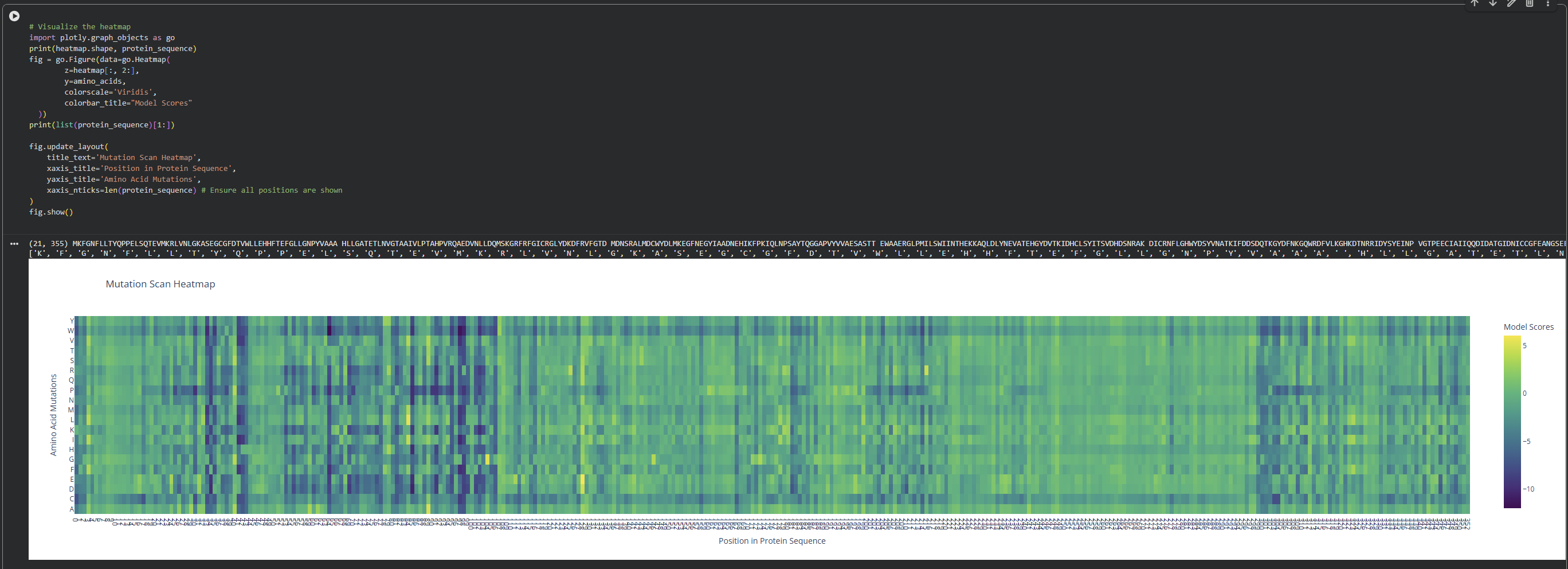
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
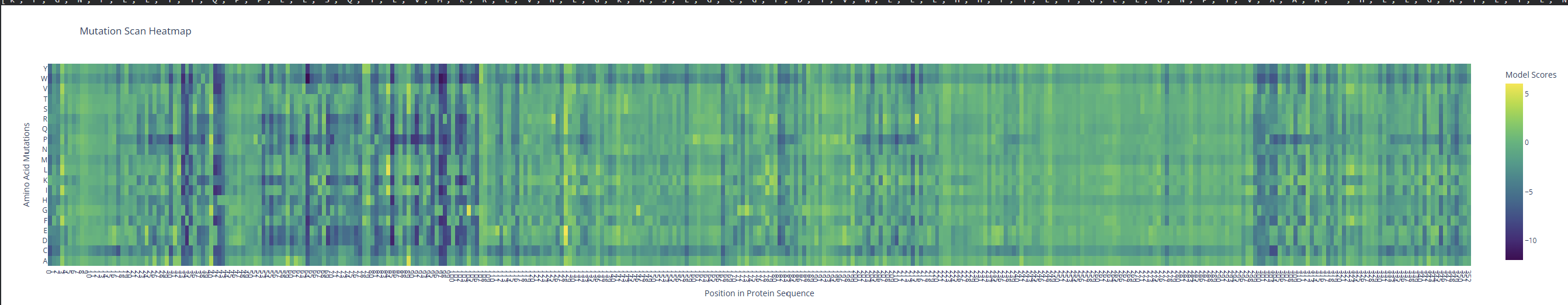

I observed a pattern of high compatibility in the Valine (V) row. Unlike Proline, which shows dark blue (low likelihood) across most positions, Valine remains consistently light-colored. This suggests that the luciferase structure is robust to Valine substitutions, likely because Valine’s small, hydrophobic side chain can be easily accommodated in various positions of the protein’s fold without causing major structural disruptions
2. Latent Space Analysis
Use the provided sequence dataset to embed proteins in reduced dimensionality.
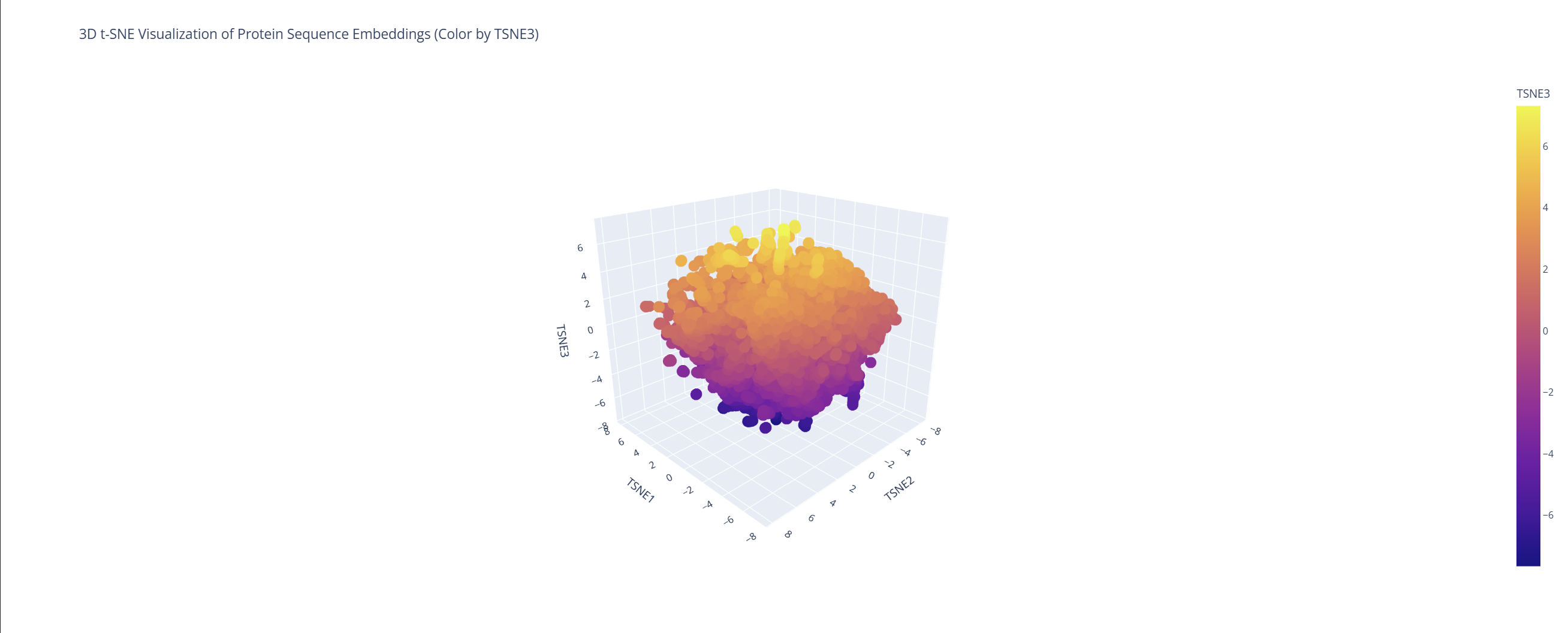
Analyze the different formed neighborhoods: do they approximate similar proteins?
Based on this visualization, we can clearly see the existence of different groupings or clusters that create these ’neighborhoods’ of proteins. This shows that the ESM2 model is effectively generating these connections by grouping proteins that share similar characteristics.
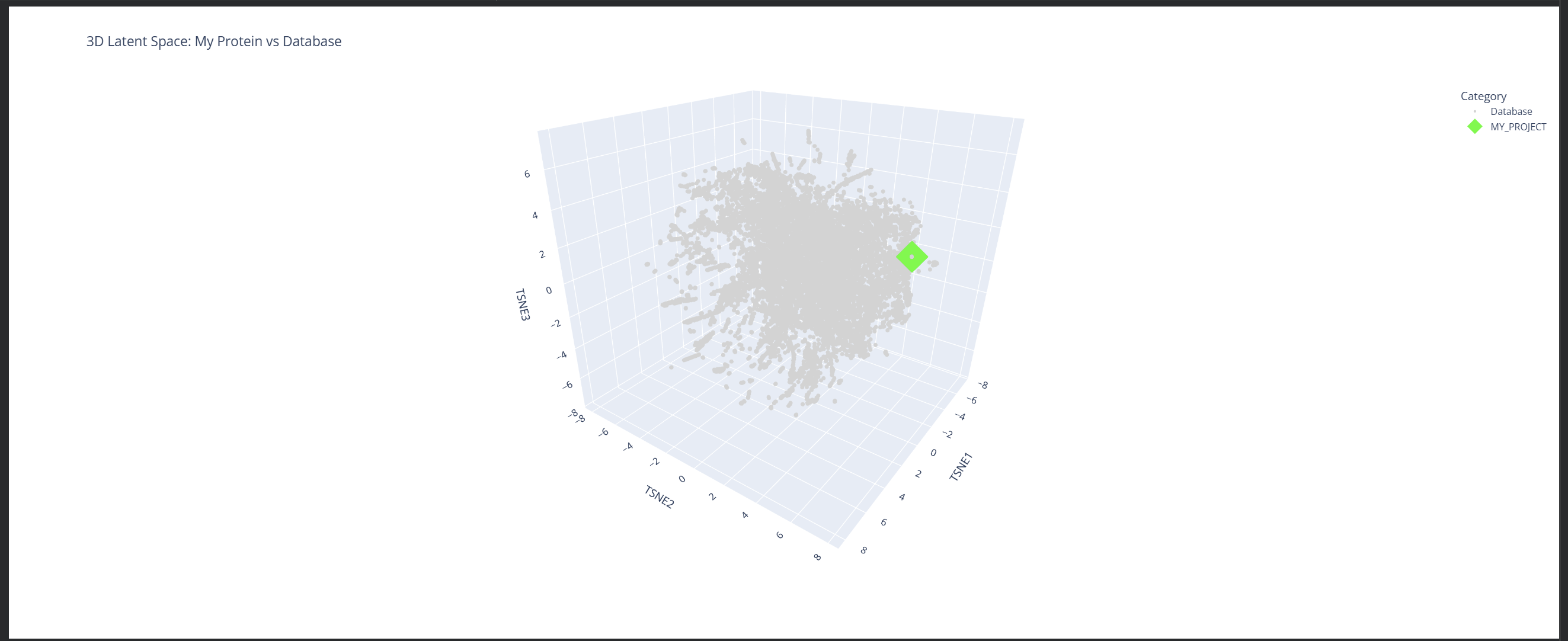
Place your protein in the resulting map and explain its position and similarity to its neighbors.
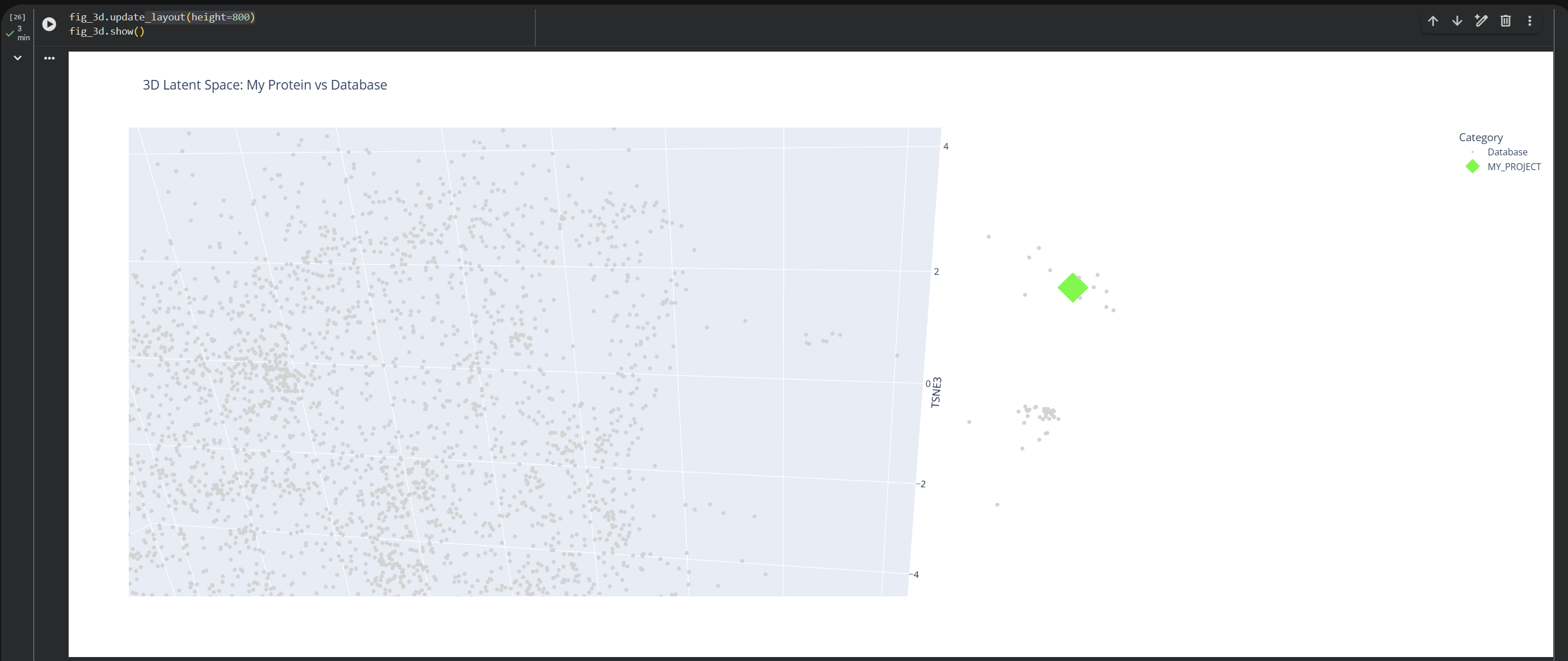
My protein (1LUC) landed in a specific neighborhood within the map. Being positioned next to other proteins from the database shows that they share similar characteristics. Given its biological nature, this confirms the model successfully grouped it with its functional relatives.
C2. Protein Folding
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
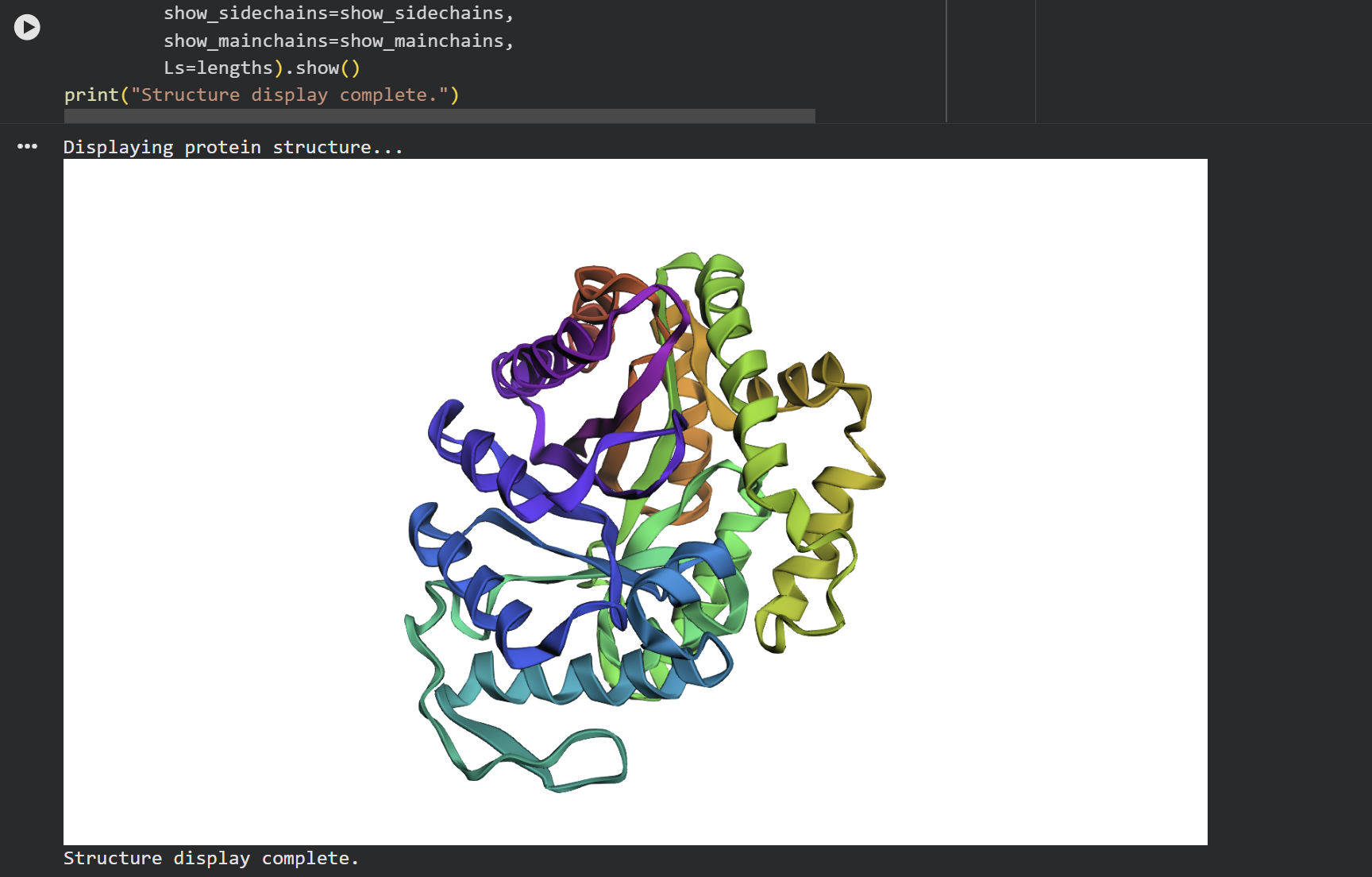
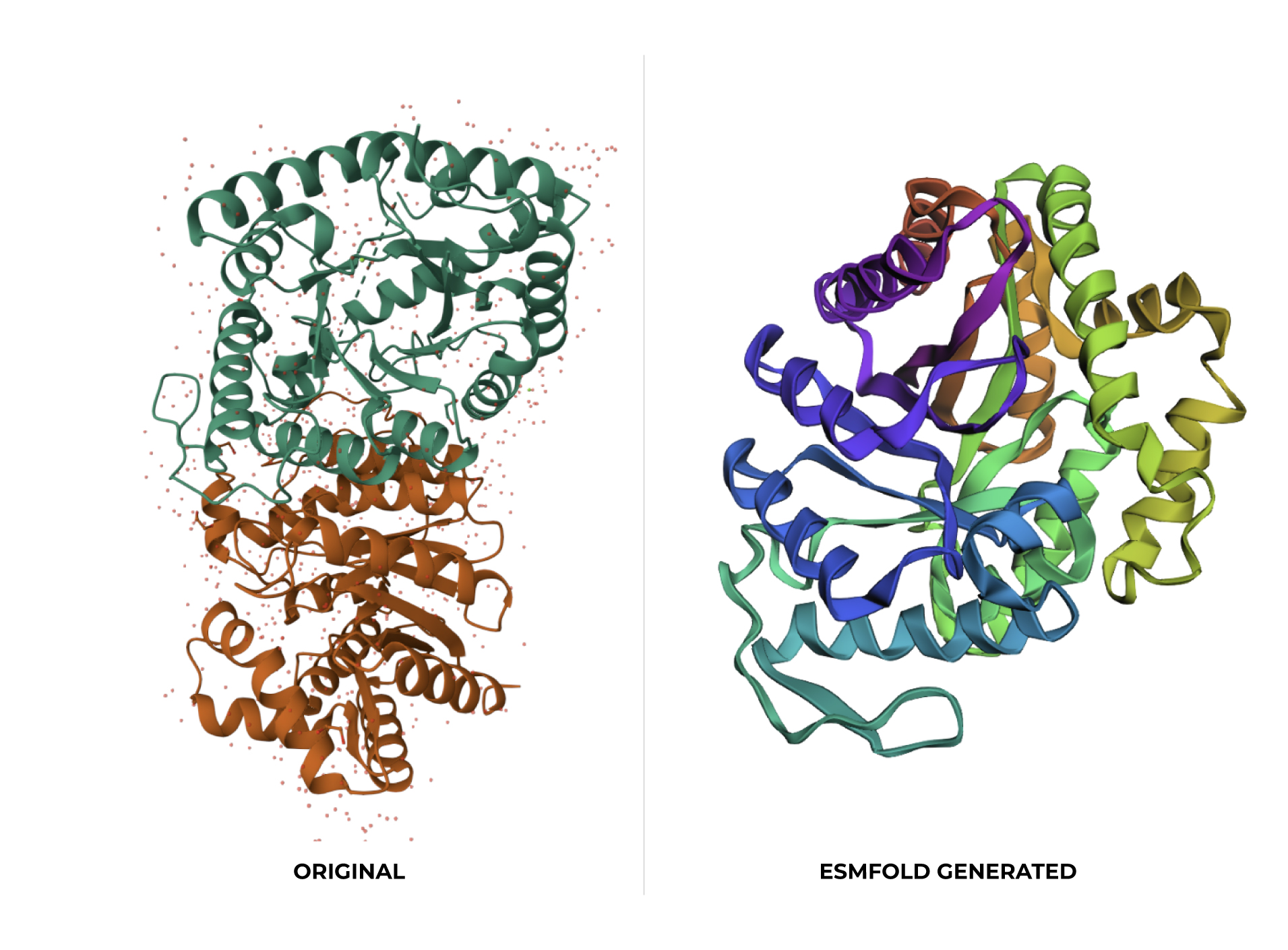
At first glance, the predicted coordinates do not seem to match the original structure, as the overall complexity is different. However, upon closer inspection, there is a clear structural resemblance to one of the specific domains of the protein. The original 1LUC is composed of two different chains, while the simulation produced a single-domain fold. It appears the model successfully captured the architecture of one of these subunits, even if it couldn’t replicate the full multi-domain assembly of the real protein.
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
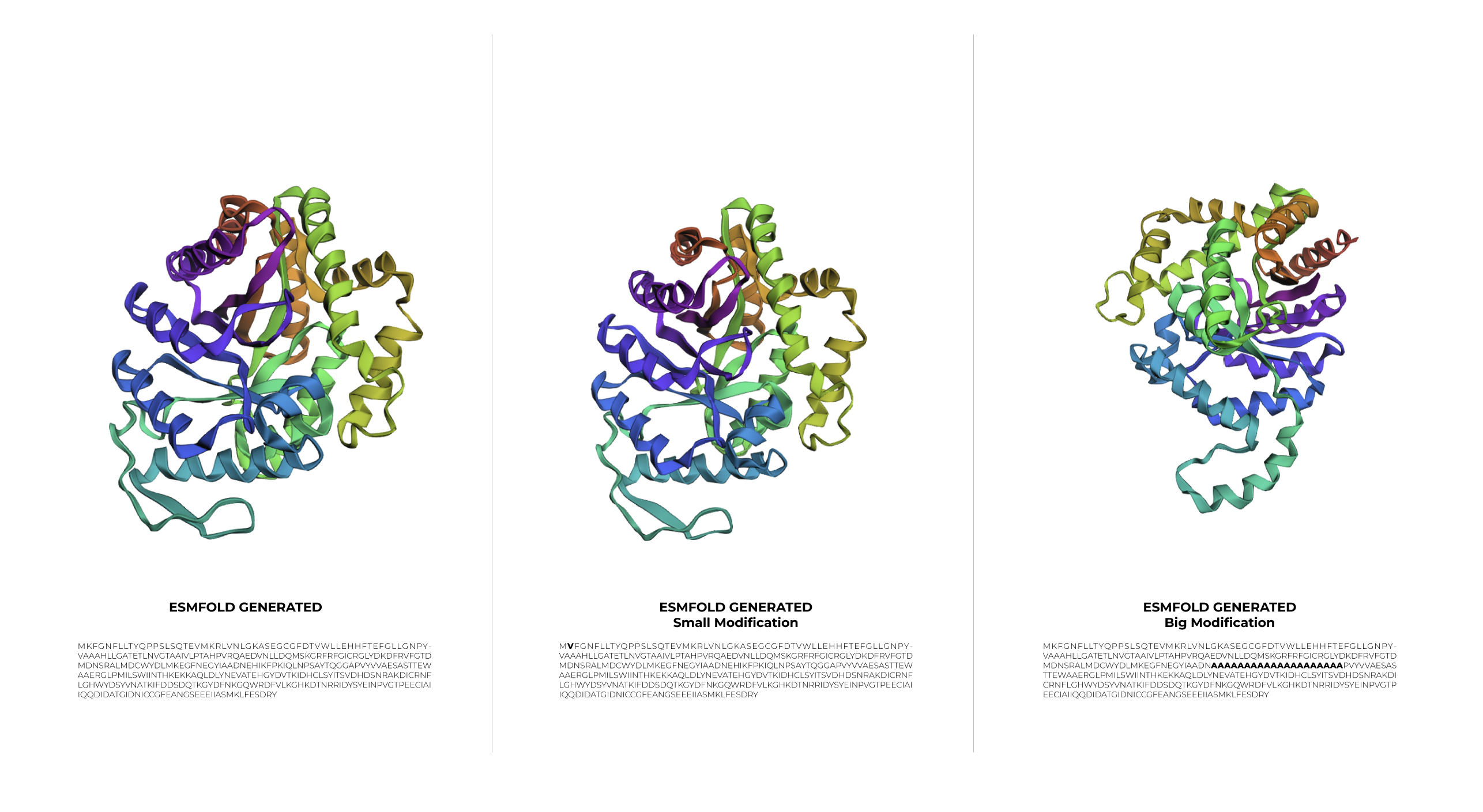
During the first stage of experimentation, a minimal change was made to the protein sequence by swapping Lysine for Valine. The result was a structure basically identical to the original one. This outcome demonstrates that 1LUC is highly resilient to small modifications, as the point mutation did not compromise the structural integrity of the 1LUC.
In the second stage, a larger modification was introduced. Although the sequence was significantly altered, the protein showed a surprising level of resilience, maintaining its general folded shape even if the symmetry was slightly affected.
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN

Results
Original LuxA Secuence
MKFGNFLLTYQPPELSQTEVMKRLVNLGKASEGCGFDTVWLLEHHFTEFGLLGNPYVAAA HLLGATETLNVGTAAIVLPTAHPVRQAEDVNLLDQMSKGRFRFGICRGLYDKDFRVFGTD MDNSRALMDCWYDLMKEGFNEGYIAADNEHIKFPKIQLNPSAYTQGGAPVYVVAESASTT EWAAERGLPMILSWIINTHEKKAQLDLYNEVATEHGYDVTKIDHCLSYITSVDHDSNRAK DICRNFLGHWYDSYVNATKIFDDSDQTKGYDFNKGQWRDFVLKGHKDTNRRIDYSYEINP VGTPEECIAIIQQDIDATGIDNICCGFEANGSEEEIIASMKLFQSDVMPYLKEKQ
Inverse Folding with ProteinMPNN
MKFGYHIHGYQPPHLTREEVLARIEKLAAAAPEQGFDTVWIHEGYFHPHNLIADPFATAEKFLAASKDLKVGTEALDLPARDPLEVAKKANTLDNKSKGRFIFGISRGAFEYVFKVFGKDFSKRFELTKYKLELIEEGFKTGTMSADSEYLKFPKTKVTPASYTPGGPPIYVVARTDEEVEFAAERGIPLVLSPLITVEEKKAMLDKYKEVAAKNGFDVSKIEHVLSYFGVVDEDEELAKKKMLAYLKEYAASLEEAKNLLXXXXXXXXXXXXXXXXXXXXXXXXXXXXXGLEKAPEINPVGTPEHAVEVIQKDIDATGITYINVNFEAAGTVEEILKSMKLFKEEVMPKLKPKK
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
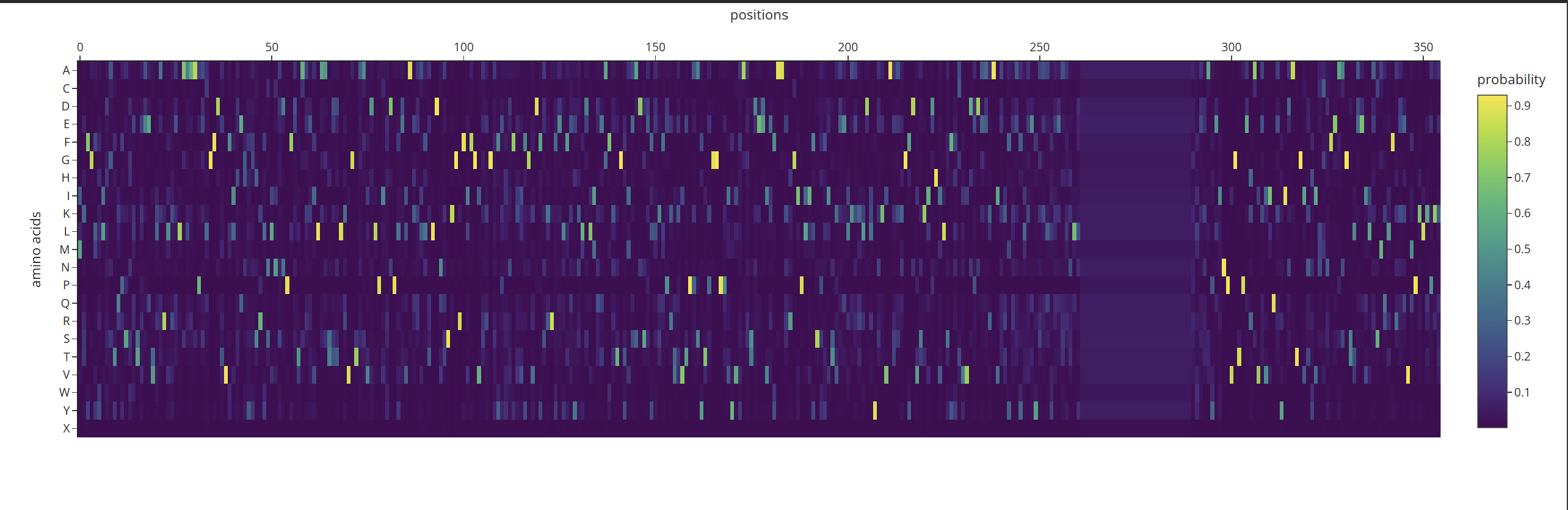
According to what I can understand by analyzing the probability heatmap, we can identify the critical points of the structure. The areas marked in yellow represent the positions where amino acids must be strictly maintained so that the protein does not lose its properties or shape. Meanwhile, the dark-colored areas are zones where the AI can ‘fill in’ with different options without necessarily affecting the overall fold.
Input this sequence into ESMFold and compare the predicted structure to your original.
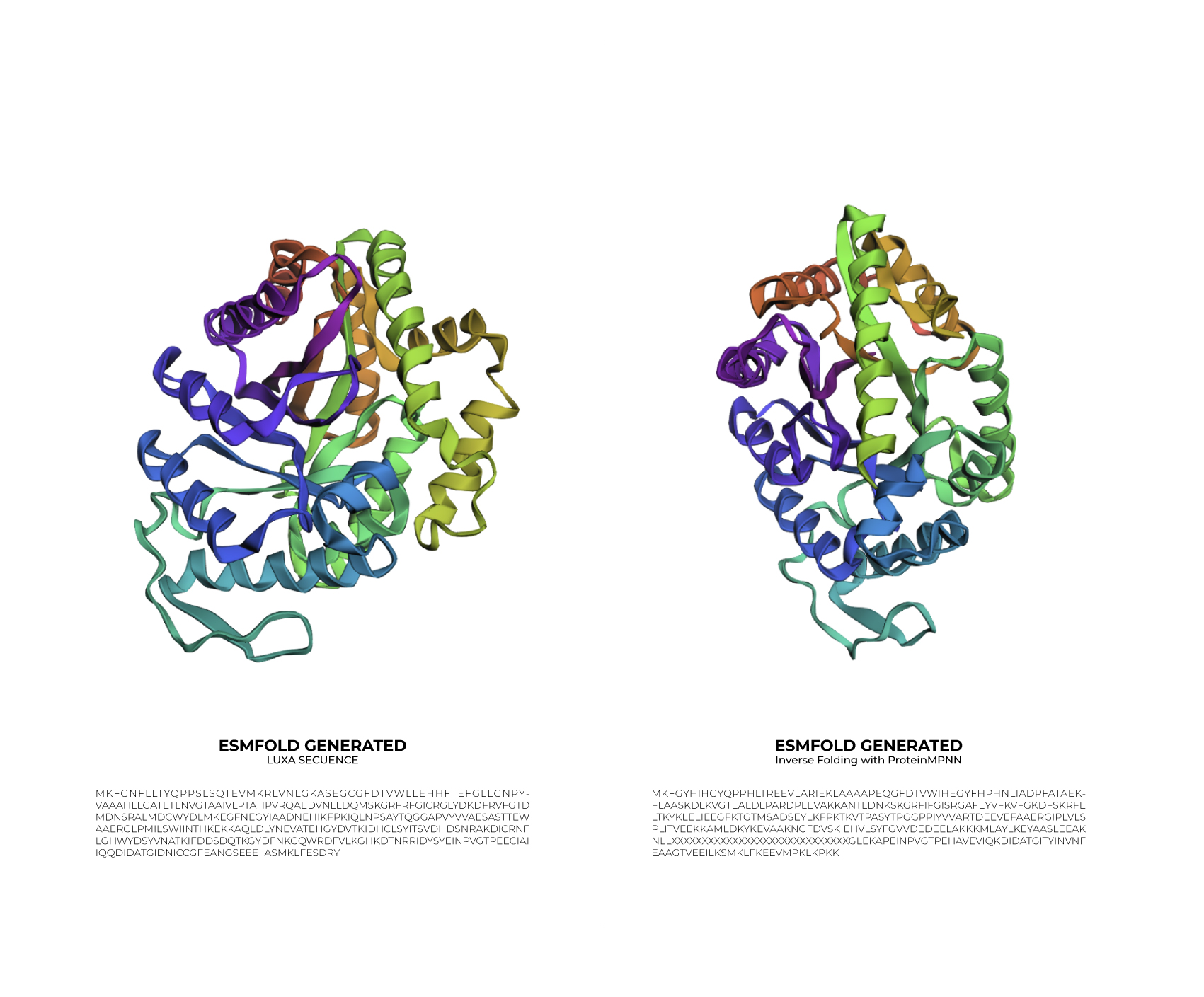
Based on the data obtained from the output (>T=0.1, sample=0, score=0.8181, seq_recovery=0.4632), it can be concluded that although the Colab changed 53% of the original protein, the final result still maintained the essential points of the structure. Even the preview remained highly faithful to the original