Subsections of Homeworks
Week 1 HW: Principles and Practices
HOMEWORK 1
1.First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
One of the main reasons I am interested in synthetic biology is because I see it as a real and accessible bridge between design and biology, as well as a key pathway toward the future of design applied to living systems. Throughout my experience as a designer, I have developed a growing interest in reducing the gap between what is created by humans and natural systems. Often, even when design draws inspiration from nature, it remains an exclusively human interpretation. This has led me to question what the true role of the designer should be when working with living systems and natural environments.
In this context, I am deeply interested in the future exploration of synthetic morphogenesis, understood as the possibility of creating biological frameworks that allow materials or living systems to develop their own form and function autonomously through natural processes. From this perspective, the designer does not necessarily define the final outcome but rather facilitates the initial conditions that allow nature to actively participate in the creative process. I believe synthetic biology can offer tools to advance toward these kinds of practices, where our role is to design conditions rather than impose forms. However, while recognizing that this concept still requires deeper development, specific areas already exist where certain approaches and principles can begin to be explored and consolidated.
As a design professional, I have always felt a strong call to action to create solutions that not only improve people’s quality of life but also contribute to a broader ecosystemic perspective on wellbeing. My interest is not limited to human benefit; rather, I aim to explore how design can help care for, regenerate, and strengthen the natural environments and living systems upon which we depend.
This vision has led me to explore fields such as bio-inspired design, biomimetics, bionics, and ultimately biodesign. Throughout this trajectory, one of my main interests has been design oriented toward conservation and ecological remediation, particularly within marine ecosystems. My recent work has focused on exploring design- and material-based solutions that contribute to the regeneration of marine environments affected by climate change.
In recent years, I have become particularly interested in developing proposals to mitigate the impact of ocean acidification. Ocean acidification is a global issue caused by increasing atmospheric CO₂ levels, which alter seawater chemistry and reduce pH levels. This phenomenon decreases the availability of calcium carbonate (CaCO₃), an essential component for calcifying organisms such as corals, mollusks, and crustaceans, affecting their ability to form skeletal structures through biomineralization and reducing their chances of survival. Many of these organisms play a fundamental role at the base of marine food webs and in the functional structure of ecosystems; therefore, significant alterations in their populations can trigger cascading effects, leading to systemic ecosystem losses and a progressive decline in biodiversity.
To address this problem, various strategies have been developed worldwide, including artificial reefs and aquaculture systems. However, despite their progress, most approaches have focused primarily on structural design, leaving the biological potential of materiality relatively underexplored.
For this reason, I would like to take this exploration to the next level. One of the biological engineering applications I would like to develop during the course How to Grow Almost Everything is a bioengineered living material that initially functions as a bioreceptor for CaCO₃ particles present in the water, attracting and concentrating them to facilitate the formation of artificial reefs in strategic locations. In alignment with my interest in synthetic morphogenesis, I would also like to explore the possibility that this material not only performs a capture function but is capable of growing, self-organizing, and adopting its own configurations through biological processes, contributing to the formation of structures that emerge from natural dynamics rather than from fully predetermined human designs.
Through this approach, it would be possible to locally increase the availability of calcium carbonate, support biomineralization processes, and contribute to restoring ecological balance in areas affected by ocean acidification, generating benefits for both marine ecosystems and the human communities that depend on them.
2.Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals. Below is one example framework (developed in the context of synthetic genomics) you can choose to use or adapt, or you can develop your own. The example was developed to consider policy goals of ensuring safety and security, alongside other goals, like promoting constructive uses, but you could propose other goals for example, those relating to equity or autonomy.
(1) Promote ecological restoration and Long-Term ecosystem balance
-Ensure the material supports native calcifying organisms and does not displace or outcompete existing species.
-Require ecological impact assessments prior to deployment in new marine environments.
-Prioritize deployment in degraded or climate-vulnerable areas where restoration potential is scientifically justified.
(2) Enhance ecosystem services while preserving biodiversity integrity
-Design materials to support habitat formation, biodiversity recovery, and coastal protection without creating monocultures.
-Monitor long-term ecological outcomes such as species diversity, trophic balance, and reef resilience.
(3) Strengthen Socio-Ecological resilience by enhancing ecosystem services
-Facilitate the recovery of marine food webs that sustain fisheries and coastal economies.
-Improve ecosystem services such as habitat formation, biodiversity support, and climate adaptation.
-Promote long-term ecological stability as the foundation for human wellbeing.
(4) Promote responsible and collaborative marine stewardship
Facilitate coordinated decision-making between research institutions, environmental agencies, and coastal communities involved in marine restoration efforts.
Encourage transparent monitoring, shared scientific data, and collective ecological oversight across regions.
Support equitable access to knowledge and responsible deployment practices that prioritize long-term ecosystem health and social wellbeing.
- Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”). Try to outline a mix of actions (e.g. a new requirement/rule, incentive, or technical strategy) pursued by different “actors” (e.g. academic researchers, companies, federal regulators, law enforcement, etc). Draw upon your existing knowledge and a little additional digging, and feel free to use analogies to other domains (e.g. 3D printing, drones, financial systems, etc.).
- Purpose: What is done now and what changes are you proposing?
- Design: What is needed to make it “work”? (including the actor(s) involved - who must opt-in, fund, approve, or implement, etc)
- Assumptions: What could you have wrong (incorrect assumptions, uncertainties)?
- Risks of Failure & “Success”: How might this fail, including any unintended consequences of the “success” of your proposed actions?
1.Controlled Access and Authorized Deployment Framework
While projects focused on the implementation of artificial reefs largely aim to benefit marine ecosystems, their direct interaction with complex natural environments means that inadequate planning or mismanagement could produce outcomes contrary to those intended. This concern becomes even more significant when considering the deployment of a bioengineered living material with active interaction within marine ecosystems. For this reason, the first proposed governance action is the establishment of a controlled access and regulated deployment framework, in which the use and implementation of this material would be authorized exclusively for public research institutions operating in formal collaboration and joint monitoring with governmental environmental agencies and organizations specialized in ecological restoration. Such a model would help reduce the risk of irresponsible or insufficiently supervised applications while promoting evidence-based interventions grounded in long-term ecosystem planning. Additionally, each deployment should be subject to prior ecological impact assessments and continuous monitoring systems capable of adapting interventions according to ecosystem responses over time. Nevertheless, this approach may also face challenges, including administrative processes that could slow urgent restoration efforts or institutional limitations in sustaining long-term oversight.
2.Creation of an International Marine Bioengineering Association
Oceans are not uniform and present unique ecological, chemical, and biological characteristics depending on the regions in which they are located; however, any intervention in these systems may generate transboundary ecological effects. In this context, most marine restoration initiatives continue to be managed primarily at local or national levels, which may limit a comprehensive understanding of their impacts and reduce their effectiveness at broader scales. Therefore, the creation of an international marine bioengineering governance association is proposed to coordinate shared standards, biosafety protocols, and collaborative ecological assessment frameworks across countries. As part of this initiative, it would be essential to establish an international network of authorized laboratories and research centers with regulated access to these types of materials, facilitating collaborative monitoring of their use, the exchange of scientific data, and the planning of interventions at regional and global scales. This approach would strengthen local actions through a broader ecosystem-based perspective, promoting informed decision-making and adaptive strategies grounded in shared scientific evidence. Nevertheless, this model may also face important challenges, including difficulties in political coordination between countries and inequalities in access to the scientific and technological resources necessary for its effective implementation.
3.Biosecurity & Environmentally Controlled Activation of Living Material
Beyond regulatory and institutional frameworks, a third governance action involves integrating principles of ecological responsibility directly into the functional design of the living material from its earliest stages of development. Considering that the material’s primary objective is to act as a bioreceptor capable of attracting and concentrating calcium carbonate (CaCO₃) particles present in the water to facilitate natural biomineralization processes in nearby marine organisms, it is essential to establish clear limits on its ecological behavior and its interaction with the surrounding environment. In this regard, the material could be designed to remain functionally inert under normal environmental conditions and to activate only under specific scenarios of marine acidification, responding to defined physicochemical parameters associated with ecosystem imbalance. This conditional activation would help reduce the risk of unintended alterations to local ecological dynamics, promoting a strategy of “responsibility by design” in which researchers, biological designers, and academic institutions integrate ethical and ecosystem-based considerations from the outset of technological development.
4.Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
| Does the option: | ACTION 1 | ACTION 2 | ACTION 3 |
|---|
| Promote ecological restoration & long-term ecosystem balance | 2 | 3 | 1 |
| • By Supporting calcifying organisms | 1 | 2 | 1 |
| • Require ecological impact assessments | 1 | 2 | 1 |
| • By Prioritizing degraded or climate-vulnerable areas | 1 | 2 | 1 |
| Enhance ecosystem services while preserving biodiversity integrityy | 1 | 1 | 3 |
| • By protecting biodiversity | 1 | 2 | 1 |
| • By Monitoring long-term ecological outcomes | 1 | 2 | 2 |
| Strengthen socio-ecological resilience & human wellbeing | 1 | 1 | 2 |
| • By Supporting fisheries & coastal economiess | 1 | 3 | 2 |
| • By Improving ecosystem services & climate adaptation | 1 | 3 | 1 |
| Promote responsible & collaborative marine stewardship | 1 | 1 | 3 |
| • By Controlled and authorized deployment | 1 | 1 | 1 |
| • Feasibility | 1 | 3 | 2 |
| • Ethical responsibility integrated into design | 1 | 2 | 1 |
| • Promote constructive applications | 1 | 3 | 1 |
5.Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties. For this, you can choose one or more relevant audiences for your recommendation, which could range from the very local (e.g. to MIT leadership or Cambridge Mayoral Office) to the national (e.g. to President Biden or the head of a Federal Agency) to the international (e.g. to the United Nations Office of the Secretary-General, or the leadership of a multinational firm or industry consortia). These could also be one of the “actor” groups in your matrix.
From my personal perspective, the key element of this project lies in the strategic development of materials specifically designed to minimize the impacts of ocean acidification. Building on this foundation, a comprehensive strategic plan should be developed in collaboration with governmental entities to ensure a conscious, well-structured approach directed exclusively toward ecological restoration. Such coordination would allow for responsible implementation, clear environmental oversight, and long-term sustainability aligned with conservation priorities.

Week 2 HW:DNA Read, Write, & Edit
Class Assignment Homework 2
Part1: Benchling & In-sico GelArt
See this week’s lab protocol “Gel Art: Restriction Digests and Gel Electrophoresis” for details. Overview:
Make a free account at benchling.com
Import the Lambda DNA.
Simulate Restriction Enzyme Digestion with the following Enzymes:
EcoRI
HindIII
BamHI
KpnI
EcoRV
SacI
SalI
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
You might find Ronan’s website a helpful tool for quickly iterating on designs!
This was my first time working with Benchling, as well as my first experience exploring the possibilities of DNA design. As a first step, I imported the Lambda DNA sequences to begin experimenting with the digestion using enzymes such as EcoRI, EcoRV, SalI, SacI, KpnI, among others. This process led me to explore combinations and fusions between these enzymes, resulting in a wide variety of sequence ladder patterns. From that point, I began manipulating the patterns by rearranging the order of the tabs, which allowed me to design and create my own custom sequence design.

Now behold the first robot, made with 100% dna Enzymes.
3.1. Choose your protein.
In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose.
[Example from our group homework, you may notice the particular format~~~ The example below came from UniProt]
sp|P03609|LYS_BPMS2 Lysis protein OS=Escherichia phage MS2 OX=12022 PE=2 SV=1 METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLL EAVIRTVTTLQQLLT
For the second week of the HTGAA course, I chose a molluscan shell matrix protein involved in calcium carbonate biomineralization, called mantle protein N25 (N25). This protein is found in the calcifying organism Pinctada fucata, commonly known as the Akoya pearl oyster. N25 is part of the shell matrix and helps regulate calcium carbonate biomineralization, playing a key role in crystal growth, morphology, and the formation of microscopic calcifying structures. I want to explore this protein because my goal is to engineer a material for artificial reefs that can act as a “magnet” for dissolved calcium carbonate in seawater, creating localized microenvironments that favor CaCO₃ deposition and make carbonate more accessible for calcifying organisms affected by ocean acidification.
IMAGE FROM:https://www.newscientist.com/article/2151281-oysters-can-hear-the-ocean-even-though-they-dont-have-ears/
Yang, D., Yan, Y., Yang, X., Liu, J., Zheng, G., Xie, L., & Zhang, R. (2019). A basic protein, N25, from a mollusk modifies calcium carbonate morphology and shell biomineralization. Journal of Biological Chemistry, 294(21), 8371–8384. https://doi.org/10.1074/jbc.RA118.007338
Using UniProt, I obtained the amino acid sequence of mantle protein N25 (N25):

**FASTA
tr|A0A0E3XA28|A0A0E3XA28_PINFU Mantle protein N25 OS=Pinctada fucata OX=50426 PE=2 SV=1
MKRIYVLVLLFILLVCIAEAQKKSKDSKKASSKSSSKSSGKSKSSPKSSGAKGKSPAPSA
PASKGPSEMQKLAEEMVALSNRLLKAIKAGEQMPPPMCPNGLPKADCSPLACDKWTCSNI
LNTVCKEQCHVCEPKFYIGGSEVTQFCELKPANMQPRATQSPPTSRNTATDQGPQNSGPS
SNGAPSNMPPMPGMPMMFSENPMPMGGPPGMEFMPNFENFPPGMSPMQFFHHLQNMNMPN
ENQGSRSQAN**
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
[Example: Get to the original sequence of phage MS2 L-protein from its genome phage MS2 genome - Nucleotide - NCBI]
Lysis protein DNA sequence atggaaacccgattccctcagcaatcgcagcaaactccggcatctactaatagacgccggccattcaaacatgaggattacccatgtcgaagacaacaaagaagttcaactctttatgtattgatcttcctcgcgatctttctctcgaaatttaccaatcaattgcttctgtcgctactggaagcggtgatccgcacagtgacgactttacagcaattgcttacttaa
To reverse translate the mantle protein N25 amino acid sequence to DNA, I used the online reverse translation tool at novoprolabs.com/tools/revtrans.
N25 protein DNA sequence
atgaaacgcatttatgtgctggtgctgctgtttattctgctggtgtgcattgcggaagcgcagaaaaaaagcaaagatagcaaaaaagcgagcagcaaaagcagcagcaaaagcagcggcaaaagcaaaagcagcccgaaaagcagcggcgcgaaaggcaaaagcccggcgccgagcgccggcgagcaaaggcccgagcgaaatgcagaaactggcggaagaaatggtggcgctgagcaaccgcctgctgaaagcgattaaagcgggcgaacagatgccgccgccgatgtgcccgaacggcctgccgaaagcggattgcagcccgctggcgtgcgataaatggacctgcagcaacattctgaacaccgtgtgcaaagaacagtgccatgtgtgcgaaccgaaattttatattggcggcagcgaagtgacccagttttgcgaactgaaaccggcgaacatgcagccgcgcgcgacccagagcccgccgaccagccgcaacaccgcgaccgatcagggcccgcagaacagcggcccgagcagcaacggcgcgccgagcaacatgccgccgatgccgggcatgccgatgatgtttagcgaaaacccgatgccgatgggcggcccgccgggcatggaatttatgccgaactttgaaaactttccgccgggcatgagcccgatgcagttttttcatcatctgcagaacatgaacatgccgaacgaaaaccagggcagccgcagccaggcgaac
3.3 Codon optimization.Once nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why do you need to optimize codon usage. Which organism have you chose to optimize the codon sequence for and why?
I recently discovered what codons are and the key role they have in the protein formation process. They function as coordinates to encode amino acids. In the world, there are 64 possible codons for the 20 types of amino acids, which means that different codons can encode the same amino acid.
That being said, codon optimization is necessary because different organisms prefer some codons over others due to DNA reading bias. This is a vital step to create a more effective way to reproduce genes from one organism to another. Although the genetic code is universal, certain organisms preferentially use specific codons due to differences in tRNA abundance and translation efficiency.
For this project, I optimized the codon sequence for expression in Bacillus subtilis. I selected Bacillus subtilis because:
It is genetically well-characterized
It is widely used in biotechnology
It can secrete proteins extracellularly
It tolerates moderate saline environments
This process was performed using an online codon optimization tool. The optimized sequence maintains the same amino acid sequence but improves translational efficiency in the chosen host organism.
3.4 You Have a sequence! Now What?
With the codon-optimized DNA sequence of N25, the protein can be produced using a cell-dependent system with Bacillus subtilis as the host. The optimized DNA is introduced into the bacteria through transformation. Inside the cells, the DNA is transcribed into mRNA and then translated by ribosomes into the N25 protein.
Bacillus subtilis can secrete the protein extracellularly, which simplifies collection and use. This approach leverages the host’s natural cellular machinery while ensuring that the codon optimization maximizes translation efficiency. By producing N25 in Bacillus subtilis, it is possible to generate sufficient protein for bioengineering applications, such as creating microenvironments that attract calcium carbonate for artificial reef materials.
**Part 4: Prepare a Twist DNA Synthesis Order **
This is a practice exercise, not necessarily your real Twist order!
4.1. Create a Twist account and a Benchling account
UFQ89828.1 mCherry [synthetic construct]
MVSKGEEDNMAIIKEFMRFKVHMEGSVNGHEFEIEGEGEGRPYEGTQTAKLKVTKGGPLPFAWDILSPQF
MYGSKAYVKHPADIPDYLKLSFPEGFKWERVMNFEDGGVVTVTQDSSLQDGEFIYKVKLRGTNFPSDGPV
MQKKTMGWEASSERMYPEDGALKGEIKQRLKLKDGGHYDAEVKTTYKAKKPVQLPGAYNVNIKLDITSHN
EDYTIVEQYERAEGRHSTGGMDELYK
reverse translation of UFQ89828.1 mCherry [synthetic construct] to a 708 base sequence of most likely codons.
atggtgagcaaaggcgaagaagataacatggcgattattaaagaatttatgcgctttaaa
gtgcatatggaaggcagcgtgaacggccatgaatttgaaattgaaggcgaaggcgaaggc
cgcccgtatgaaggcacccagaccgcgaaactgaaagtgaccaaaggcggcccgctgccg
tttgcgtgggatattctgagcccgcagtttatgtatggcagcaaagcgtatgtgaaacat
ccggcggatattccggattatctgaaactgagctttccggaaggctttaaatgggaacgc
gtgatgaactttgaagatggcggcgtggtgaccgtgacccaggatagcagcctgcaggat
ggcgaatttatttataaagtgaaactgcgcggcaccaactttccgagcgatggcccggtg
atgcagaaaaaaaccatgggctgggaagcgagcagcgaacgcatgtatccggaagatggc
gcgctgaaaggcgaaattaaacagcgcctgaaactgaaagatggcggccattatgatgcg
gaagtgaaaaccacctataaagcgaaaaaaccggtgcagctgccgggcgcgtataacgtg
aacattaaactggatattaccagccataacgaagattataccattgtggaacagtatgaa
cgcgcggaaggccgccatagcaccggcggcatggatgaactgtataaa
4.1 (i) What DNA would you want to sequence and why?
As an industrial designer, I’m always looking for ways to improve urban lifestyles through innovative materials. With CO₂ emissions rising in cities, I would sequence DNA from engineered E. coli bacteria embedded in “bio-bricks” designed for future buildings and urban infrastructure. These bacteria act as simple CO₂ sensors: when CO₂ levels are high, they modify a specific part of their DNA to record exposure events. By sampling bacteria from different bricks across the city and sequencing this “memory region,” I could map pollution patterns over time and create bio-integrated technologies that literally store environmental data within our built environment.

IMAGE FROM: https://www.front-materials.com/news/biomason-front-biobasedtile/
4.1 (ii) What technology would you use to perform sequencing and why?
Is your method first-, second- or third-generation or other? How so?
For this project I would choose the second-generation method, specifically Illumina, since it is very accessible and efficient and allows reading millions of short DNA fragments at the same time in parallel. In contrast, first-generation (Sanger) reads one DNA molecule at a time, which is very slow, and third-generation (Nanopore, PacBio) reads complete long molecules one by one but is less precise for my case. For my bio-bricks I only need to read a specific short region of the bacterial DNA, so second generation is more accurate and pertinent.
What is your input? How do you prepare your input? List the essential steps.
As a first step in preparing the input, I would obtain samples from the bacteria in the bio-bricks by scraping their surface. As a next step, I understand that I would use Polymerase Chain Reaction (PCR) to obtain copies only of the specific DNA region where the “CO₂ memory” is stored.
As a third step, I would add short adapter sequences to allow the fragments to enter the sequencing machine. I would also use a type of identification tag (barcode) for each bio-brick in order to distinguish the samples. Finally, I would mix the samples together so they can be sequenced simultaneously.
What are the essential steps of your chosen sequencing technology, and how does it decode the bases of your DNA sample (base calling)?
Since I chose the second-generation Illumina method, the process begins with attaching DNA fragments to a special plate called a flow cell. Once attached, the fragments are copied many times, forming small groups called clusters.
Then, the machine adds one nucleotide at a time, and each incorporation generates a fluorescent color signal (for example, A is green, T is red, C is blue, and G is yellow). After each cycle, a camera takes an image of the entire plate, and a software program interprets the color detected in each cluster to determine which base was added. By repeating this process many times, the complete DNA sequence is reconstructed.
What is the output of your chosen sequencing technology?
The result I get from Illumina is a set of files that contain millions of small DNA fragments from the region where my bacteria store the “CO₂ memory.”Each sequence comes from a different group of DNA that was read by the machine, and thanks to the labels I added earlier, I can tell which bio-brick each fragment came from. With all this information, I can compare patterns between different bricks and analyze the history of CO₂ exposure in different parts of the city
4.2 DNA Write
(i) What DNA would you want to synthesize (write) and why?
As a designer, I have always been interested in understanding why things have specific shapes. For that reason, I would like to synthesize simple genetic circuits related to morphogenesis — how living tissues or cell populations grow into particular forms. During the first class, the possibility of inoculating bacteria with DNA fragments to make them generate shapes or patterns inspired by much larger organisms was mentioned, but within an in vitro environment.
In the long term, and in a more speculative way, I would like to explore applying this principle to the creation of architectural modules — potentially shaping urban landscapes, or even designing products that are grown by nature itself, but guided by human intention.I am aware that this is not currently a mature technology, so I see it more as an exploratory and future-oriented design question rather than something that can be fully built today.
(ii) What technology would you use to perform this DNA synthesis and why?
What are the essential steps of your chosen sequencing methods?
From what I’ve understood, DNA synthesis is done by adding one base at a time in a controlled way. First, the last added base is protected, then a new base is incorporated, the bond is chemically stabilized, the remaining reagents are washed away, and the cycle is repeated until the full sequence is completed. I understand that it is a repetitive process that requires a lot of precision to obtain the correct sequence.
4.2 (ii) What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
One of the main limitations is the turnaround time of 2-4 weeks since chemical synthesis happens cycle by cycle, plus limitations on chain length up to 2000 bases and occasional errors of ~1 every 100-300 bases.
4.3 DNA Edit
(i) What DNA would you want to edit and why?
I would like to edit a specific gene in the Peruvian scallop (Argopecten purpuratus) to make its shells more resistant to ocean acidification during the larval stage. Ocean acidification dissolves calcium carbonate shells of shellfish, and scallop larvae are especially vulnerable. This species is important for Peruvian aquaculture and coastal economies, so helping them survive better would support sustainable fisheries. The principle could also apply to oysters, mussels, and other shellfish facing the same threat.
(ii) What technology would you use and why?
I understand that one of the most precise and accessible gene editing tools available today is CRISPR-Cas9, and also because it’s precise and has been used successfully in shellfish for targeted edits.
https://www.frontiersin.org/journals/marine-science/articles/10.3389/fmars.2022.912409/full
How does your technology of choice edit DNA? What are the essential steps?
I still find it hard to fully understand the principles of genetic modification, but I understand that CRISPR-Cas9 is a tool that allows cutting and editing DNA at a specific location. This tool uses an RNA molecule as a guide to select the sequence we want to modify, and then the Cas9 protein works like scissors to cut the DNA. Once the DNA is cut, it is possible to deactivate a gene or insert new sequences with different genetic information.
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
To change a gene that makes oysters vulnerable, you first need to identify which gene it is. Then, you use CRISPR-Cas9 with a guide RNA to find that specific gene and Cas9 to cut it like scissors. After the cut, the oyster cell can repair the DNA, and at that moment you can insert a new version of the gene that doesn’t cause vulnerability.
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
CRISPR-Cas9 is not perfect. I understand that sometimes it could cut in the wrong places and damage healthy genes. Also, to carry out this process, it is necessary to do many tests to make sure it actually worked. In the case of oysters, the larvae are very fragile and some could die during the injection. In addition, it takes a lot of time, even months, to raise them and check the results.
Week 3 HW: Lab Automation
Assignment: Python Script for Opentrons Artwork
Your task this week is to Create a Python file to run on an Opentrons liquid handling robot.
- Review this week’s recitation and this week’s lab for details on the Opentrons and programming it.
- Generate an artistic design using the GUI at opentrons-art.rcdonovan.com.
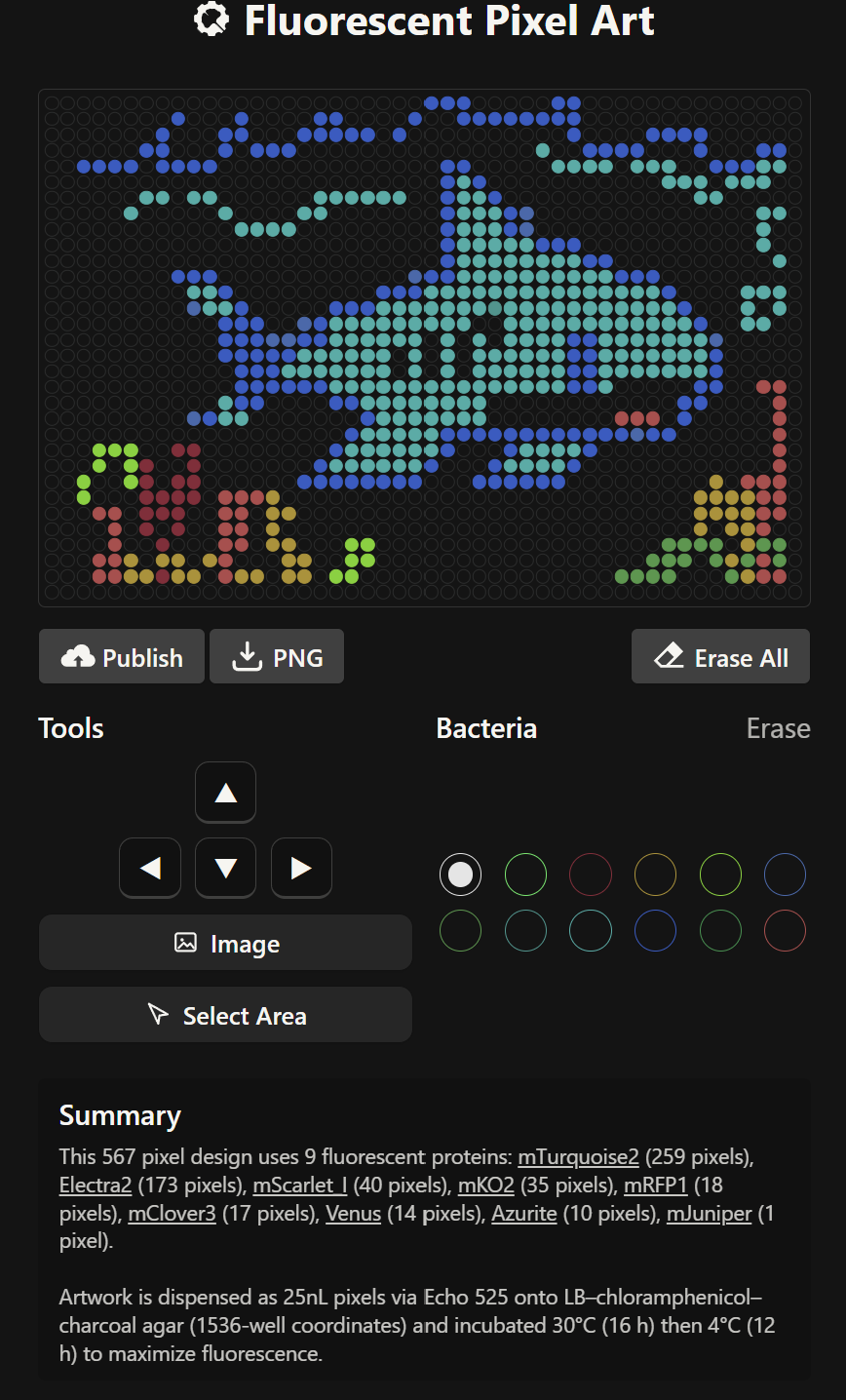
During this stage, the Fluorescent Pixel Art tool was used to create the desired design. However, after completing the design, I encountered significant difficulties when trying to download its coordinates. As a result, the coordinate mapping had to be carried out manually, with the assistance of artificial intelligence. To make the process more manageable, I ultimately opted for a simpler design.
Final Coordinates
mko2_points = [‘E20’,‘E21’,‘E22’,‘E23’,‘E24’,‘E25’,‘E26’,‘E27’,‘F18’,‘F19’,‘F20’,‘F21’,‘F22’,‘F23’,‘F24’,‘F25’,‘F26’,‘F27’,‘F28’,‘F29’,‘G17’,‘G18’,‘G19’,‘G20’,‘G21’,‘G22’,‘G23’,‘G24’,‘G25’,‘G26’,‘G27’,‘G28’,‘G29’,‘G30’,‘H16’,‘H17’,‘H18’,‘H19’,‘H20’,‘H21’,‘H22’,‘H23’,‘H24’,‘H25’,‘H26’,‘H27’,‘H28’,‘H29’,‘H30’,‘H31’,‘I15’,‘I16’,‘I17’,‘I21’,‘I22’,‘I23’,‘I24’,‘I25’,‘I26’,‘I27’,‘I28’,‘I32’,‘I33’,‘I34’,‘J15’,‘J16’,‘J17’,‘J21’,‘J22’,‘J23’,‘J24’,‘J25’,‘J26’,‘J27’,‘J28’,‘J32’,‘J33’,‘J34’,‘K15’,‘K16’,‘K17’,‘K21’,‘K22’,‘K23’,‘K24’,‘K25’,‘K26’,‘K27’,‘K28’,‘K32’,‘K33’,‘K34’,‘L14’,‘L15’,‘L16’,‘L17’,‘L21’,‘L22’,‘L23’,‘L24’,‘L25’,‘L26’,‘L27’,‘L28’,‘L32’,‘L33’,‘L34’,‘L35’,‘M14’,‘M15’,‘M16’,‘M17’,‘M21’,‘M22’,‘M23’,‘M24’,‘M25’,‘M26’,‘M27’,‘M28’,‘M32’,‘M33’,‘M34’,‘M35’,‘N14’,‘N15’,‘N16’,‘N17’,‘N21’,‘N22’,‘N23’,‘N24’,‘N25’,‘N26’,‘N27’,‘N28’,‘N32’,‘N33’,‘N34’,‘N35’,‘O14’,‘O15’,‘O16’,‘O17’,‘O21’,‘O22’,‘O23’,‘O24’,‘O25’,‘O26’,‘O27’,‘O28’,‘O32’,‘O33’,‘O34’,‘O35’,‘P14’,‘P15’,‘P16’,‘P17’,‘P18’,‘P19’,‘P20’,‘P21’,‘P22’,‘P23’,‘P24’,‘P25’,‘P26’,‘P27’,‘P28’,‘P29’,‘P30’,‘P31’,‘P32’,‘P33’,‘P34’,‘P35’,‘Q14’,‘Q15’,‘Q16’,‘Q17’,‘Q18’,‘Q19’,‘Q20’,‘Q21’,‘Q22’,‘Q23’,‘Q24’,‘Q25’,‘Q26’,‘Q27’,‘Q28’,‘Q29’,‘Q30’,‘Q31’,‘Q32’,‘Q33’,‘Q34’,‘Q35’,‘R15’,‘R16’,‘R17’,‘R18’,‘R19’,‘R20’,‘R21’,‘R22’,‘R23’,‘R24’,‘R25’,‘R26’,‘R27’,‘R28’,‘R29’,‘R30’,‘R31’,‘R32’,‘R33’,‘R34’,‘S15’,‘S16’,‘S17’,‘S32’,‘S33’,‘S34’,‘T16’,‘T17’,‘T18’,‘T31’,‘T32’,‘T33’,‘U17’,‘U18’,‘U19’,‘U20’,‘U29’,‘U30’,‘U31’,‘U32’,‘V18’,‘V19’,‘V20’,‘V21’,‘V22’,‘V27’,‘V28’,‘V29’,‘V30’,‘V31’,‘W19’,‘W20’,‘W21’,‘W22’,‘W23’,‘W24’,‘W25’,‘W26’,‘W27’,‘W28’,‘W29’,‘W30’,‘X20’,‘X21’,‘X22’,‘X23’,‘X24’,‘X25’,‘X26’,‘X27’,‘X28’,‘X29’,‘Y21’,‘Y22’,‘Y23’,‘Y24’,‘Y25’,‘Y26’,‘Y27’,‘Y28’,‘Z22’,‘Z23’,‘Z24’,‘Z25’,‘Z26’,‘Z27’,‘AA23’,‘AA24’,‘AA25’,‘AA26’]
- Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script which draws your design using the Opentrons.
You may use AI assistance for this coding — Google Gemini is integrated into Colab (see the stylized star bottom center); it will do a good job writing functional Python, while you probably need to take charge of the art concept.
If you’re a proficient programmer and you’d rather code something mathematical or algorithmic instead of using your GUI coordinates, you may do that instead.
During this stage of the task, we were asked to develop code capable of simulating the application of our design within a controlled environment in a Petri dish. However, this required prior knowledge of Python, and since this was my first experience working with the language, I found it quite challenging. To overcome this difficulty, I decided to rely on the assistance of artificial intelligence to help generate the code needed for the simulation. I provided the necessary parameters and requirements to ensure that the experiment could be carried out successfully and that the simulation would run without errors.
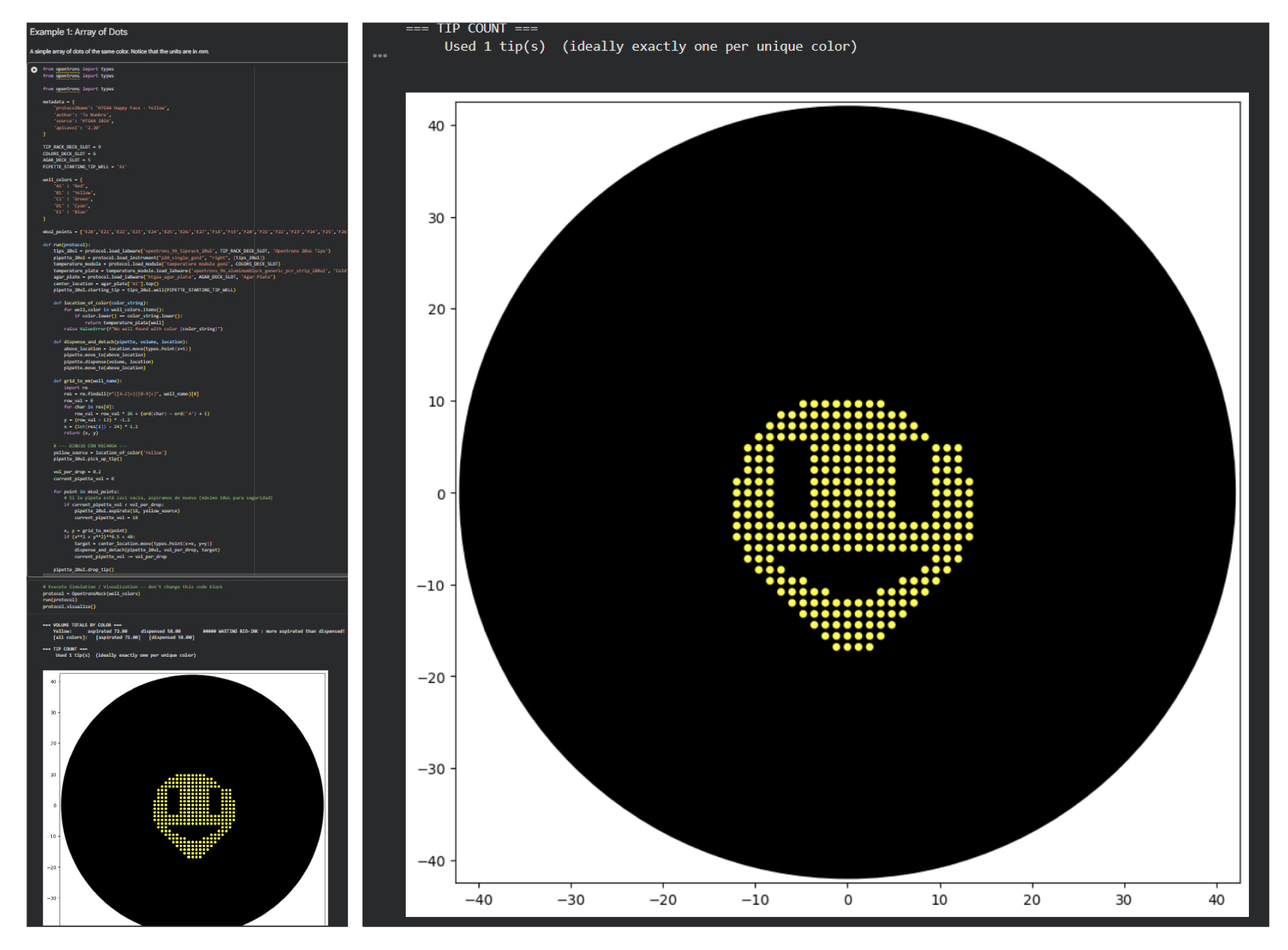
Considering that the coordinates had to be obtained manually, some misalignments were present in the final design. However, the overall morphology and core structure were preserved, which suggests that the intended design was successfully captured. This outcome also opens the possibility of progressively experimenting with more complex designs in the future.
Post-Lab Questions
For this week, we’d like for you to do the following:
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
While your description/project idea doesn’t need to be set in stone, we would like to see core details of what you would automate. This is due at the start of lecture and does not need to be tested on the Opentrons yet.
Project Name: Ocean Acidification Bioluminescent Biosensor.
Description: My project proposal is about a pH-responsive hydrogel with genetically engineered bacteria (Vibrio fischeri). When the ocean pH drops below a threshold, the bacteria should emit bioluminescent light. The core purpose is to have visually intuitive biosensors for local ocean acidification events.
I intend to use the Opentrons robot to automate the testing of this material. First, I will have a petri dish with the hydrogel and the bacteria already inside. Then, I will use the robot to:
Prepare pH Samples: The robot will create a set of small tubes with different seawater pH levels (like 8.1, 7.8, 7.5, 7.2, 6.9).
Add Tiny Drops: The robot will pick up a tiny drop (0.2 µL) from each tube and dispense it onto different spots of the hydrogel.
Create a Map of Light: Since the robot knows the exact location and pH of each drop, I can observe exactly where the hydrogel glows and where it does not. This will help me find the best pH threshold for my material.
Final Project Ideas



Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
1. Briefly describe the protein you selected and why you selected it.
Vibrio fischeri LuxA
2. Identify the amino acid sequence of your protein.
sp|P19907|LUXA_ALIFS Alkanal monooxygenase alpha chain OS=Aliivibrio fischeri OX=668 GN=luxA PE=3 SV=1
MKFGNICFSYQPPGETHKLSNGSLCSAWYRLRRVGFDTYWTLEHHFTEFGLTGNLFVAAA
NLLGRTKTLNVGTMGVVIPTAHPVRQLEDVLLLDQMSKGRFNFGTVRGLYHKDFRVFGVD
MEESRAITQNFYQMIMESLQTGTISSDSDYIQFPKVDVYPKVYSKNVPTCMTAESASTTE
WLAIQGLPMVLSWIIGTNEKKAQMELYNEIATEYGHDISKIDHCMTYICSVDDDAQKAQD
VCREFLKNWYDSYVNATNIFNDSNQTRGYDYHKGQWRDFVLQGHTNTNRRVDYSNGINPV
GTPEQCIEIIQRDIDATGITNITCGFEANGTEDEIIASMRRFMTQVAPFLKEPK
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
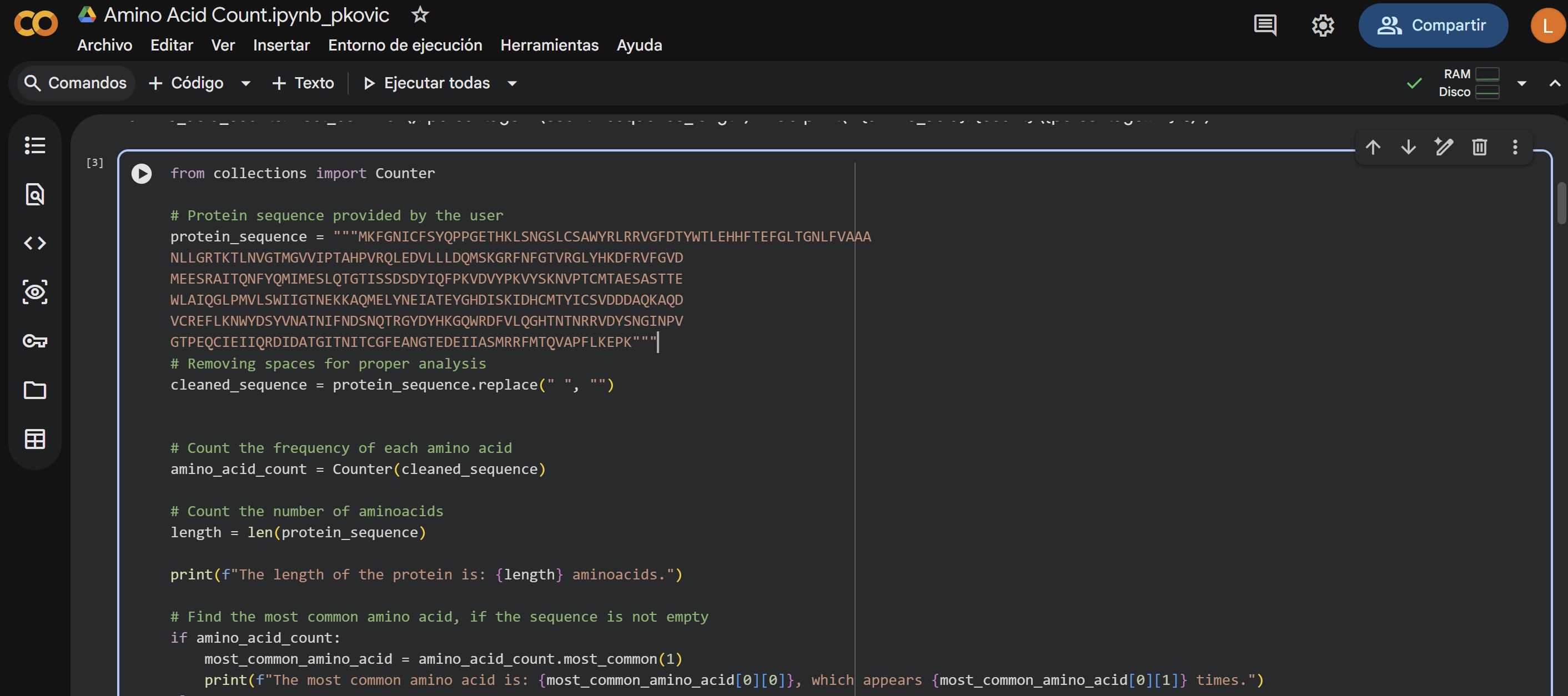
The length of the protein is: 359 aminoacids.
The most common amino acid is: T, which appears 30 times.
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
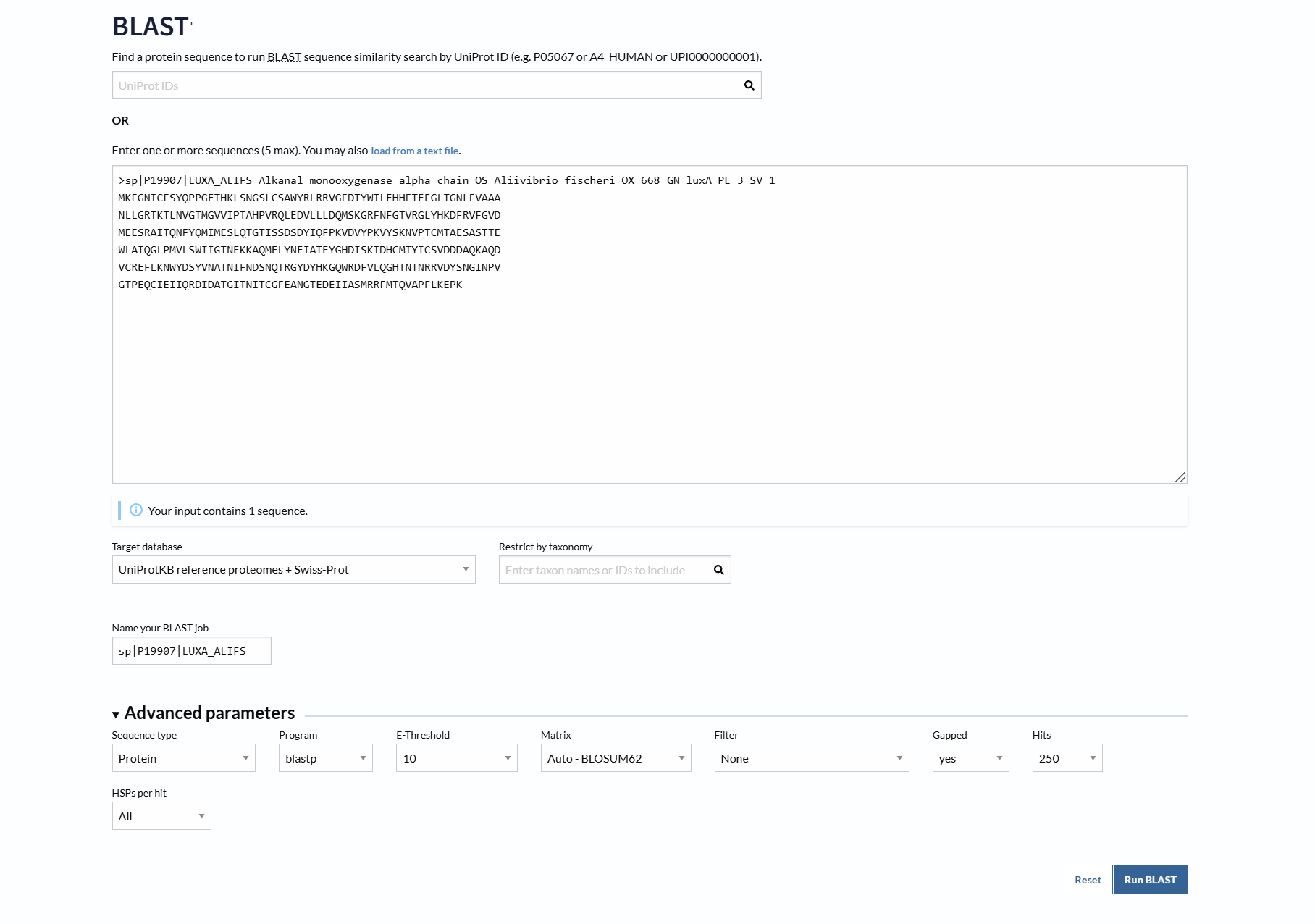
Using UniProt BLAST, 250 sequence homologs were found in UniProtKB (17 reviewed in Swiss-Prot and 233 unreviewed in TrEMBL).
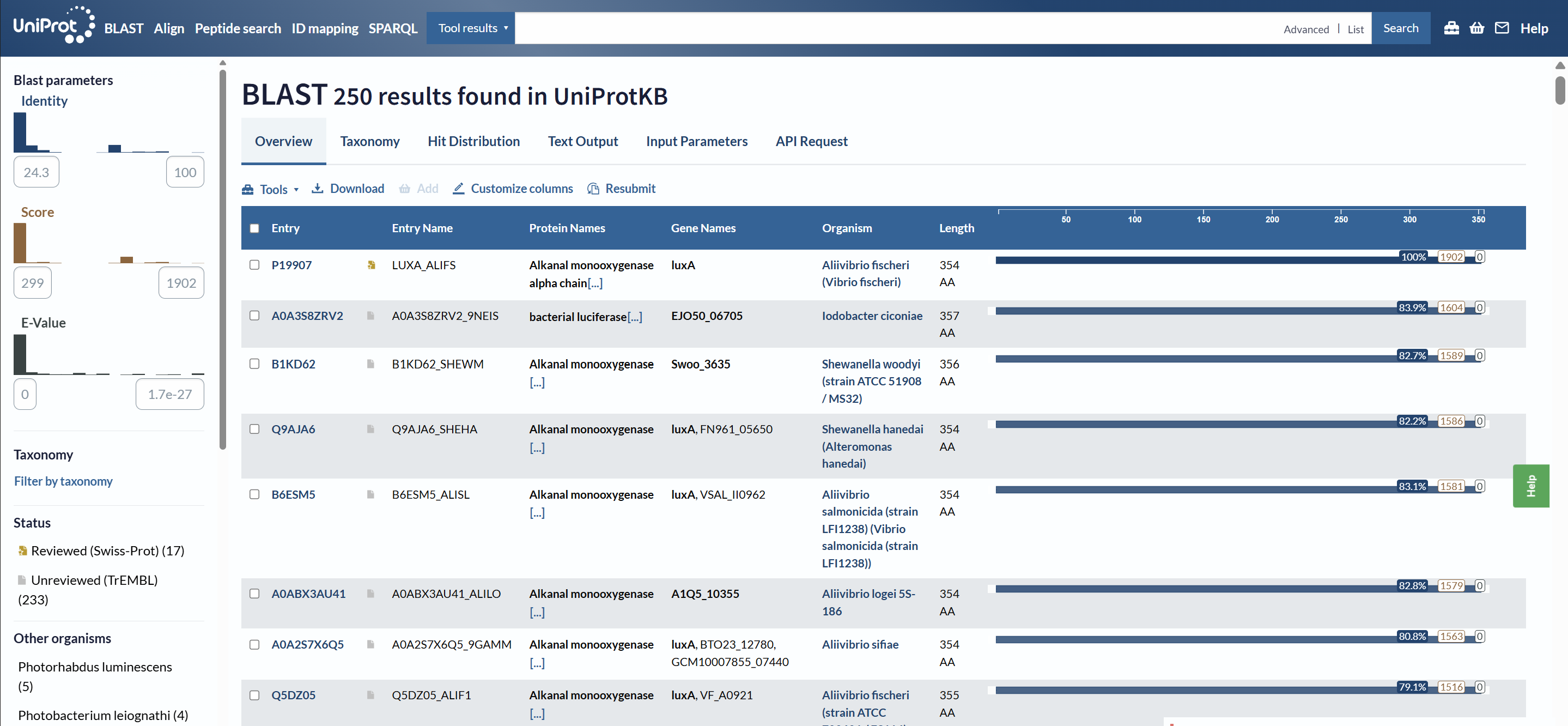
3.Identify the structure page of your protein in RCSB
A direct crystal structure for LuxA from Aliivibrio fischeri was not found in the RCSB PDB database. Therefore, a highly homologous bacterial luciferase structure (PDB ID: 1LUC) from Vibrio harveyi was used as a structural reference.
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
The structure was solved in 1996 with a resolution of 1.50 Å, which is considered high quality because lower resolution values (<2.0 Å) indicate more detailed atomic information.
Are there any other molecules in the solved structure apart from protein?
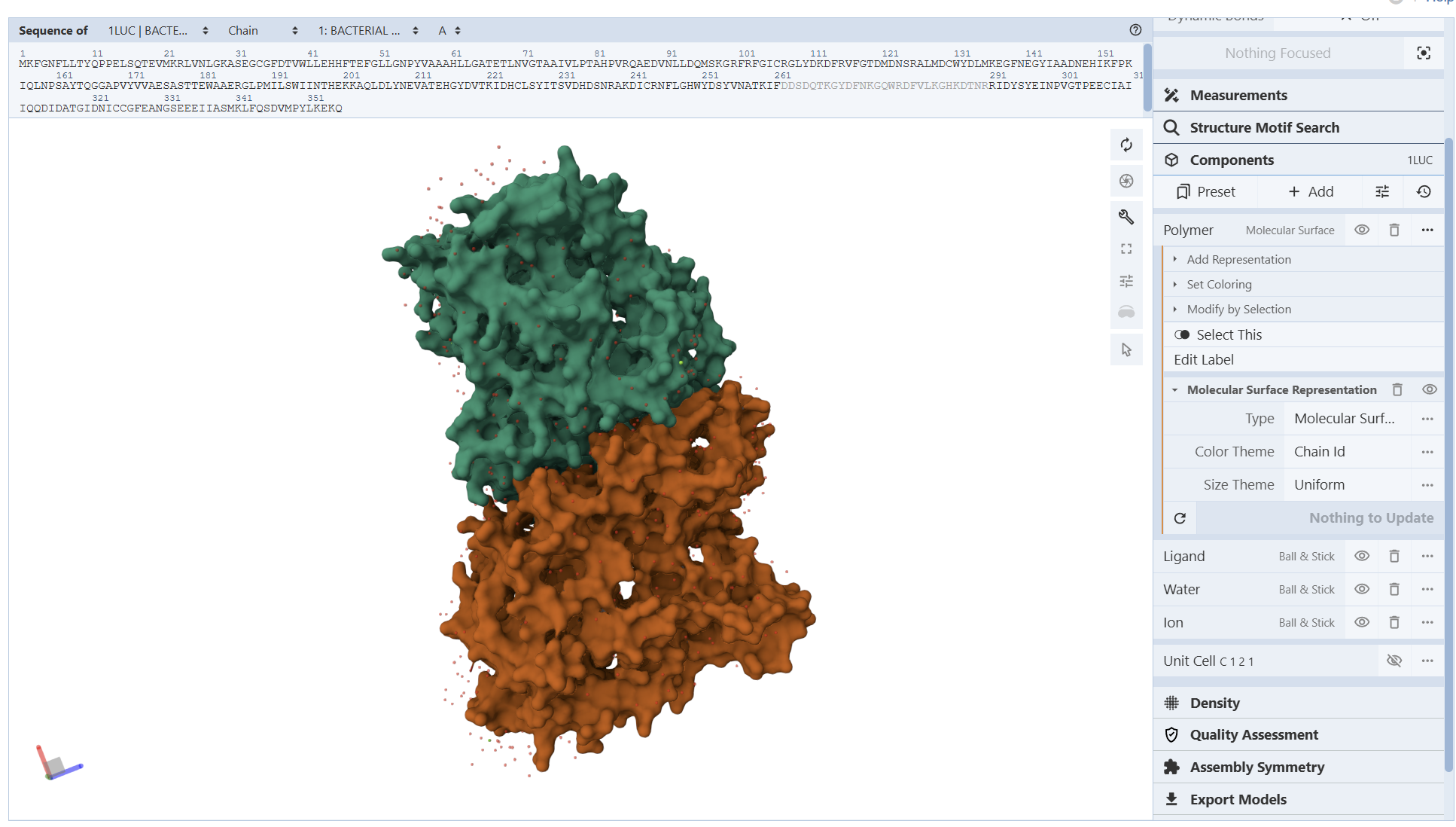
Yes. In addition to the protein chains, the structure contains 1,2-ethanediol (EDO) and a magnesium ion (Mg²⁺).
Does your protein belong to any protein family?
Yes. LUXA belongs to the bacterial luciferase family and is a flavin-dependent monooxygenase involved in bioluminescence.
4. Open the structure of your protein in any 3D molecule visualization software
- PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
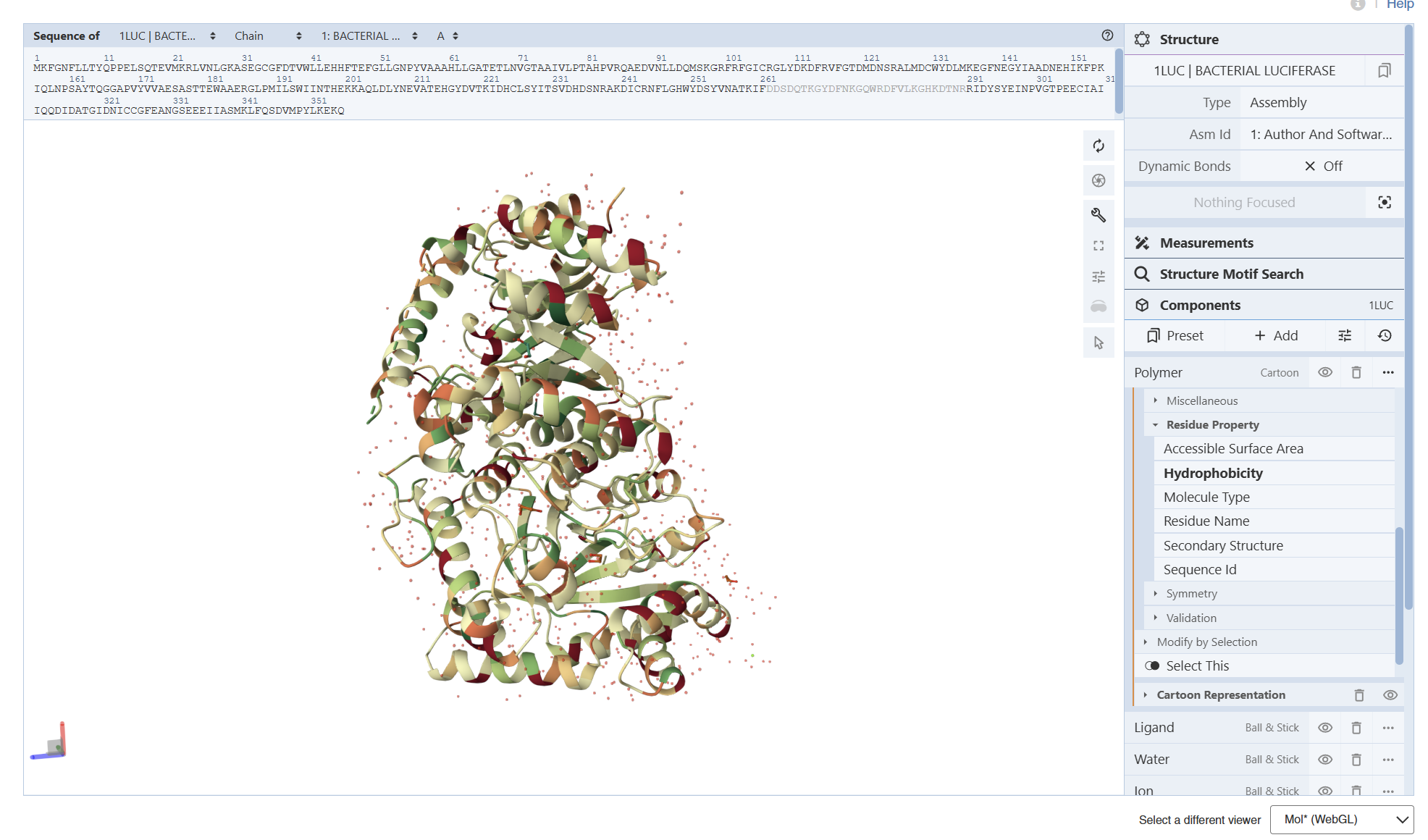
I encountered technical difficulties while attempting to install PyMOL on my local system. To complete the assignment, I opted to use online molecular visualization software (such as the RCSB PDB 3D Viewers). These web-based tools allowed me to accurately generate and analyze the required protein structures, including the Cartoon, Spacefill, and Surface representations shown in my results
- Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
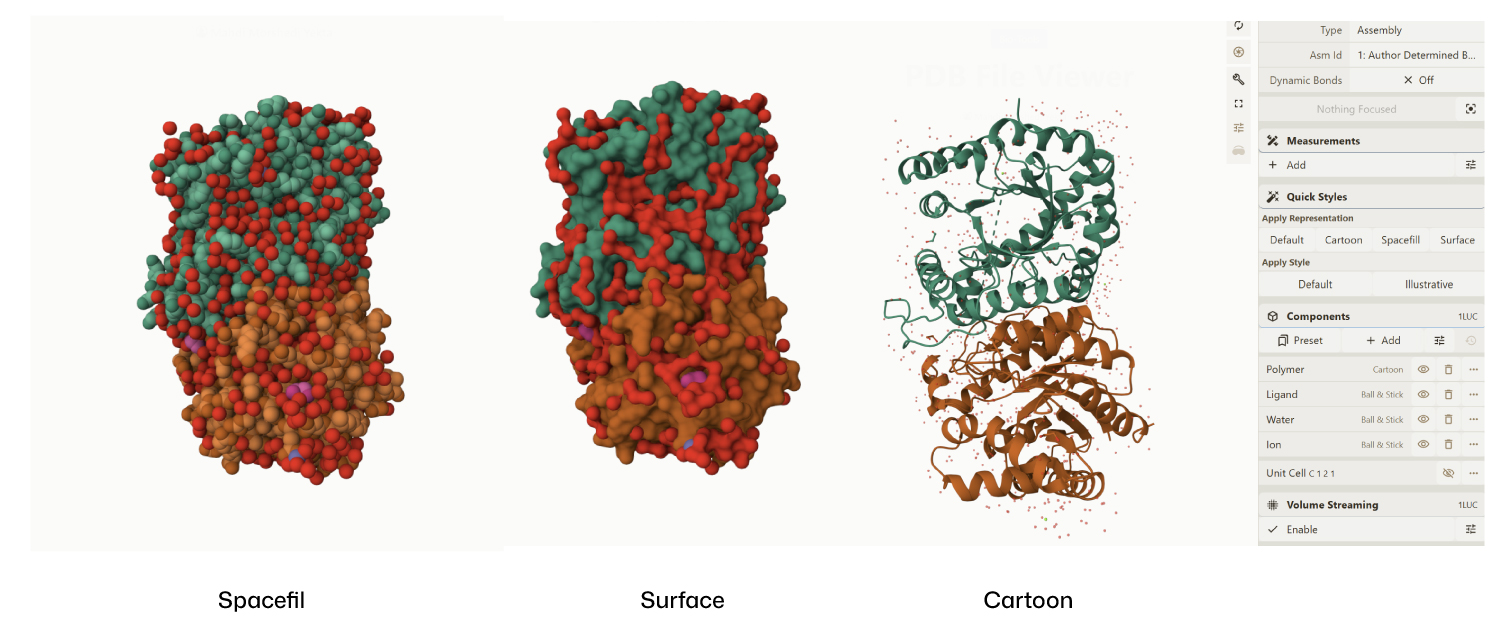
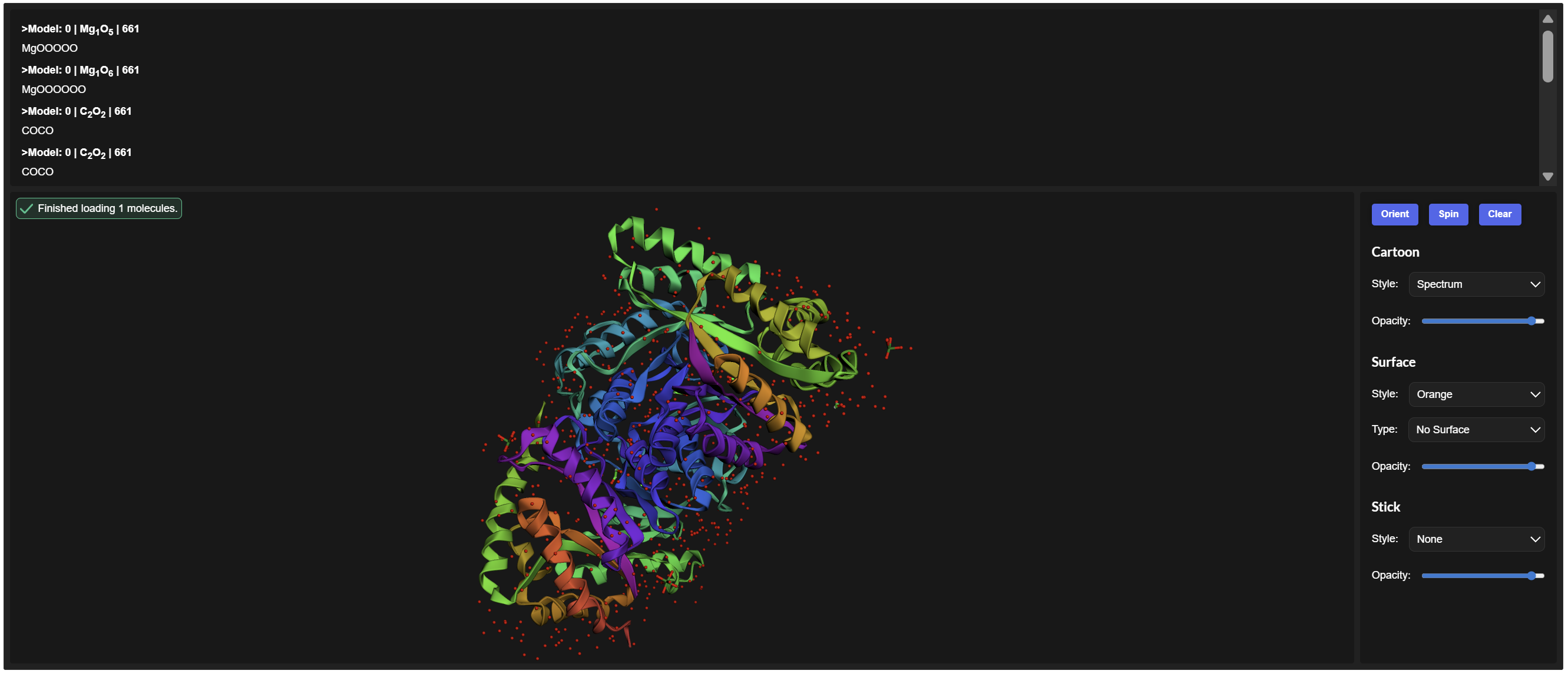
- Color the protein by secondary structure. Does it have more helices or sheets?
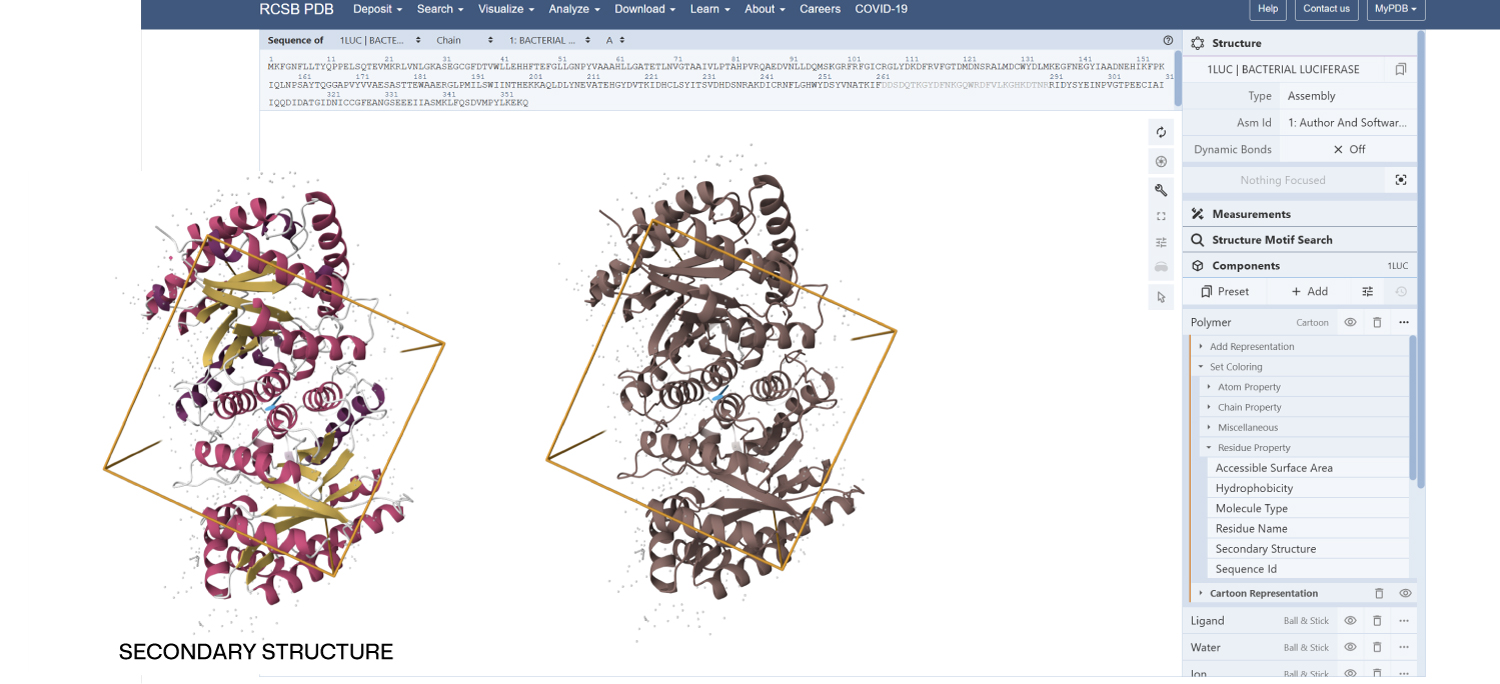
Secondary Structure: According to the previsualization,the protein has more helices than sheets. The alpha-helices are prominent and surround the internal beta-strands
I am not entirely sure, but based on what I can observe in the visualization, the hydrophobic residues (represented by the darker/reddish tones) appear to be predominantly located in the interior of the protein. From what I understand, these residues ‘hide’ in the core, and as shown in the model, they are mostly tucked away from the surface
From these visualization results, it can be observed that the protein structure features various holes and cavities, which demonstrates the presence of several prominent binding pockets. As I understand it, these spaces are necessary for molecules to enter and fit into the protein, allowing the chemical reaction that produces light to occur.
Part C. Using ML-Based Protein Design Tools
- Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
- Choose your favorite protein from the PDB.
- We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
C1. Protein Language Modeling
- Deep Mutational Scans
- Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
PDB ID: 1LUC
sp|P07740|LUXA_VIBHA Alkanal monooxygenase alpha chain OS=Vibrio harveyi OX=669 GN=luxA PE=1 SV=1
MKFGNFLLTYQPPELSQTEVMKRLVNLGKASEGCGFDTVWLLEHHFTEFGLLGNPYVAAA
HLLGATETLNVGTAAIVLPTAHPVRQAEDVNLLDQMSKGRFRFGICRGLYDKDFRVFGTD
MDNSRALMDCWYDLMKEGFNEGYIAADNEHIKFPKIQLNPSAYTQGGAPVYVVAESASTT
EWAAERGLPMILSWIINTHEKKAQLDLYNEVATEHGYDVTKIDHCLSYITSVDHDSNRAK
DICRNFLGHWYDSYVNATKIFDDSDQTKGYDFNKGQWRDFVLKGHKDTNRRIDYSYEINP
VGTPEECIAIIQQDIDATGIDNICCGFEANGSEEEIIASMKLFQSDVMPYLKEKQ
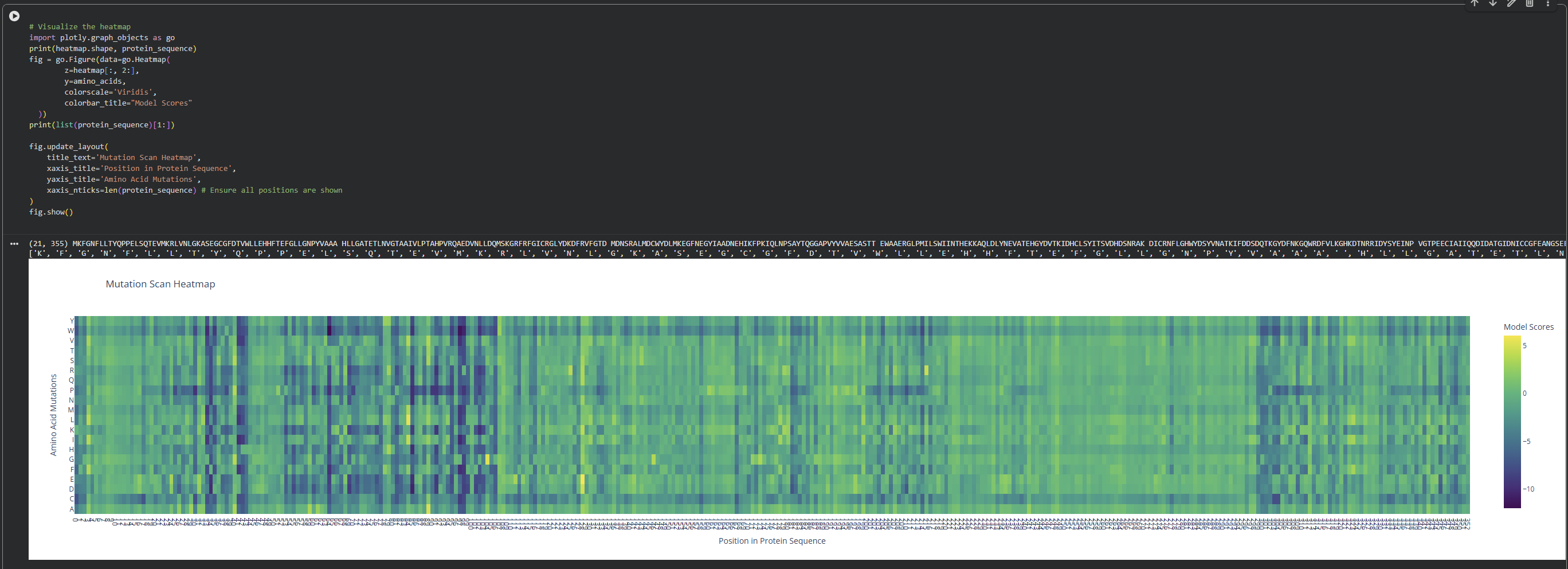
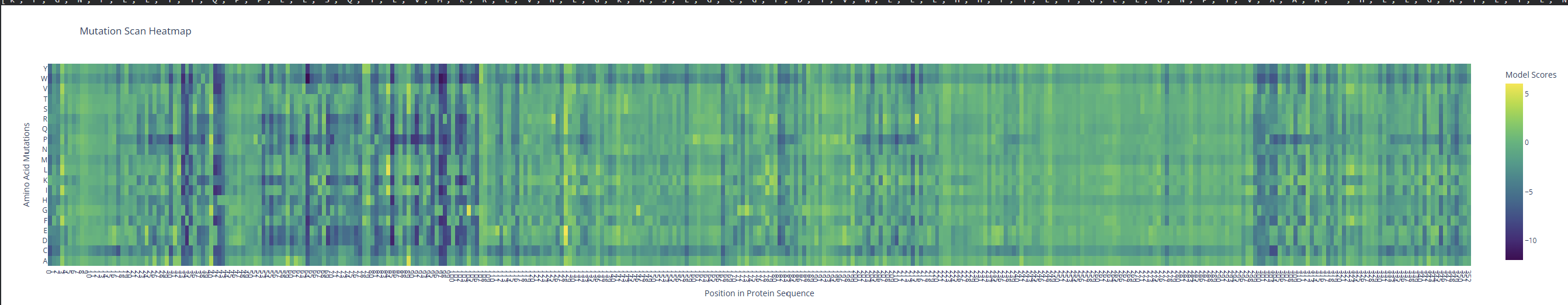
I observed a pattern of high compatibility in the Valine (V) row. Unlike Proline, which shows dark blue (low likelihood) across most positions, Valine remains consistently light-colored. This suggests that the luciferase structure is robust to Valine substitutions, likely because Valine’s small, hydrophobic side chain can be easily accommodated in various positions of the protein’s fold without causing major structural disruptions
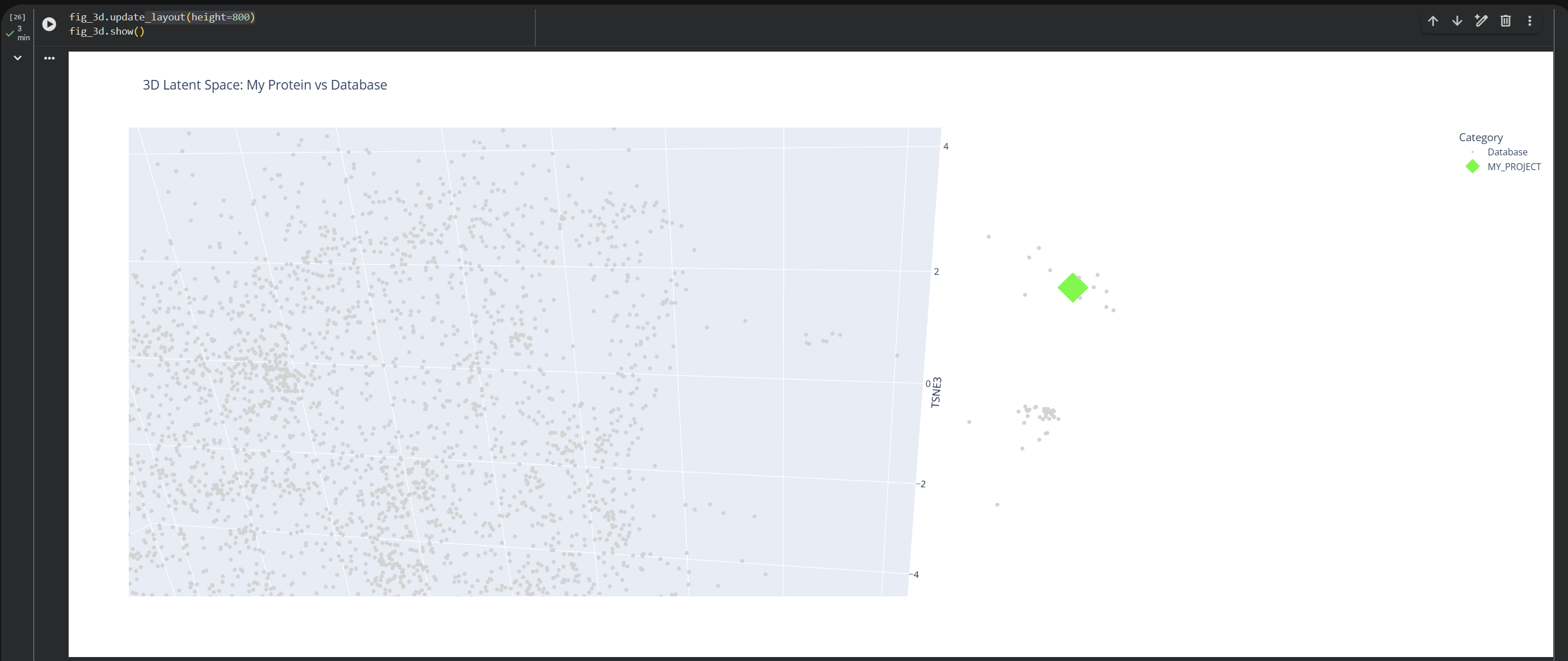
2. Latent Space Analysis
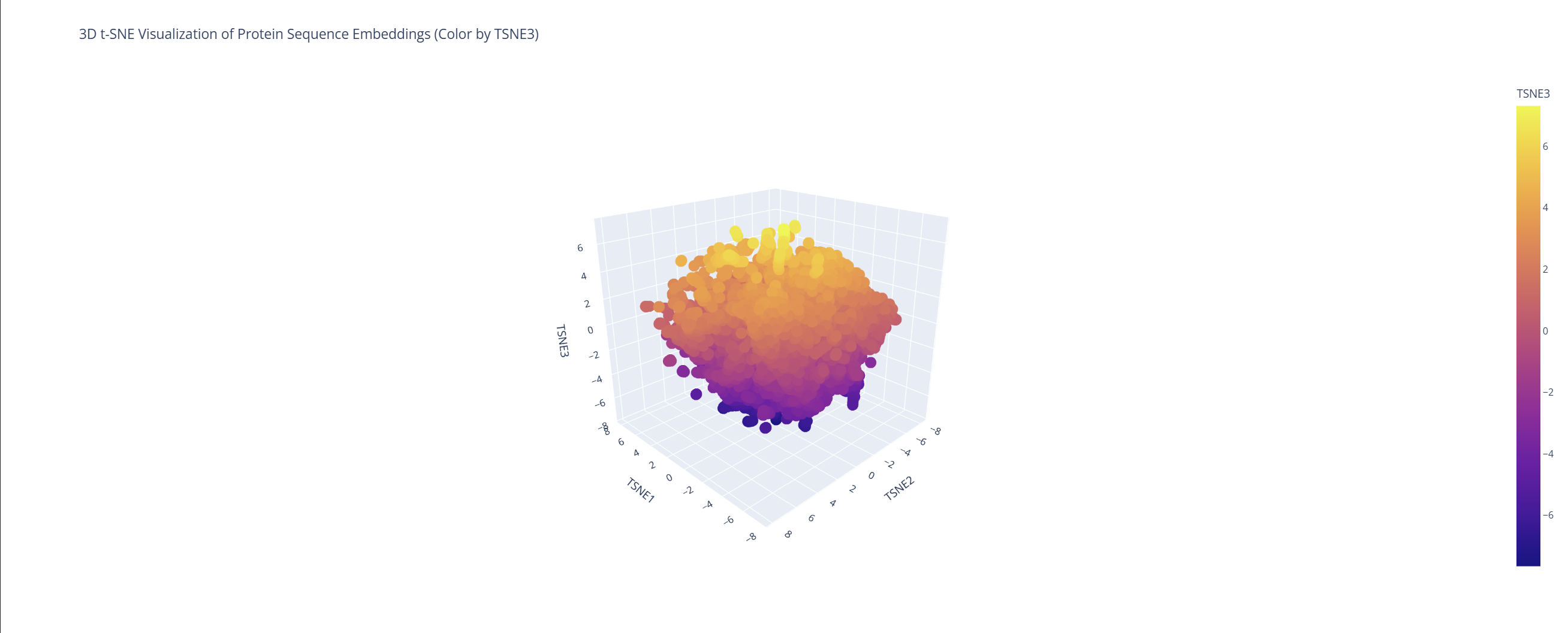
Based on this visualization, we can clearly see the existence of different groupings or clusters that create these ’neighborhoods’ of proteins. This shows that the ESM2 model is effectively generating these connections by grouping proteins that share similar characteristics.
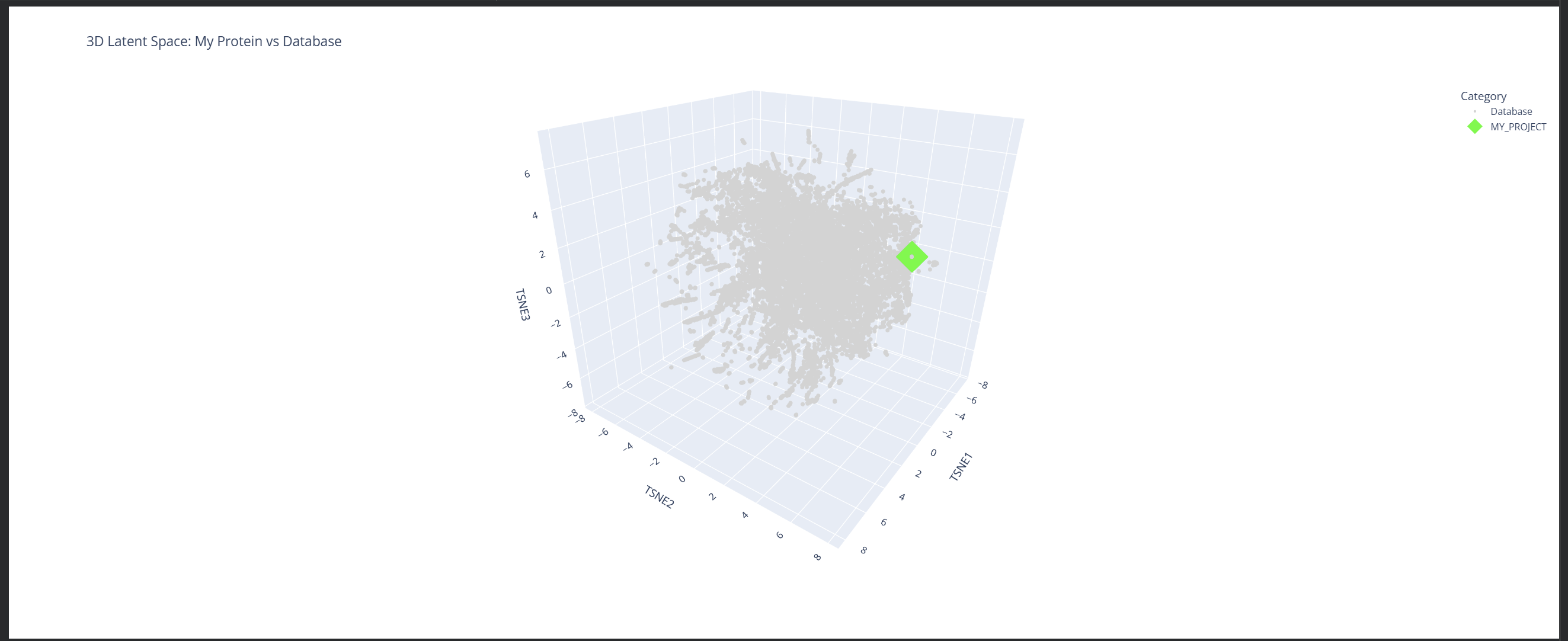
My protein (1LUC) landed in a specific neighborhood within the map. Being positioned next to other proteins from the database shows that they share similar characteristics. Given its biological nature, this confirms the model successfully grouped it with its functional relatives.
C2. Protein Folding
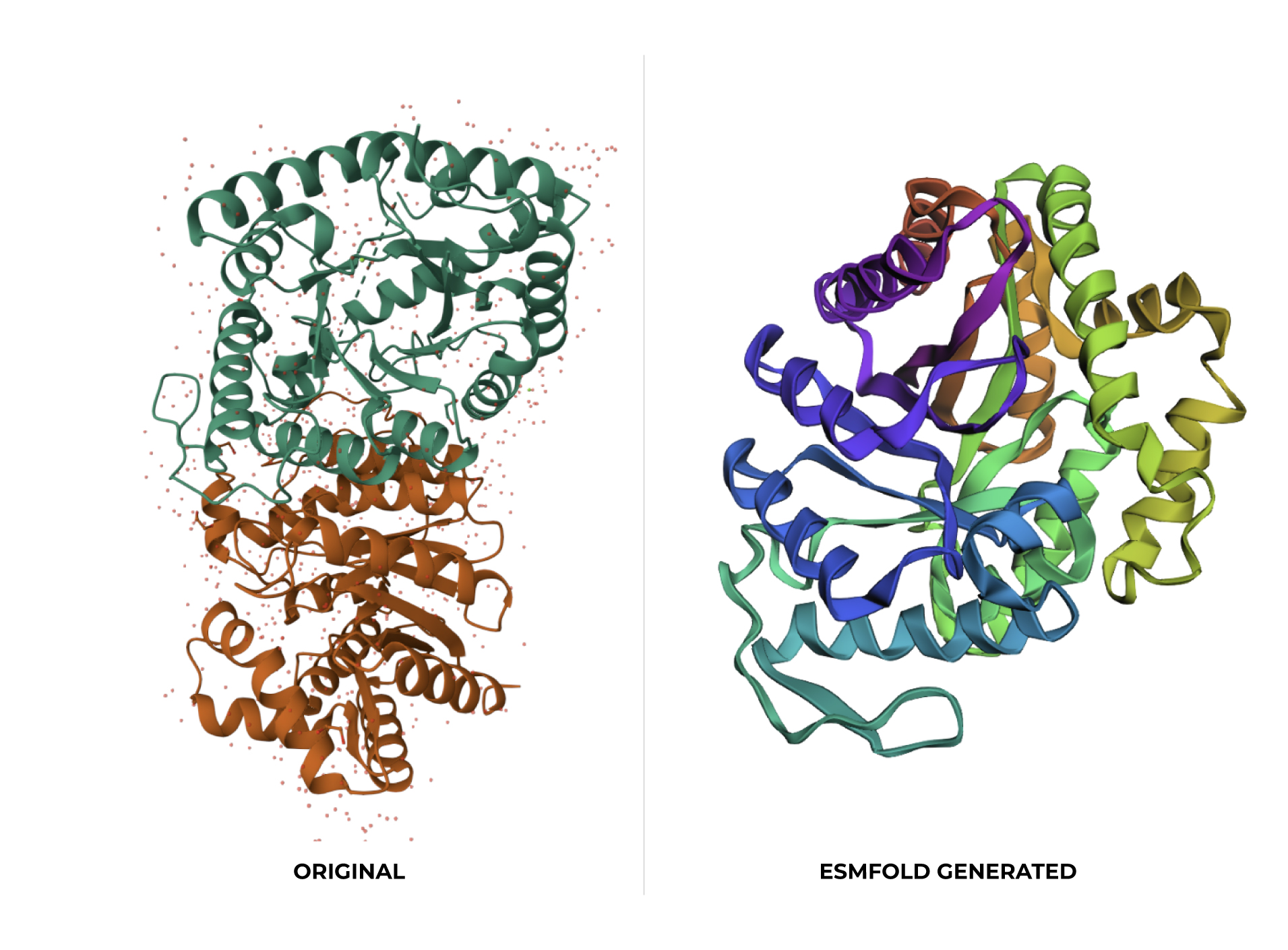
At first glance, the predicted coordinates do not seem to match the original structure, as the overall complexity is different. However, upon closer inspection, there is a clear structural resemblance to one of the specific domains of the protein. The original 1LUC is composed of two different chains, while the simulation produced a single-domain fold. It appears the model successfully captured the architecture of one of these subunits, even if it couldn’t replicate the full multi-domain assembly of the real protein.
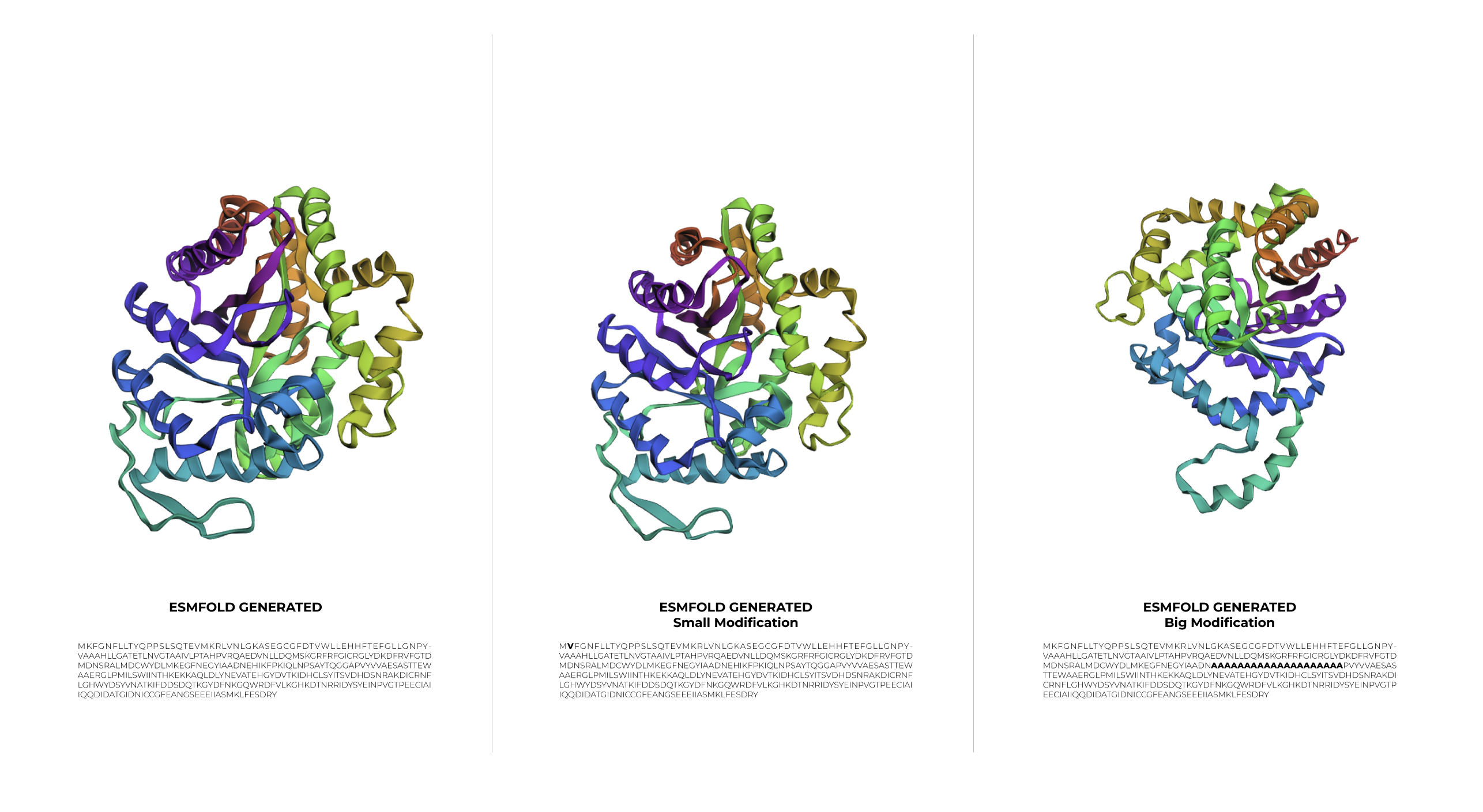
During the first stage of experimentation, a minimal change was made to the protein sequence by swapping Lysine for Valine. The result was a structure basically identical to the original one. This outcome demonstrates that 1LUC is highly resilient to small modifications, as the point mutation did not compromise the structural integrity of the 1LUC.
In the second stage, a larger modification was introduced. Although the sequence was significantly altered, the protein showed a surprising level of resilience, maintaining its general folded shape even if the symmetry was slightly affected.
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN

Results
Original LuxA Secuence
MKFGNFLLTYQPPELSQTEVMKRLVNLGKASEGCGFDTVWLLEHHFTEFGLLGNPYVAAA HLLGATETLNVGTAAIVLPTAHPVRQAEDVNLLDQMSKGRFRFGICRGLYDKDFRVFGTD MDNSRALMDCWYDLMKEGFNEGYIAADNEHIKFPKIQLNPSAYTQGGAPVYVVAESASTT EWAAERGLPMILSWIINTHEKKAQLDLYNEVATEHGYDVTKIDHCLSYITSVDHDSNRAK DICRNFLGHWYDSYVNATKIFDDSDQTKGYDFNKGQWRDFVLKGHKDTNRRIDYSYEINP VGTPEECIAIIQQDIDATGIDNICCGFEANGSEEEIIASMKLFQSDVMPYLKEKQ
Inverse Folding with ProteinMPNN
MKFGYHIHGYQPPHLTREEVLARIEKLAAAAPEQGFDTVWIHEGYFHPHNLIADPFATAEKFLAASKDLKVGTEALDLPARDPLEVAKKANTLDNKSKGRFIFGISRGAFEYVFKVFGKDFSKRFELTKYKLELIEEGFKTGTMSADSEYLKFPKTKVTPASYTPGGPPIYVVARTDEEVEFAAERGIPLVLSPLITVEEKKAMLDKYKEVAAKNGFDVSKIEHVLSYFGVVDEDEELAKKKMLAYLKEYAASLEEAKNLLXXXXXXXXXXXXXXXXXXXXXXXXXXXXXGLEKAPEINPVGTPEHAVEVIQKDIDATGITYINVNFEAAGTVEEILKSMKLFKEEVMPKLKPKK
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
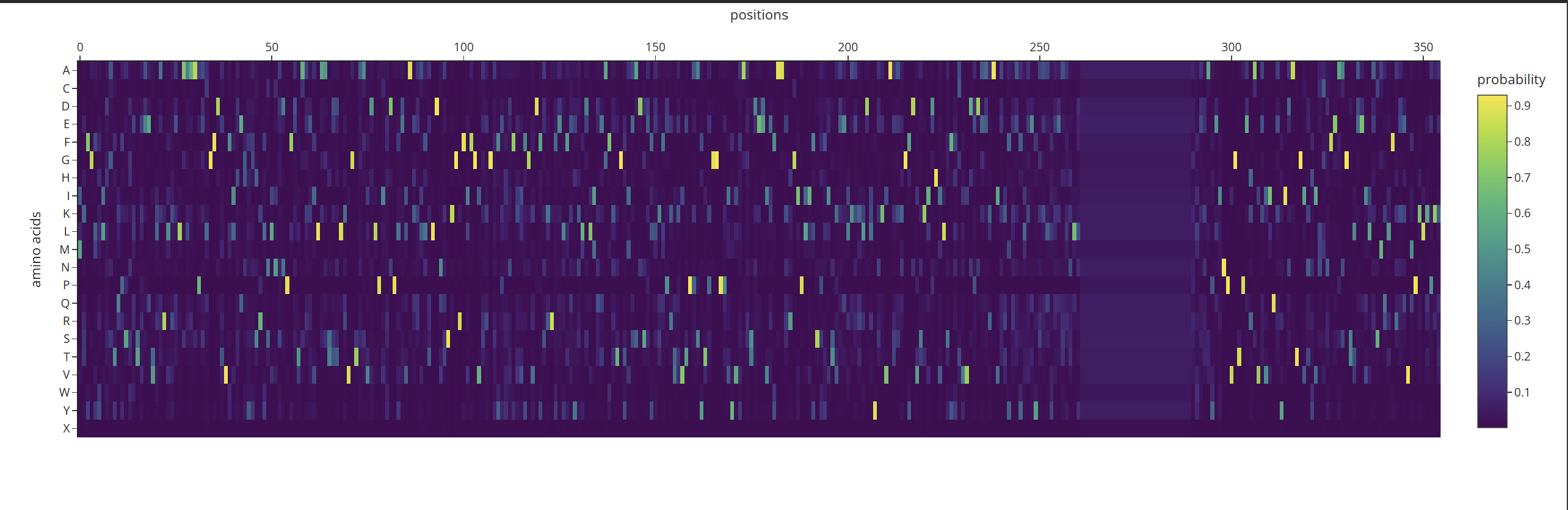
According to what I can understand by analyzing the probability heatmap, we can identify the critical points of the structure. The areas marked in yellow represent the positions where amino acids must be strictly maintained so that the protein does not lose its properties or shape. Meanwhile, the dark-colored areas are zones where the AI can ‘fill in’ with different options without necessarily affecting the overall fold.
Input this sequence into ESMFold and compare the predicted structure to your original.
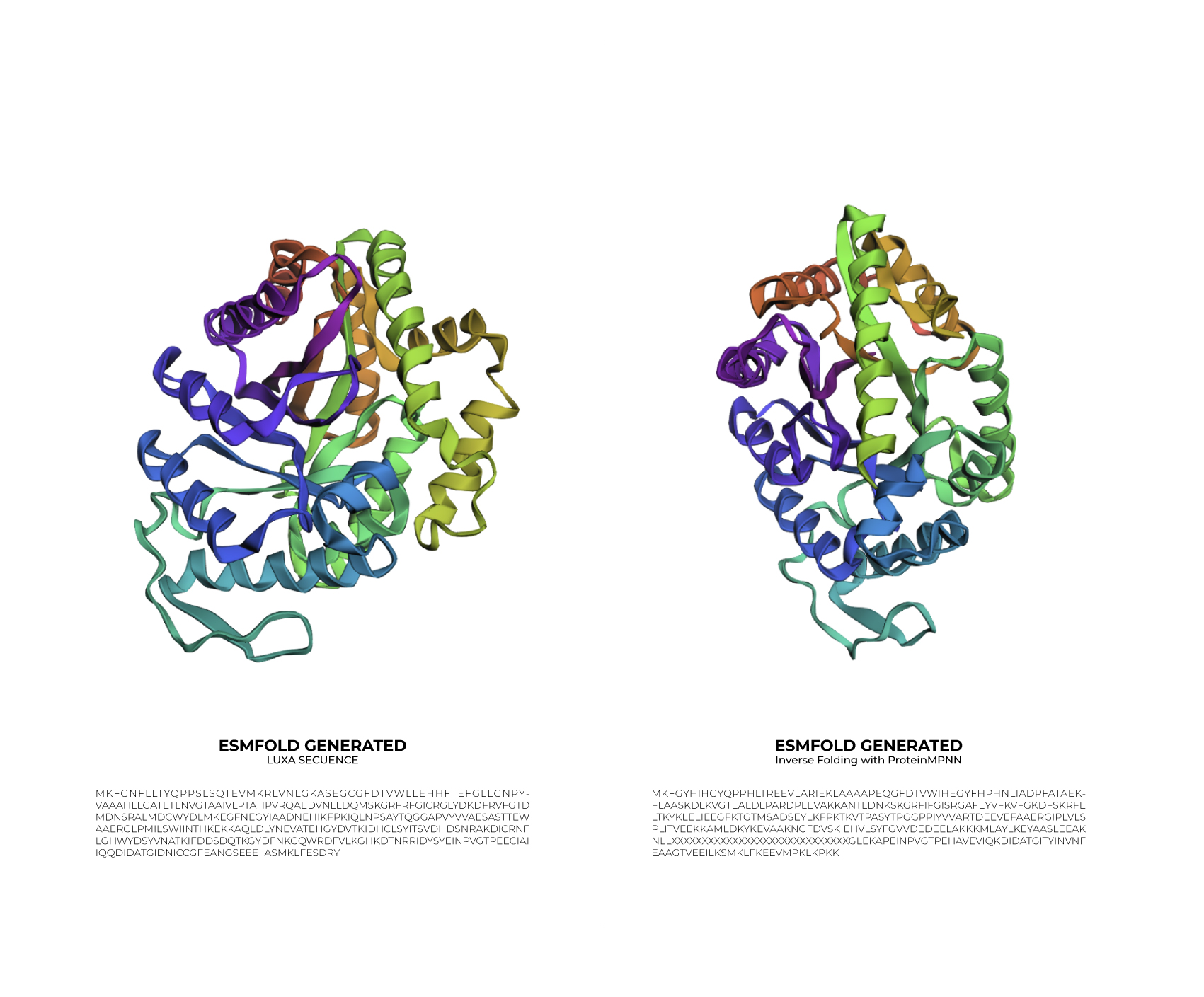
Based on the data obtained from the output (>T=0.1, sample=0, score=0.8181, seq_recovery=0.4632), it can be concluded that although the Colab changed 53% of the original protein, the final result still maintained the essential points of the structure. Even the preview remained highly faithful to the original
Week 5 HW: Protein Design Part II
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Your challenge:
Design short peptides that bind mutant SOD1.
Then decide which ones are worth advancing toward therapy.
You will use three models developed in our lab:
PepMLM: target sequence-conditioned peptide generation via masked language modeling
PeptiVerse: therapeutic property prediction
moPPIt: motif-specific multi-objective peptide design using Multi-Objective Guided Discrete Flow Matching (MOG-DFM)
Part 1: Generate Binders with PepMLM
1.Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
Human SOD1 sequence
sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS
AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV
HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Human SOD1 sequence A4V mutation.
MATVAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS
AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV
HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
2.Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
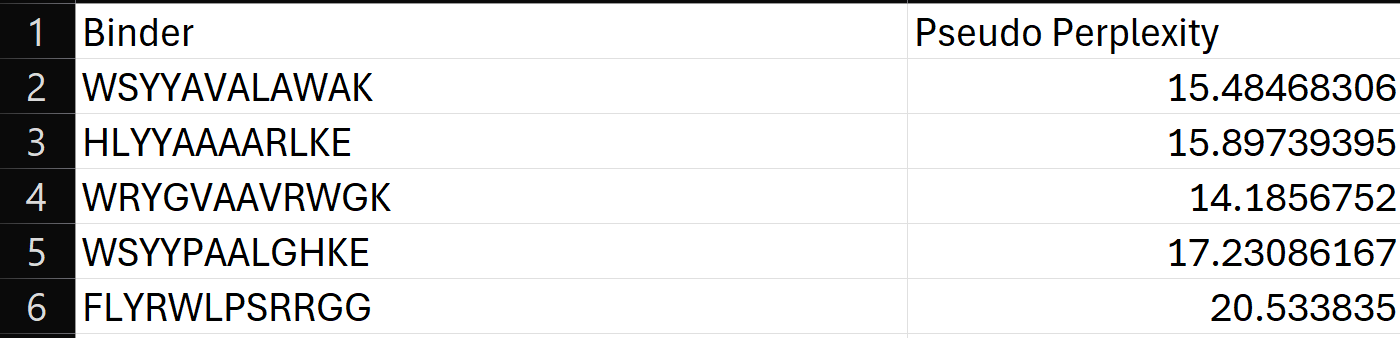
Record the perplexity scores that indicate PepMLM’s confidence in the binders.
3.Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
Using the PepMLM-650M model conditioned on the human SOD1 sequence (UniProt P00441) carrying the A4V mutation, I generated four de novo 12-residue peptides predicted to bind mutant SOD1.
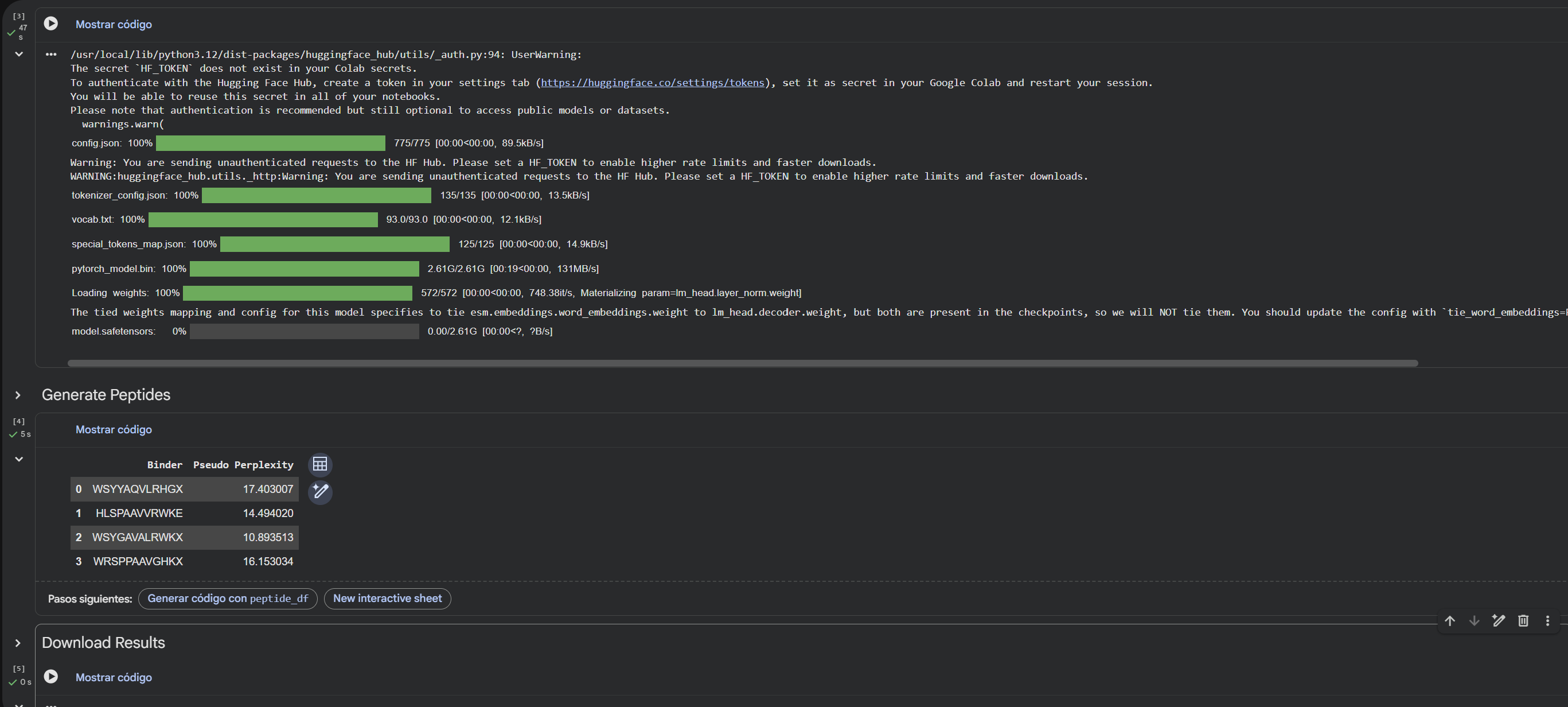
4.To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
Record the perplexity scores that indicate PepMLM’s confidence in the binders.
5.Record the perplexity scores that indicate PepMLM’s confidence in the binders.
Part 2: Evaluate Binders with AlphaFold3
1.Navigate to the AlphaFold Server: alphafoldserver.com
2.For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
3.Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
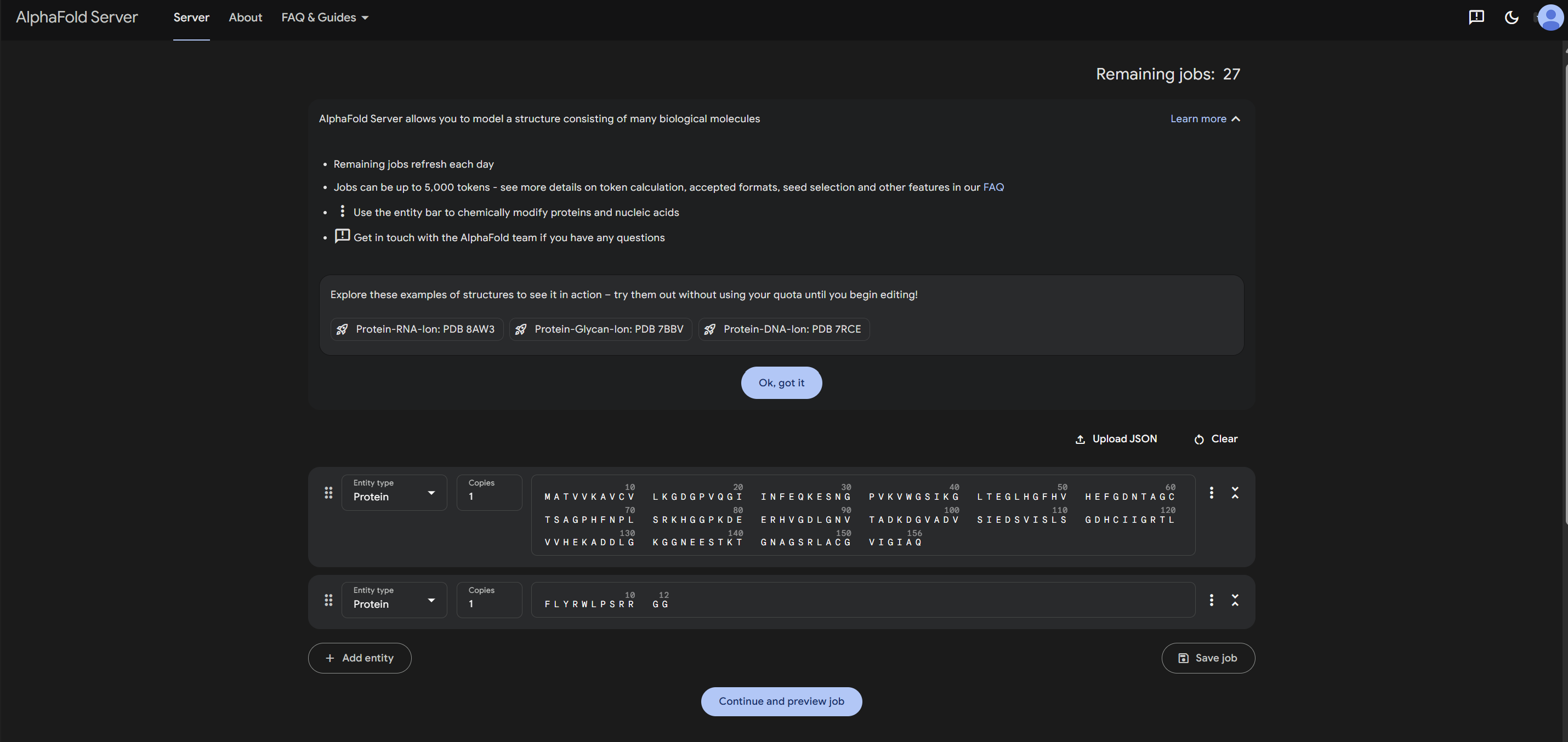
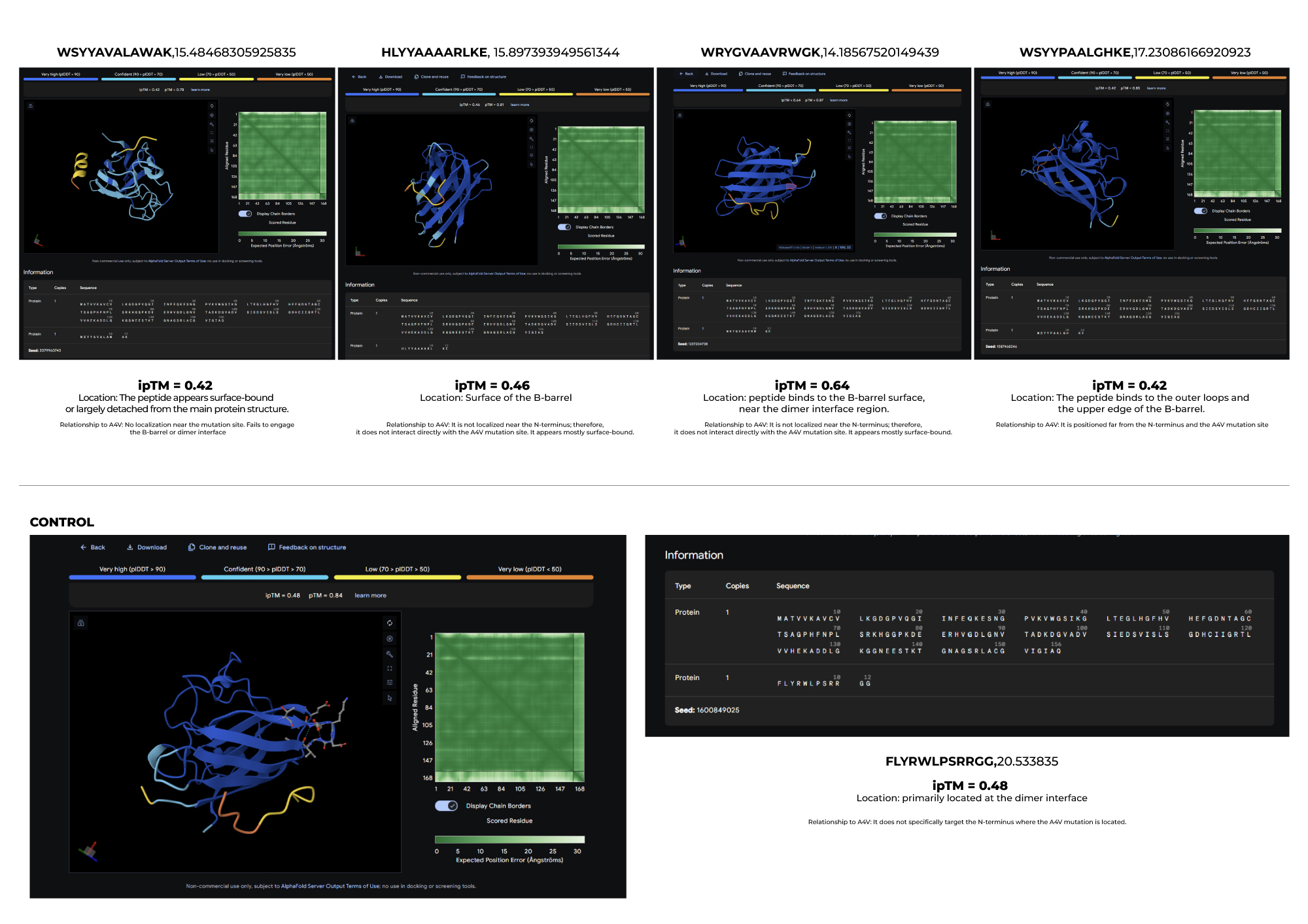
4.In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
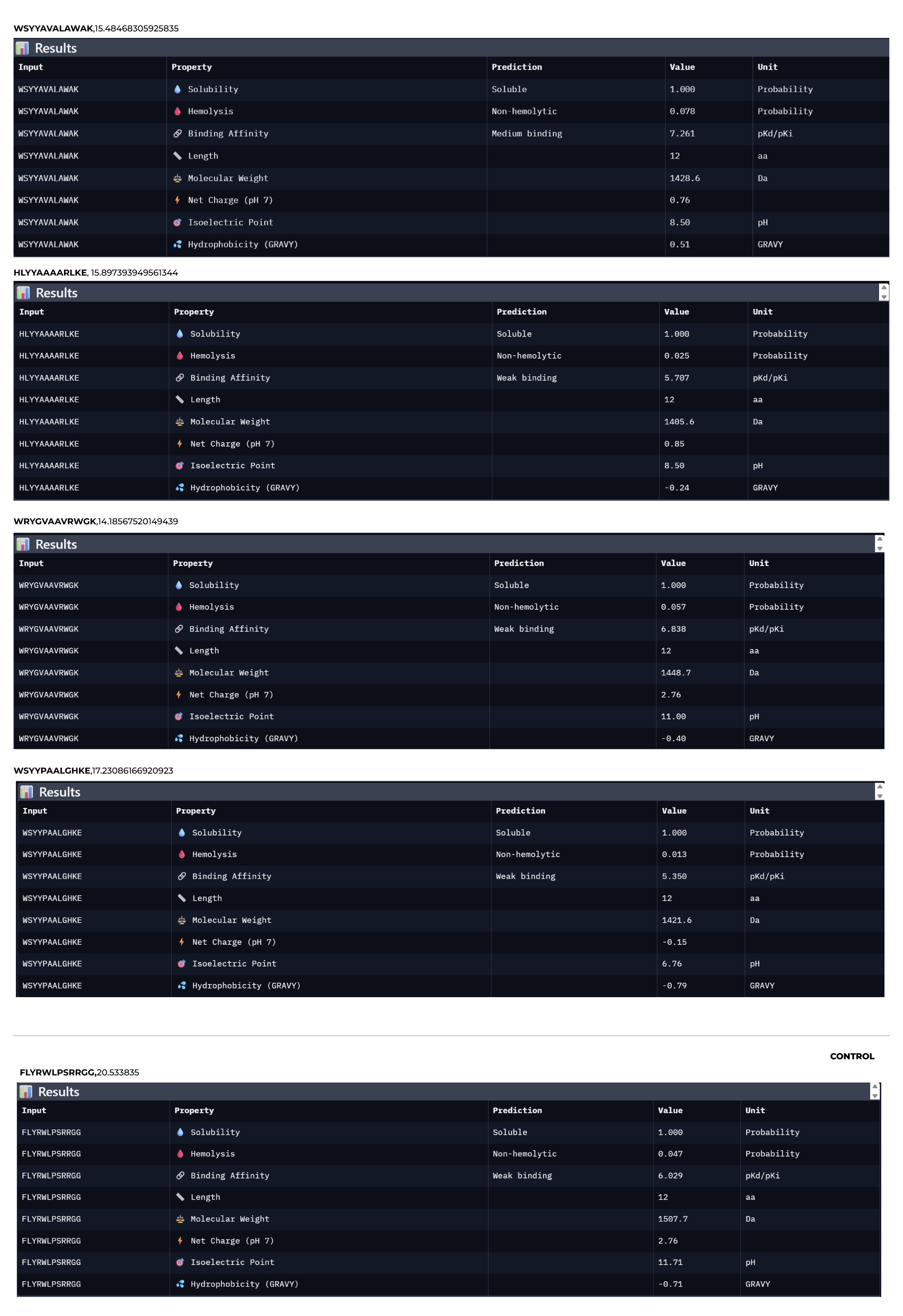
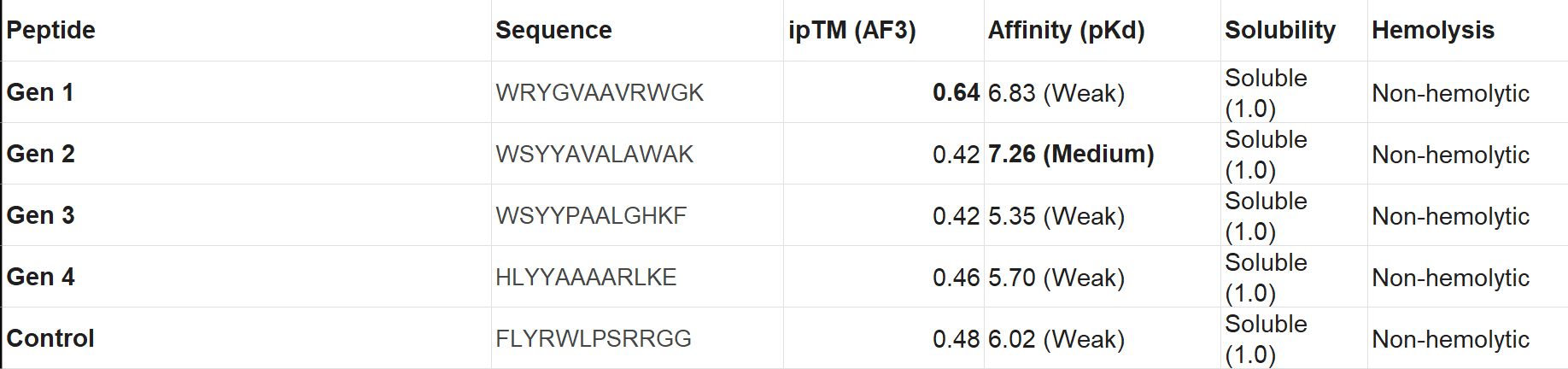
Based on the results I obtained, I find that PepMLM shows a remarkable ability to generate de novo peptide sequences that not only match but can even surpass the predicted binding confidence of known ligands. In particular, the higher ipTM score observed for Peptide 2 leads me to think that it may be a particularly promising candidate, worthy of further biochemical validation as a potential stabilizer of the SOD1-A4V mutant.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
1.Paste the peptide sequence.
2.Paste the A4V mutant SOD1 sequence in the target field.
3.Check the boxes
Predicted binding affinity
Solubility
Hemolysis probability
Net charge (pH 7)
Molecular weight
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Choose one peptide you would advance and justify your decision briefly.
I would advance Peptide 1 for further development. Although Peptide 2 shows a slightly higher chemical affinity score, Peptide 1 is the only candidate that demonstrates high structural confidence (ipTM 0.64) and a clear binding orientation near the A4V mutation site and the dimer interface. Its excellent solubility and low hemolysis probability confirm that it balances superior structural complementarity with the necessary therapeutic properties to act as a potential SOD1 stabilize
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
- Open the moPPit Colab linked from the HuggingFace moPPIt model card
- Make a copy and switch to a GPU runtime.
- In the notebook:Paste your A4V mutant SOD1 sequence.
- Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
- Set peptide length to 12 amino acids.
- Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
- After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
Results:
RYCTSAQLTQKD
GYPCGYSIGTYL
TGKLREIYIEQE
GCGFKKKGQFEP
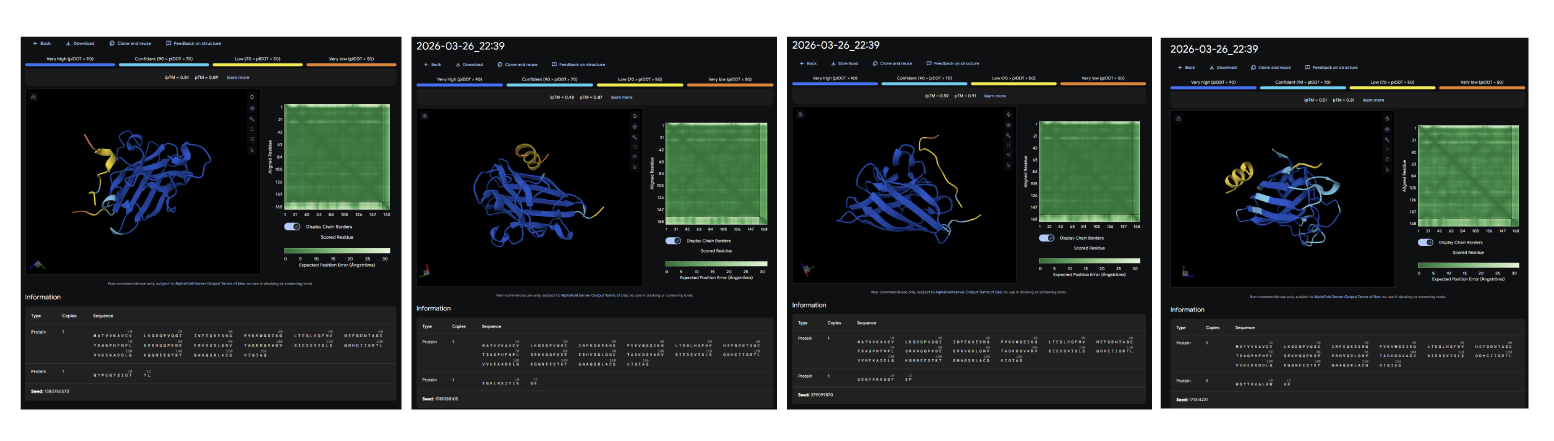
After generating the peptides, I used AlphaFold Server to look at how they interact with the target protein. Compared to the PepMLM peptides, which were mainly designed based on sequence prediction and properties using tools like PeptiVerse, the moPPIt peptides seem to show better binding at specific points, although sometimes their structure is a bit less compact overall and conected with the main structure. From what I have learned, before moving to clinical studies, I would first test things like how stable they are, how easily they degrade, and whether they actually reduce harmful protein aggregation.
Week 6 HW: Genetic Circuits Part I: Assembly Technologies
Assignment: DNA Assembly
1.What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
According to the protocol, the Phusion High-Fidelity PCR Master Mix is used to amplify specific DNA regions and prepare the necessary fragments for Gibson Assembly with high precision and minimal errors.
This pre-mixed solution is composed of four key elements. First, the high-fidelity DNA polymerase, which is responsible for copying the DNA and ensuring accurate replication with minimal errors. Second, dNTPs, which function as the building blocks to assemble the new DNA strands. Then, the reaction buffer, which maintains the chemical environment, such as pH, at optimal levels. And finally, magnesium an essential component that activates the polymerase so it can start joining the blocks together.
2.What are some factors that determine primer annealing temperature during PCR?
Following what was mentioned in class, several factors determine the primer annealing temperature, primarily the length and specific sequence of the primers. According to the protocol, primers should be 18–22 bp long with a GC content of 40–60%, including a GC-clamp to ensure specific binding. These elements define the melting temperature, and the is Ta is typically set 2–5°C below that value. In practice, this results in temperatures that typically range between 50°C and 65°C for a duration of 5 to 30 seconds, allowing the primers to bind accurately to the template DNA.
3.There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR and restriction enzyme digests both generate linear DNA fragments, but they do so in different ways and at different points in the cloning workflow. In my view, one of the most important differences is that PCR can be carried out entirely in a small PCR tube, inside a thermocycler, as a cell‑free reaction that directly copies and amplifies the DNA. In contrast, restriction enzyme digestion only cuts the DNA at specific sites; it does not copy it. The actual replication of the fragment occurs only after the digested DNA is ligated into a vector and introduced into a living host (such as E. coli), which then replicates the plasmid during cell growth.
PCR: is a method to copy a specific piece of DNA many times. It is done in a small tube inside a machine called a thermocycler, where you add the original DNA (the template), two primers that are short DNA sequences marking the start and end of the region you want to copy, a polymerase enzyme that copies the DNA, and the nucleotides that are the “letters” of DNA needed to make the new strands.
This process is divided in 3 steps:
- Denaturation: it heats the DNA so that it opens up and separates into two strands.
- Annealing: it cools down a bit so that the primers can stick to their matching sites on the DNA.
- Extension: it heats up a little more so that the polymerase can copy the region between the primers.
RED: Restriction enzyme digestion is a method that cuts DNA at very specific places. It is done in a small tube, where the DNA is mixed with a restriction enzyme and a buffer that has the right conditions, such as pH and salt. The enzyme recognizes a short sequence of letters in the DNA and cuts it at that site, generating linear fragments. But the enzyme only cuts the DNA; it does not copy it. The actual copying happens later, when the cut fragment is introduced into a bacterium (like E. coli) and the cell replicates it as it divides.
Now that I understand it better, I think restriction enzyme digestion is “easier to replicate” in the sense that, once the DNA is inside a living cell like E. coli, the cell copies the plasmid by itself while it grows, and I don’t have to control temperature cycles like in PCR. However, the technique still needs many lab steps—cutting the DNA, ligating the insert into the vector, doing the transformation, and growing the bacteria—and uses expensive materials like restriction enzymes and ligase, so it is not always simpler or cheaper than it first seems. In terms of when to use each method, PCR is better when you want to amplify a specific region and introduce mutations. While restriction enzyme digestion is better when the insert and the vector already have compatible restriction sites and you want to do simple cut‑and‑ligate cloning.
4.How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To ensure that the DNA sequences are appropriate for Gibson Assembly, three main requirements from the protocol must be met.
Mechanism: To ensure the assembly works, the DNA must be compatible with the four-step enzymatic process
1.Exonuclease
2.Annealing
3.Polymerase
4.Ligase
Overlaps: The fragments must have 20–40 bp of sequence identity at their ends.
Molar Ratios: You must use a 2:1 (insert:vector) molar ratio
5.How does the plasmid DNA enter the E. coli cells during transformation?
During transformation, the plasmid DNA enters the E. coli cells thanks to a heat shock step. The cells are first made more able to take up DNA in the cold, then briefly heated to 42°C and cooled again. This creates temporary openings in the cell membrane so the plasmid can get inside by diffusion. After transformation, only the cells that have taken up the plasmid can survive on the antibiotic‑containing plates and start to grow.
6.Describe another assembly method in detail (such as Golden Gate Assembly)
I’m still trying to understand this, but I understood that Golden Gate Assembly is another method to join pieces of DNA into a plasmid, and it uses a special type of restriction enzyme (like BsaI) together with a ligase in the same tube. In this method, the BsaI enzyme cuts the DNA a little beyond its recognition site, leaving ends that can be custom‑designed. The thermocycler repeats cycles of cutting and ligating, and at the end you get a circular plasmid with all the fragments in the correct order, without the original restriction sites.
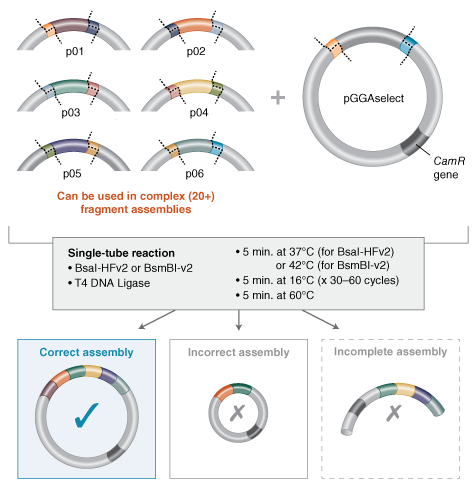
Source:https://www.neosynbio.com/golden-gate-assembly
7.Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Model this assembly method with Benchling or Asimov Kernel!
First, I design the DNA fragments and the vector so that each fragment has a unique end (overhang) that will only fit the next fragment, like puzzle pieces in a fixed order.
Prepare the DNA fragments and make sure each one has the BsaI cut site with the custom overhang on each side.
In a small tube, I mix all the fragments, the circular vector, the restriction enzyme BsaI, DNA ligase, ATP, and the reaction buffer.
I put the tube in a thermocycler and run many cycles where the enzyme cuts the DNA a little outside its site and the ligase rejoins the compatible ends.
After the reaction, the main product is a circular plasmid where all fragments are joined in the correct order, and the BsaI sites disappear from the final construct.
Then I transform this plasmid into competent E. coli cells, select the colonies on antibiotic plates, and check the correct assembly by PCR or digestion.
Week 7 HW: Genetic Circuits Part II: Neuromorphic Circuits
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
I found really interesting and maybe usefull for my final proyect. IANNs have an important advantage over traditional genetic circuits because while traditional circuits act like simple on/off switches, IANNs can process multiple input signals and produce a gradual and smooth response. This type of circuit can generate intermediate levels of expression, such as weak, medium, or strong fluorescence, depending on the value of the signal. This allows the cell to make finer and more complex decisions, such as better distinguishing between closely related conditions, something Boolean circuits cannot do with the same level of precision.
2. Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
Following this idea that this type of circuit could be useful for my final project, a useful application for an IANN that comes to mind is a pH sensor inside a bacterium, where the intensity of a color (for example, fluorescence) indicates the pH value. In this case, the inputs of the IANN would be the molecular signals associated with pH, and the output would be the amount of fluorescent protein produced. With a traditional circuit, the color would only turn on once the pH passes a certain threshold, and you would not see the difference between a moderately high pH and a very high pH. In contrast, with an IANN the color becomes progressively more intense as the pH changes, allowing you to see whether the value is continuously increasing or decreasing.
3.Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
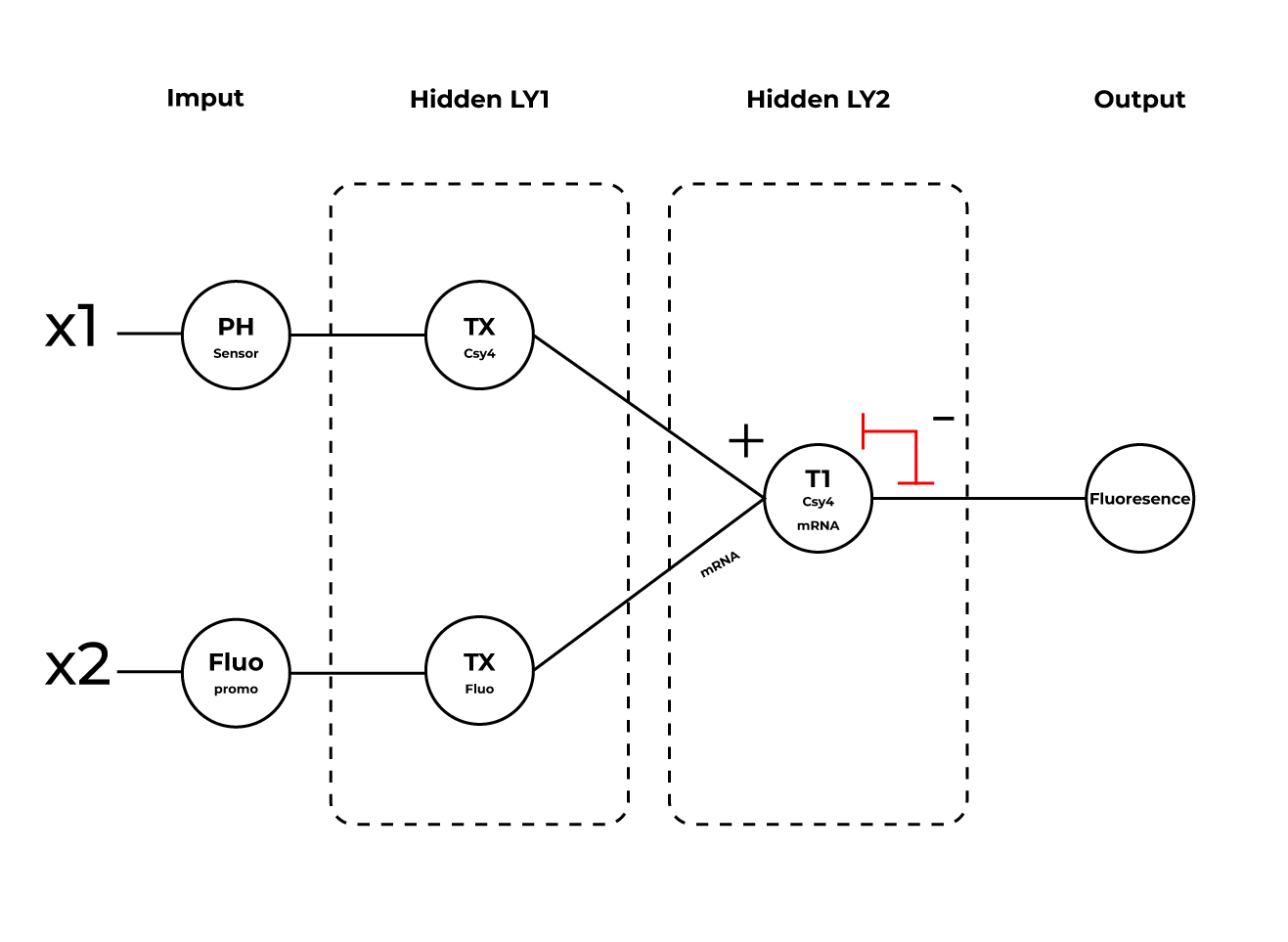
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Assignment Part 2: Fungal Materials
- What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
In recent years, materials made from living organisms, especially fungi, have started to gain a lot more visibility in everyday life. Most of these materials are based on mycelium, which is basically the root system of fungi. It acts as a natural binder, feeding on organic waste and growing into dense, compact structures.
These materials are already being used in different ways. For example, they can be used to create biodegradable packaging that replaces traditional plastic or cardboard. They also have good sound insulation properties, which makes them useful for things like acoustic panels.
More recently, this type of material has started to appear in the fashion industry as well. Companies like MycoWorks are working on developing leather-like materials made from mycelium, offering an alternative that avoids the environmental and ethical costs of animal leather.

image source: https://vegnews.com/south-carolina-sustainable-vegan-mushroom-leather
Advantages:
One of the clear advantages of mycelium-based materials compared to traditional ones is that they are sustainable and biodegradable, so they don’t create long-term waste. Moreover, they can be grown using organic waste matter, which makes the production process more environmentally friendly. Another benefit is that they require less energy and fewer resources than conventional materials.
Disadvantages:
However, there are still some limitations. One of them is that these materials are not always as strong or durable as conventional ones, especially for heavy-duty uses. Another major disadvantage is that the production process can be slower, since it depends on biological growth, which can increase costs and require more storage space. Finally, scalability is also an issue, as it can be difficult to produce these materials on a large industrial scale.
- What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
Fungi are already some of nature’s best decomposers, but what I find really interesting is the idea of taking that a step further through genetic engineering. You could imagine a fungal material that stays stable and strong while it’s being used, but once it ends up in a landfill or a compost environment, some kind of genetic “switch” activates. Instead of just breaking itself down, it could be engineered to release enzymes that also help degrade surrounding waste, like microplastics or even certain toxic substances, much faster than they would normally break down.
From what I understand, and also just based on how I see it, doing this with fungi could actually be more effective than using bacteria in some cases. Bacteria are great, but they usually stay at a very small scale, like in lab conditions or Petri dishes. Fungi, on the other hand, grow as mycelium, which means they can form much larger, three-dimensional structures.