First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about. I am curious aboutI the develop of engineered biological decoys systems designed to reduce infections by preventing bacteria or viruses from attaching to human cells. Instead of destroying pathogens these systems would work by mimicking the cellular receptors that microorganisms need to initiate infection. It would consist of synthetic type of cells that display molecules similar to those found on real cell surfaces. Pathogens would bind to these decoys rather than to human tissues, thereby reducing the likelihood of infection and lowering the initial pathogen load. This is inspired by natural protective mechanisms such as mucosal barriers and could help reduce antibiotic use. By not directly killing microorganisms, it would reduce evolutionary pressure and potentially slow the development of antimicrobial resistance.
HW 3: Lab automation Assignment: Python Script for Opentrons Artwork Generate an artistic design using the GUI at opentrons-art.rcdonovan.com. Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script which draws your design using the Opentrons. You may use AI assistance for this coding — Google Gemini is integrated into Colab (see the stylized star bottom center); it will do a good job writing functional Python, while you probably need to take charge of the art concept. If you’re a proficient programmer and you’d rather code something mathematical or algorithmic instead of using your GUI coordinates, you may do that instead.
Part A. Conceptual Questions How many molecules of amino acids do you take with a piece of 500 grams of meat? (On average an amino acid is ~100 Daltons) The calculation would be to use an equation like moles = mass / molar mass, then multiply by Avogadro’s number. Roughly ~3 × 10²⁴ molecules.
Why do humans eat beef but do not become a cow, eat fish but do not become fish? We absorb the nutrients, don’t imitate the animal, and slowly turn them into our own molecules.
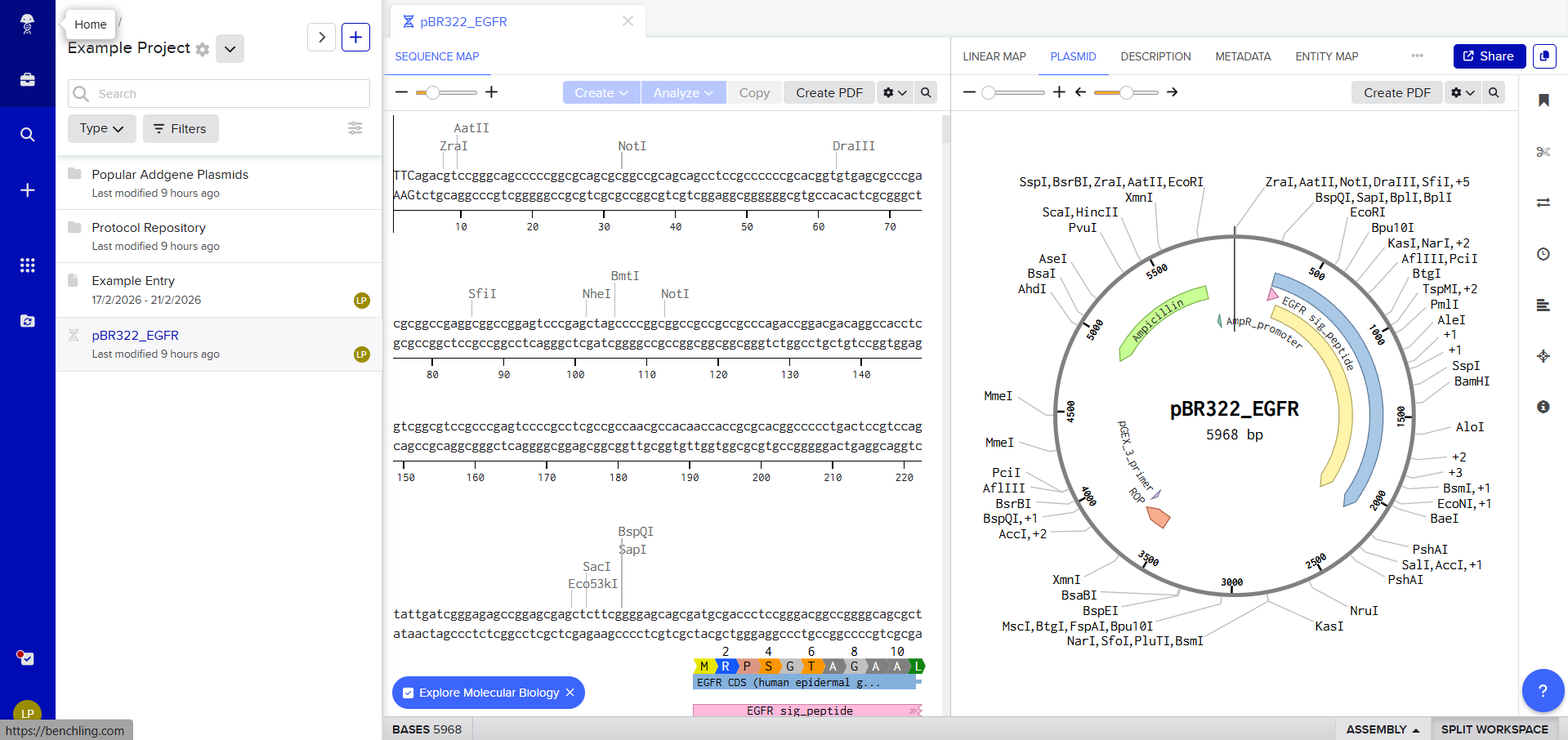
HW 2: DNA Read, Write, & Edit Part 1: Benchling & In-silico Gel Art Own experience on: Make a free account at benchling.com Import the Lambda DNA.
The first view of Benchling workspace Then i transfered de Lambda DNA, here we have two option for DNA document type, either we can use Genbank or, but Benchlink does have more options. Upload to Benchling So we can now view it on Benchling and start exploring the different options available
Subsections of Homework
Week 1 HW: Principles and Practices
First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
I am curious aboutI the develop of engineered biological decoys systems designed to reduce infections by preventing bacteria or viruses from attaching to human cells. Instead of destroying pathogens these systems would work by mimicking the cellular receptors that microorganisms need to initiate infection.
It would consist of synthetic type of cells that display molecules similar to those found on real cell surfaces. Pathogens would bind to these decoys rather than to human tissues, thereby reducing the likelihood of infection and lowering the initial pathogen load.
This is inspired by natural protective mechanisms such as mucosal barriers and could help reduce antibiotic use. By not directly killing microorganisms, it would reduce evolutionary pressure and potentially slow the development of antimicrobial resistance.
Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals. Below is one example framework (developed in the context of synthetic genomics) you can choose to use or adapt, or you can develop your own. The example was developed to consider policy goals of ensuring safety and security, alongside other goals, like promoting constructive uses, but you could propose other goals for example, those relating to equity or autonomy.
Ensure biosafety and biosecurity by preventing misuse or unintended biological effects of decoy technologies.
Promote constructive medical use as an alternative or complement to antibiotics, rather than as an unregulated biomedical product.
Support equity and responsible access and ensuring that these tools are deployed based on public health needs and not only commercial interests.
Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”). Try to outline a mix of actions (e.g. a new requirement/rule, incentive, or technical strategy) pursued by different “actors” (e.g. academic researchers, companies, federal regulators, law enforcement, etc). Draw upon your existing knowledge and a little additional digging, and feel free to use analogies to other domains (e.g. 3D printing, drones, financial systems, etc.).
Purpose: What is done now and what changes are you proposing?
Design: What is needed to make it “work”? (including the actor(s) involved - who must opt-in, fund, approve, or implement, etc)
Assumptions: What could you have wrong (incorrect assumptions, uncertainties)?
Risks of Failure & “Success”: How might this fail, including any unintended consequences of the “success” of your proposed actions?
Action 1: Controlled clinical and environmental regulation of bioengineered decoys
Purpose:
Currently, many decoy-based technologies fall between medical devices and biological products. This action proposes clear regulatory pathways before clinical use.
Design:
Health regulators including lab testing, clinical trials, and post-deployment monitoring.
Assumptions:
This assumes existing regulatory institutions can adapt to novel bioengineered tools.
Risks of Failure and “Success”:
If regulation is too strict, innovation may be slow. If successful then early deployment may still miss rare long-term effects.
Action 2: Mandatory impact assessments focused on resistance and evolution
Purpose:
Unlike antibiotics, decoys aim to reduce selective pressure, but this is not guaranteed. This action proposes assessing evolutionary and ecological risks.
Design:
Researchers would be required to model pathogen adaptation and submit monitoring plans as part of approval.
Assumptions:
It assumes resistance can be predicted and monitored with current scientific models.
Risks of Failure and “Success”:
Pathogens may evolve in unexpected ways as that its their purpose, success could lead to overconfidence and reduced surveillance.
Action 3: Limits on non-medical and non-approved uses
Purpose:
This action aims to prevent decoy technologies from being used outside regulated medical or public health contexts.
Design:
Governments and funding agencies restrict access to production protocols and require licensing for manufacturing and distribution.
Assumptions:
It assumes enforcement is possible and effective.
Risks of Failure and “Success”:
Over-restriction could push development underground and success could concentrate control among few institutions.
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
1
1
2
• By helping respond
1
2
1
Foster Lab Safety
• By preventing incident
2
1
1
• By helping respond
n/a
1
n/a
Other considerations
• Minimizing costs and burdens to stakeholders
2
3
1
• Feasibility?
2
2
1
• Not impede research
2
3
3
• Promote constructive applications
1
2
3
Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties. For this, you can choose one or more relevant audiences for your recommendation, which could range from the very local (e.g. to MIT leadership or Cambridge Mayoral Office) to the national (e.g. to President Biden or the head of a Federal Agency) to the international (e.g. to the United Nations Office of the Secretary-General, or the leadership of a multinational firm or industry consortia). These could also be one of the “actor” groups in your matrix.
Regulated deployment of non-living biological decoys is prioritized because it allows these tools to reduce infection risk without directly modifying human immune systems or releasing living organisms. The main trade-off is that strict regulation may slow innovation and limit rapid deployment during outbreaks, but this is balanced by increased biosafety and public trust, impact assessments are prioritized alongside regulation to address uncertainties about long-term ecological and evolutionary effects, such as whether pathogens could adapt to the decoys over time. The trade-off here is added cost and administrative burden, but this is justified by the need to anticipate unintended consequences before large-scale use.
Key assumptions include the belief that pathogens will preferentially bind to decoys instead of host cells, and that resistance will evolve more slowly than with traditional antibiotics. There is also uncertainty about how these tools might interact with existing immune responses or medical treatments prioritizing regulated use with strong assessment frameworks balances innovation with precaution, helping ensure that bioengineered decoys contribute to public health without creating new biological risks.
Trade-offs:
Innovation vs. safety:
Strong regulation increases safety but may limit early-stage research capacity.
Cost vs. oversight:
Surveillance systems improve accountability but require sustained funding and coordination.
Equity vs. speed:
Incentive-based approaches promote broader participation but may not prevent all risks.
Reflection
While watching the class on Principles and Practices and learning about interventions involving Indigenous communities, I was reminded of a concern from my own community. In my community, there is a plant that is culturally important because it is used to decorate a symbol with strong meaning in our Indigenous traditions. For many years, part of the tradition involved walking long distances to collect this plant. However it is now becoming scarce due to its uncareful extraction from outsiders .
To protect the plant, some people have started growing it at home, and there are discussions about conserving it in new ways. While this may help preserve the species, it could also change or weaken the tradition connected to how the plant was originally collected. This raised an ethical question for me: how can we protect living organisms without losing the cultural practices linked to them?
One possible governance response is to combine conservation efforts with cultural participation, instead of fully replacing traditional practices. Regulations could focus not only on protecting the plant but also on supporting community led conservation that allows traditions to adapt while still being respected.
Assignment
(Week 2 Lecture Prep)
Professor Jacobson - Gene Synthesis
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
DNA polymerase has an error rate of 1 in 10⁶ bases. The human genome is about 3.2 × 10⁹ base pairs so errors would be tend to be frecuent if no proofreading and DNA repair mechanisms existed, so these dramatically reduce the final error rate and keep replication a part of biology.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
An average human protein with around 345 amino acids can be encoded in 4 eleveted to the amino acids to have different DNA sequences, because most amino acids are coded by more than one codon this makes the number of possible DNA codes very large.
However, not all of these codes work in practice. Cells prefer certain codons, and different DNA sequences can affect how stable the mRNA is, how efficiently the protein is made, and how the gene is regulated. Also, while many DNA changes do not affect the amino acid or only change it to a similar one, some changes can still disrupt protein production. As a result, only a subset of DNA sequences can properly code for a functional protein.
Dr.LeProust - DNA Synthesis Development and Application
What’s the most commonly used method for oligo synthesis currently?
The most commonly used method for oligo synthesis is phosphoramidite chemical synthesis. However Twist approach: silicon-based synthesis platforms are starting to outperform traditional methods by offering better scalability and control.
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Because errors and inefficiencies add up at each step, making long, error-free sequences very unlikely.
Why can’t you make a 2000bp gene via direct oligo synthesis?
The error rate and low yield make long sequences impractical. Instead genes are built by assembling many shorter, verified oligos.
George Church - Reading & Writing Life
[Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
10 essential amino acids in all animals
Arginine
Histidine
Isoleucine
Leucine
Lysine
Methionine
Phenylalanine
Threonine
Tryptophan
Valine
All animals require the same set of essential amino acids, including lysine, because they cannot synthesize them on their own. This means animals already depend on getting lysine from their diet. As discussed in the Reddit thread, this makes the “Lysine Contingency” from Jurassic Park biologically weak: removing lysine wouldn’t act as a special genetic control, it would simply cause general protein failure, not a precise safety switch. So while the idea works for science fiction, it doesn’t make much sense as a real biological containment strategy.
References
Use of gemini AI on double checking english grammar and help on website style view
Acevedo-Rocha, C. G., & Budisa, N. (2016). Xenomicrobiology: a roadmap for genetic code engineering. Microbial biotechnology, 9(5), 666–676. https://doi.org/10.1111/1751-7915.12398
Generate an artistic design using the GUI at opentrons-art.rcdonovan.com.
Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script which draws your design using the Opentrons.
You may use AI assistance for this coding — Google Gemini is integrated into Colab (see the stylized star bottom center); it will do a good job writing functional Python, while you probably need to take charge of the art concept.
If you’re a proficient programmer and you’d rather code something mathematical or algorithmic instead of using your GUI coordinates, you may do that instead.
Here are some drafts from my review of the website:
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
What they did:
The researchers developed an automated method to manufacture “synthetic cells” (microscopic fat-based bubbles known as Giant Unilamellar Vesicles, or GUVs) using an Opentrons OT-2 liquid-handling robot. Previously, these synthetic cells were made by hand in a slow, tedious process that often produced inconsistent results.
Why it matters:
By using a robot, the team was able to produce hundreds of identical synthetic cells in a single experiment with high precision. This allows researchers to study complex biological processes or test new medicines in a controlled environment that mimics the membrane structure of living cells, but without the complications of using complex living organisms.
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
While your description/project idea doesn’t need to be set in stone, we would like to see core details of what you would automate. This is due at the start of lecture and does not need to be tested on the Opentrons yet.
Final Project Description: Autonomous Fabrication of Synthetic Immune-Sensing Protocells
Project Vision:
My project aims to automate the production of synthetic cells (Protocells) that function as “sentinels” for the immune system. Instead of using living cells, which are difficult to standardize and maintain, I will build lipid-based vesicles from scratch. These synthetic cells will be “programmed” to detect specific antigens in a blood-like environment and produce a fluorescent signal in response.
Implementation Plan:
DNA Circuit Design (Ginkgo Nebula):
I will use the Ginkgo Nebula platform to design a cell-free genetic circuit. This circuit will consist of a specialized promoter that triggers the expression of Green Fluorescent Protein (GFP) only when a specific immune marker (like a cytokine) is present.
Automated Assembly (Opentrons OT-2):
I will utilize the Opentrons robot to perform a Phase Transfer Assembly. The robot will precisely pipette aqueous “cytoplasm” (containing the DNA and cell-free machinery) into a lipid-oil phase. The automation is critical to ensure that thousands of vesicles are produced with a uniform size and encapsulation efficiency.
Custom Hardware (3D Printing):
I plan to design and 3D print a custom aluminum block adapter or a “Vesicle Collection Rack.” This holder will be optimized for the Opentrons deck to keep the lipid-oil mixtures at a stable temperature, preventing the lipids from aggregating during the delicate phase-transfer process.
Example Automation Pseudocode:
This script represents the logic for creating different “batches” of synthetic cells with varying sensitivity levels.
# 2. Preparation of the "Synthetic Cytoplasm"
# Mixing Ginkgo-designed DNA with Cell-Free TX-TL system
pipette.pick_up_tip()
pipette.aspirate(10, dna_source)
pipette.aspirate(10, cell_free_mix)
pipette.dispense(20, mixing_vial)
pipette.mix(5, 15)
# 3. Vesicle Formation (Phase Transfer)
# Carefully layering the aqueous mix over the lipid-oil phase
for well in target_wells:
pipette.aspirate(2, mixing_vial)
# Slow dispense at the interface to form GUVs
pipette.dispense(2, lipid_plate[well].top(-2), rate=0.1)
pipette.drop_tip()
How many molecules of amino acids do you take with a piece of 500 grams of meat? (On average an amino acid is ~100 Daltons)
The calculation would be to use an equation like moles = mass / molar mass, then multiply by Avogadro’s number. Roughly ~3 × 10²⁴ molecules.
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
We absorb the nutrients, don’t imitate the animal, and slowly turn them into our own molecules.
Why are there only 20 natural amino acids?
This is due to evolution and chemical stability; these 20 cover the needed protein functions efficiently.
Can you make other non-natural amino acids? Design some new amino acids.
This is a fascinating idea, and chemists can create synthetic amino acids with new side chains for special functions.
Where did amino acids come from before enzymes that make them, and before life started?
They come from prebiotic chemistry, like reactions in early Earth’s atmosphere, meteorites, or hydrothermal vents.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
By the rules of stereochemistry, a D-amino acid helix forms a left-handed α-helix.
Can you discover additional helices in proteins?
Yes, protein structures keep revealing new helical motifs beyond the standard α-helix and 3₁₀-helix.
Why are most molecular helices right-handed?
Right-handed helices are more stable with L-amino acids due to steric and energetic favorability.
Why do β-sheets tend to aggregate?
Because of hydrogen bonding between sheets and flat surfaces that stick together.
What is the driving force for β-sheet aggregation?
Hydrogen bonds and hydrophobic interactions drive the sheets to stack.
Why do many amyloid diseases form β-sheets?
Misfolded proteins form stable β-sheet aggregates that resist degradation.
Can you use amyloid β-sheets as materials?
Yes, they can be used in nanomaterials, fibers, and hydrogels due to their stability.
Design a β-sheet motif that forms a well-ordered structure.
Use alternating hydrophobic and polar residues to promote sheet stacking with hydrogen bonds and prevent disorder.
Part B: Protein Analysis and Visualization
Aquí tienes la tarea completa y estructurada, lista para que la copies y pegues en tu documento. He seleccionado la estructura PDB 1ZHI, que es la versión humana del grupo sanguíneo A (Glicosiltransferasa A), por su altísima calidad de resolución.
Protein Analysis: ABO Glycosyltransferase
Protein Selection
Protein Name: ABO glycosyltransferase (Human).
Why I selected it: I chose this protein because of its critical role in human immunohematology. It is the enzyme responsible for determining an individual’s blood type (A, B, or AB) by modifying the H antigen on red blood cells. Understanding its structure is essential for my interests in synthetic cells and blood-related biotechnologies.
Amino Acid Sequence & Homology
Sequence Length: The canonical sequence (Isoform A) is 354 amino acids long.
Most Frequent Amino Acid: Leucine (L) is the most frequent residue, appearing approximately 35-40 times (approx. 10-11% of the total sequence).
Homologs: Using Uniprot’s BLAST tool, I identified over 500 homologs. Significant sequence identity is found in primates (Chimpanzees, Macaques) and other mammals like pigs and cows, reflecting the evolutionary conservation of the ABO system.
Protein Family: It belongs to the Glycosyltransferase 6 family (GT6).
RCSB Structure Page (PDB ID: 1ZHI)
The structure was published in 2005.
Quality: It is an excellent quality structure with a resolution of 1.50 Å. This is significantly better than the 2.70 Å threshold mentioned in the homework.
Other Molecules: Yes, the solved structure contains UDP (Uridine-5’-Diphosphate), GalNAc (N-acetylgalactosamine), and Manganese ions (Mn2+), which act as essential cofactors for the enzymatic reaction.
Structure Classification: It belongs to the GT-B fold structural superfamily.
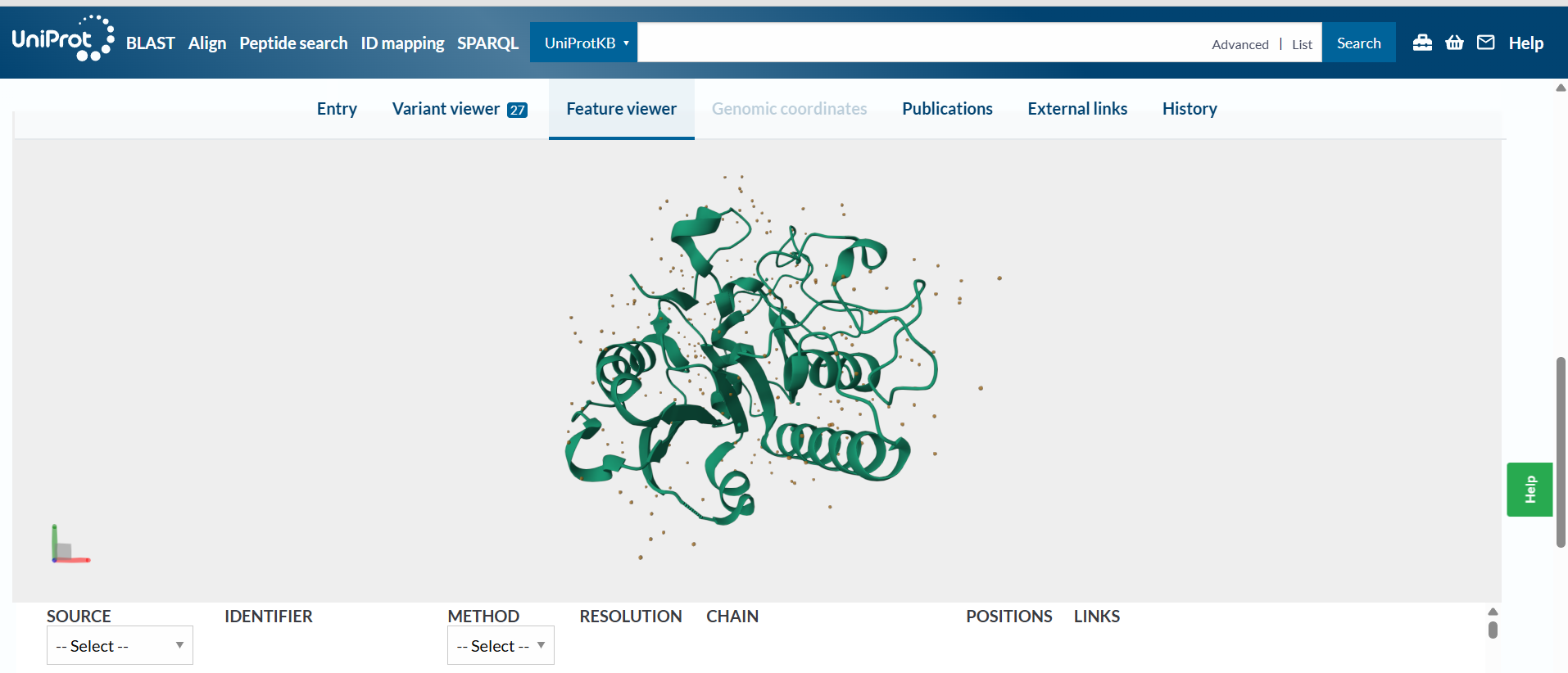
Here we can see it:
3D Visualization (PyMol Analysis)
Visualization Styles:
Cartoon: Shows the overall folding and path of the polypeptide chain.
Ribbon: Highlights the main backbone flow.
Ball and Stick: Useful for seeing the specific orientation of side chains in the active site.
Secondary Structure: After coloring by secondary structure (color red, ss h for helices; color yellow, ss s for sheets), the protein displays a classic Rossmann-like fold. It has a balanced distribution but features a prominent core of beta-sheets surrounded by several alpha-helices.
Residue Type (Hydrophobicity): When colored by residue type, there is a clear hydrophobic core (non-polar residues tucked inside) and a hydrophilic surface (polar residues facing the solvent). This distribution is typical for a globular protein found in the cytoplasm or Golgi apparatus.
Surface & Binding Pockets: Visualizing the surface (show surface) reveals a deep binding pocket (the active site). This “hole” is where the Manganese ion and the UDP-sugar donor bind to perform the catalytic transfer to the H-antigen.
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
Deep Mutational Scans
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
A clear pattern is that mutations at conserved residues strongly decrease the model likelihood. For example, when a hydrophobic residue located in the protein core is mutated to a charged amino acid (e.g., Leu → Asp), the likelihood drops significantly, suggesting the mutation would destabilize the structure.
(Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
Language model predictions generally correlate with experimental results: mutations predicted with low likelihood often correspond to experimentally observed decreases in stability or function, although some discrepancies appear for mutations affecting specific functional interactions.
Latent Space Analysis
Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins?
Yes. The neighborhoods tend to cluster proteins belonging to similar families or with similar functions, suggesting that the embeddings capture meaningful evolutionary and structural information.
Place your protein in the resulting map and explain its position and similarity to its neighbors.
The ABO protein appears close to other glycosyltransferases in the embedding space. This is consistent with its biological role in transferring sugar residues during blood group antigen synthesis, indicating that the model groups it with proteins performing similar enzymatic functions.
C2. Protein Folding
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
The predicted structure from ESMFold largely matches the original experimental structure. The core structural elements align well, while minor differences appear mainly in flexible loop regions.
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
Single point mutations usually do not significantly alter the global fold. However, larger sequence changes or mutations in core structural regions can destabilize the fold and lead to noticeable structural deviations.
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Many positions show high probability for amino acids similar to the original sequence, especially in structurally important regions. Surface positions tend to show higher variability, which is expected because they are less constrained structurally.
Input this sequence into ESMFold and compare the predicted structure to your original.
When the generated sequence is folded with ESMFold, the predicted structure remains highly similar to the original backbone, indicating that the designed sequence is compatible with the same structural fold.
Week 5 HW: Protein Design Part 2
Part A: SOD1 Binder Peptide Design (From Pranam)
Part C: Final Project: L-Protein Mutants
Week 2 HW: DNA Read, Write, & Edit
HW 2: DNA Read, Write, & Edit
Part 1: Benchling & In-silico Gel Art
Own experience on:
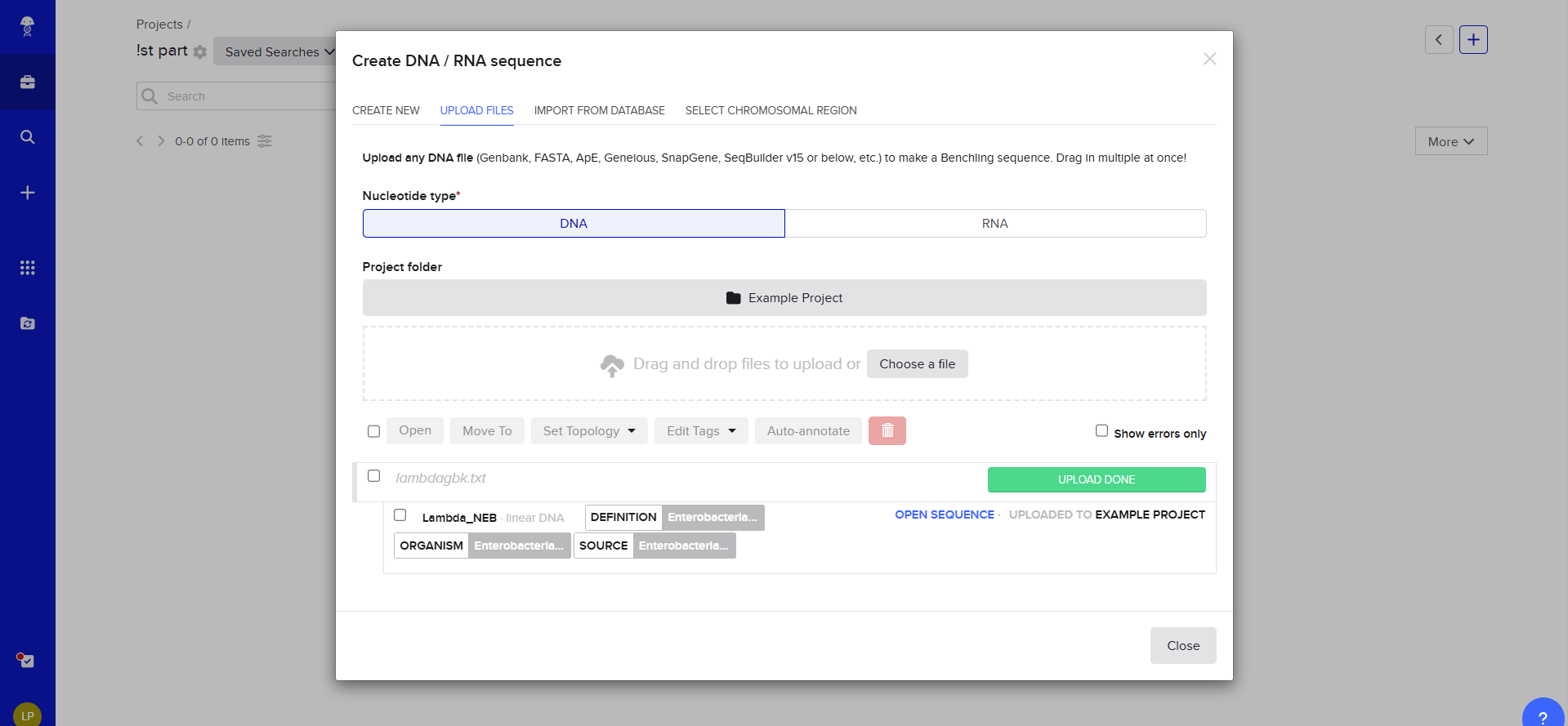
Make a free account at benchling.com
Import the Lambda DNA.
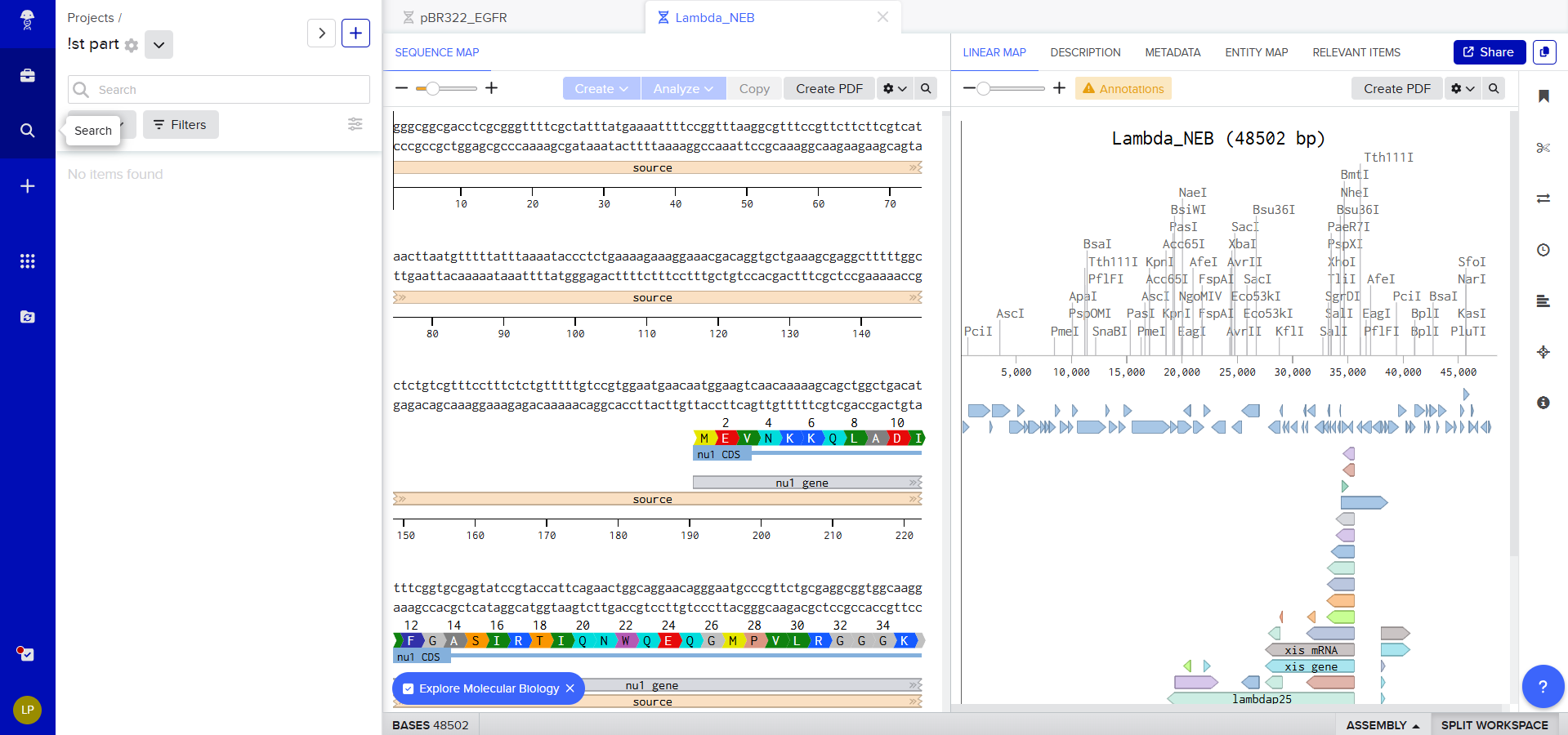
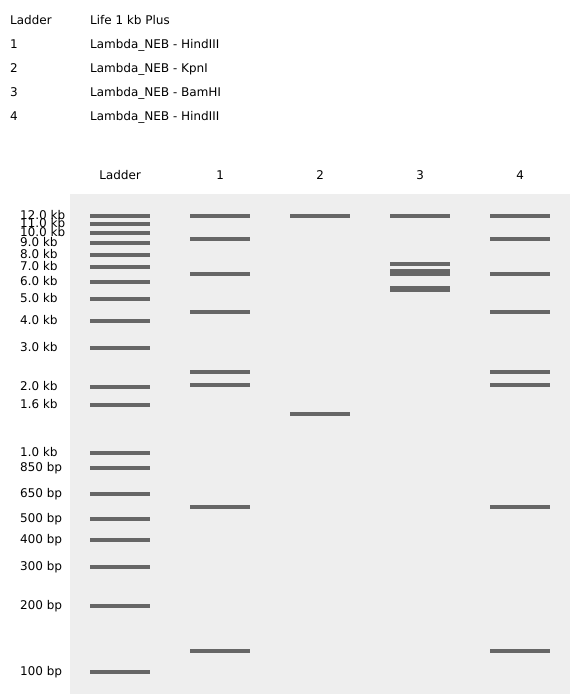
The first view of Benchling workspace
Then i transfered de Lambda DNA, here we have two option for DNA document type, either we can use Genbank or, but Benchlink does have more options.
Upload to Benchling
So we can now view it on Benchling and start exploring the different options available
Journey on:
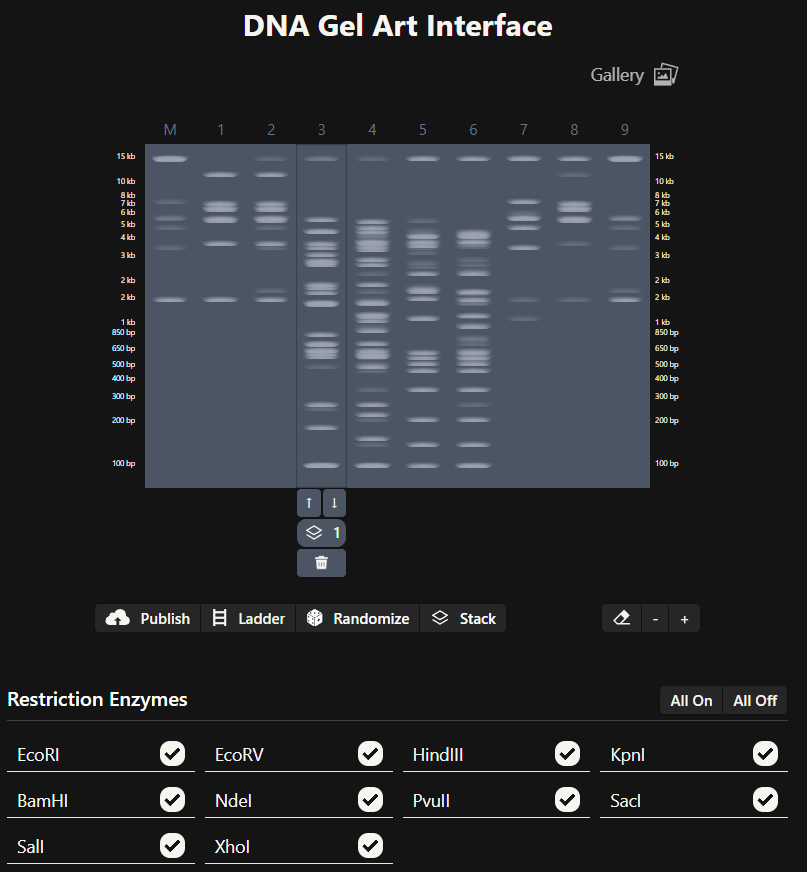
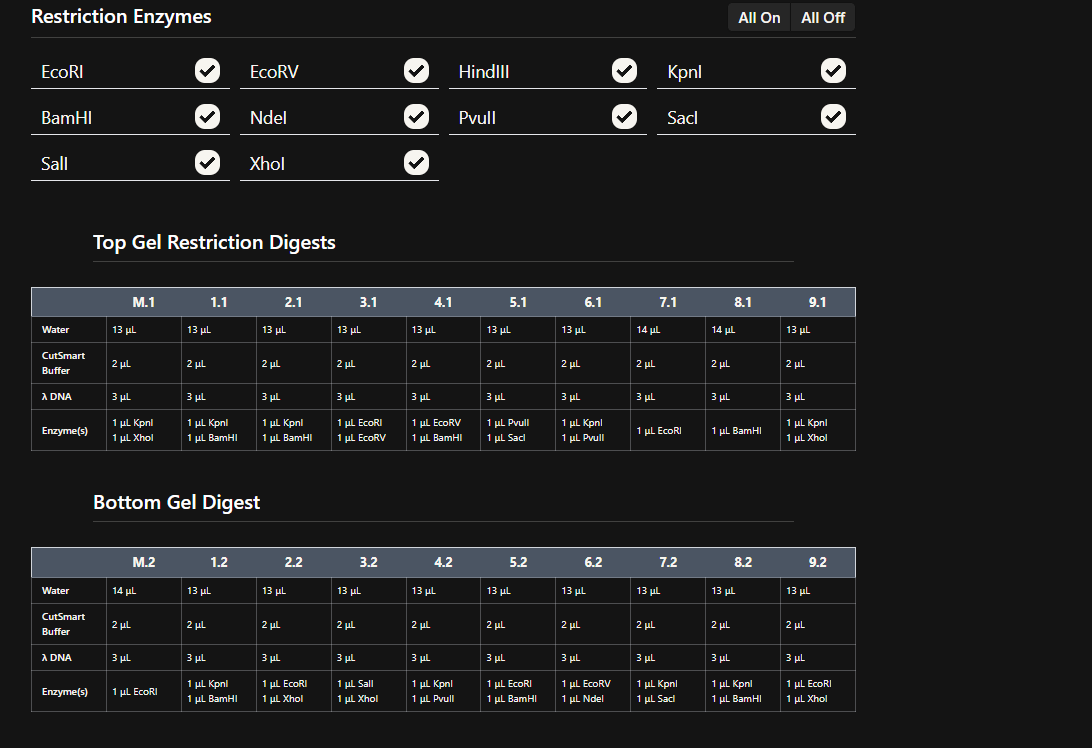
Simulate Restriction Enzyme Digestion with the following Enzymes:
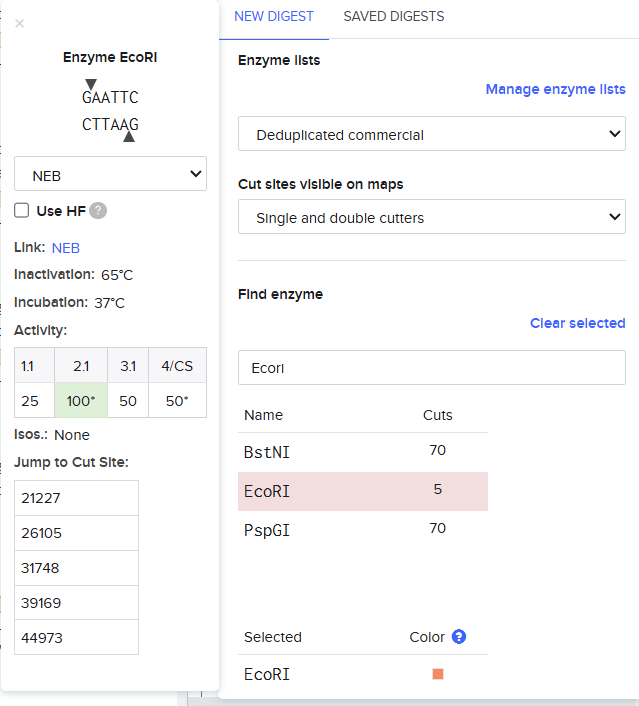
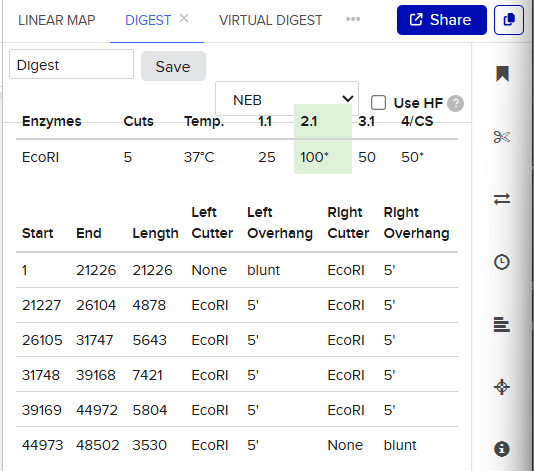
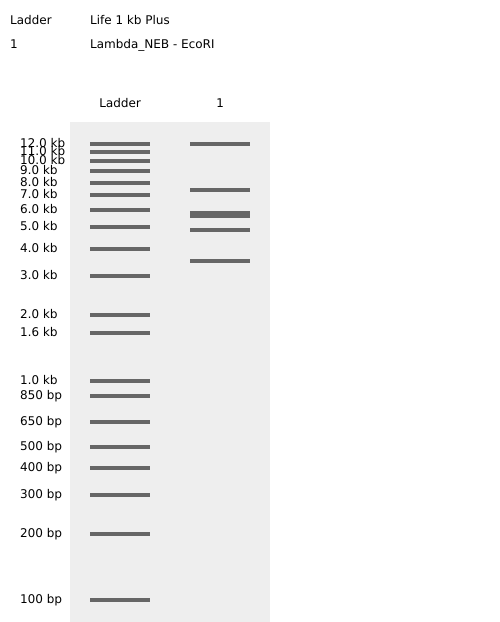
EcoRI
Finding the enzyme is the first step, the description displayed on the left shows improtant information regarding the enzyme and on the top the place where a cut is going to be made is also shown.
So when we procede to run the digestion we get Digest and the virtual digest, both simulated ways of the final product view.
Digest generals
Virtual Digest
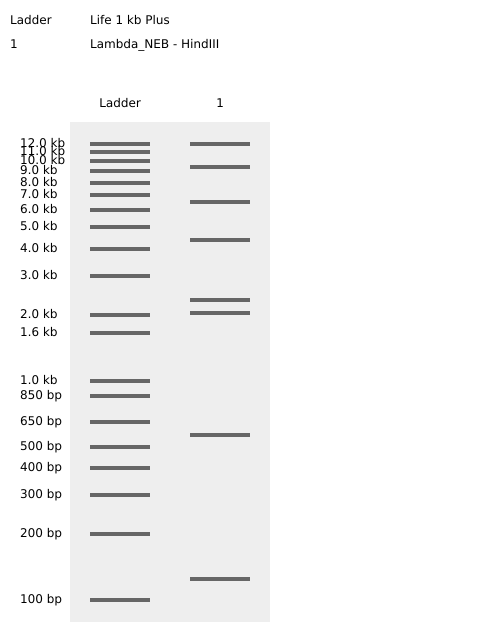
HindIII
BamHI
KpnI
EcoRV
SacI
SalI
Journey on:
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
You might find Ronan’s website a helpful tool for quickly iterating on designs!
First attempt was a dog:
Some about the dog settings:
Since i am not doing a lab i have plenty of freedom to choose what enzymes to use on the DNA Gel Art Interface
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
Lab only
Part 3: DNA Design Challenge
3.1. Choose your protein.
The protein I chose is the Histo-blood group ABO system transferase from Homo sapiens. This protein is responsible for determining human blood types (A, B, AB, or O). It is a glycosyltransferase enzyme that modifies carbohydrate antigens on the surface of red blood cells by adding specific sugar residues. The differences in this enzyme’s activity result in the different ABO blood groups.
The sequence was obtained from UniProt: P16442
The protein:
sp|P16442|BGAT_HUMAN Histo-blood group ABO system transferase OS=Homo sapiens OX=9606
MAEVLRTLAGKPKCHALRPMILFLIMLVLVLFGYGVLSPRSLMPGSLERGFCMAVREPDH
LQRVSLPRMVYPQPKVLTPCRKDVLVVTPWLAPIVWEGTFNIDILNEQFRLQNTTIGLTV
FAIKKYVAFLKLFLETAEKHFMVGHRVHYYVFTDQPAAVPRVTLGTGRQLSVLEVRAYKR
WQDVSMRRMEMISDFCERRFLSEVDYLVCVDVDMEFRDHVGVEILTPLFGTLHPGFYGSS
REAFTYERRPQSQAYIPKDEGDFYYLGGFFGGSVQEVQRLTRACHQAMMVDQANGIEAVW
HDESHLNKYLLRHKPTKVLSPEYLWDQQLLGWPAVLRKLRFTAVPKNHQAVRNP
Length: 354 amino acids
Organism: Homo sapiens
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
I chose the ABO blood group transferase because I find it biologically interesting and medically relevant. It determines human blood type, which is important for transfusions and healthcare. I also selected it because there is a lot of available information about it, making it a good protein to start practicing sequence analysis and better understand how genes relate to protein function.
The optimized gene can be chemically synthesized and inserted into a plasmid designed with regulatory elements such as a strong promoter, a ribosome binding site, and a transcription terminator. These components function like biological switches, controlling when and how much protein is produced. The engineered plasmid is then introduced into E. coli, where the synthetic gene is transcribed into mRNA and translated by ribosomes into the ABO transferase protein. Because the sequence was codon-optimized, the bacteria can produce the protein more efficiently.
In synthetic biology, DNA is treated as modular and programmable, allowing us to design regulatory elements and optimize sequences to precisely control protein production and convert a natural human gene into an engineered system for research or biotechnology applications.
Part 4: Prepare a Twist DNA Synthesis Order
Due to my location (Mexico) i was not able to perform this point.
Part 5: DNA Read/Write/Edit
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I would sequence the human ABO gene from genomic DNA samples. My main interest would be investigating how specific genetic variants in this gene are associated with disease risk, particularly cardiovascular disease, clotting disorders, and susceptibility to certain infections. By sequencing the ABO gene across different individuals, researchers could identify mutations or patterns that correlate with clinical outcomes and better understand how genetic variation contributes to disease mechanisms.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Also answer the following questions:
Is your method first-, second- or third-generation or other? How so?
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
What is the output of your chosen sequencing technology?
I would use Illumina sequencing technology because it is highly accurate and well suited for detecting small mutations such as single nucleotide changes.
This method is considered second-generation sequencing because it sequences millions of short DNA fragments in parallel rather than reading single long molecules in real time.
The input would be extracted human genomic DNA. The DNA would first be fragmented into short pieces, and synthetic adapter sequences would be attached to their ends. These fragments would then be amplified on a flow cell to create clusters.
The essential step of the sequencing technology is sequencing-by-synthesis. During this process, fluorescently labeled nucleotides are incorporated one base at a time into a growing DNA strand. Each incorporated base emits a specific signal that is detected by the instrument. The machine converts these signals into a digital sequence through base calling.
The output consists of short DNA sequence reads with associated quality scores, which can then be analyzed computationally to identify variants in the ABO gene and examine their relationship to disease.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
See some famous examples of DNA design
I would synthesize specific variants of the ABO gene that have been associated with altered disease risk. For example, I would design versions containing mutations suspected to influence clotting or inflammatory pathways. By synthesizing these variants and expressing them in a controlled system, researchers could directly compare how different DNA sequences affect protein function and potentially contribute to disease-related phenotypes.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Also answer the following questions:
What are the essential steps of your chosen sequencing methods?
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
I would use commercial gene synthesis technology, where short DNA fragments are chemically synthesized and assembled into a full-length gene.
The essential steps involve synthesizing short oligonucleotides, assembling them into the complete coding sequence, inserting the gene into a plasmid, and verifying the sequence through quality control sequencing.
The input is a digitally designed DNA sequence file. The output is a physical DNA construct, typically delivered in a plasmid vector.
Limitations include cost for longer or complex sequences, potential synthesis errors that require verification, and turnaround time for manufacturing and validation.
5.3 DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
I would edit the ABO gene directly in cultured human cells to introduce specific disease-associated mutations. Editing the gene in its natural genomic context would allow researchers to study how those precise genetic changes influence cellular processes related to clotting, vascular biology, or immune responses. This approach moves beyond correlation and allows functional testing of suspected disease-related variants. But of course this part has its limitations.
(ii) What technology or technologies would you use to perform these DNA edits and why?
Also answer the following questions:
How does your technology of choice edit DNA? What are the essential steps?
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
I would probably use CRISPR-Cas9 genome editing technology because it allows precise targeting of specific DNA sequences within the genome.
This system works by designing a guide RNA that matches the target sequence in the ABO gene. The guide RNA directs the Cas9 enzyme to that location, where it creates a double-strand break in the DNA. The cell then repairs the break either by introducing small insertions or deletions or, if a repair template is provided, by incorporating a specific desired mutation.
The preparation involves designing the guide RNA sequence, preparing the Cas9 protein or plasmid, and optionally designing a repair template containing the intended mutation. These components are delivered into cultured cells as the input material.
Limitations include the possibility of off-target edits at similar DNA sequences, variable editing efficiency, and the fact that precise edits using repair templates may not occur in all cells.