Week 2 HW: Read, Write, Edit DNA
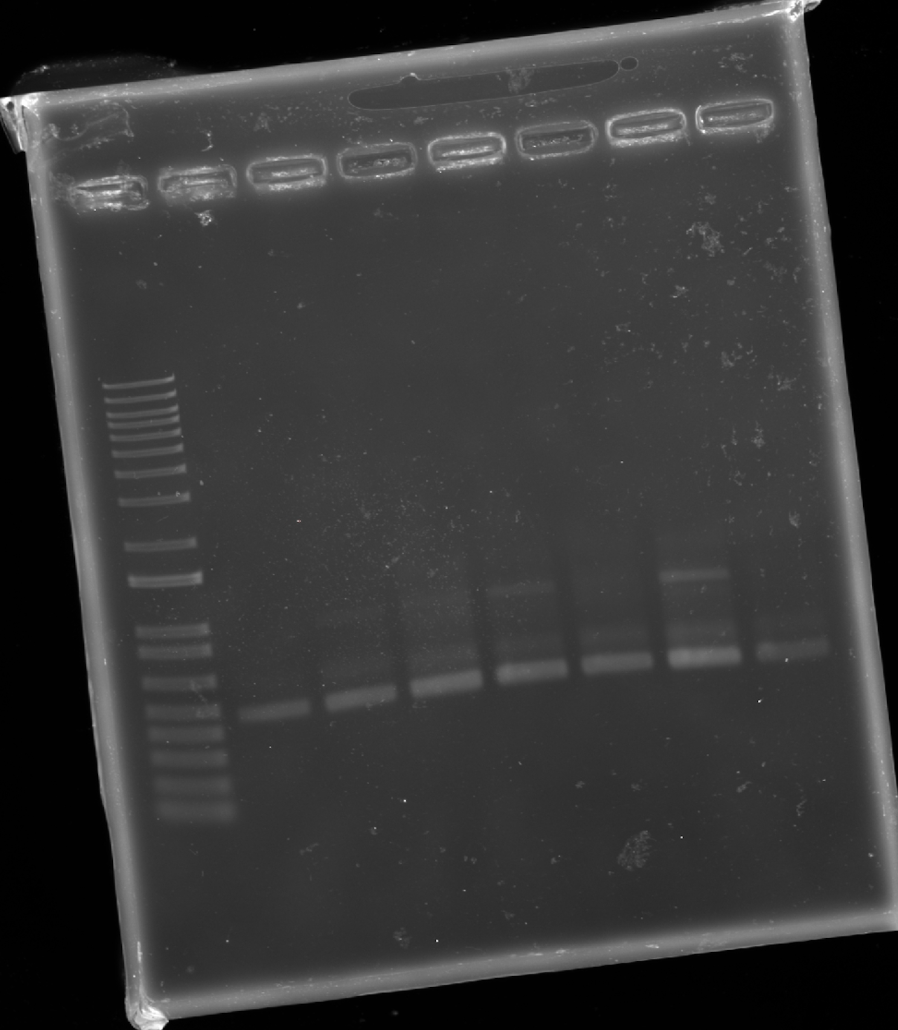
DNA Design Challenge
3.1. Choose your protein.
I’ve chosen PprA, a protein that contributes to radiation resistence in the extremophile bacterium Deinococcus radiodurans, which can survive exposure to space conditions, via DNA repair mechanisms. PprA and other proteins involved in D. radiodurns’s space response could have space biotechnology applications–e.g., engineering space-tolerant food sources and terraforming Martian soil for agriculture. (Note that other research groups have successfully expressed PprA in E. coli before. I’m interested in eventually engineering PprA into a different chassis with direct relevance to space travel or exploring alternative proteins that enhance space (radiation, microgravity, vacuum, etc.) tolerance.)
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
PprA DNA sequence, from D. radiodurns refseq NC_001264.1, below:
(http://ncbi.nlm.nih.gov/nuccore/NC_001264.1?from=381165&to=382067) (Note that the above nt sequence is the forward strand, but the protein is encoded by the reverse. See the AA seq for the product that it encodes. Note also that D. radiodurans sometimes uses non-standard start codons.)
3.3. Codon optimization.
Codon optimization ensures that the codons in an engineered construct’s nucleotide sequence correspond with tRNAs that the host species commonly uses and that maximize the efficiency of translation (without changing the resulting amino acid sequence). To account for the NCBI nt sequence’s reverse strand location and D. radiodurans’s non-standard start codons, I optimized directly from the amino acid sequence using the IDT codon optimization tool. (Note that I needed to add a stop codon (TAA) manually because, given the lack of a corresponding AA, the protein seq does NOT encode one.)
I optimized the sequence for use in E. coli. E. coli could be engineered to exhibit extremophile characteristics in order to enhance its viability as a chassis for space applications. Note also that I avoided BbsI, BsaI, BsmBI cut sites. Below is the optimized sequence:
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? The most straightforward way to transcribe and translate this DNA sequence into protein would be to engineer it into a plasmid, or order it as part of a plasmid, and then transform it into a model bacterium such as E. coli, which could then produce the protein.
Prepare a Twist DNA Synthesis Order
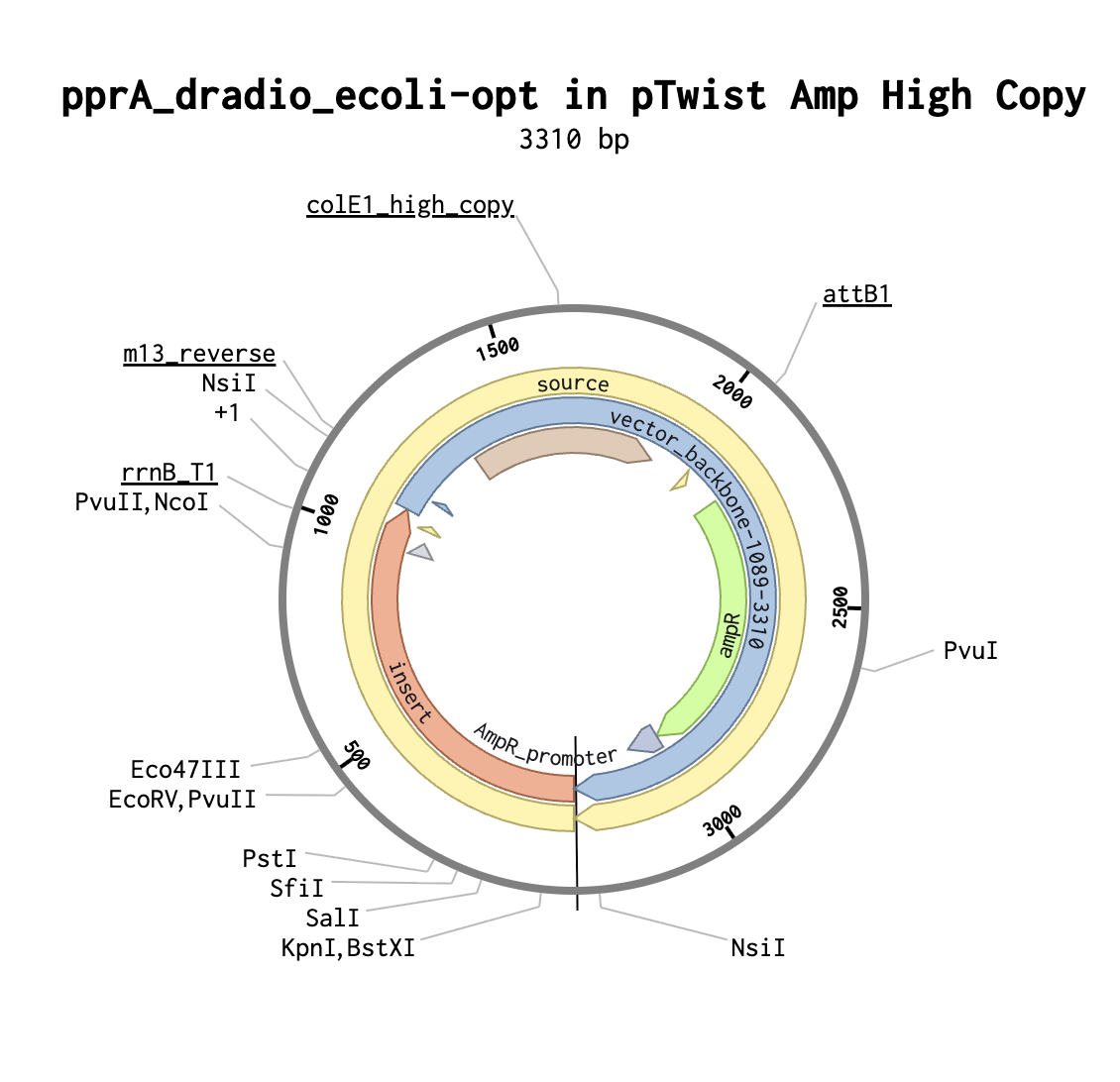
I’ve included the BBa_J23106 promoter for high constitutive expression in E. coli, the BBa_B0034 RBS, and BBa_B0015 Terminator. If I were to order this construct, I might look into E. coli promoters that are UV or general radiation or DNA damage-inducible, as this might reduce the burden of constitutive expression and ensure that the cell only diverts resources to producing the protein under conditions where it is useful.
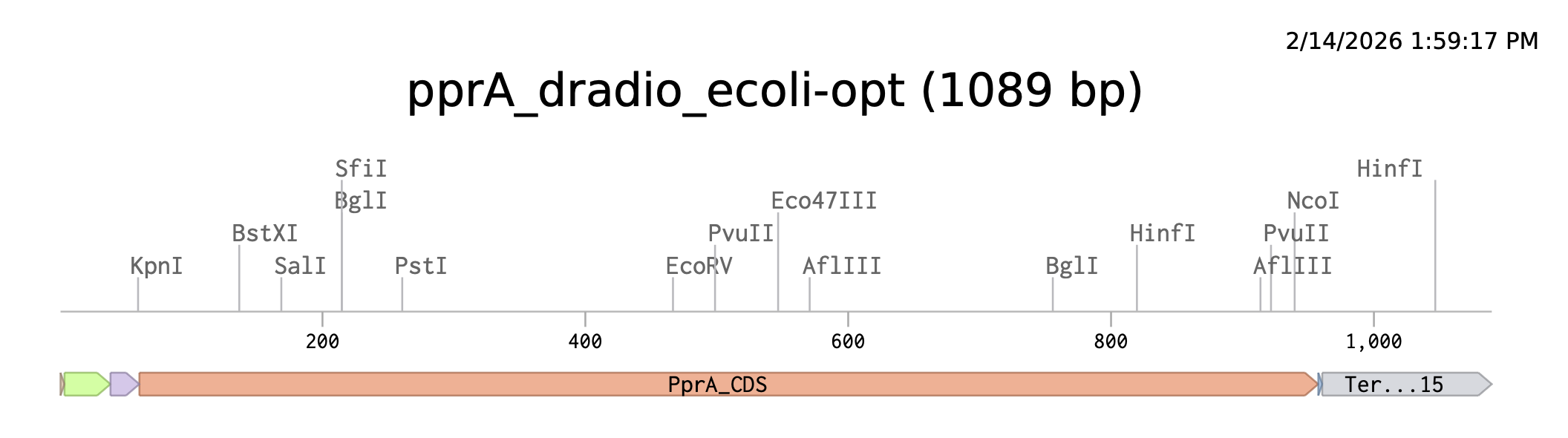
Selected pTwist Amp high copy vector on Twist
DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why?
I would like to perform metagenomic sequencing (or metatranscriptomics, though this would be RNAseq) on one of the harmful algal blooms (HABs) that frequently affect local lakes in Virginia. I studied HABs as part of the William & Mary iGEM project last year and am currently working with transcriptomic data from HAB microcosms. Local stakeholders told me that we still cannot fully predict when these blooms might happen and what factors contribute to their duration and severity. Understanding the Virginia blooms’ taxonomic composition (via metagenomics) and functional roles of the relevant microbes (via metatranscriptomics) would inform public safety measures and bloom mitigation strategies.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
1) Is your method first-, second- or third-generation or other? How so?
I would use PacBio HiFi sequencing, which is a third-generation/long read method. Longer reads make it easier to accurately identify the taxa present in the sample and to de novo assemble metagenomes, while shorter, fragmented reads provide less information and and may be more likely to yield false matches.
2) What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
My input would be DNA extracted from the lakewater (probably via an extraction kit designed for water samples) and prepared via the following steps:
- DNA fragmentation
- Adapter ligation - in this case, capping of DNA fragments with “ligated hairpin adapters”, which basically turn the sequence into a loop around which the RNA polymerase can move.
3) What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
In PacBio HiFi sequencing 1) the scientist performs library prep (see above), 2) DNA molecules stick to “zero-mode waveguides,” which are wells in the sequencing device, the “SMRT Cell”, and 3) DNA polymerase adds fluorescent nucleotides to each individual DNA molecule, emitting light that indicates the sequence.
4) What is the output of your chosen sequencing technology?
PacBio HiFi sequencing produces long-read raw sequence data as output.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
The cyanobacterium N. commmune has a number of qualities–such as relative tolerance to vacuum conditions, radiation, dry environments, and extreme heat, anti-inflammatory properties, and the ability to promote plant growth–that make it promising for space travel applications, including terraforming Martian soil and providing a sustainable food source. However, the species is extremely slow-growing, difficult to engineer, and may produce toxins harmful to human health–so on its own may be difficult to deploy/apply. I would like to either engineer Nostoc to be faster-growing and more manageble as a chassis–or would synthesize relevant Nostoc genes and pathways (e.g., extracellular polymeric substance production mechanisms, nutrient content, and survival under extreme conditions) and express them in model organisms, which we could then more-easily manipulate and use as chassis for space, nutrition, agriculture, wastewater-management, and more.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
I would use clonal gene sythesis methods (e.g., from Twist) to order the relevant genes and/or circuits in plasmid form so that I could easily express them in a chassis of choice. Producing the plasmids would involve synthesizing the relevant fragments (e.g., with the phosphoramidite method) and then assembling them together (e.g., via Gibson or Golden Gate).
5.3 DNA Edit
(i) What DNA would you want to edit and why?
I would like to modify genes and pathways that regulate bacterial growth, particularly in the cyanobacterium N. commune. The bacterium’s slow growth rate is a major barrier to engineering, deployment as a chassis, and basic science. Modifying growth rate may be difficult because of potential off-target effects on bacterial performance (e.g., the species may need to grow slowly in order to produce an essential compound) and would likely require me to edit regulatory components of genes involved in metabolism and nutrient uptake–and potentially could involve knocking out non-essential genes that might slow down bacterial growth.
(ii) What technology or technologies would you use to perform these DNA edits and why?
I might use a combination of CRISPR and TALENs to potentially 1) replace sequences relevant to metabolic regulation (e.g., that promote nutrient uptake and processing) with more-active counterparts via homology-directed repair with template sequences of interest and 2) knock out sequences that are not essential to bacterial survival/performance and that may impose a burden on bacterial growth.