Week 4 HW: Protein Design Part I

A. Conceptual Questions
Answer any NINE of the following questions from Shuguang Zhang:
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
There are about 20 grams of protein per 100 grams of chicken. If you eat 500 grams of chicken, you’ve eaten 100 grams of protein. Looking only at the amino acids contained within proteins, this is 6.022x1025 daltons, so 6.022x1023 amino acids.
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
We repurpose the proteins, amino acids, etc contained in other organisms for our own biological needs. Since we have human genetic material that instructs our bodies on how to produce human proteins in contexts that are relevant for human development and survival, we don’t become cows when we eat beef.
3. Why are there only 20 natural amino acids?
20 natural amino acids provides enough structural and chemical variation to produce a vast number of diverse proteins when combined in different arrangements.
4. Can you make other non-natural amino acids? Design some new amino acids.
Yes, this is possible and involves complex chemical synthesis pathways.
5. Where did amino acids come from before enzymes that make them, and before life started?
Amino acids can be formed from inorganic matter in reactions that are triggered by the irradiation of specific compounds. This process is generates amino acids in space, and scientists have replicated it in the lab.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
You would expect a left handed alpha-helix.
7. Can you discover additional helices in proteins?
Yes, AI and ML methods make it possible to uncover novel structural information (such as additional helices) about a given protein.
8. Why are most molecular helices right-handed?
Most amino acids are left-handed. The most chemically favorable combination of left-handed amino acids into a helix is a right-handed helix.
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
Beta-sheets have the ability to hydrogen-bond at the edges and have hydrophobic regions. Beta-sheets can hydrogen bond with each other and their hydrophobic regions clump to avoid water.
10. Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Misfolded proteins often naturally naturally aggregate in a beta-sheet-like structure because of the structure’s stability and energetic favorability. Amyloid beta-sheets are very “sticky” and difficult to get rid of and could be good building materials for this reason.
11. Design a β-sheet motif that forms a well-ordered structure.
A beta-sheet motif with alternating hydrophilic and hydrophobic amino acids will form a well-ordered structure because the hydrophobic sides will group strongly together. E.g., LTINLTIN
B. Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
1. Briefly describe the protein you selected and why you selected it.
I selected the NpDps4 protein from Nostoc punctiforme (Howe et al., 2019, https://pmc.ncbi.nlm.nih.gov/articles/PMC6675082/#abstract1). Nostoc is interesting because of its nutritional relevance, high tolerance to extreme conditions, and ability to uptake toxic environmental compounds. A number of proteins (particularly, those involved in pathways that synthesize Nostoc’s extrapolymeric substances, or EPS) contribute to these qualities, but few have a structure available on PDB. One of N. punctiforme’s Dps proteins, a group of DNA-binding proteins involved in iron uptake and the oxidative stress response across bacteria, is available on PDB and may contribute to Nostoc’s resilience in extreme conditions. Nostoc’s has a higher number of Dps proteins than other species, suggesting that has a complex system for managing oxidative stress and iron homeostasis. NpDps4 has an unusual cyanobacteria-specific structure and has some similarities to the equivalent protein in Deinococcus radiodurans, an extremophile bacterium that, like Nostoc, can survive space-like conditions (Howe et al., 2019).
2. Identify the amino acid sequence of your protein.
- How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
- How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
- Does your protein belong to any protein family?
PDB link: https://www.rcsb.org/structure/5HJH
NpDps4 is 178 amino acids long and its most frequent amino acid is Leucine (L). The protein belongs to the Dps family, a group of DNA-binding proteins involved in iron homeostasis and the oxidative stress response. A BLAST against UniProtKB yielded 27 results with 90% identity or higher and 250 results with 36.9% identity or higher.
3. Identify the structure page of your protein in RCSB
- When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
- Are there any other molecules in the solved structure apart from protein?
- Does your protein belong to any structure classification family?
The structure was solved in 2017 with a resolution of 1.88 Å. The solved structure includes iron 3+ ions to which the protein is bound in a complex. The page also includes a 2D diagram of 4-(2-HYDROXYETHYL)-1-PIPERAZINE ETHANESULFONIC ACID, which is a ligand for the protein. NpDps4 belongs to the Dps protein family, which share a similar structure.
4. Open the structure of your protein in any 3D molecule visualization software:
- PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
- Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
- Color the protein by secondary structure. Does it have more helices or sheets?
- Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
- Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Ball and stick visualization:
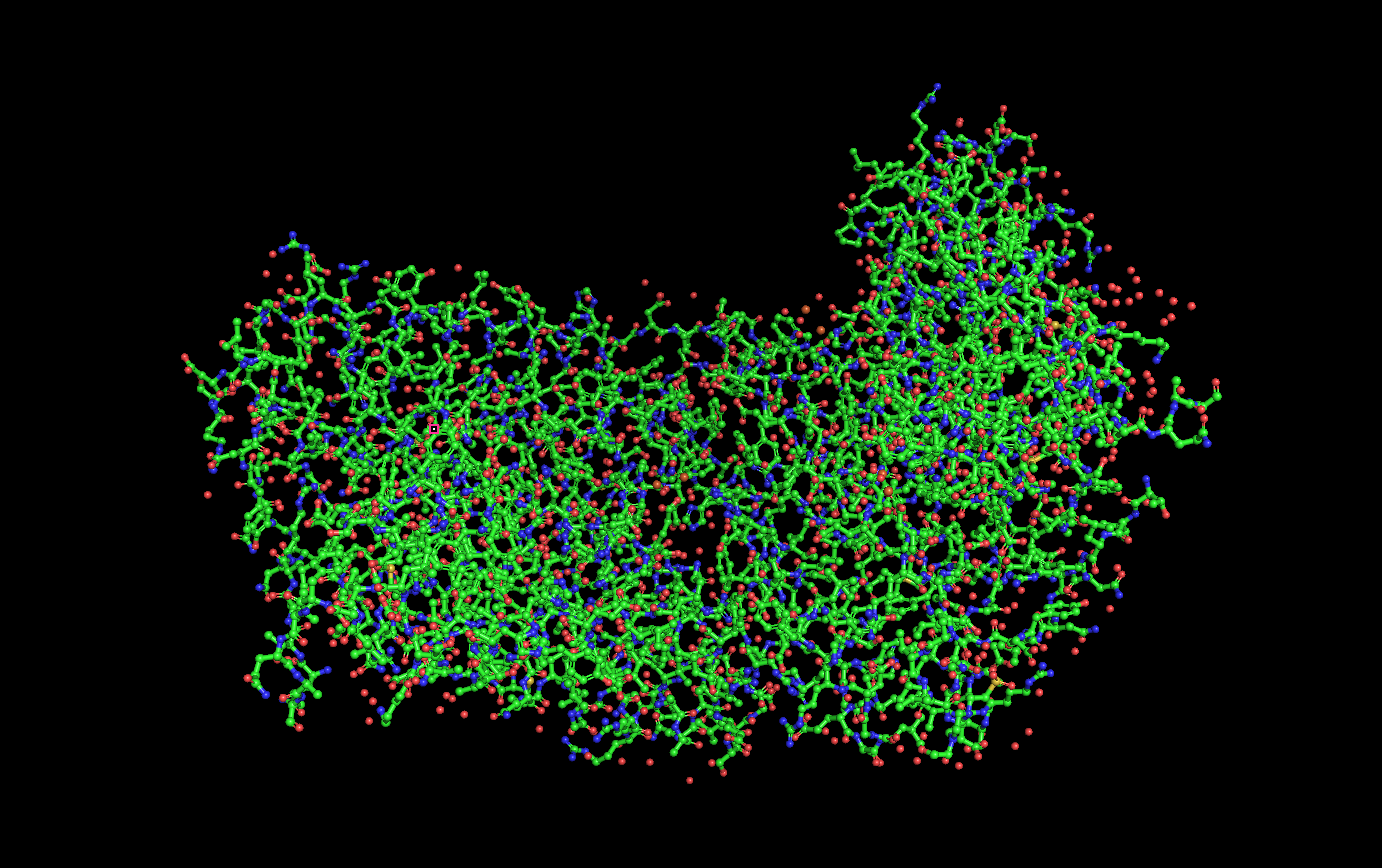
Secondary structure visualization:
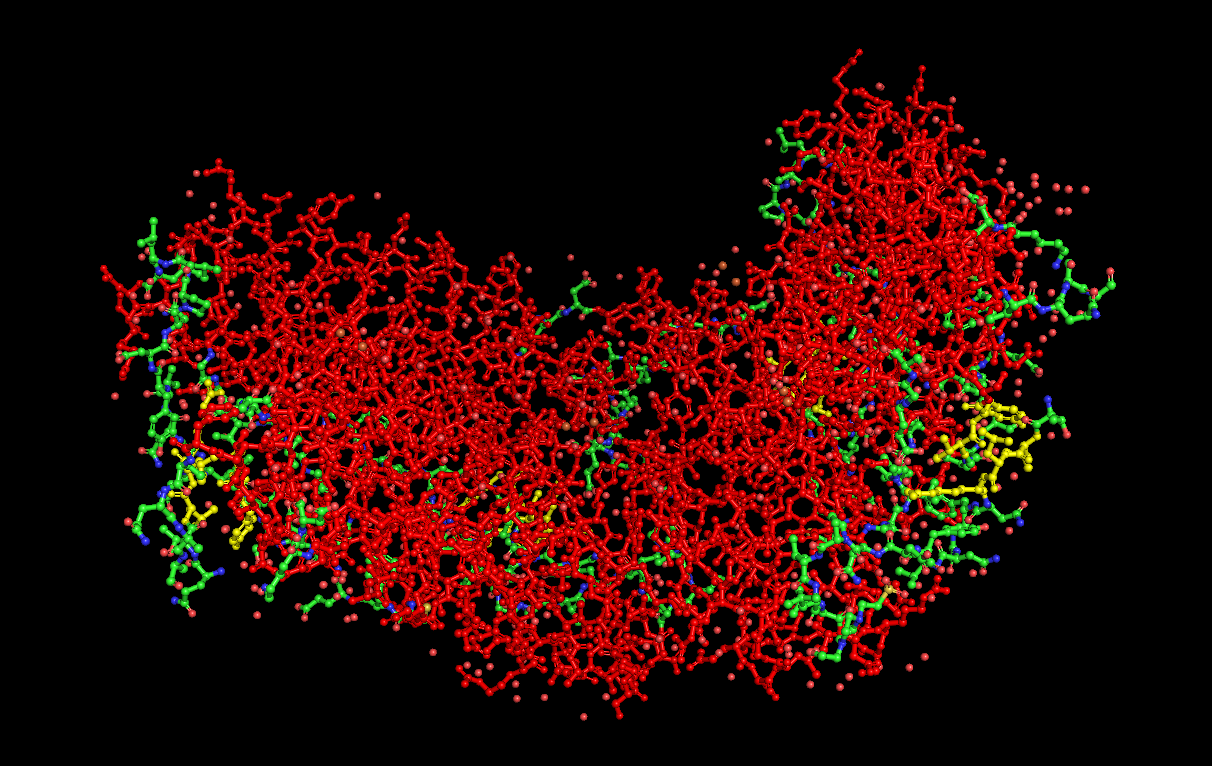
(Helices = Red, Sheets = Yellow, Loops/Coils = Green)
Residue hydrophilicity visualization:

(Hydrophobic = Purple, Hydrophilic = Pink)
Surface visualization
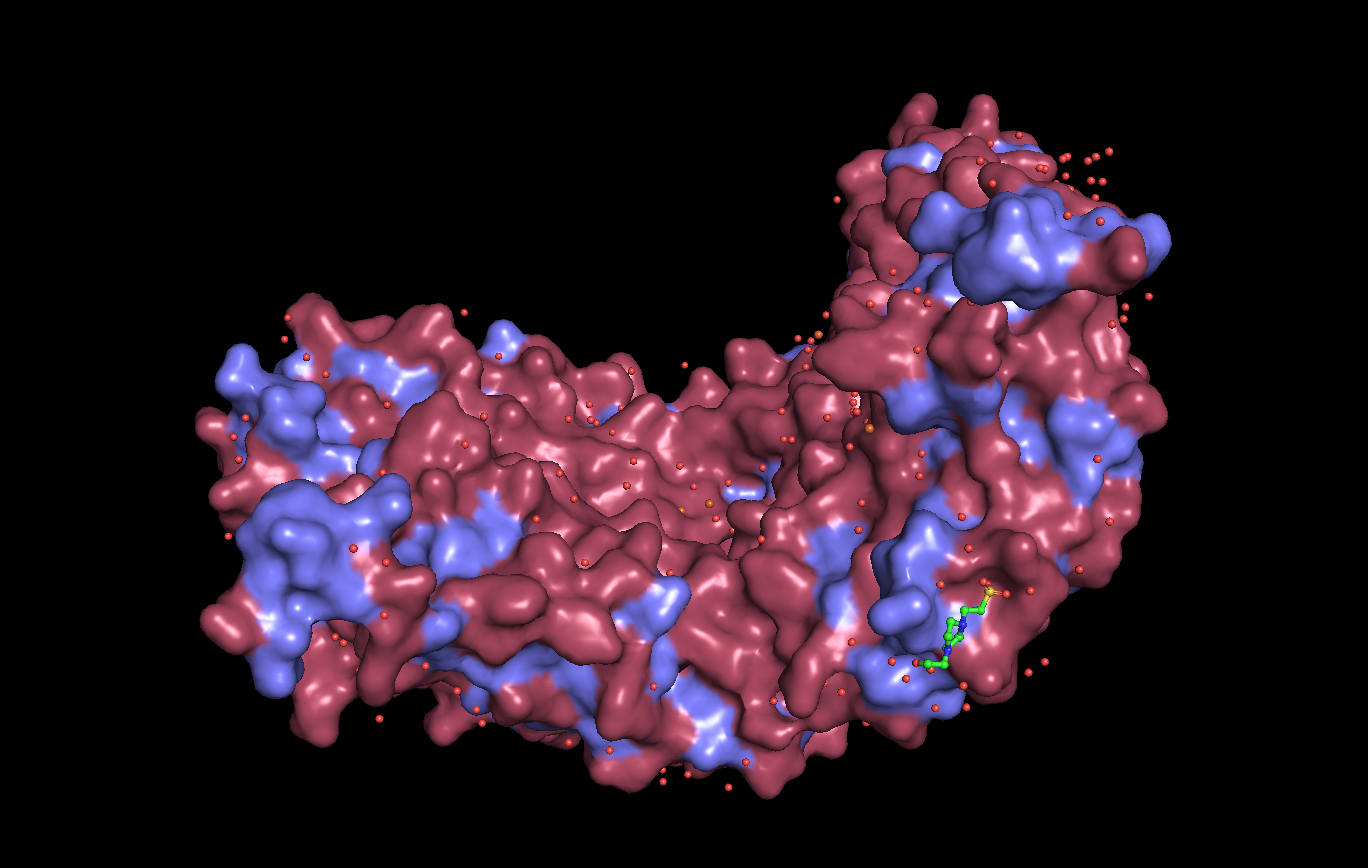
Analysis: Dps4 is mostly composed of helices. Most of its hydrophilic residues are on the outside of the protein, while the hydrophobic residues are on the interior. The protein does not appear to have any major “holes,” but has a concave center.
C. Using ML-Based Protein Design Tools
In this section, we will learn about the capabilities of modern protein AI models and test some of them in your chosen protein.
- Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
- Choose your favorite protein from the PDB.
- We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
C1) Protein Language modeling 1. Deep Mutational Scans
- Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
- Can you explain any particular pattern? (choose a residue and a mutation that stands out)
Mutational scan heatmap
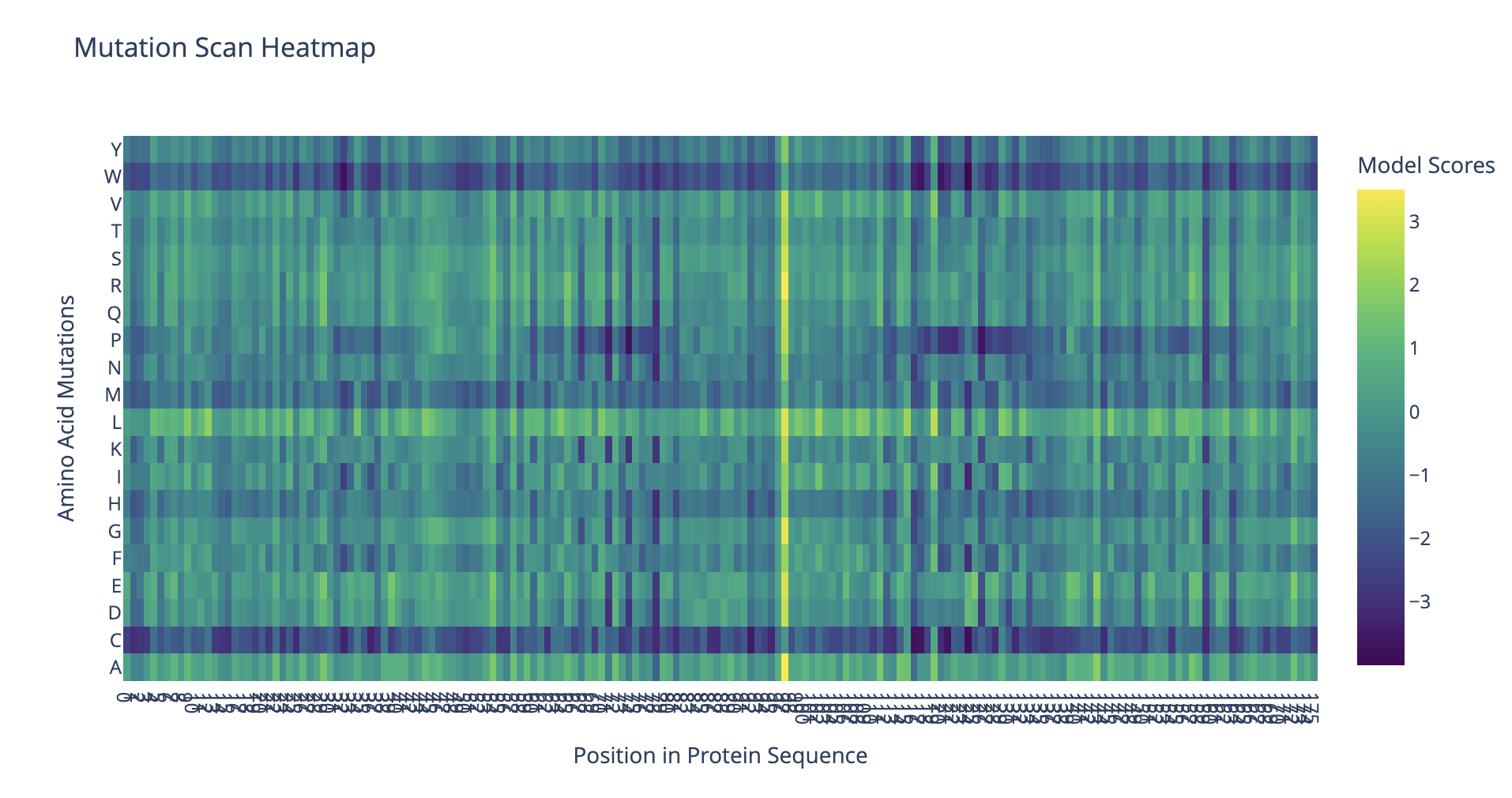
The protein seems to be highly tolerant to mutations at residue 97, which is leucine in the original sequence. This location probably is not critical for protein structure and function.
2. Latent Space Analysis
- Use the provided sequence dataset to embed proteins in reduced dimensionality.
- Analyze the different formed neighborhoods: do they approximate similar proteins?
- Place your protein in the resulting map and explain its position and similarity to its neighbors.
Latent space visualization
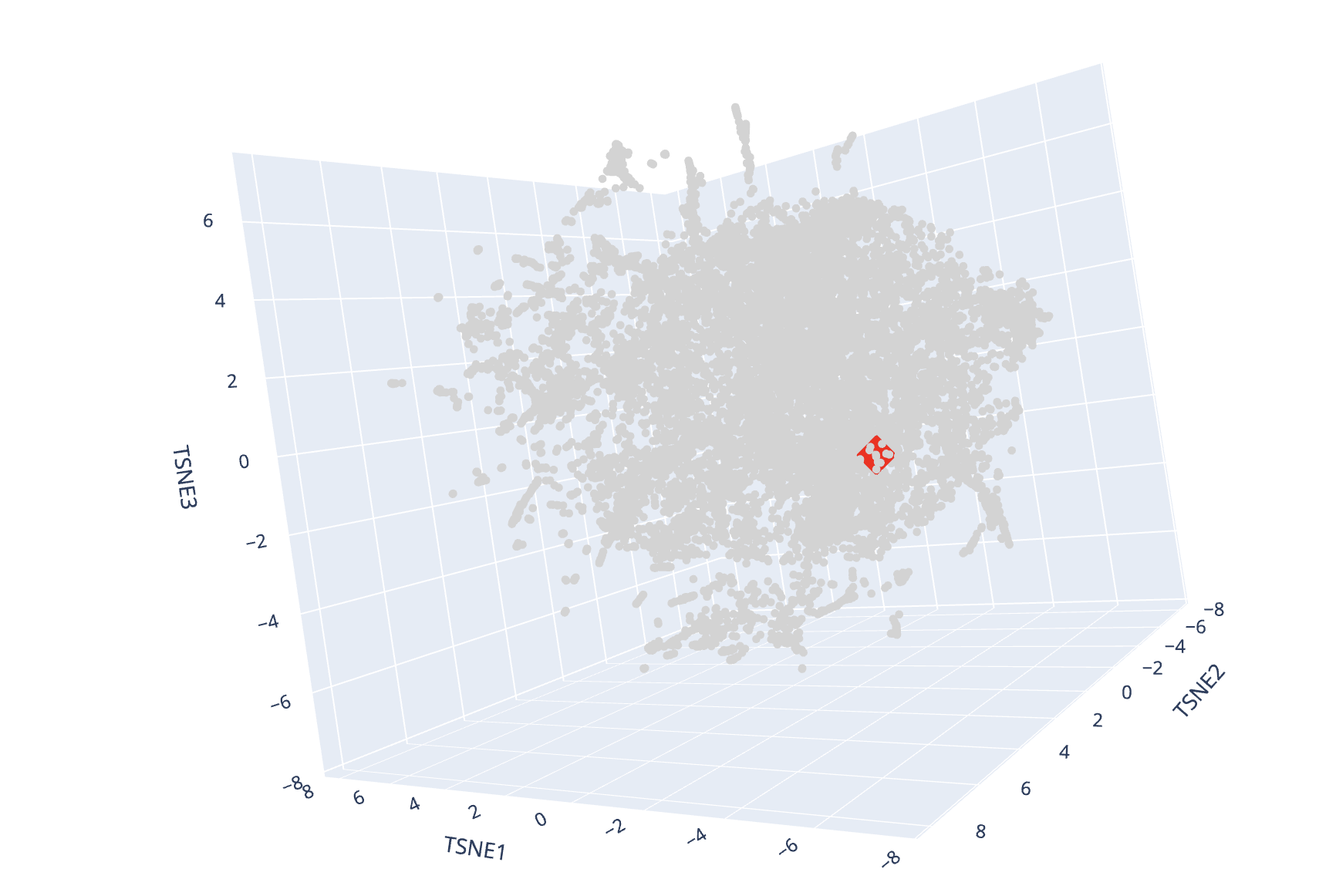
(Red diamond = Dps4, Gray circles = Other proteins)
I found some groups of proteins in the map that were similar in function. For example, there was a cluster of proteins involved in respiration and another that appeared related to oxidateive stress. Enzymes of similar types grouped together. For example, I found a cluster of transferases.
My protein was surrounded by proteins of a variety of different functions and species of origin. One of the closer proteins was a DNA binding protein, which makes sense given that NpDps4 is a DNA-binding protein.
C2) Protein Folding
1. Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
ESMFold Prediction
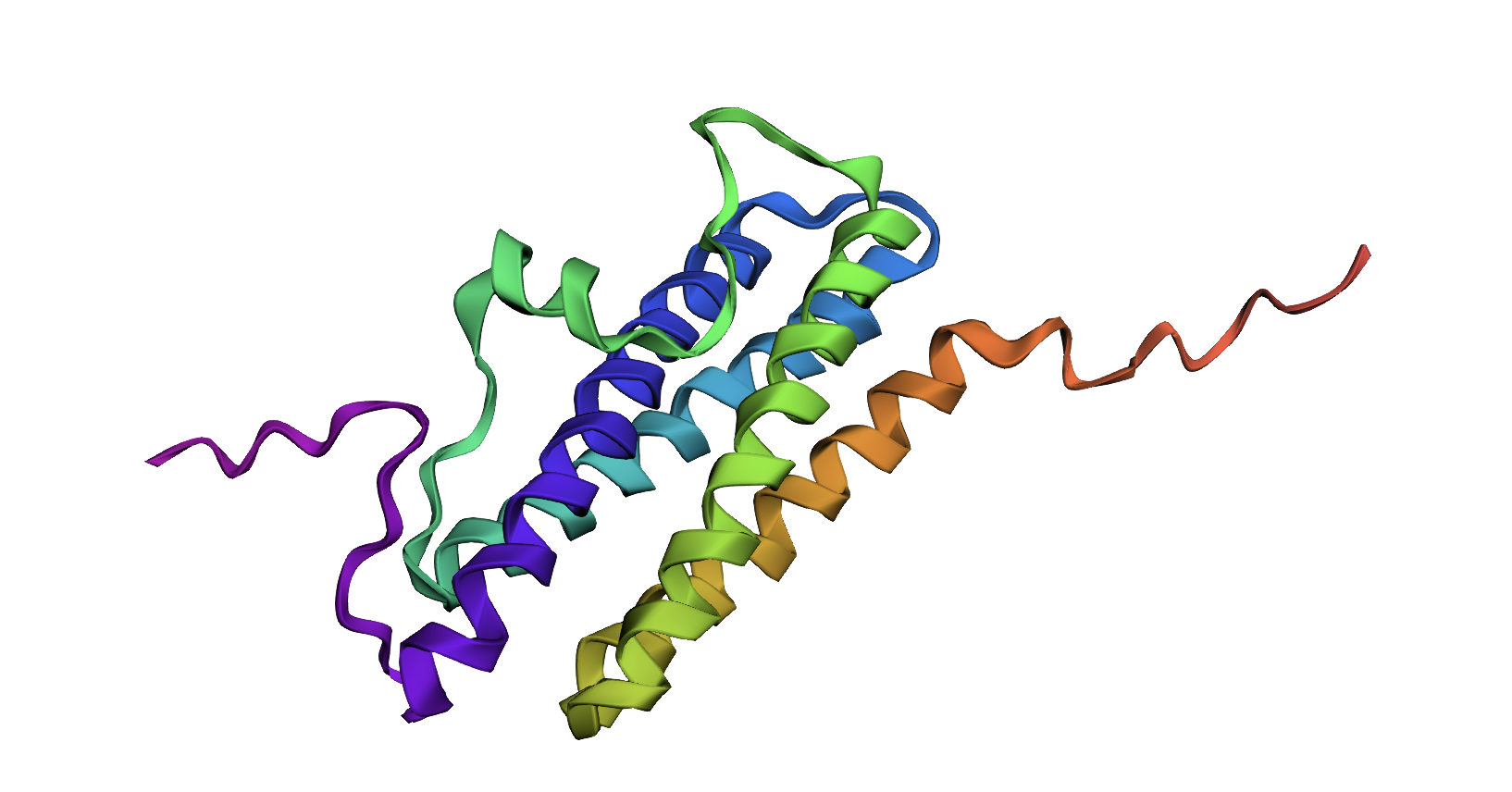
The ESMFold prediction looks significantly different from my original structure as visualized in PyMOL. In this example, the AI prediction may not have generated a structure accurate to the experimentally-validated one, potentially because the experimental structure shows the protein interacting with iron ions and a ligand.
2. Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
Changing just a few amino acids did not make a major difference, suggesting that the structure is relatively resilient to mutations. Randomly changing the amino acids in large segments of the sequence generated variants with new structural components (e.g., an alpha helix where there was previously an intrinsically disordered region), but mostly retained the same overall shape.
C3) Protein Generation Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN 1. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Predicted sequence:
Protein MPNN assigned this a score of 0.9236.
At a glance, this sequence looks significantly different from the original.
- Input this sequence into ESMFold and compare the predicted structure to your original.
ESMFold prediction for inverse-folded sequence
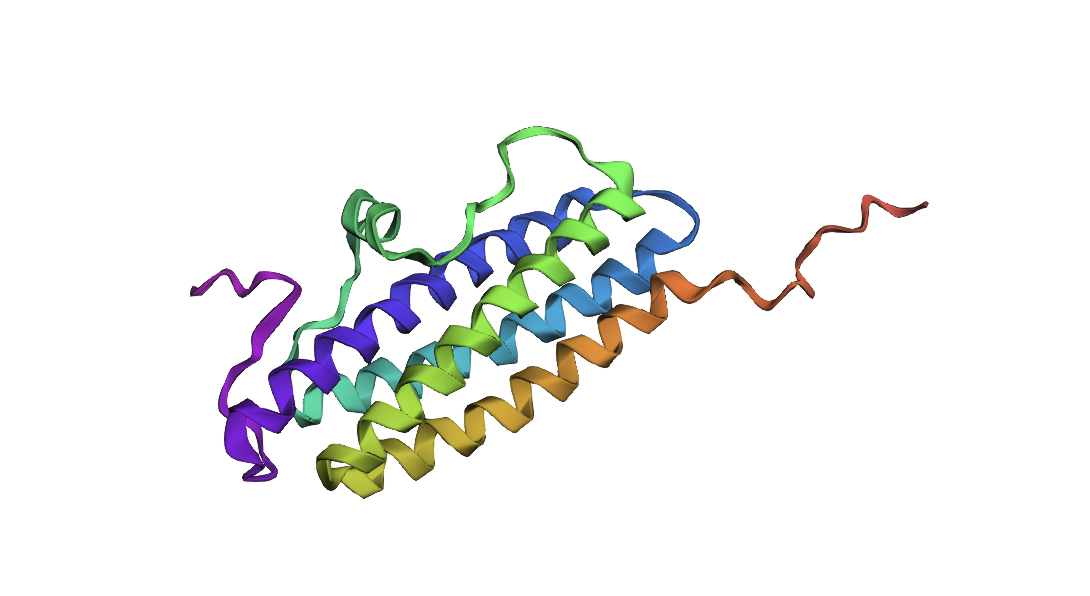
The result looks like it has the same domains as the original, but a slightly different arrangement.
D. Group Brainstorm on Bacteriophage Engineering
Brainstorm/Options for increasing MS2 titer and lysis protein toxicity:
Overall workflow idea:
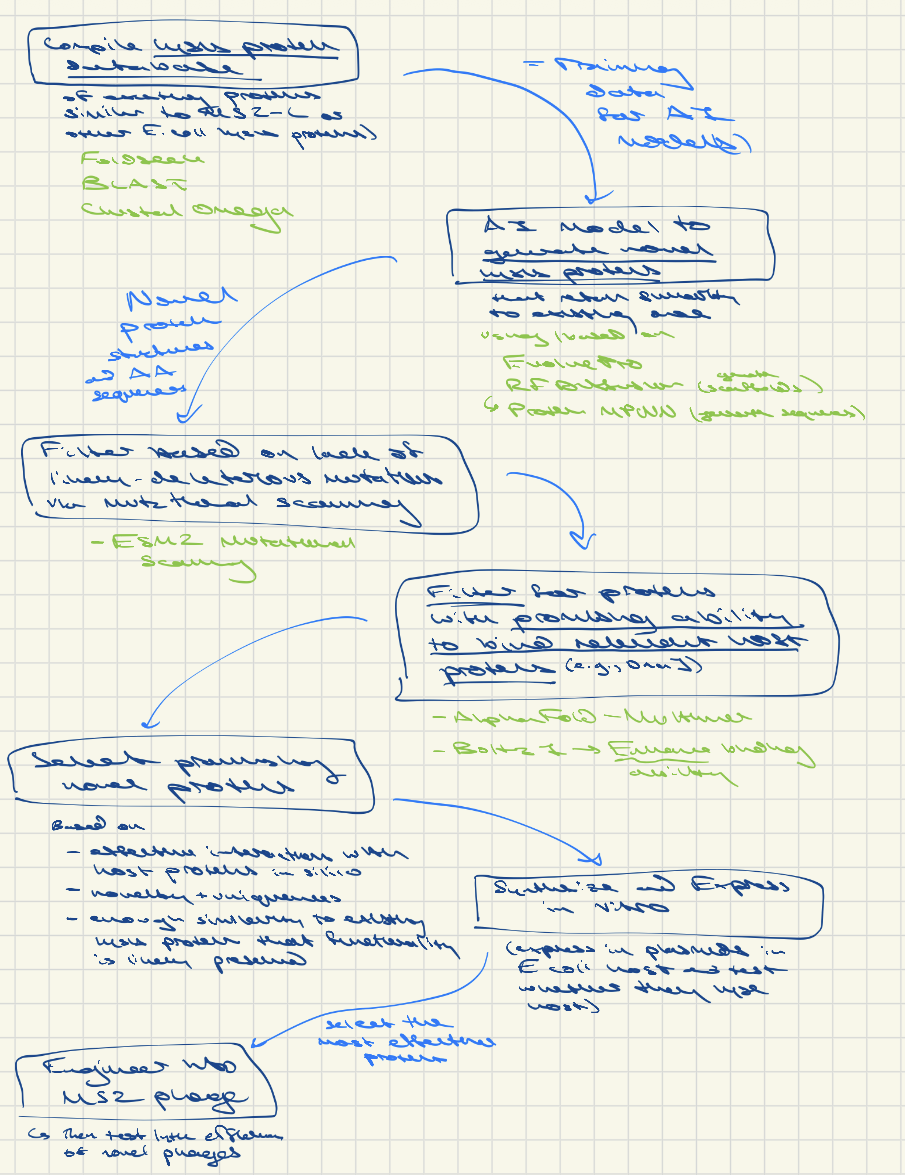
Option 1: Compile known lysis proteins of a variety of phage (or maybe just E coli ones given high specificity of phage) and info on their lytic efficiency → then use AI models (possibly similar to Evo 1 and Evo 2, as described in the King et al. preprint, though these are whole genome models) to find structural or sequence patterns that are related to enhanced vs. reduced lytic efficiency across all phage and across E. coli phage in particular. Synthesize the supposedly more-optimal E. coli phage protein variants and/or synthesize modified MS2 L proteins based off of them. Identify novel proteins that are effective, then engineer them into MS2.
- Could compile a database of lysis proteins that are similar in structure and/or sequence to MS2 L (and therefore more likely to work in E. coli) using FoldSeek, BLAST, and Clustal Omega
- (Unsure to what extent lysis proteins are host-specific. They likely have less of an effect on host range than tail fibers, which affect ability to bind to host, however, if a lysis protein is dependent on binding to a certain host protein, it would likely be highly host-specific.)
- Train an AI with the above database and generate new lysis proteins that retain enough similarity to existing MS2 L or other E. coli lysis proteins that they might still function in MS2 / E. coli (or look at all lysis proteins in general). This could involve or be based on a tool like EvolvePro or a combination of RFDiffusion and ProteinMPNN (RFDiffusion to generate novel scaffolds and ProteinMPNN to generate sequences for them)
- Possibly, test the novel proteins’ ability to bind to relevant host proteins (e.g., DnaJ) in silico using AlphaFold-Multimer or Boltz-1 (, which we could also use to enhance the binding interactions)
- Determine which residues are more likely to be essential to MS2 or other lysis protein functioning via ESM2 mutational scanning. Filter out novel sequence options that include likely detrimental mutations at these locations. (Alternatively, incorporate this step into the AI algorithm somehow and instruct the model to avoid mutations that ESM2 classifies as deleterious.)
- Based on the above, select novel in silico-designed proteins that display the most promising interactions with host proteins in silico and that have unusual sequences and/or structures (to increase the chances of enhancing lytic capabilities beyond those of the existing proteins) but that retain a level of fundamental similarity to MS2 L and other lysis proteins at the essential locations as determined through ESM2.
- Test the lytic ability of the most promising ones by synthesizing them into plasmids, transforming into E. coli, and inducing them on a plate to measure whether and how effectively they lyse host cells
- Engineer the most effective ones into MS2
- Limitations: there may be a high chance of developing novel proteins that are non-functional because we lack knowledge about the exact function that they need to perform and the interactions with host proteins that we would need to retain in order to preserve lytic abilities. Additionally, to have the highest possible chance of obtaining proteins that function in an E. coli phage, we may need to limit the database to E. coli phage lysis proteins, which may not be enough training data for an AI model.
Option 2: Alternatively, random mutagenesis of MS2 L until we get a higher titer or more lytic version. I.e., perform random mutagenesis on MS2 phages and then run infection assays to identify mutants that are higher titer or lyse the host faster than the wildtype.
- This is the “traditional” method and is time-consuming and likely limited in the variety of functional novel phages that it can generate