Week 5 HW: Protein Design Part II
A. SOD1 Binder Peptide Design
Part 1: Generate Binders with PepMLM
Mutant SOD sequence:
Binder peptides generated with PepMLM, evaluated with AlphaFold:
| Peptide | Perplexity | ipTM | Location |
|---|---|---|---|
| WRVPAAGAELGX | 7.415045 | 0.41 | Over disordered region |
| WHYYAAAVRWKX | 15.435134 | 0.37 | Across 2 rows in sheet |
| WLYPAAGLRHWX | 16.586151 | 0.26 | Over small helix |
| WRYYAAALALGX | 7.577719 | 0.38 | Over sheet region, peptide is a helix |
For comparison, here is a known SOD1-binder peptide:
| Peptide | Perplexity | ipTM | Location |
|---|---|---|---|
| FLYRWLPSRRGG | N/A | 0.31 | Across 2 rows in sheet |
Part 2: Evaluate Binders with AlphaFold3
All but one of the new binder peptides had an intrinsically disordered structure, and all binders varied in their location of binding. All binders appeared to “float” above the SOD protein in alphafold’s visualization, suggesting that none would directly integrate into the structure. All but one of the new peptides had a higher ipTM value than the known binder. (See previous section for specific stats and binding locations.)
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
PeptiVerse results for all new peptide options:
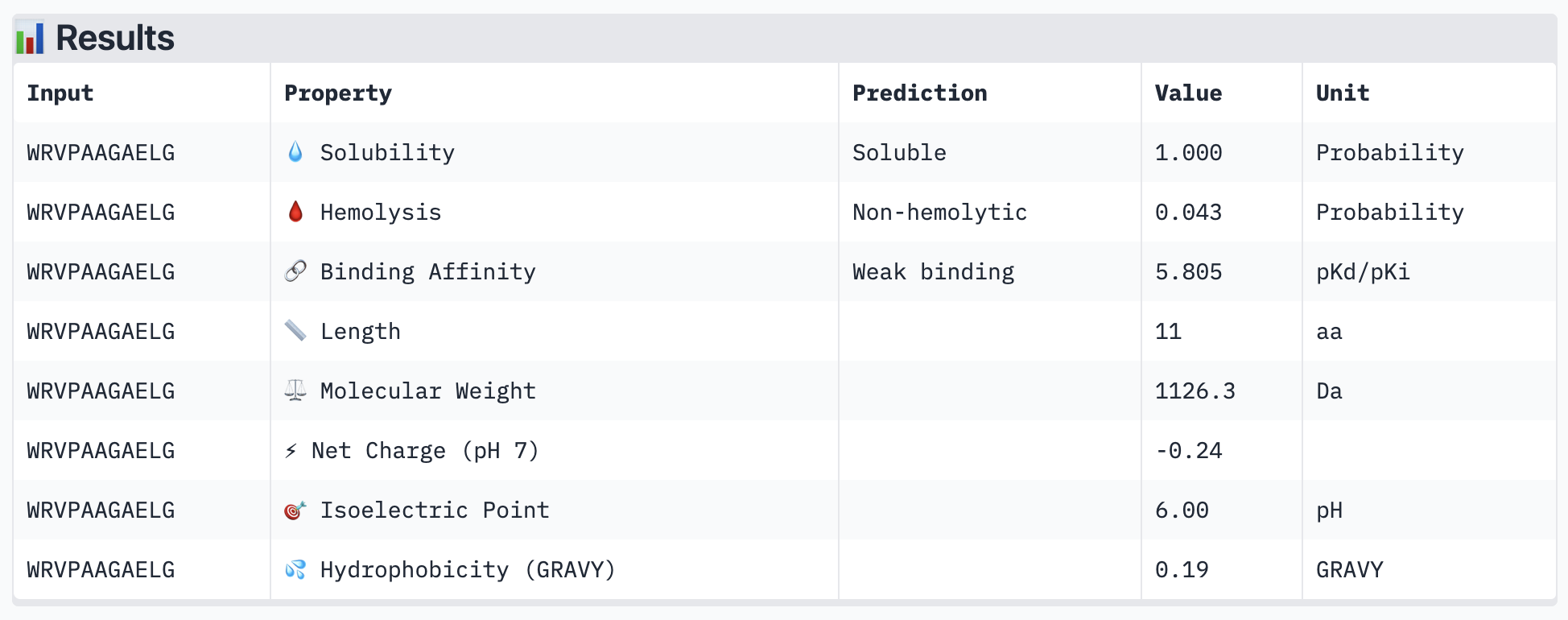
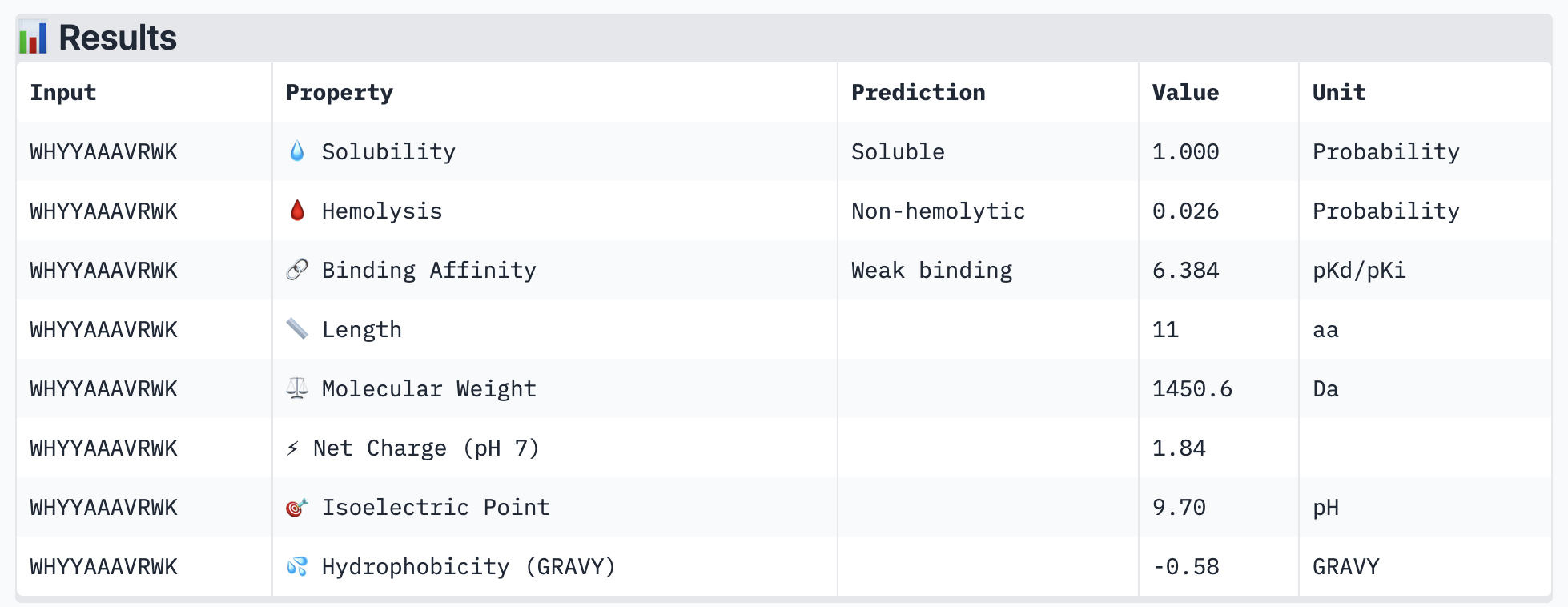
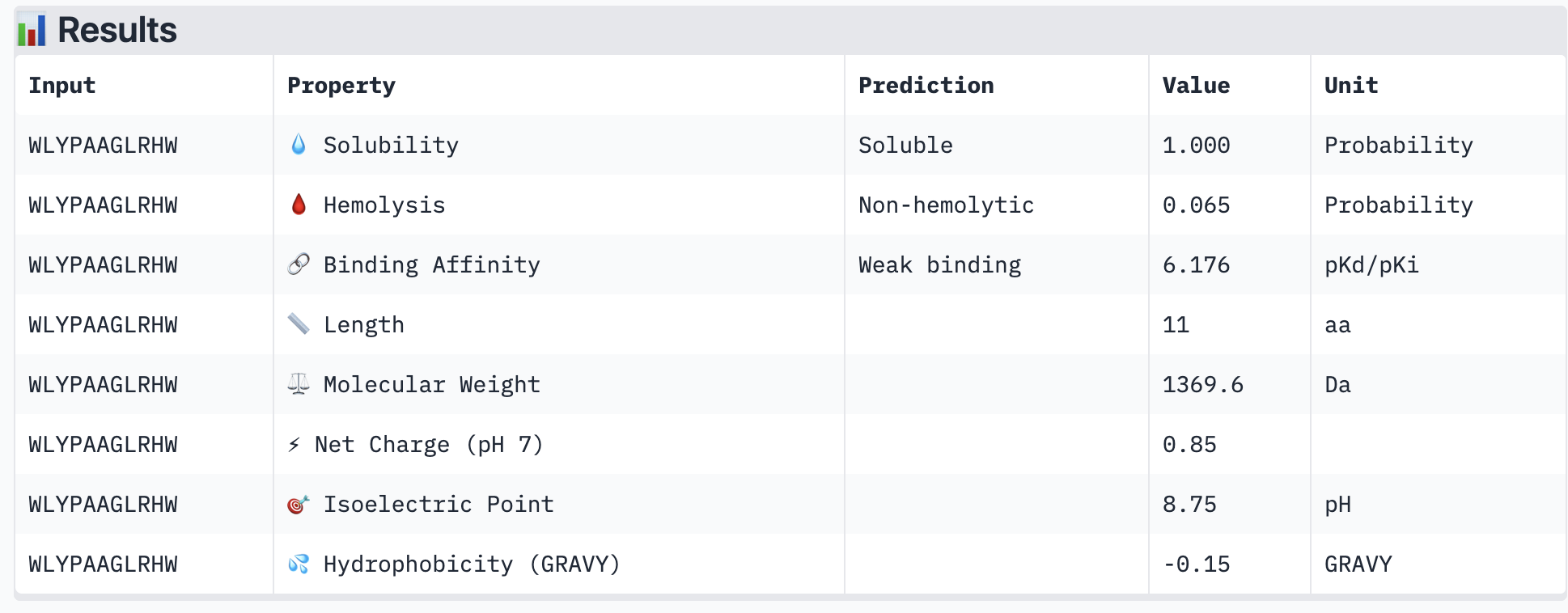
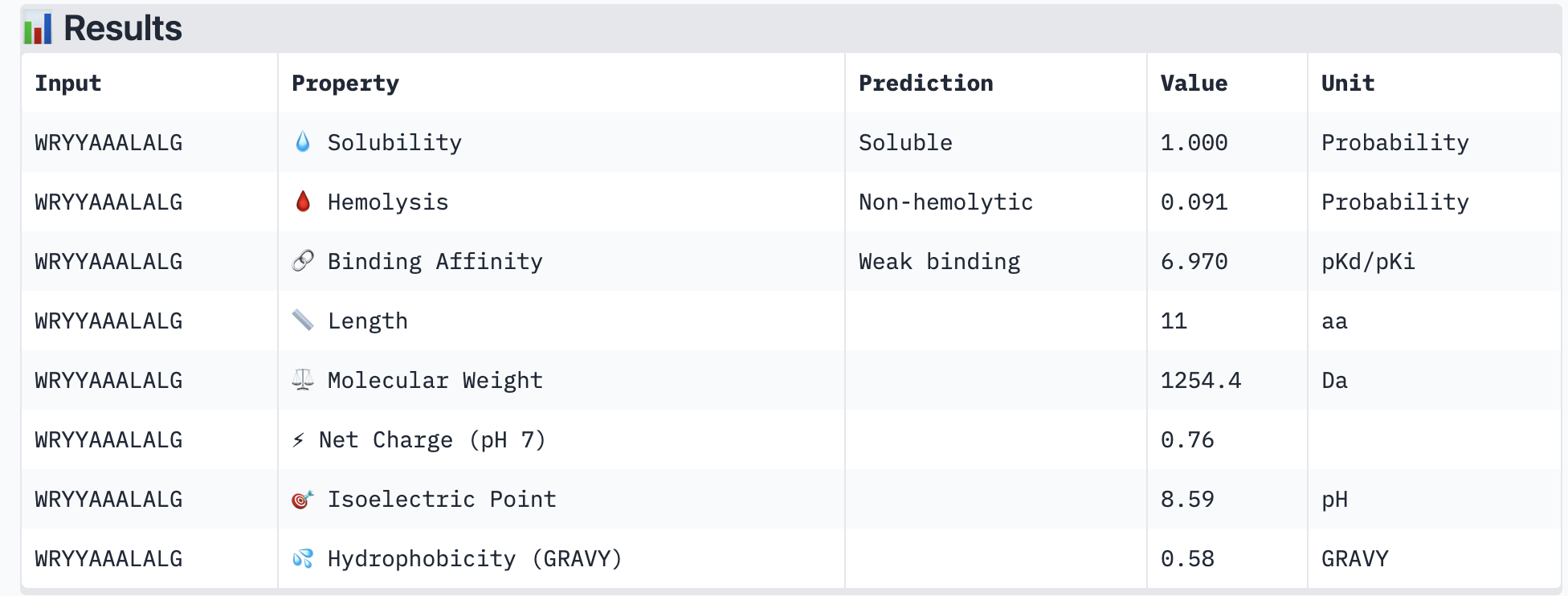
All of the resulting peptides have similarly weak binding affinities according to PeptiVerse. All were also soluble and non-hemolytic. If I were to advance one of the peptides, I would choose the fourth one – it had the strongest predicted binding affinity of the four (per PeptiVerse) and, according to AlphaFold, had a more-interesting structure: helical rather than disordered.
Part 4: Generate Optimized Peptides with moPPIt
Getting CUDA Out of Memory Errors even though using L4 GPU as recommended. Tried to debug, but the program keeps crashing.
B. BRD4 Drug Discovery Platform Tutorial
Part 1: Structural Predictions in the Sandbox
| Compound | Binding Confidence | Optimization Score | Structure Confidence |
|---|---|---|---|
| Hit | 0.42 | 0.22 | 0.98 |
| Lead | 0.75 | 0.27 | 0.98 |
| JQ1 | 0.96 | 0.44 | 0.98 |
The predicted binding confidence in Boltz won’t change as a potential ligand advances from “hit” to candidate for clinical treatment. However, it is safer to advance ligands that have favorable binding scores to clinical candidates.
The JQ1 ligand appears to bind between three alpha helices in BRD4. JQ1 has a higher optimization score than the Lead and Hit.
Part 2: Setting Up a BRD4 Design Project
Generated target region for BRD4 based on JQ1 binding site.
Part 3: Running Your Virtual Screen
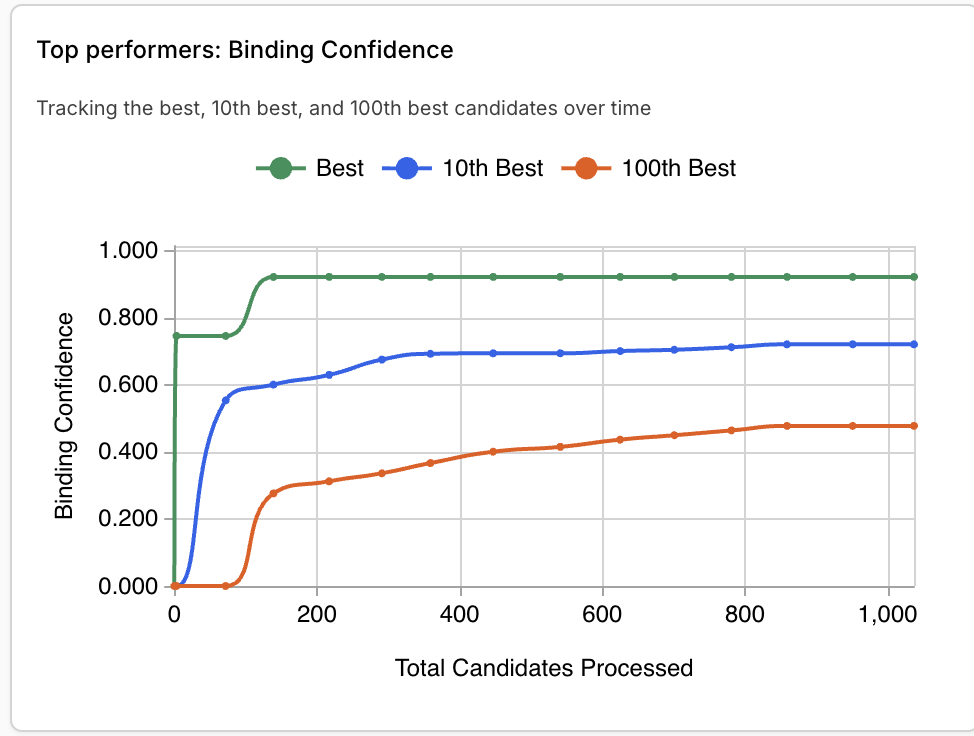
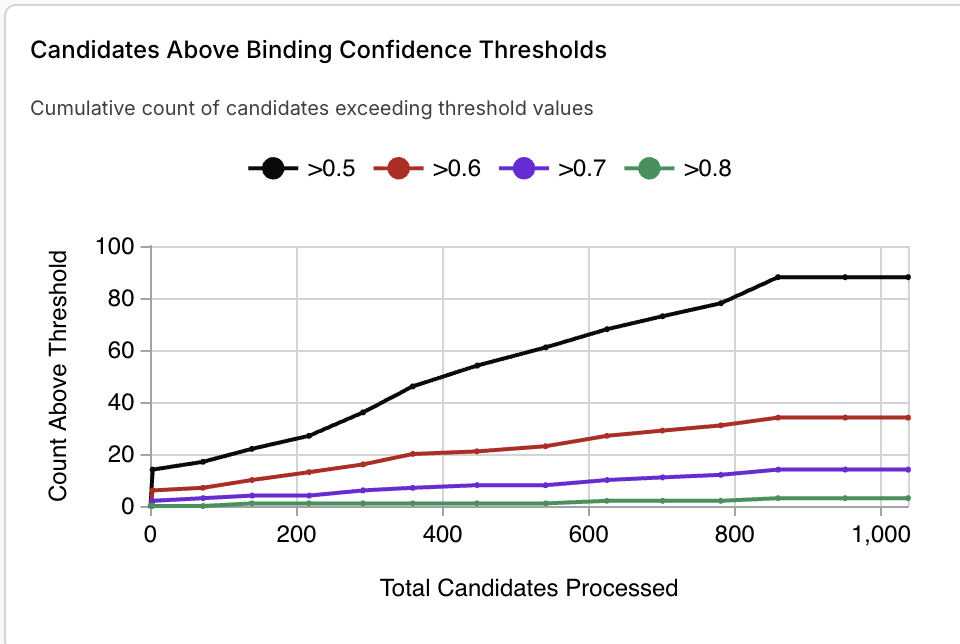
Part 4: Analysis and Discussion
JQ1 is the top compound, with a binding confidence of 0.96. The second closest new binder had a score of 0.92. Other top binders had confidence levels between 0.7-0.8.
I ran the top 8 BRD4 binders against the target BRD2. All had comparable or higher binding confidence scores with BRD2.
C. Final Project: L-Protein Mutants
Heatmap scoring possible mutations
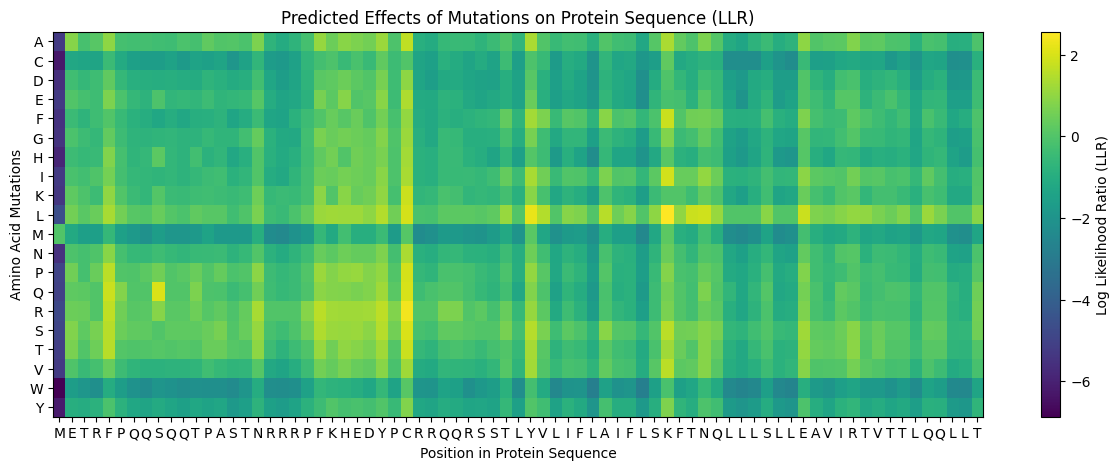
Positive log likelihoods on the heatmap tend to correspond with tolerable mutations based on experimental data. Negative log likelihoods tend to correspond with deleterious mutations.
L-protein seq:
| Mutation | Region | Benefit | Multimer (AF-2) | Comments |
|---|---|---|---|---|
| Y -> L, AA 39 | Transmembrane | Heatmap score is about 2, indicating that this would be highly tolerable and potentially beneficial. Swapping a given amino acid with leucine appears to be more tolerable than alternative swaps across the residues. |  | Retained pore structure |
| I -> F, AA 46 | Transmembrane | Experimental data showed that the lysis protein functioned after this mutation. The heatmap shows a moderate log likelihood ratio, indicating that this mutation might not occur evolutionarily but probably is not deleterious. | 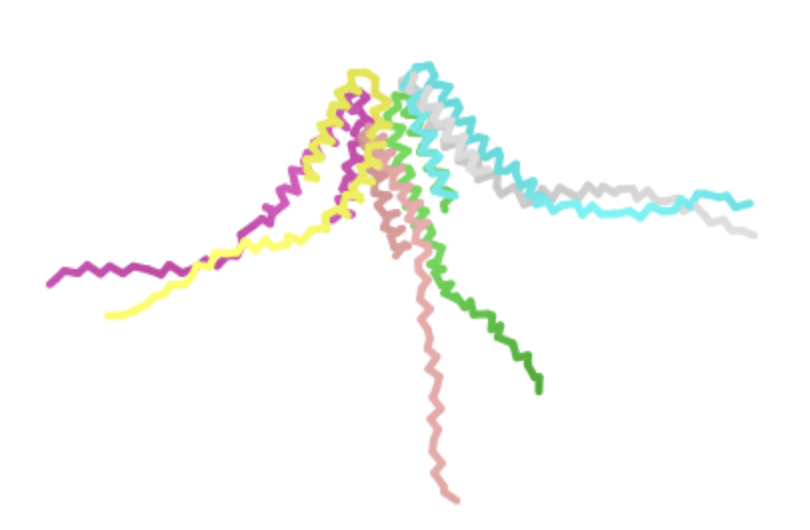 | Also retained pore structure, though AlphaFold displayed it at a different angle. See its second visualization for a better comparison 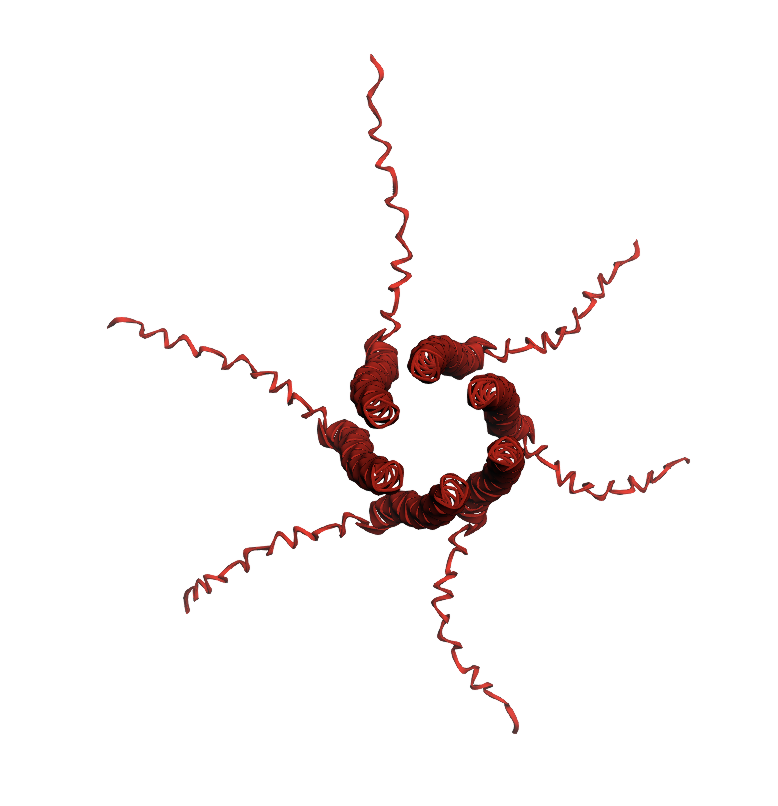 |
| L -> P, AA 44 | Transmembrane | Experimentally validated but interestingly appears to have a relatively low log likelihood ratio on the heatmap | 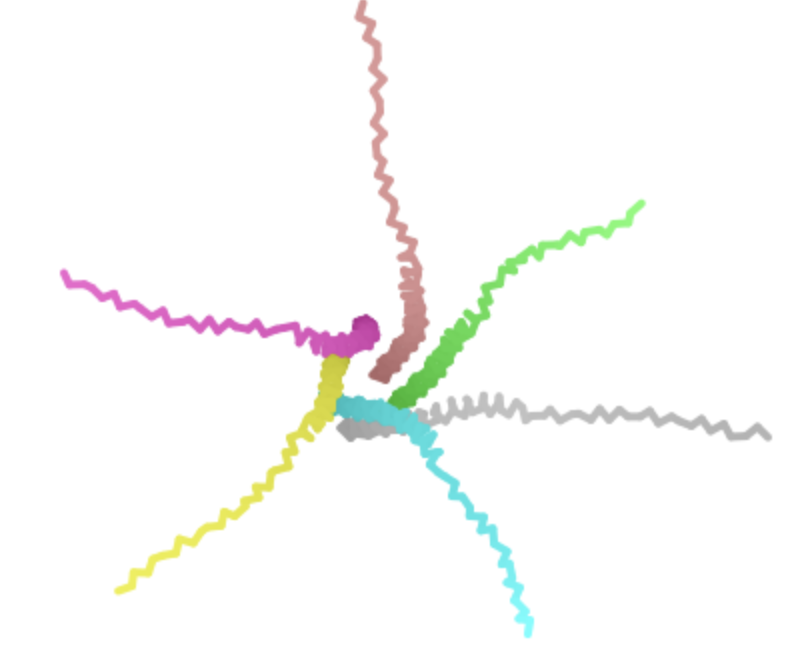 | Retains pore structure, but the disordered portions of the proteins appear to be associating more with each other in the complex and/or appear misshapen. Not sure why changing the transmembrane domain would directly affect the disordered regions, so suspect that this is an artifact of AF-2’s chosen display angle |
| P -> L, AA 13 | Soluble | Experiemntally validated and the heatmap gives a relatively high log likelihood ratio. We know that the mutated protein works in practice and could occur naturally due to evolution. | 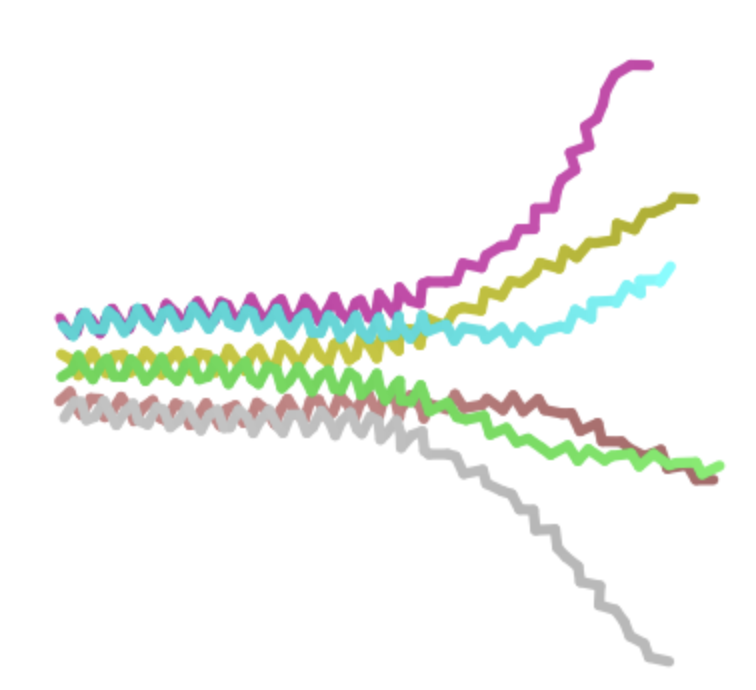 | Retains pore structure. Note AF-2 visualization shows a side view. |
| S -> Q, AA 9 | Soluble | Unlikely to have a major deleterious effect given that serine and glutamine are both polar and uncharged. The heatmap shows a high log likelihood ratio. |  | Similar to above, retains pore structure. AF-2 visualization shows side view again. |