Week 04: HW protein design part I
This week will focus on how sequence, structure, and energetics can be modeled and manipulated to create or optimize proteins with specified functions…
Objective:
- Learn basic concepts:
- amino acid structure
- 3D protein visualization
- the variety of ML-based design tools
- Brainstorm as a group how to apply these tools to engineer a better bacteriophage (setting the stage for the final project).
Part A. Conceptual Questions
Assignees for the following sections
| MIT/Harvard students | Required |
| Committed Listeners | Required |
I answered nine of the conceptual questions below.
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
It is about 3 *10^{24} amino acid molecules.
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
We digest proteins into amino acids and then rebuild them into human proteins.
3. Why are there only 20 natural amino acids?
Life evolved to use a small set that is enough for many protein functions.
5. Where did amino acids come from before enzymes that make them, and before life started?
They may have formed through prebiotic chemistry on early Earth or arrived from space.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
I would expect a left-handed helix.
8. Why are most molecular helices right-handed?
Most natural proteins use L-amino acids, which usually favor right-handed helices.
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheets aggregate because their backbones can make many hydrogen bonds. Hydrophobic interactions also help drive aggregation.
10. Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Amyloid proteins often stack into stable β-sheet-rich fibers. Yes, amyloid β-sheets can also be used as strong biomaterials.
11. Design a β-sheet motif that forms a well-ordered structure.
A simple design is an alternating pattern of hydrophobic and polar residues, such as Val-Lys-Val-Glu-Val-Lys-Val-Glu. This can help form a stable β-sheet.
Part B: Protein Analysis and Visualization
Assignees for the following sections
| MIT/Harvard students | Required |
| Committed Listeners | Required |
In this part of the homework, I used online resources and 3D visualization software to analyze a protein of interest that has a known 3D structure.
1. Briefly describe the protein you selected and why you selected it.
From now on I have decided to focus on paclitaxel for my final project and to optimize a crucial bottleneck in its current biosynthesis. Thus, the protein I wanted to visualize is beta-tubulin, which is the target of paclitaxel in cancer treatment.
2. Identify the amino acid sequence of your protein.
I used the UniProt entry for Beta-tubulin 1 subunit:
https://www.uniprot.org/uniprotkb/Q9H4B7/entry
The Beta-tubulin 1 subunit is 451 amino acids long.
Sequence:
MREIVHIQIGQCGNQIGAKFWEMIGEEHGIDLAGSDRGASALQLERISVYYNEAYGRKYVPRAVLVDLEPGTMDSIRSSKLGALFQPDSFVHGNSGAGNNWAKGHYTEGAELIENVLEVVRHESESCDCLQGFQIVHSLGGGTGSGMGTLLMNKIREEYPDRIMNSFSVMPSPKVSDTVVEPYNAVLSIHQLIENADACFCIDNEALYDICFRTLKLTTPTYGDLNHLVSLTMSGITTSLRFPGQLNADLRKLAVNMVPFPRLHFFMPGFAPLTAQGSQQYRALSVAELTQQMFDARNTMAACDLRRGRYLTVACIFRGKMSTKEVDQQLLSVQTRNSSCFVEWIPNNVKVAVCDIPPRGLSMAATFIGNNTAIQEIFNRVSEHFSAMFKRKAFVHWYTSEGMDINEFGEAENNIHDLVSEYQQFQDAKAVLEEDEEVTEEAEMEPEDKGH
The most frequently occurring amino acid is alanine (A), with a total of 34 occurrences.
Based on the UniProt BLAST results, I found approximately 250 homologs for this protein:
https://www.uniprot.org/blast/uniprotkb/ncbiblast-R20260305-180445-0890-48978074-p2m/overview
This protein belongs to the tubulin family.
3. Identify the structure page of your protein in RCSB.
I used the following RCSB structure page:
https://www.rcsb.org/structure/7QUC
The structure was solved in 2022 using electron microscopy, with a resolution of 3.20 Å. This is a reasonable quality structure, although it is not as high resolution as some crystal structures.
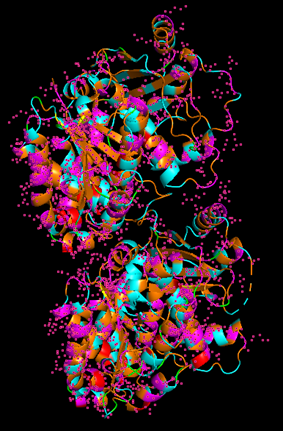
There are other molecules in the solved structure apart from the protein. Specifically, the structure is a dimer of the beta and alpha tubulin subunits, so this structure contains one beta subunit and one alpha subunit.
Based on what I found, it does not appear to belong to a structure classification family in the way asked by the prompt.
4. Open the structure of your protein in any 3D molecule visualization software.
Because the resolved structure contains two proteins from the microtubule, I colored the alpha-tubulin yellow and the beta-tubulin green, since paclitaxel binds to beta-tubulin in cancer treatment.
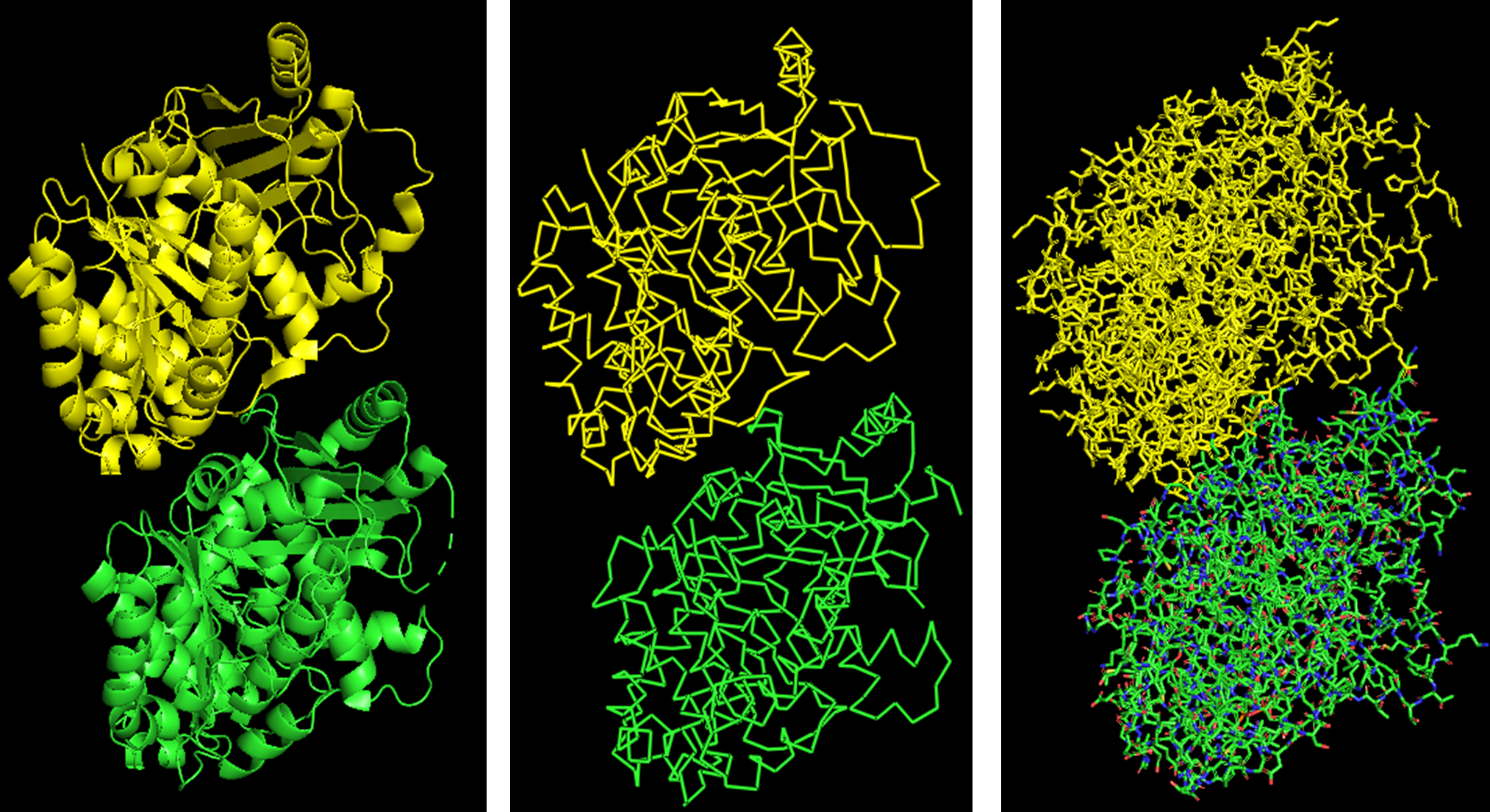
Cartoon representation of the alpha- and beta-tubulin structure.
When coloring the protein by secondary structure, the structure seemed to have more helices than sheets, with a ratio of approximately 3:1 (3501:1074).
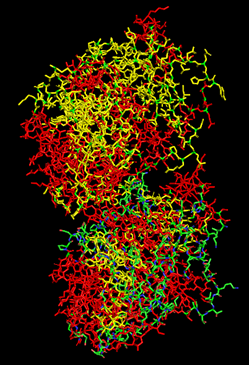
When coloring the protein by residue type, the visualization showed hydrophobic residues with 2704 atoms in orange, and hydrophilic residues with 3732 atoms in cyan/magenta. The hydrophilic residues seemed to be distributed more on the outer parts of the structure, while the hydrophobic parts were more concentrated on the inner regions.
When visualizing the surface of the protein, it looked like there is a binding pocket between the two proteins. I compared my image to the known paclitaxel binding pocket and indicated it with an arrow. However, this visualization alone is not completely clear, and other methods would analyze this more reliably.
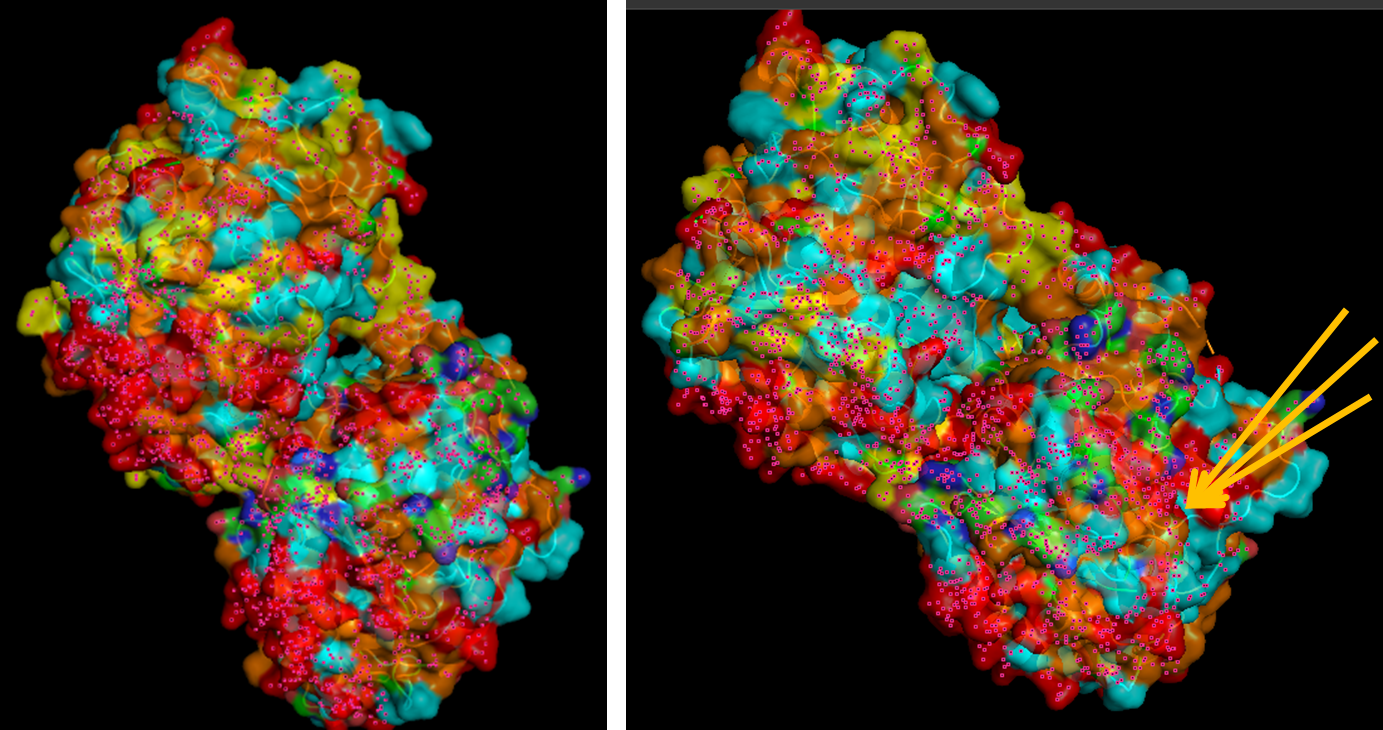
Part C. Using ML-Based Protein Design Tools
Assignees for the following sections
| MIT/Harvard students | Required |
| Committed Listeners | Required |
In this section, I tested several modern protein AI models on my chosen protein.
I selected the following PDB sequence, which was slightly different from the UniProt sequence:
MREIVHIQAGQCGNQIGAKFWEIISDEHGIDATGAYHGDSDLQLERINVYYNEASGGKYVPRAVLVDLEPGTMDSVRSGPFGQIFRPDNFVFGQSGAGNNWAKGHYTEGAELVDSVLDVVRKEAESCDCLQGFQLTHSLGGGTGSGMGTLLISKIREEYPDRIMNTYSVVPSPKVSDTVVEPYNATLSVHQLVENTDETYCIDNEALYDICFRTLKLTTPTYGDLNHLVSLTMSGVTTCLRFPGQLNADLRKLAVNMVPFPRLHFFMPGFAPLTSRGSQQYRALTVPELTQQMFDAKNMMAACDPRHGRYLTVAAIFRGRMSMKEVDEQMLNIQNKNSSYFVEWIPNNVKTAVCDIPPRGLKMSATFIGNSTAIQELFKRISEQFTAMFRRKAFLHWYTGEGMDEMEFTEAESNMNDLVSEYQQYQEATADEDAEFEEEQEAEVDEN
C1. Protein Language Modeling
1. Deep Mutational Scans
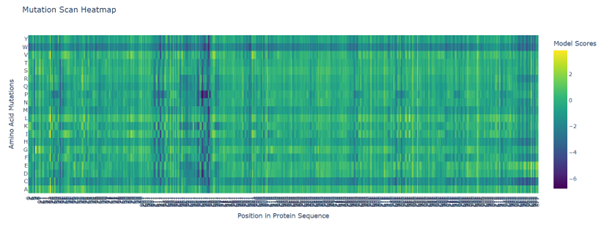
b. Can you explain any particular pattern? (choose a residue and a mutation that stands out)
Positions 140–160 form a structurally constrained region where most substitutions are harmful. In particular, position 158 is intolerant to bulky hydrophobic residues such as tryptophan, tyrosine, and valine, while position 153 tolerates only flexible or polar residues such as serine, arginine, glycine, proline, and asparagine. This suggests loop-specific flexibility requirements.
c. Place your protein in the resulting map and explain its position and similarity to its neighbors.
2. Latent Space Analysis
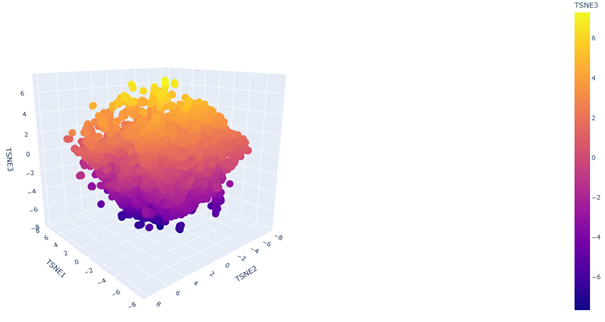
b. Analyze the different formed neighborhoods: do they approximate similar proteins?
Yes. In the 3D latent space, nearby points form local neighborhoods of proteins with similar sequence features, indicating that the embedding groups related proteins together.
C2. Protein Folding
Folding a protein
1. Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
The structure predicted by ESMFold aligns very well with the experimentally determined β-tubulin structure. The low RMSD of 0.782 Å after alignment indicates that the predicted atomic coordinates closely match the original structure.
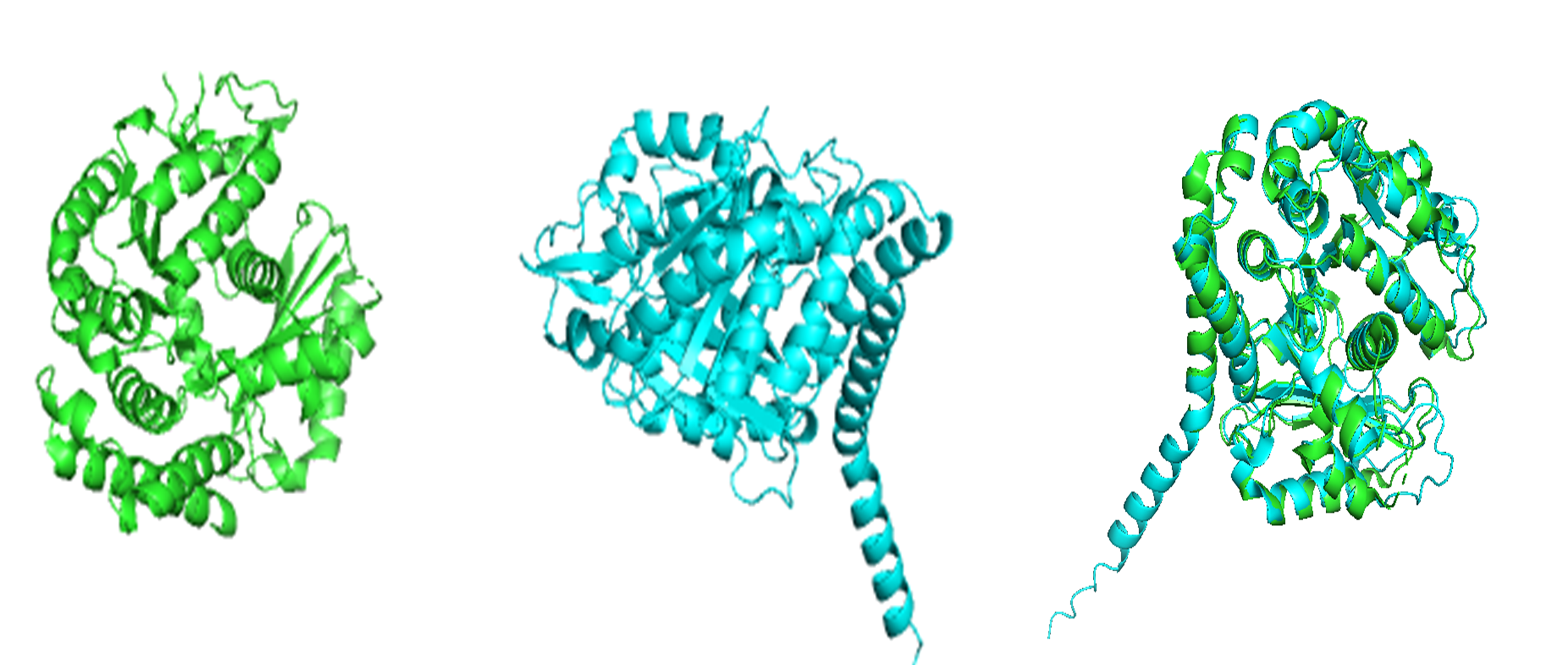
Result: RMSD = 0.782
2. Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
For small mutations, I tested the following sequence:
MREIVHIQAGQCGNQIGAKFWEIISDEHGIDATGAYHGDSDLQLERINVYYNEASGGKYVPRAVLVDLEPGTMDSVRSGPFGQIFRPDNFVFGQSGAGNNWAKGHYTEGAELVDSVLDVVRKEAESCDCLQGFQLTHSLMMMTGSGMGTLLISKIREEYPDRIMNTYSVVPSPKVSDTVVEPYNATLSVHQLVENTDETYCIDNEALYDICFRTLKLTTPTYGDLNHLVSLTMSGVTTCLRFPGQLNADLRKLAVNMVPFPRLHFFMPGFAPLTSRGSQQYRALTVPELTQQMFDAKNMMAACDPRHGRYLTVAAIFRGRMSMKEVDEQMLNIQNKNSSYFVEWIPNNVKTAVCDIPPRGLKMSATFIGNSTAIQELFKRISEQFTAMFRRKAFLHWYTGEGMDEMEFTEAESNMNDLVSEYQQYQEATADEDAEFEEEQEAEVDEN
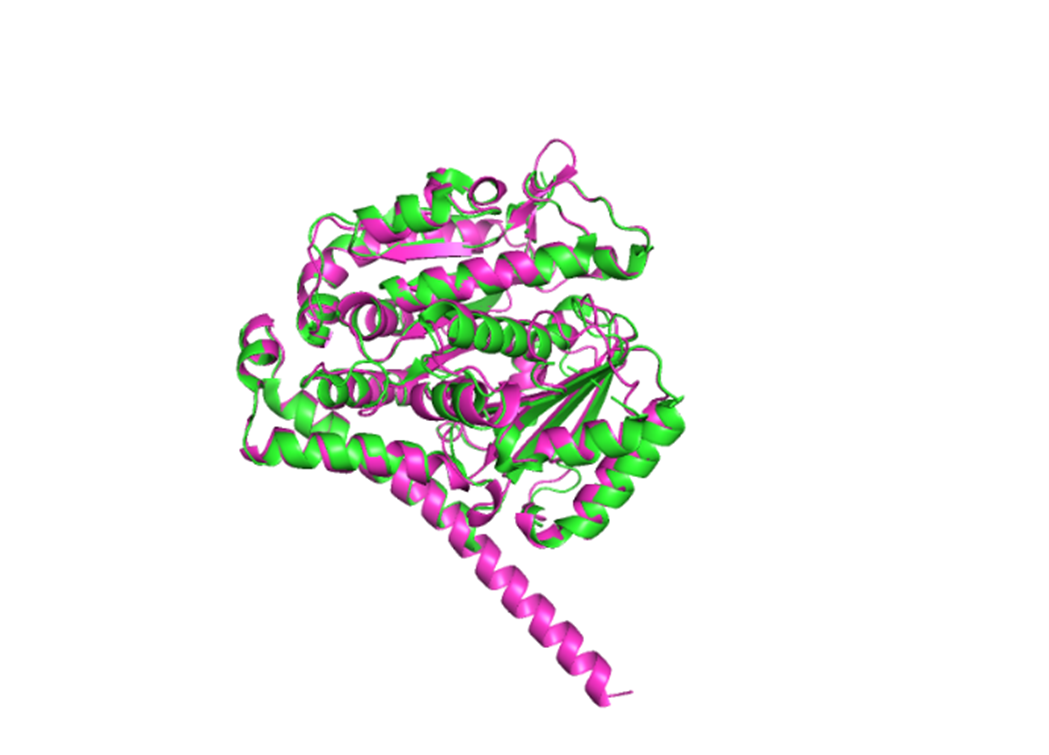
RMSD: 0.750
For larger mutations, I tested the following sequence:
MREIVHIQAEEQGTLIGAKFWEIISDEHGIDATGAYHGDSDLQLINVYYNEASGGKYVPRAVLVDLEPGTMDSVRSGPFGQIFRPDNFVFGQSGAGNNWAKGHYTEGAELVDSVLDVVRKEAESCDCLQGFQLTHSLMMMTGSGMGTLLISKIREEYPDRIMNTYSVVPSPKVSDTVVEPYNATLSVHQLVENTDETYCIDNEALYDICFRTLKLTTPTYGDLNHLVSLTMSGVTTCLRFPGQLNADLRKLAYLTVACIFRGKMSTKGFAPLTSRGSQQYRALTVPELTQQMFDAKNMMAACDPRHGRVNMVPFPRLHFFMPEVDEQMLNIQNKNSSYFVEWIPNNVKTAVCDIPPRGLKMSATFIGNSTAIQELFKRISEQFTAMFRRKAFLHWYTGEGMDEMEFTEAESNMNDLVSEYQQYQEATADEDAEFEEEQEAEVDEN
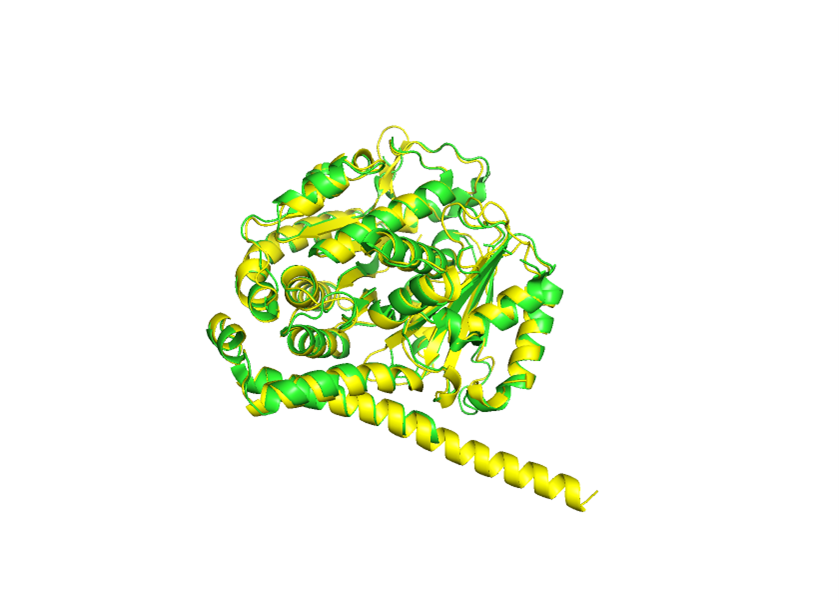
RMSD: 0.873
This was quite surprising, because I expected a much larger variance to occur. Based on these results, the overall fold appears to be fairly resilient to both small and somewhat larger sequence changes.
C3. Protein Generation
Inverse-Folding a protein
1. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
The predicted sequence has a better ProteinMPNN score (0.8587) than the original sequence (1.8495), with a sequence recovery of 0.3783. This means ProteinMPNN proposed a substantially different sequence that it still predicts will fit the same backbone.
2. Input this sequence into ESMFold and compare the predicted structure to your original.
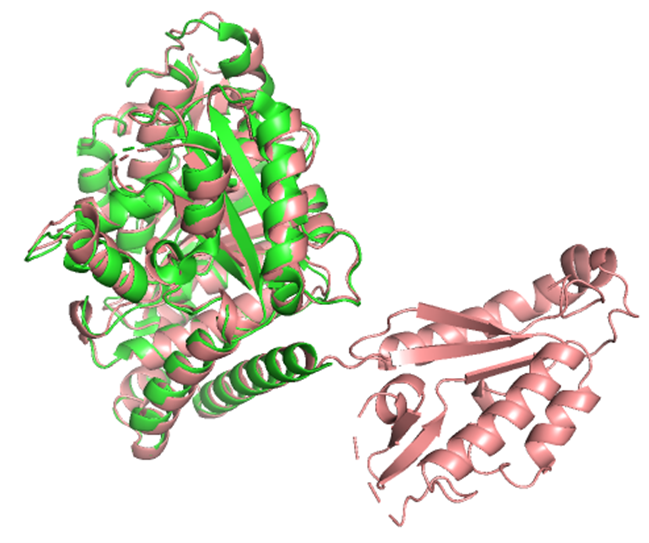
The predicted structure had an RMSD of 0.756 Å relative to the original. The main structure seems to be well preserved, but there appears to be one extra part that is not present in the original structure, which is quite strange.
Part D. Group Brainstorm on Bacteriophage Engineering
Assignees for the following sections
| MIT/Harvard students | Optional |
| Committed Listeners | Required |
Proposal: Computational engineering of the MS2 L protein for increased stability and higher titers
For my project, I will focus on two goals for the MS2 L protein: increased stability and higher phage titers. I chose these because the literature suggests that L contains a small but sensitive functional core, and that lysis timing likely influences how many phage particles are produced before the host cell breaks open. In simple terms, I want a version of L that is more reliable as a protein, but not so aggressive that it causes lysis too early.
My main approach would be to combine sequence conservation analysis, in silico mutagenesis, protein language model scoring, and structure/topology prediction. I would first identify residues that are likely too important to mutate, especially around the conserved LS motif, since mutational studies show that this region is highly sensitive and likely involved in an essential interaction. I would also treat the basic N-terminal region carefully, because it regulates activity through interaction with DnaJ rather than acting as the main lytic domain itself.
Next, I would computationally test mutations that might improve folding robustness or membrane association while preserving the protein’s core lytic features. This seems reasonable because recent work suggests that MS2-L forms oligomeric assemblies in membrane-like environments, and that the transmembrane/C-terminal region is central to this behavior. In plain language, I am not only asking whether the protein folds, but whether it can still adopt the right shape and assembly state to work properly.
For the higher titer goal, I would not try to predict titers directly. Instead, I would use a proxy strategy and prioritize variants that are predicted to be more stable while still preserving the regulatory features that may prevent premature lysis. This is important because N-terminal truncations can bypass DnaJ and trigger earlier lysis, which may actually reduce phage output if the host is killed before assembly is complete.
Planned pipeline
- Collect sequence, mutational, and structural/topology information for L.
- Mark function-sensitive residues and conserved regions.
- Run in silico mutagenesis and rank variants with language-model or sequence-based scores.
- Filter variants using membrane topology and structural plausibility.
- Prioritize candidates that may improve stability while preserving productive lysis timing.
Potential pitfalls
One pitfall is that higher titers are not controlled by L alone, so even a better L variant may not improve total phage yield. Another is that MS2 has overlapping genes and RNA-level regulation, so a mutation that looks good for the protein might still be harmful in the native phage genome.