Week 5: HW protein design part II
This week we learn how cutting-edge AI and protein language models are used to design functional proteins and peptides “in silico”.
Objective:
- Design short peptides that bind mutant SOD1.
- Decide which peptides are worth advancing toward therapy.
- Evaluate generated peptides using structural and therapeutic-property prediction tools.
- Think about how computational protein design can be applied to the final project on L-protein mutants.
Part A: SOD1 Binder Peptide Design
Assignees for the following sections
| MIT/Harvard students | Required |
| Committed Listeners | Required |
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Your challenge:
- Design short peptides that bind mutant SOD1.
- Then decide which ones are worth advancing toward therapy.
You will use three models developed in our lab:
- PepMLM: target sequence-conditioned peptide generation via masked language modeling
- PeptiVerse: therapeutic property prediction
- moPPIt: motif-specific multi-objective peptide design using Multi-Objective Guided Discrete Flow Matching (MOG-DFM)
Part 1: Generate Binders with PepMLM
1. Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
I used the following A4V mutant SOD1 sequence:
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQFLYRWLPSRRGG
2. Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
- https://colab.research.google.com/drive/1u0i-LBog_lvQ5YRKs7QLKh_RtI-tV8qM?usp=sharing
- https://huggingface.co/ChatterjeeLab/PepMLM-650M
3. Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
4. To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
5. Record the perplexity scores that indicate PepMLM’s confidence in the binders.
| Binder | Sequence | Pseudo Perplexity |
|---|---|---|
| Binder 1 | WLYGATGLRLKK | 17.92783425 |
| Binder 2 | WRSGAVALELGX | 6.975748166 |
| Binder 3 | WRYYAVAAEWKX | 11.07016835 |
| Binder 4 | WRYGPAALAHKE | 10.98033022 |
| Binder 5 (example) | FLYRWLPSRRGG | / |
Part 2: Evaluate Binders with AlphaFold3
1. Navigate to the AlphaFold Server: http://alphafoldserver.com
2. For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
3. Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
4. In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
0.70–1.00 → strong, specific binding (good binder)
0.50–0.70 → moderate binding (possible binder, but uncertain)
0.30–0.50 → weak surface contact (likely not a real binder)
Below 0.30 → basically no meaningful binding
Across all five peptides, the ipTM values fell between 0.28 and 0.37, which places every binder in the “weak surface interaction” range (0.30–0.50) rather than showing strong or specific binding. Binder 4 slightly exceeded the ipTM of the known binder, with an ipTM of 0.37 compared to 0.35, but all showed similarly low-confidence, superficial binding. These results indicate that while the peptides contact SOD1’s surface, none form a stable or well defined interface, and no generated peptide displays stronger predicted binding than the known binder.
Binder 1: ipTM = 0.32, pTM = 0.79
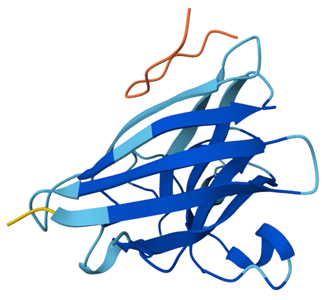
Binder 1 interacts only weakly with SOD1, remaining positioned on the outer surface without engaging the N terminus, β-barrel, or dimer interface.
Binder 2: ipTM = 0.36, pTM = 0.86
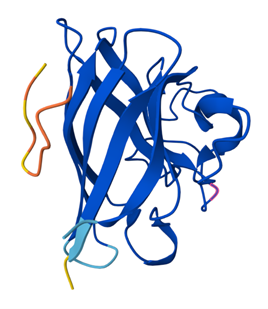
In the AlphaFold3 model, the peptide binds loosely on the surface of SOD1 rather than near the N terminus, β-barrel, or dimer interface.
Binder 3: ipTM = 0.28, pTM = 0.72
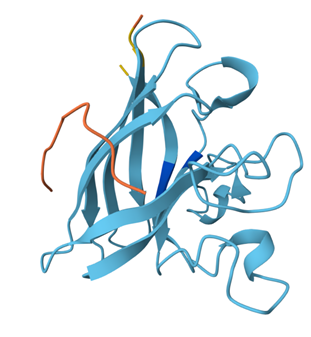
Similarly, this one also binds to the surface and does not penetrate or bind near anything meaningful.
Binder 4: ipTM = 0.37, pTM = 0.84
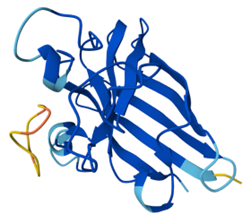
Binder 4 shows another weak binding on the surface; it doesn’t engage the N-terminus, β-barrel, or dimer interface.
Binder 5 (the example): ipTM = 0.35, pTM = 0.83
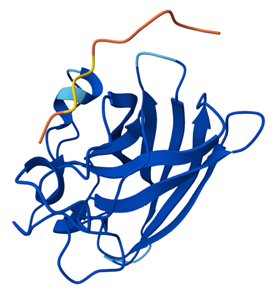
The known binder (Binder 5) shows a slightly more defined and closer interaction with the SOD1 surface compared to the generated peptides, but still binds only shallowly and does not specifically target the N terminus, β-barrel core, or dimer interface.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using https://huggingface.co/spaces/ChatterjeeLab/PeptiVerse, let’s evaluate the therapeutic properties of the peptide.
For each PepMLM-generated peptide:
- Paste the peptide sequence.
- Paste the A4V mutant SOD1 sequence in the target field.
- Check the boxes:
- Predicted binding affinity
- Solubility
- Hemolysis probability
- Net charge (pH 7)
- Molecular weight
The target sequence I used was:
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
| Binder (Sequence) | Binding Affinity (pKd/pKi) | Solubility (Probability) | Hemolysis Risk (Probability) | Net Charge (pH 7) | Molecular Weight (Da) | Hydrophobicity (GRAVY) |
|---|---|---|---|---|---|---|
| Binder 1 (WLYGATGLRLKK) | 5.961 (Weak) pKd/pKi | 1.000 | 0.061 (Non hemolytic) | +2.76 | 1405.7 Da | -0.23 |
| Binder 2 (WRSGAVALELGX) | 5.993 (Weak) pKd/pKi | 1.000 | 0.042 (Non hemolytic) | -0.24 | 1140.5 Da | +0.41 |
| Binder 3 (WRYYAVAAEWKX) | 6.612 (Weak) pKd/pKi | 1.000 | 0.041 (Non hemolytic) | +0.76 | 1424.7 Da | -0.56 |
| Binder 4 (WRYGPAALAHKE) | 5.336 (Weak) pKd/pKi | 1.000 | 0.014 (Non hemolytic) | +0.85 | 1398.6 Da | -0.84 |
| Binder 5 (FLYRWLPSRRGG) | 5.968 (Weak) pKd/pKi | 1.000 | 0.047 (Non hemolytic) | +2.76 | 1507.7 Da | -0.71 |
The PeptiVerse results show that all peptides had perfect predicted solubility and low hemolysis probability, meaning none are predicted to be especially toxic or poorly soluble. However, the predicted binding affinities were still weak across all peptides, which matches the AlphaFold3 observation that all binders only showed weak surface interactions. Peptides with higher ipTM did not necessarily show stronger predicted affinity. For example, Binder 4 had the highest ipTM, but one of the weakest affinity predictions, while Binder 3 had the strongest predicted affinity but the lowest ipTM. This means structural confidence and affinity prediction did not clearly align here. Overall, the peptides appear relatively safe, but none showed convincing strong binding behavior.
Choose one peptide you would advance and justify your decision briefly.
I would advance Binder 2 (WRSGAVALELGX) because it had one of the better ipTM scores among the generated peptides, while also showing perfect solubility, low hemolysis probability, and a relatively balanced net charge. Even though its affinity is still weak, it seems to offer the best overall trade-off between predicted binding and therapeutic properties.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. https://www.biorxiv.org/content/10.1101/2024.07.31.606098v2 uses Multi-Objective Guided Discrete Flow Matching (https://openreview.net/forum?id=8YIMLoHP9J) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
- Open the https://colab.research.google.com/drive/16n8PIwKwAiG-oDLm171BWvv-lQH0dHMg?usp=sharing linked from the https://huggingface.co/ChatterjeeLab/moPPIt.
- Make a copy and switch to a GPU runtime.
- In the notebook:
- Paste your A4V mutant SOD1 sequence.
- Choose specific residue indices on SOD1 that you want your peptide to bind.
- Set peptide length to 12 amino acids.
- Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
- After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
The moPPit peptides are quite different in their structure and thus, their properties. Many of the PepMLM peptides started with tryptophan and generally had very similar amino acids. Although the PepMLM peptides differed slightly in their binding, affinity, charge, and hydrophobicity, it is not particularly noteworthy. On the other hand the moPPit peptides varied in structure a lot relative to each other and the PepMLM, generally their affinity was a bit higher but still awfully low. What is interesting is that the hemolysis probability is roughly 20x higher for nearly all moPPit peptides compared to PepMLM. This would be very toxic as it likely will rupture blood cells, making the peptides very dangerous. Furthermore, before advancing moPPit generated peptides to clinical studies, further evaluation through structural docking validation to confirm stable protein–peptide interactions, toxicity and hemolysis screening to assess potential cellular damage, and in vitro stability assays to ensure peptide integrity under physiological conditions would be required to support therapeutic safety and effective binding to mutant SOD1.
| Binder (Sequence) | Binding Affinity (pKd/pKi) | Solubility (Probability) | Hemolysis Risk (Probability) | Net Charge (pH 7) | Molecular Weight (Da) | Hydrophobicity (GRAVY) |
|---|---|---|---|---|---|---|
| Binder 1 (WLYGATGLRLKK) | 5.961 (Weak) pKd/pKi | 1 | 0.061 (Non hemolytic) | 2.76 | 1405.7 Da | -0.23 |
| Binder 2 (WRSGAVALELGX) | 5.993 (Weak) pKd/pKi | 1 | 0.042 (Non hemolytic) | -0.24 | 1140.5 Da | 0.41 |
| Binder 3 (WRYYAVAAEWKX) | 6.612 (Weak) pKd/pKi | 1 | 0.041 (Non hemolytic) | 0.76 | 1424.7 Da | -0.56 |
| Binder 4 (WRYGPAALAHKE) | 5.336 (Weak) pKd/pKi | 1 | 0.014 (Non hemolytic) | 0.85 | 1398.6 Da | -0.84 |
| Binder 5 (FLYRWLPSRRGG) | 5.968 (Weak) pKd/pKi | 1 | 0.047 (Non hemolytic) | 2.76 | 1507.7 Da | -0.71 |
| moPPit Binder 1 (YVCYSYNYCVCH) | 7.878 | 0.833 | 0.911 | |||
| moPPit Binder 2 (TEKTTQAKKYCV) | 6.274 | 0.833 | 0.978 | |||
| moPPit Binder 3 (GDMTRYSYYKKC) | 6.790 | 0.916 | 0.964 |
Part B: BRD4 Drug Discovery Platform Tutorial
Assignees for the following sections
| MIT/Harvard students | Optional |
| Committed Listeners | Optional |
I did not include a written response for Part B.
Part C: Final Project: L-Protein Mutants
Assignees for the following sections
| MIT/Harvard students | Required |
| Committed Listeners | Required |
High level summary: The objective is to improve the stability and autofolding of the lysis protein.
More specifically, we want to engineer the lysis protein to increase the ability of MS2 to overcome a common E. coli resistance mechanism: a single point mutation in DnaJ prevents the binding of the lysis protein. We can attempt this by mutating the lysis protein to change its properties. Together, we aim for finding mutations that change the lysis protein in one of the following ways:
- an independence of lysis protein processing from DnaJ or other bacterial chaperones
- a faster or more efficient killing of E. coli to reduce the window in which the host can acquire resistance
- higher lysis protein expression
L-Protein Engineering | Option 1: Mutagenesis
Designing these mutants with good computational confidence is hard. It will show you limitations of some of the structure based models. Ultimately, you can pick various combinations of mutations and get lab results and then decide to pick the next round of mutations, but this assay will not be easy to run at scale in this class.
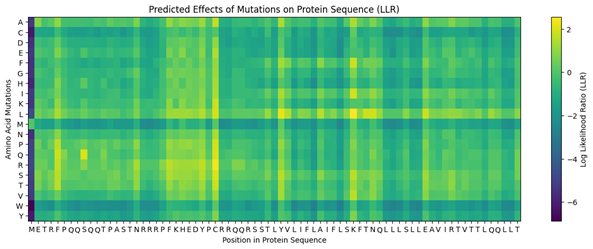
Run this notebook to generate for each position in the amino acid sequence, a “score” for what would happen to the protein if you mutated into another amino acid. It can be positive or negative for the protein. We want to identify possible mutations that are “positive”. If you run this notebook, you will see a .csv file in the sidebar. You can download it and look at it in google sheets if that’s easier.
- Use the experimental data here. This dataset contains information about mutants of the L-Protein and their effect on lysis in the lab.
4. First check, does the experimental data correlate with the scores from the notebook in (b)? This should give you a clue on how well these language embeddings capture information about this protein sequence.
The experimental data seems to correlate exactly with the scores from the notebook.
5. Using information about the effect of protein mutations at these sites - both the scores and the experimental data in the drive, come up with 5 mutations for each student along with how you came up with them and why you believe they would work. 2 of the variants you submit must have mutations in the transmembrane region (refer to notes above on what amino acid positions these are) and 2 of them must be in the soluble region. Remember that you can also use the pBLAST to see which residues are conserved and not mutate them if you want to.
One easy way to generate sequence mutations could be to look for residue positions and mutations that have a positive mutational effect either in the experimental or have a positive score from step 1. And pick a combination of those mutations.
The MS2 L-protein consists of a short N-terminal soluble domain (residues 1–37) and a single transmembrane α-helix located at residues 38–60.
https://www.uniprot.org/uniprotkb/P03609/entry
To select the final mutation set, I first focused on mutations that were positive in both the experimental dataset and the computational scoring results, thereby prioritising candidates supported by both experimental evidence and predicted mutational benefit. From this overlap, I chose the two highest-scoring mutations in the soluble region (E25G and E25V) and the only positive candidate from the transmembrane region (A45P). For the final two mutations, I selected the highest-scoring computational candidates that were not experimentally tested: K50L and Y39L; both are located in the transmembrane region.
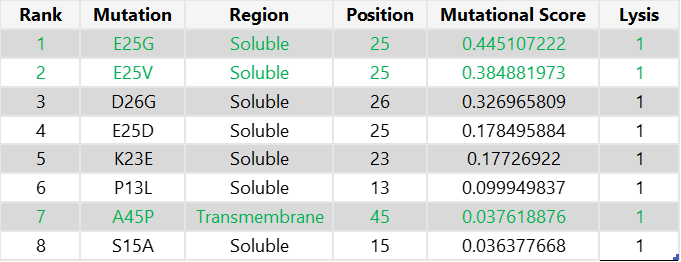
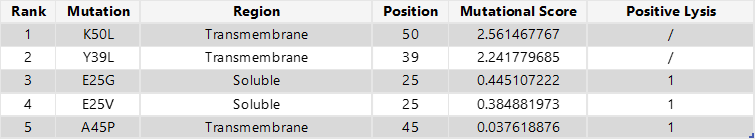
Final 5 mutations:
- E25G
- E25V
- A45P
- K50L
- Y39L
6. You can utilize Af2_Multimer to generate a Multimeric Assembly; you can do this by making your query sequence as. We want to do this because - A running hypothesis for how this protein functions is that it assembles to make a perforation in the bacterial membrane.
Figure generated using the following multimeric assembly (where each chain is separated from the other with a :):
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT:METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT