Biological Engineering Application Proposed application: Engineered microbial biofactories for small-molecule drug production.
Having a background in Biomedical Science and current training in translational physiology and pharmacology, I am particularly interested in small-molecule drug development. This project proposes engineering commercially viable cells capable of producing small-molecule drugs that are difficult or costly to synthesize using traditional chemical methods.
Part 1: Benchling & In-silico Gel Art
This is the Lambda Sequence
This is the Lambda sequence with the cuts
First, I tried to use Ronan’s website to get a template, and then I made it in Benchling
Template:
Benchling:
3.1. Choose your protein.
I decided to use the Kappa Opioid GPCR, as this is the target for my biofactory, which is for my final project. It comes from the OPKR1 gene, and UniProt entry P41145 (OPRK_HUMAN) lists the canonical human kappa opioid receptor as 380 amino acids with this sequence:
Homework Submission Design Explanation For the art, I first sketched ideas on my tablet and then tried to upload them as an image on OpenTrons. The image with birds flying through the sky was pleasing to me aesthetically, but there were limited colours available to us. I chose the following palm tree and adapted some of the colours.
Bio Design & Bio Fabrication (Michael Chen, Christina Agapakis) Lab: Final Project work
Subsections of Homework
Week 1 HW: Principles and Practices
1. Biological Engineering Application
Proposed application: Engineered microbial biofactories for small-molecule drug production.
Having a background in Biomedical Science and current training in translational physiology and pharmacology, I am particularly interested in small-molecule drug development. This project proposes engineering commercially viable cells capable of producing small-molecule drugs that are difficult or costly to synthesize using traditional chemical methods.
Inspired by microbial production of compounds such as penicillin, this approach would use bacterial or alternative host cells to generate either full drug molecules or high-value intermediates, depending on chemical feasibility. Using CRISPR or prime editing, metabolic pathways would be modified to enhance yield and specificity, similar in concept to the work by Paddon et al. (Nature, 2013).
As a proof of concept, the kappa opioid receptor (KOR) is selected as the biological target, with Salvinorin A as the compound of interest. Chemical synthesis of Salvinorin A suffers from extremely low yields (~0.15–5%), making it expensive and impractical for large-scale research. Improving yield through microbial biosynthesis would reduce costs, accelerate KOR research, and support the development of novel analgesics.
2. Governance & Policy Goals
Goal 1: Safety
1a. Prevent misuse of engineered microbes to produce psychoactive or harmful substances
1b. Prevent harmful exposure to laboratory personnel
Goal 2: Equal Opportunity
2a. Maintain low production costs to ensure global accessibility
2b. Avoid monopolization of the technology and promote open access
Goal 3: Ethical Innovation
3a. Encourage transparent reporting of methods, yields, and failures
3b. Align research incentives with public health goals, particularly analgesic development
3. Governance Actions
Action 1: Biosafety Review (DURC)
Purpose: Identify misuse and safety risks in engineered microbes producing bioactive compounds
Design: Mandatory dual-use and toxicity assessments by IBCs; compliance tied to funding and regulatory approval
Assumptions: Honest reporting; predictable risks
Risks & Success:
Failure: Bureaucratic burden slows research
Success: Improved biosecurity and public trust
Action 2: Genetic Kill Switches
Purpose: Prevent environmental escape or uncontrolled proliferation
Design: Engineered auxotrophy and kill-switch mechanisms; incentives via funding and approvals
Assumptions: Stability and affordability of safeguards
Risks & Success:
Failure: Mutation or safeguard failure
Success: Reduced environmental risk
Action 3: Pharmacovigilance
Purpose: Monitor production and use of KOR-targeted molecules
Design: Controlled distribution; adverse-event reporting by clinicians
Assumptions: Reliable detection and reporting
Risks & Success:
Failure: Under-reporting or diversion
Success: Safe translation without blocking research
4. Governance Scoring Matrix
Policy Goal
Option 1
Option 2
Option 3
Prevent biosecurity incidents
1
1
2
Respond to incidents
2
2
1
Prevent lab safety incidents
1
1
n/a
Environmental protection
2
1
n/a
Minimize burden
2
2
3
Feasibility
1
2
2
Avoid impeding research
2
2
3
Promote constructive use
1
1
2
5. Prioritization & Ethical Reflection
Based on the scoring, Options 1 (DURC biosafety review) and 2 (genetic kill switches) are the highest priorities. These address the most immediate ethical risks associated with misuse, environmental contamination, and accidental exposure.
While pharmacovigilance is important, it becomes more relevant at later translational stages. Trade-offs include increased upfront costs and longer development timelines; however, these are justified by improved safety, transparency, and public trust.
The primary ethical concern identified during this week’s coursework is dual-use misuse of engineered microbes, including unauthorized production or environmental release. Strong oversight, transparency, and adherence to biosafety protocols should sufficiently mitigate these risks.
Homework – Lecture 2 Questions
George Church Question
Question: What are the 10 essential amino acids in all animals, and how does this affect the “Lysine Contingency”?
Answer: The 10 essential amino acids in animals are:
Histidine
Isoleucine
Leucine
Lysine
Methionine
Phenylalanine
Threonine
Tryptophan
Valine
Arginine
Animals cannot synthesize lysine endogenously and must obtain it from their environment. This weakens the concept of the lysine contingency as a biosafety mechanism for engineered organisms. Many natural environments already contain lysine, meaning deprivation is unreliable. Additionally, organisms can evolve around this dependency, making lysine-based containment a fragile and insufficient safety strategy on its own.
Week 2 HW: DNA Read, Write, & Edit
Part 1: Benchling & In-silico Gel Art
This is the Lambda Sequence
This is the Lambda sequence with the cuts
First, I tried to use Ronan’s website to get a template, and then I made it in Benchling
Template:
Benchling:
3.1. Choose your protein.
I decided to use the Kappa Opioid GPCR, as this is the target for my biofactory, which is for my final project. It comes from the OPKR1 gene, and UniProt entry P41145 (OPRK_HUMAN) lists the canonical human kappa opioid receptor as 380 amino acids with this sequence:
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
A convenient “answer key” for the corresponding human CDS is provided in KEGG for hsa:4986 (OPRK1), showing a 1143 nt coding region (380 aa + stop) https://www.genome.jp/dbget-bin/www_bget?hsa:4986
3.3. Codon optimisation.
Codon optimisation is done to match the host codon usage to improve the translation efficiency and protein yield.
For the sake of this protein, I chose E. coli for simplicity so I can practice this according to the homework
Technology 1: Clone codon-optimized CDS into a bacterial plasmid (T7/lac promoter), transform into E. coli. Codon optimization is used to match host codon bias to improve expression.
Technology 2: Induce expression; proteinproduction follows the same central dogma (DNA to RNA to protein), but membrane insertion/folding for 7TM proteins is a key challenge in bacteria.
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why?
This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I think it could be interesting to sequence small genetic regions related to caffeine metabolism and sensitivity; CYP1A2, AHR, ADORA2A. This is relevant as many individuals experience coffee and caffene differently and do not enjoy it as much as others, or experience more abhorrent side effects than others. By analysing the metabolism of caffeine from CYP1A2 and investigating the adenosine receptors, a specialised suggestion of caffeine intake, bean type, and coffee type can be permutated to give people the best experience with minimal side effects.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Also answer the following questions:
Is your method first-, second- or third-generation or other? How so?
Sanger sequencing is 1st-generation sequencing.
It reads DNA by creating DNA fragments terminated by special nucleotides (ddNTPs) and separating them by capillary electrophoresis to infer the base order
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
The input would be genomic DNA from a cheek swab or saliva sample. The steps to prepare them would be to Extract DNA from the sample, PCR amplify the short region(s) containing the SNP(s), to purify the PCR product and then set up Sanger sequencing reaction
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
In Sanger sequencing, you first make many DNA copies, but the copying sometimes stops when a special “terminator” base (a fluorescent ddNTP) is added. This creates lots of DNA fragments of different lengths, each ending in a colored base. The fragments are then separated by capillary electrophoresis, and a detector reads the color signal as fragments pass by. The sequencing software converts the color peaks into A, C, G, T letters
What is the output of your chosen sequencing technology?
The output of Sanger sequencing is usually a chromatogram/trace file (often .ab1) showing colored peaks, plus a text DNA sequence that the software called from those peaks
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
I want to synthesize plasmid DNA vectors that turn bacteria into biofactories for making human-useful therapeutics, specifically:
a therapeutic hormone for metabolic disease (example: human insulin)
a small-molecule product relevant to cardiovascular/metabolic health (example: Coenzyme Q10 (CoQ10) as a medically used antioxidant supplement with cardiovascular relevance)
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Also answer the following questions:
What are the essential steps of your chosen sequencing methods?
The essential steps are: (1) computational design of the DNA construct (promoters/RBS/genes/terminators plus plasmid features), (2) chemical DNA writing using cyclic solid-phase phosphoramidite synthesis to generate oligos, (3) assembly of oligos into longer gene-length fragments when needed, (4) cloning/packaging into a plasmid backbone for bacterial expression, and (5) sequence verification/quality control so the final plasmid matches the intended design. These steps reflect the standard phosphoramidite cycle used for oligo construction and the gene/plasmid workflow offered by commercial synthesis providers
What are the limitations of your writing method (if any) in terms of speed, accuracy, scalability?
A key limitation is that errors accumulate as DNA length increases, because each chemical base-addition step is not perfectly efficient; this means long constructs are more likely to contain substitutions or deletions and often require assembly from shorter pieces plus verification. In addition, although high-throughput synthesis platforms scale very well for many sequences in parallel, overall cost and turnaround time can still be bottlenecks for very large libraries or long, complex constructs, and certain sequence patterns (like repeats or extreme GC content) can reduce synthesis success and increase the need for troubleshooting or redesign
5.3 DNA Edit
(i) What DNA would you want to edit and why?
In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
I would want to edit human DNA SNPs that cause metabolic/cardiovascular disease, especially those that lead to very high LDL cholesterol and early heart disease risk, such as variants involved in familial hypercholesterolemia (FH). FH is commonly linked to harmful variants in LDLR (and sometimes related genes like APOB or PCSK9), and editing these could lower lifelong LDL exposure and reduce cardiovascular
(ii) What technology or technologies would you use to perform these DNA edits and why?
Also answer the following questions:
How does your technology of choice edit DNA? What are the essential steps?
In prime editing, a Cas9 nickase fused to a reverse transcriptase is guided to a specific DNA site by a pegRNA that also contains the template for the desired change; the system nicks DNA and then “writes” the corrected sequence, which cellular repair processes finalize into a stable edit. This avoids relying on the same double-strand-break repair competition that often makes precise HDR edits difficult
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
The main preparation is selecting the exact SNP to fix (e.g., an LDLR pathogenic variant) and designing the appropriate guide/pegRNA to target it. The key inputs are the editor components (prime editor or base editor), the guide RNA(s), and the target human cells/tissue context (for cholesterol disorders this is often discussed in relation to the liver because it controls LDL metabolism).
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
Major limitations include variable editing efficiency, possible off-target changes or unintended byproducts, and the practical challenge of safe, effective delivery of the editing system to the correct tissue in humans.
Week 3 HW: Lab Automation
Homework Submission
Design Explanation
For the art, I first sketched ideas on my tablet and then tried to upload them as an image on OpenTrons. The image with birds flying through the sky was pleasing to me aesthetically, but there were limited colours available to us. I chose the following palm tree and adapted some of the colours.
Furthermore, after receiving feedback on my design, I minimised colours and removed an outer edge to ensure the image is clearer and to prevent spillage
–
Homework Questions
1. Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Norton-Baker B, Denton MCR, Murphy NP, Fram B, Lim S, Erickson E, et al. Enabling high-throughput enzyme discovery and engineering with a low-cost, robot-assisted pipeline. Sci Rep. 2024;14(1):14449. http://dx.doi.org/10.1038/s41598-024-64938-0
This article by Baker et al. describes a robot-assisted pipeline for enzyme engineering. The article explains how, with the advances of AI and genome data, it is extremely taxing to manually test every permutation. There is simply too much to test using the standard laboratory methods. The main bottleneck they have found is the production, purification and characterisation of the proteins. The author’s attempt to make a generalisable protocol that is cost-effective and high-throughput to streamline enzyme discovery. Their idea is to use robot-assisted pipelines using the opentrons OT-2 liquid handling robot on a 96-well plate to scale to hundreds of proteins per week.
The workflow specifically uses the OT-2 to automate time-consuming steps like transforming E. coli with plasmids, inoculation, lysing, and protein purification using Ni-charged magnetic beads. The workflow also cleaves proteins with a protease instead of eluting to make it easier to use in downstream assays. The authors did this for PET-degrading proteins to screen for enzymes to degrade plastics. They did this using 23 published hydrolases, which were expressed and purified and then measured their thermostability and activity.
Using this pipeline, they were able to have repeatable results after doing 3 separate trials. Across replicate wells and runs, they obtained reproducible enzyme yields reaching up to 400 μg for some proteins, and verified that the samples were sufficiently pure and correctly sized using SDS-PAGE. Finally, the authors use the purified enzymes to generate a benchmark dataset by testing stability (via DSF melting temperatures) and activity across a large matrix of conditions (including different pH values, temperatures, substrates, and timepoints), which lets them rank enzyme performance in a standardised way. In their side-by-side benchmark, LCC-A2 consistently generated the largest amounts of PET breakdown products confirmed by UV-Vis and HPLC ratios, making it the strongest overall performer under their assay setup.
2. Write a description about what you intend to do with automation tools for your final project.
For my final project involving decaffeination using synthetic bacteria, automation could help in testing many enzymes and/or bacterial strains simultaneously. Similar to the article I described in the previous question, it can be helpful to use automation, such as OpenTrons, to identify the best enzyme rather than manually testing each prospective enzyme. Similar to the article by Baker et al., a 96-well screen could be done to test a different enzyme/strain condition (dose, pH, temperature, time). The OT-2 would automate all pipetting by adding tea/coffee, buffers/cofactors, enzyme/strain inputs, and pulling timed samples into a quench plate making screening faster.To ensure this doesn’t create unwanted flavour-related byproducts, I’d measure not only caffeine reduction but also methylxanthine byproducts, which can vary depending on the enzyme/strain used.
Furthermore, as for my idea regarding the biological synthesis of Salvinorin A or Paclitaxel, a common class of molecules among them is terpenes. This is a type of molecule usually derived via plant extraction. Thus, this could be a good target to screen using automation. The principle is similar to decaffeination, where an array of enzymes and pathways could be tested at the same time with an OT-2 workflow with P450 enzymes, followed by analytical tools like HPLC readouts to examine successful synthesis.
Week 04: HW protein design part I
This week will focus on how sequence, structure, and energetics can be modeled and manipulated to create or optimize proteins with specified functions…
Objective:
Learn basic concepts:
amino acid structure
3D protein visualization
the variety of ML-based design tools
Brainstorm as a group how to apply these tools to engineer a better bacteriophage (setting the stage for the final project).
Part A. Conceptual Questions
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
I answered nine of the conceptual questions below.
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
It is about 3 *10^{24} amino acid molecules.
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
We digest proteins into amino acids and then rebuild them into human proteins.
3. Why are there only 20 natural amino acids?
Life evolved to use a small set that is enough for many protein functions.
5. Where did amino acids come from before enzymes that make them, and before life started?
They may have formed through prebiotic chemistry on early Earth or arrived from space.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
I would expect a left-handed helix.
8. Why are most molecular helices right-handed?
Most natural proteins use L-amino acids, which usually favor right-handed helices.
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheets aggregate because their backbones can make many hydrogen bonds. Hydrophobic interactions also help drive aggregation.
10. Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Amyloid proteins often stack into stable β-sheet-rich fibers. Yes, amyloid β-sheets can also be used as strong biomaterials.
11. Design a β-sheet motif that forms a well-ordered structure.
A simple design is an alternating pattern of hydrophobic and polar residues, such as Val-Lys-Val-Glu-Val-Lys-Val-Glu. This can help form a stable β-sheet.
Part B: Protein Analysis and Visualization
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
In this part of the homework, I used online resources and 3D visualization software to analyze a protein of interest that has a known 3D structure.
1. Briefly describe the protein you selected and why you selected it.
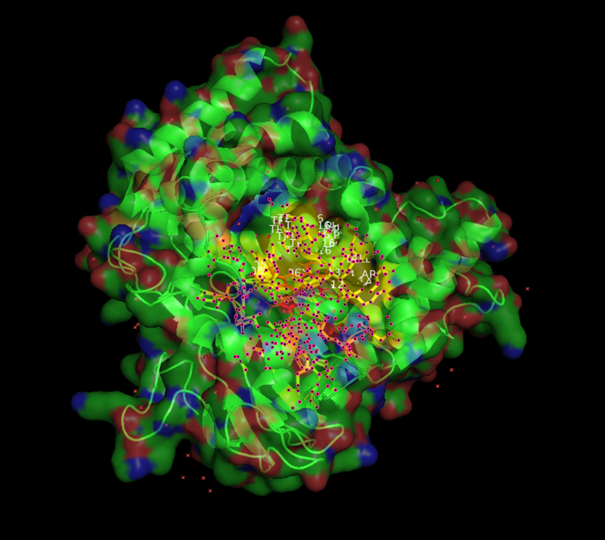
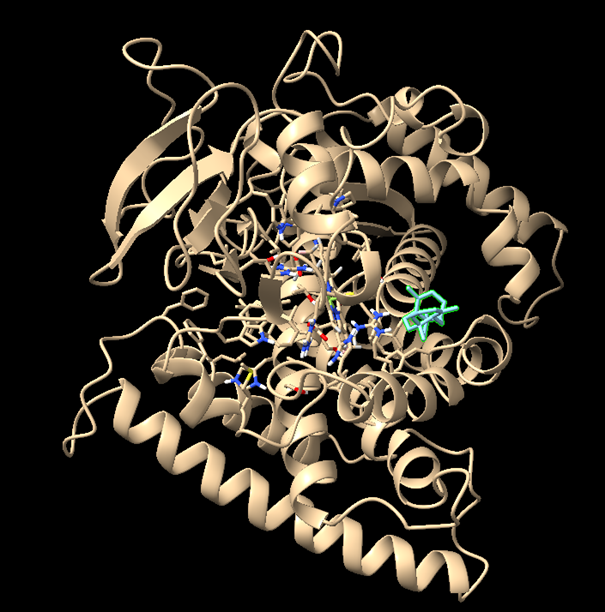
From now on I have decided to focus on paclitaxel for my final project and to optimize a crucial bottleneck in its current biosynthesis. Thus, the protein I wanted to visualize is beta-tubulin, which is the target of paclitaxel in cancer treatment.
2. Identify the amino acid sequence of your protein.
The structure was solved in 2022 using electron microscopy, with a resolution of 3.20 Å. This is a reasonable quality structure, although it is not as high resolution as some crystal structures.
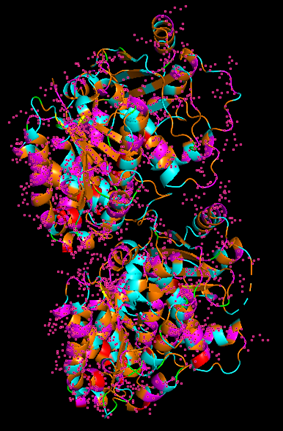
There are other molecules in the solved structure apart from the protein. Specifically, the structure is a dimer of the beta and alpha tubulin subunits, so this structure contains one beta subunit and one alpha subunit.
Based on what I found, it does not appear to belong to a structure classification family in the way asked by the prompt.
4. Open the structure of your protein in any 3D molecule visualization software.
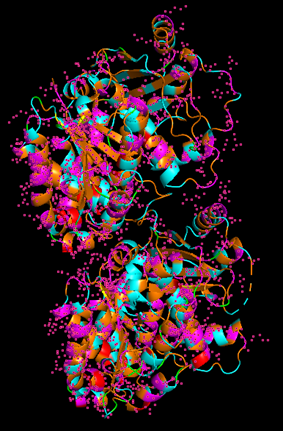
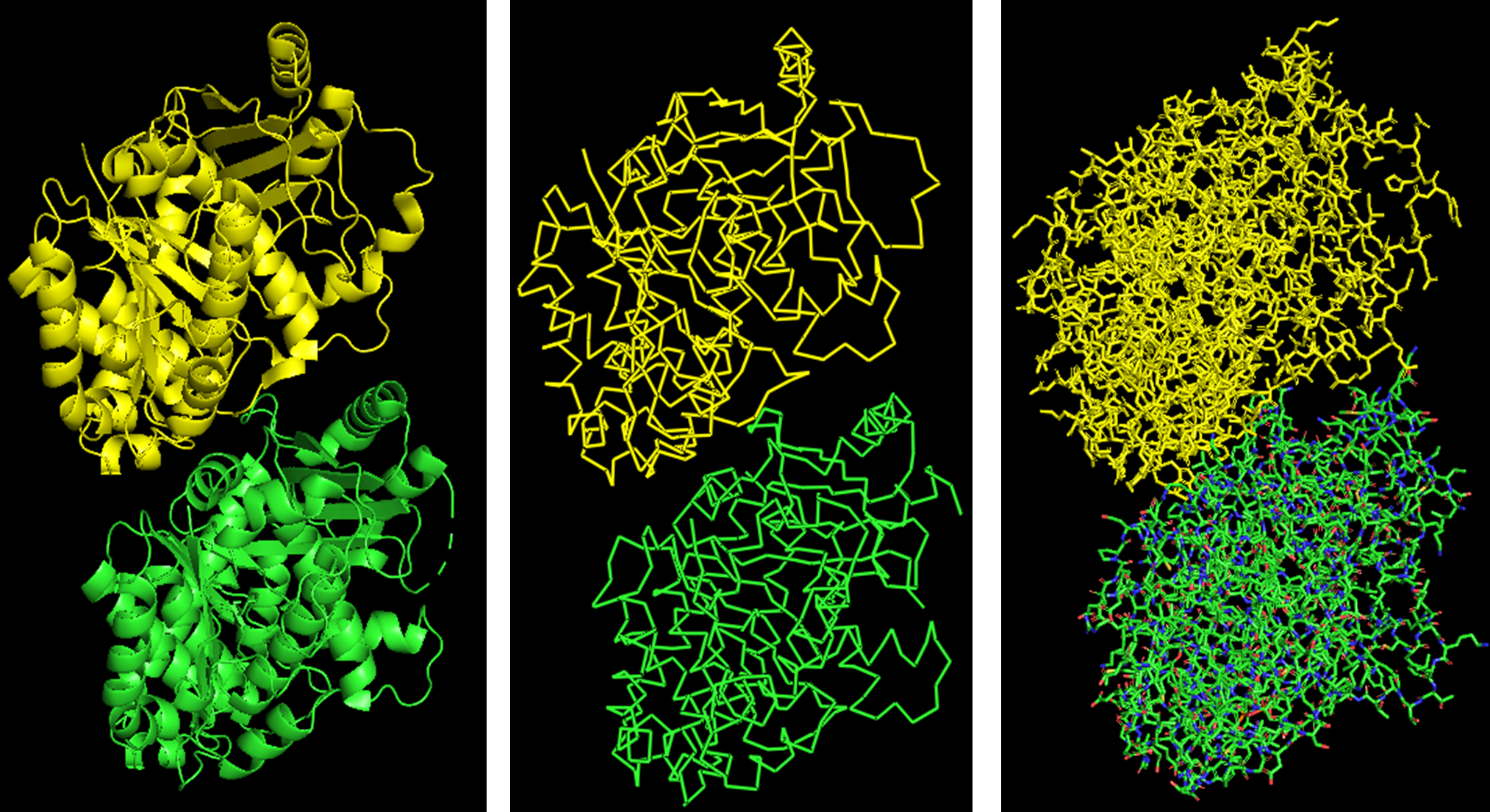
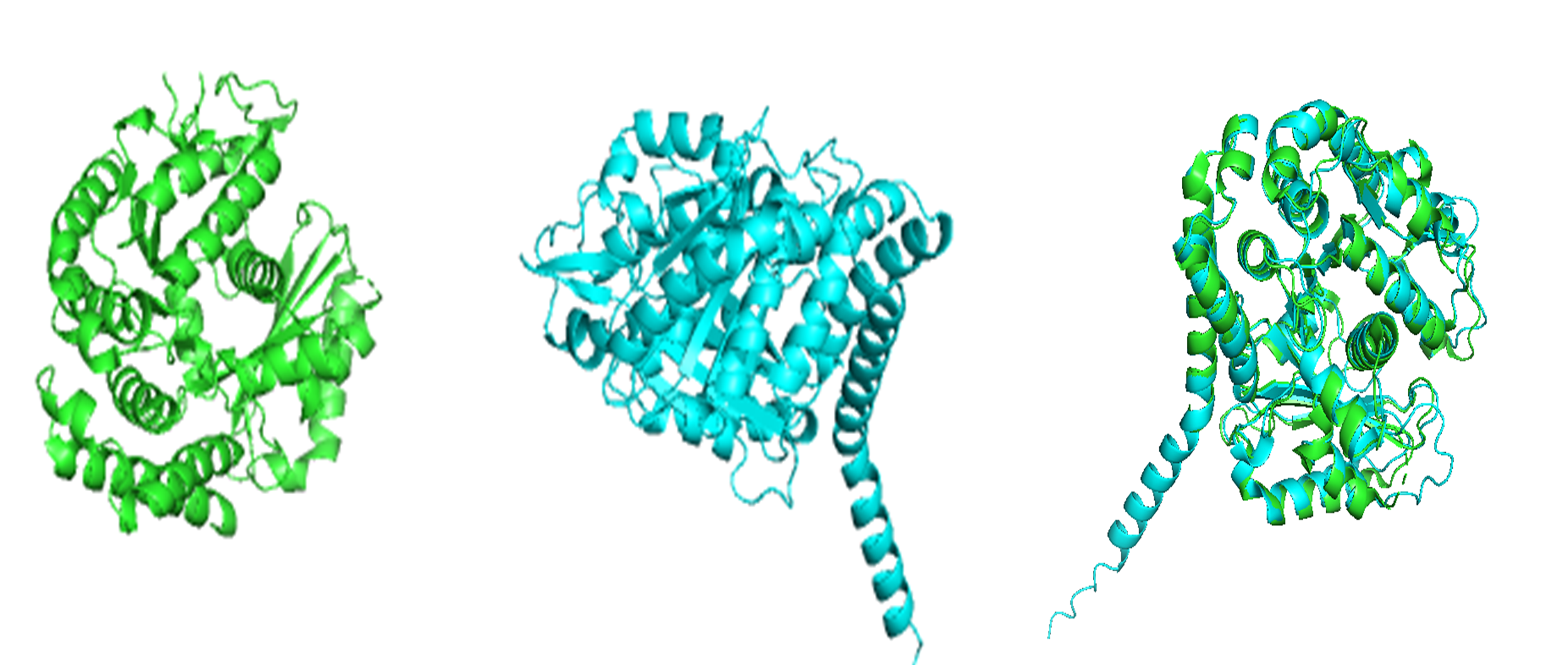
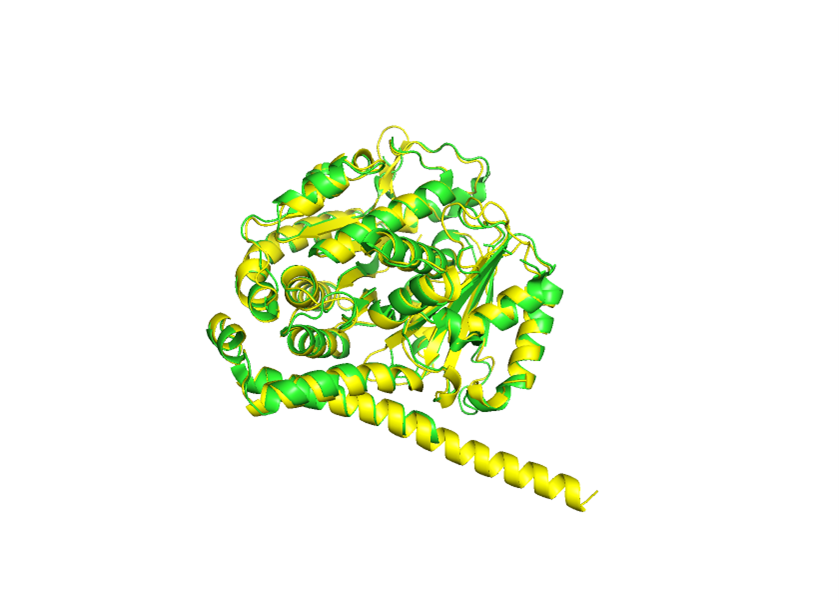
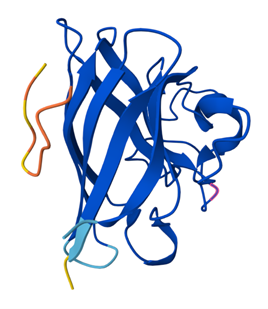
Because the resolved structure contains two proteins from the microtubule, I colored the alpha-tubulin yellow and the beta-tubulin green, since paclitaxel binds to beta-tubulin in cancer treatment.
Cartoon representation of the alpha- and beta-tubulin structure.
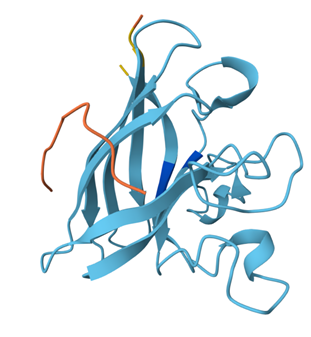
When coloring the protein by secondary structure, the structure seemed to have more helices than sheets, with a ratio of approximately 3:1 (3501:1074).
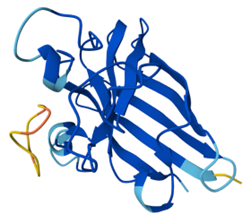
When coloring the protein by residue type, the visualization showed hydrophobic residues with 2704 atoms in orange, and hydrophilic residues with 3732 atoms in cyan/magenta. The hydrophilic residues seemed to be distributed more on the outer parts of the structure, while the hydrophobic parts were more concentrated on the inner regions.
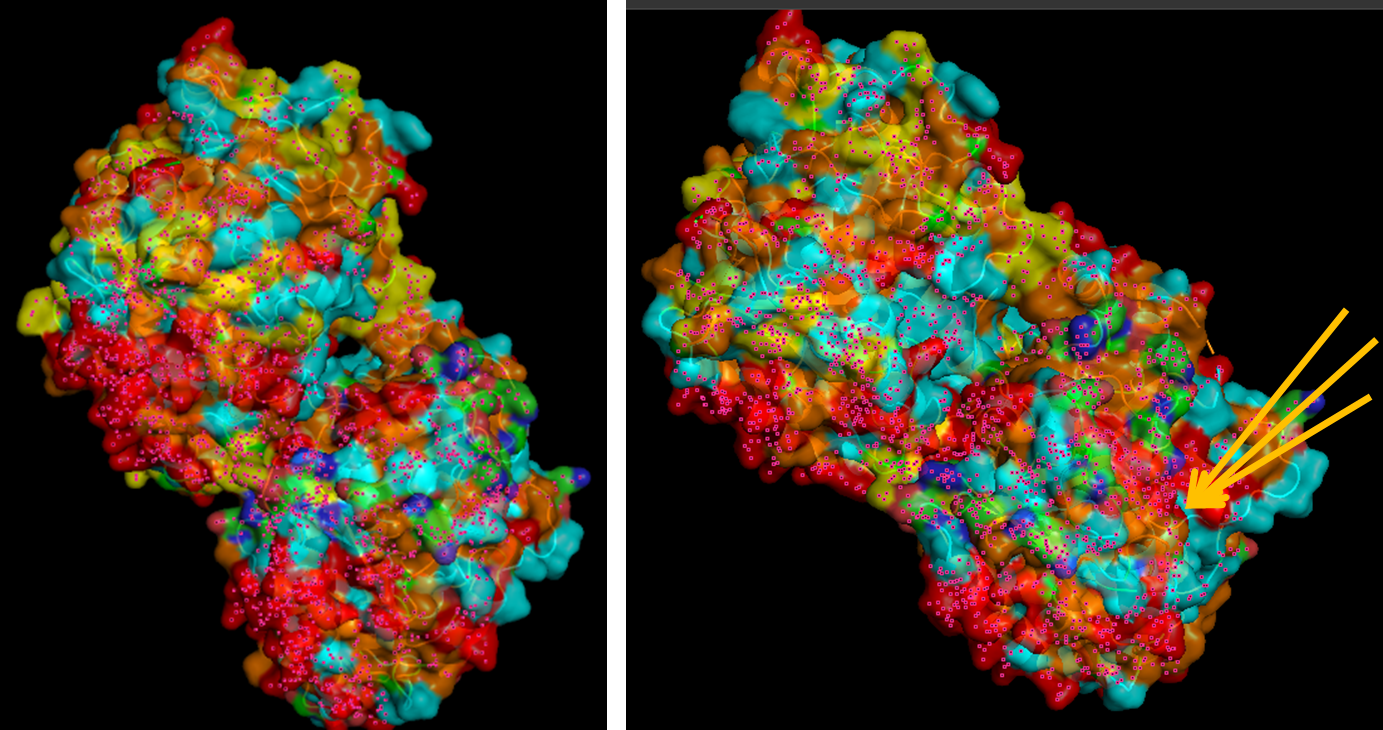
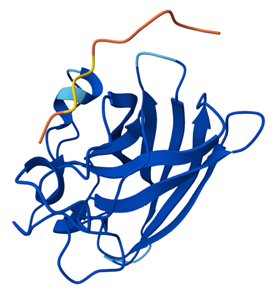
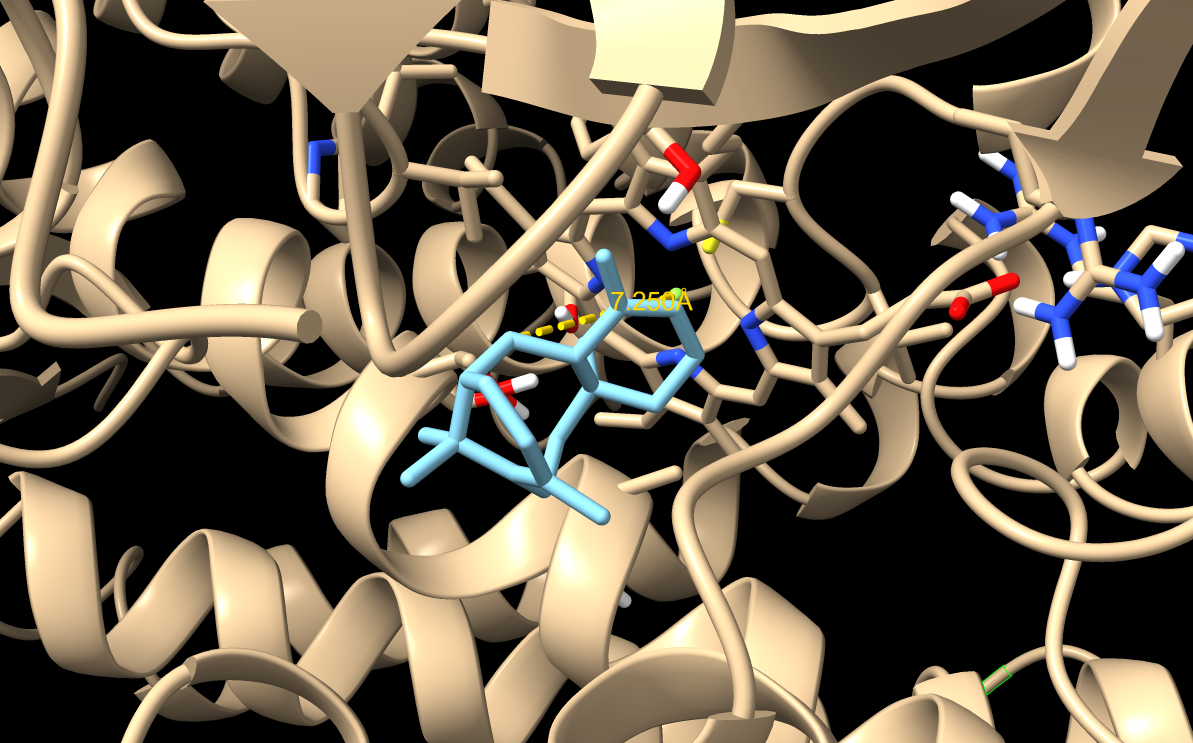
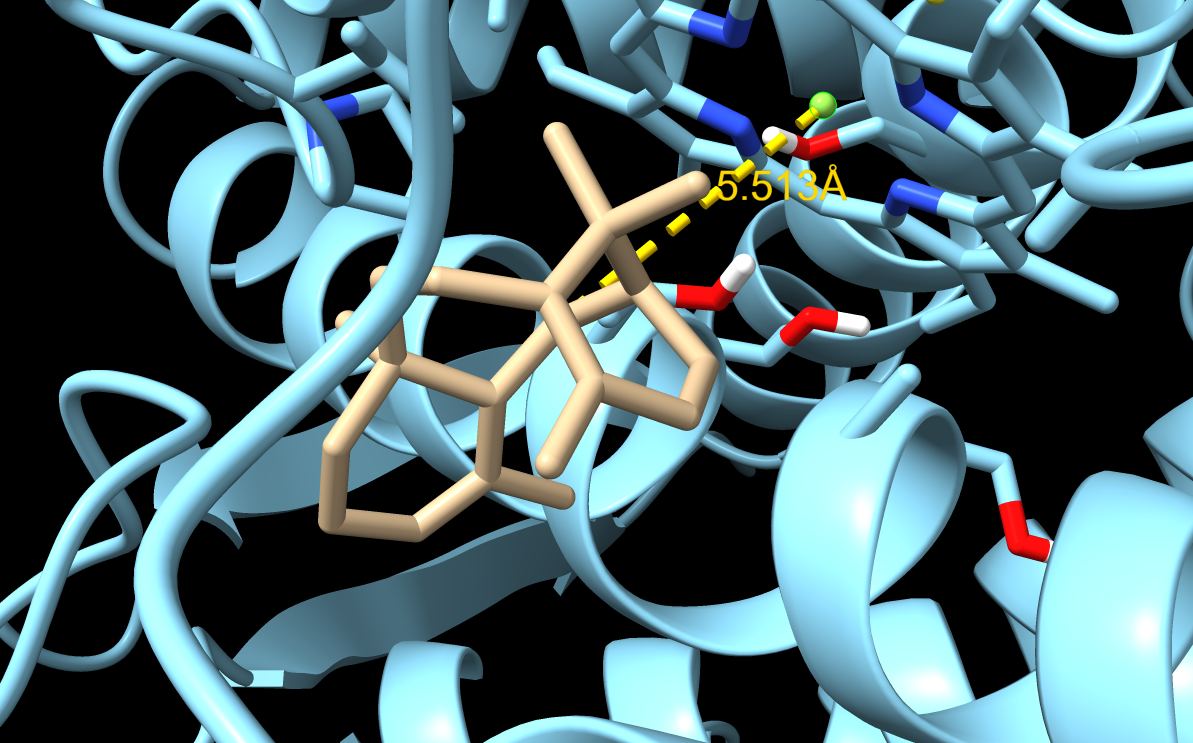
When visualizing the surface of the protein, it looked like there is a binding pocket between the two proteins. I compared my image to the known paclitaxel binding pocket and indicated it with an arrow. However, this visualization alone is not completely clear, and other methods would analyze this more reliably.
Part C. Using ML-Based Protein Design Tools
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
In this section, I tested several modern protein AI models on my chosen protein.
I selected the following PDB sequence, which was slightly different from the UniProt sequence:
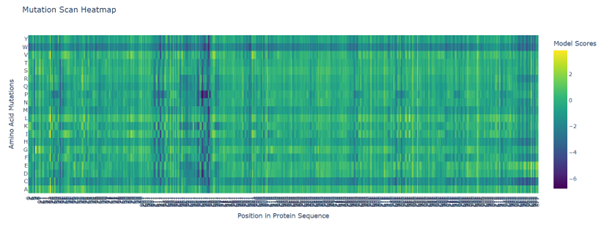
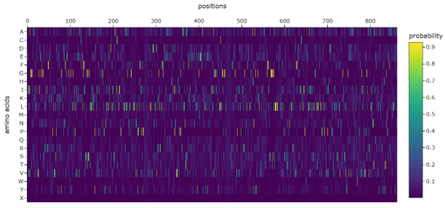
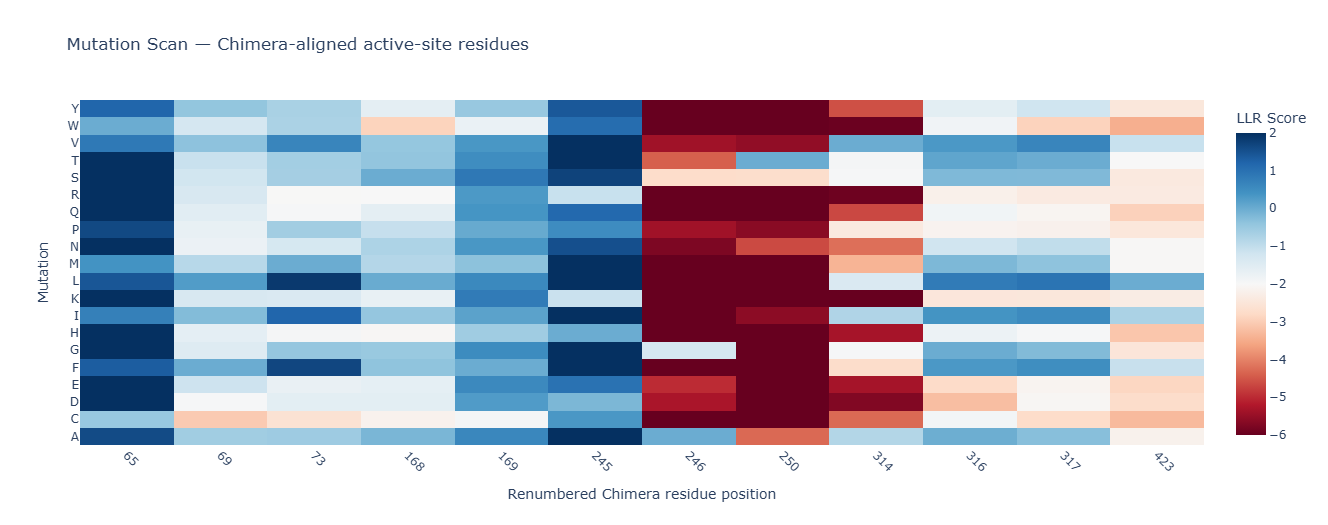
b. Can you explain any particular pattern? (choose a residue and a mutation that stands out)
Positions 140–160 form a structurally constrained region where most substitutions are harmful. In particular, position 158 is intolerant to bulky hydrophobic residues such as tryptophan, tyrosine, and valine, while position 153 tolerates only flexible or polar residues such as serine, arginine, glycine, proline, and asparagine. This suggests loop-specific flexibility requirements.
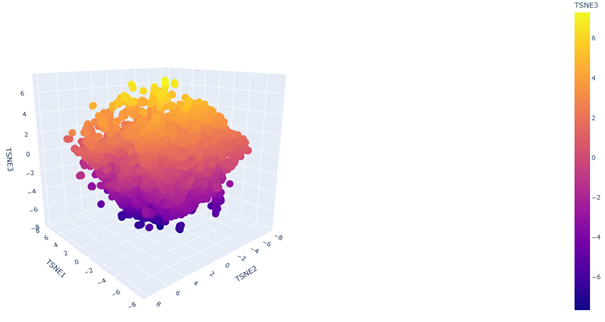
c. Place your protein in the resulting map and explain its position and similarity to its neighbors.
2. Latent Space Analysis
b. Analyze the different formed neighborhoods: do they approximate similar proteins?
Yes. In the 3D latent space, nearby points form local neighborhoods of proteins with similar sequence features, indicating that the embedding groups related proteins together.
C2. Protein Folding
Folding a protein
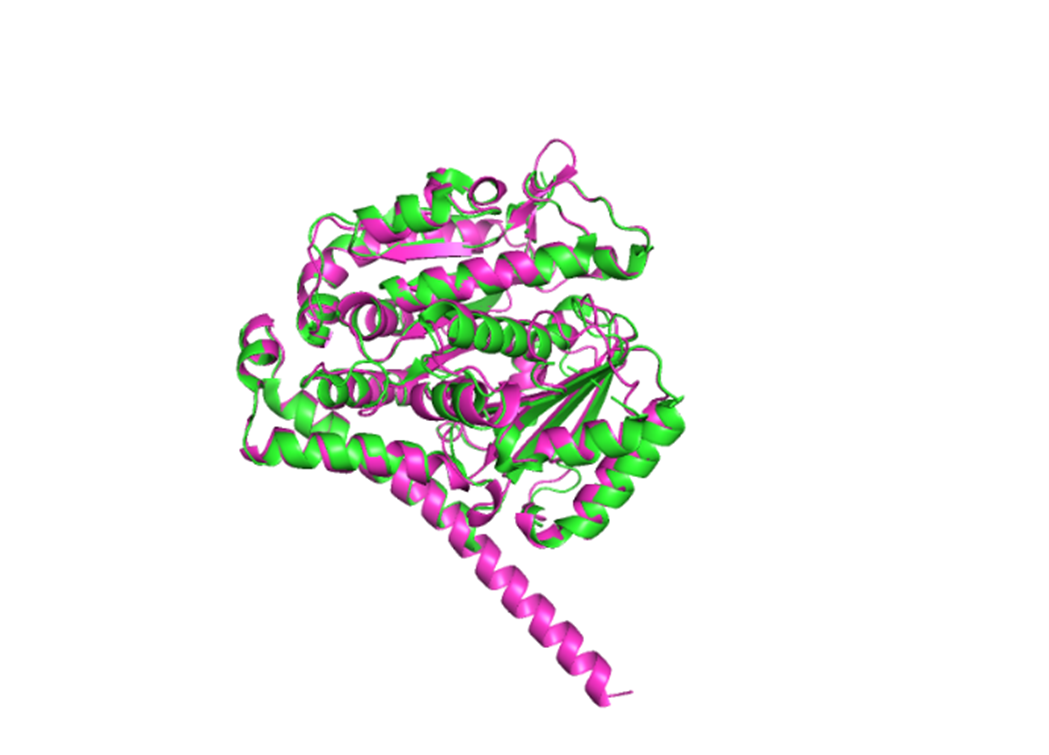
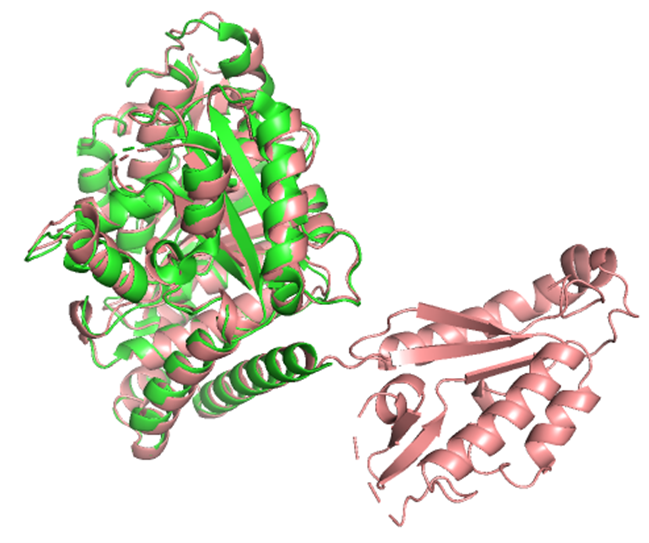
1. Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
The structure predicted by ESMFold aligns very well with the experimentally determined β-tubulin structure. The low RMSD of 0.782 Å after alignment indicates that the predicted atomic coordinates closely match the original structure.
Result:RMSD = 0.782
2. Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
For small mutations, I tested the following sequence:
This was quite surprising, because I expected a much larger variance to occur. Based on these results, the overall fold appears to be fairly resilient to both small and somewhat larger sequence changes.
C3. Protein Generation
Inverse-Folding a protein
1. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
The predicted sequence has a better ProteinMPNN score (0.8587) than the original sequence (1.8495), with a sequence recovery of 0.3783. This means ProteinMPNN proposed a substantially different sequence that it still predicts will fit the same backbone.
2. Input this sequence into ESMFold and compare the predicted structure to your original.
The predicted structure had an RMSD of 0.756 Å relative to the original. The main structure seems to be well preserved, but there appears to be one extra part that is not present in the original structure, which is quite strange.
Part D. Group Brainstorm on Bacteriophage Engineering
Assignees for the following sections
MIT/Harvard students
Optional
Committed Listeners
Required
Proposal: Computational engineering of the MS2 L protein for increased stability and higher titers
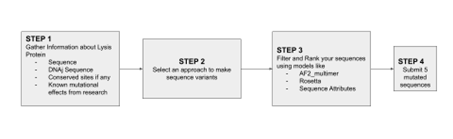
For my project, I will focus on two goals for the MS2 L protein: increased stability and higher phage titers. I chose these because the literature suggests that L contains a small but sensitive functional core, and that lysis timing likely influences how many phage particles are produced before the host cell breaks open. In simple terms, I want a version of L that is more reliable as a protein, but not so aggressive that it causes lysis too early.
My main approach would be to combine sequence conservation analysis, in silico mutagenesis, protein language model scoring, and structure/topology prediction. I would first identify residues that are likely too important to mutate, especially around the conserved LS motif, since mutational studies show that this region is highly sensitive and likely involved in an essential interaction. I would also treat the basic N-terminal region carefully, because it regulates activity through interaction with DnaJ rather than acting as the main lytic domain itself.
Next, I would computationally test mutations that might improve folding robustness or membrane association while preserving the protein’s core lytic features. This seems reasonable because recent work suggests that MS2-L forms oligomeric assemblies in membrane-like environments, and that the transmembrane/C-terminal region is central to this behavior. In plain language, I am not only asking whether the protein folds, but whether it can still adopt the right shape and assembly state to work properly.
For the higher titer goal, I would not try to predict titers directly. Instead, I would use a proxy strategy and prioritize variants that are predicted to be more stable while still preserving the regulatory features that may prevent premature lysis. This is important because N-terminal truncations can bypass DnaJ and trigger earlier lysis, which may actually reduce phage output if the host is killed before assembly is complete.
Planned pipeline
Collect sequence, mutational, and structural/topology information for L.
Mark function-sensitive residues and conserved regions.
Run in silico mutagenesis and rank variants with language-model or sequence-based scores.
Filter variants using membrane topology and structural plausibility.
Prioritize candidates that may improve stability while preserving productive lysis timing.
Potential pitfalls
One pitfall is that higher titers are not controlled by L alone, so even a better L variant may not improve total phage yield. Another is that MS2 has overlapping genes and RNA-level regulation, so a mutation that looks good for the protein might still be harmful in the native phage genome.
Week 5: HW protein design part II
This week we learn how cutting-edge AI and protein language models are used to design functional proteins and peptides “in silico”.
Objective:
Design short peptides that bind mutant SOD1.
Decide which peptides are worth advancing toward therapy.
Evaluate generated peptides using structural and therapeutic-property prediction tools.
Think about how computational protein design can be applied to the final project on L-protein mutants.
Part A: SOD1 Binder Peptide Design
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Your challenge:
Design short peptides that bind mutant SOD1.
Then decide which ones are worth advancing toward therapy.
You will use three models developed in our lab:
PepMLM: target sequence-conditioned peptide generation via masked language modeling
2. For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
3. Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
4. In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
0.70–1.00 → strong, specific binding (good binder)
0.50–0.70 → moderate binding (possible binder, but uncertain)
0.30–0.50 → weak surface contact (likely not a real binder)
Below 0.30 → basically no meaningful binding
Across all five peptides, the ipTM values fell between 0.28 and 0.37, which places every binder in the “weak surface interaction” range (0.30–0.50) rather than showing strong or specific binding. Binder 4 slightly exceeded the ipTM of the known binder, with an ipTM of 0.37 compared to 0.35, but all showed similarly low-confidence, superficial binding. These results indicate that while the peptides contact SOD1’s surface, none form a stable or well defined interface, and no generated peptide displays stronger predicted binding than the known binder.
Binder 1: ipTM = 0.32, pTM = 0.79
Binder 1 interacts only weakly with SOD1, remaining positioned on the outer surface without engaging the N terminus, β-barrel, or dimer interface.
Binder 2: ipTM = 0.36, pTM = 0.86
In the AlphaFold3 model, the peptide binds loosely on the surface of SOD1 rather than near the N terminus, β-barrel, or dimer interface.
Binder 3: ipTM = 0.28, pTM = 0.72
Similarly, this one also binds to the surface and does not penetrate or bind near anything meaningful.
Binder 4: ipTM = 0.37, pTM = 0.84
Binder 4 shows another weak binding on the surface; it doesn’t engage the N-terminus, β-barrel, or dimer interface.
Binder 5 (the example): ipTM = 0.35, pTM = 0.83
The known binder (Binder 5) shows a slightly more defined and closer interaction with the SOD1 surface compared to the generated peptides, but still binds only shallowly and does not specifically target the N terminus, β-barrel core, or dimer interface.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
The PeptiVerse results show that all peptides had perfect predicted solubility and low hemolysis probability, meaning none are predicted to be especially toxic or poorly soluble. However, the predicted binding affinities were still weak across all peptides, which matches the AlphaFold3 observation that all binders only showed weak surface interactions. Peptides with higher ipTM did not necessarily show stronger predicted affinity. For example, Binder 4 had the highest ipTM, but one of the weakest affinity predictions, while Binder 3 had the strongest predicted affinity but the lowest ipTM. This means structural confidence and affinity prediction did not clearly align here. Overall, the peptides appear relatively safe, but none showed convincing strong binding behavior.
Choose one peptide you would advance and justify your decision briefly.
I would advance Binder 2 (WRSGAVALELGX) because it had one of the better ipTM scores among the generated peptides, while also showing perfect solubility, low hemolysis probability, and a relatively balanced net charge. Even though its affinity is still weak, it seems to offer the best overall trade-off between predicted binding and therapeutic properties.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. https://www.biorxiv.org/content/10.1101/2024.07.31.606098v2 uses Multi-Objective Guided Discrete Flow Matching (https://openreview.net/forum?id=8YIMLoHP9J) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
Choose specific residue indices on SOD1 that you want your peptide to bind.
Set peptide length to 12 amino acids.
Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
The moPPit peptides are quite different in their structure and thus, their properties. Many of the PepMLM peptides started with tryptophan and generally had very similar amino acids. Although the PepMLM peptides differed slightly in their binding, affinity, charge, and hydrophobicity, it is not particularly noteworthy. On the other hand the moPPit peptides varied in structure a lot relative to each other and the PepMLM, generally their affinity was a bit higher but still awfully low. What is interesting is that the hemolysis probability is roughly 20x higher for nearly all moPPit peptides compared to PepMLM. This would be very toxic as it likely will rupture blood cells, making the peptides very dangerous. Furthermore, before advancing moPPit generated peptides to clinical studies, further evaluation through structural docking validation to confirm stable protein–peptide interactions, toxicity and hemolysis screening to assess potential cellular damage, and in vitro stability assays to ensure peptide integrity under physiological conditions would be required to support therapeutic safety and effective binding to mutant SOD1.
Binder (Sequence)
Binding Affinity (pKd/pKi)
Solubility (Probability)
Hemolysis Risk (Probability)
Net Charge (pH 7)
Molecular Weight (Da)
Hydrophobicity (GRAVY)
Binder 1 (WLYGATGLRLKK)
5.961 (Weak) pKd/pKi
1
0.061 (Non hemolytic)
2.76
1405.7 Da
-0.23
Binder 2 (WRSGAVALELGX)
5.993 (Weak) pKd/pKi
1
0.042 (Non hemolytic)
-0.24
1140.5 Da
0.41
Binder 3 (WRYYAVAAEWKX)
6.612 (Weak) pKd/pKi
1
0.041 (Non hemolytic)
0.76
1424.7 Da
-0.56
Binder 4 (WRYGPAALAHKE)
5.336 (Weak) pKd/pKi
1
0.014 (Non hemolytic)
0.85
1398.6 Da
-0.84
Binder 5 (FLYRWLPSRRGG)
5.968 (Weak) pKd/pKi
1
0.047 (Non hemolytic)
2.76
1507.7 Da
-0.71
moPPit Binder 1 (YVCYSYNYCVCH)
7.878
0.833
0.911
moPPit Binder 2 (TEKTTQAKKYCV)
6.274
0.833
0.978
moPPit Binder 3 (GDMTRYSYYKKC)
6.790
0.916
0.964
Part B: BRD4 Drug Discovery Platform Tutorial
Assignees for the following sections
MIT/Harvard students
Optional
Committed Listeners
Optional
I did not include a written response for Part B.
Part C: Final Project: L-Protein Mutants
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
High level summary: The objective is to improve the stability and autofolding of the lysis protein.
More specifically, we want to engineer the lysis protein to increase the ability of MS2 to overcome a common E. coli resistance mechanism: a single point mutation in DnaJ prevents the binding of the lysis protein. We can attempt this by mutating the lysis protein to change its properties. Together, we aim for finding mutations that change the lysis protein in one of the following ways:
an independence of lysis protein processing from DnaJ or other bacterial chaperones
a faster or more efficient killing of E. coli to reduce the window in which the host can acquire resistance
higher lysis protein expression
L-Protein Engineering | Option 1: Mutagenesis
Designing these mutants with good computational confidence is hard. It will show you limitations of some of the structure based models. Ultimately, you can pick various combinations of mutations and get lab results and then decide to pick the next round of mutations, but this assay will not be easy to run at scale in this class.
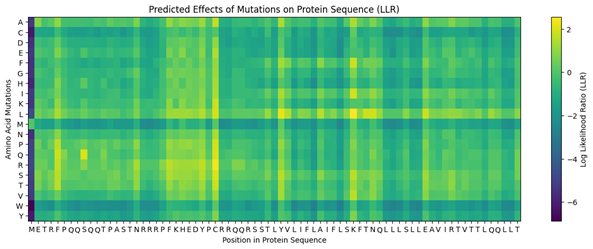
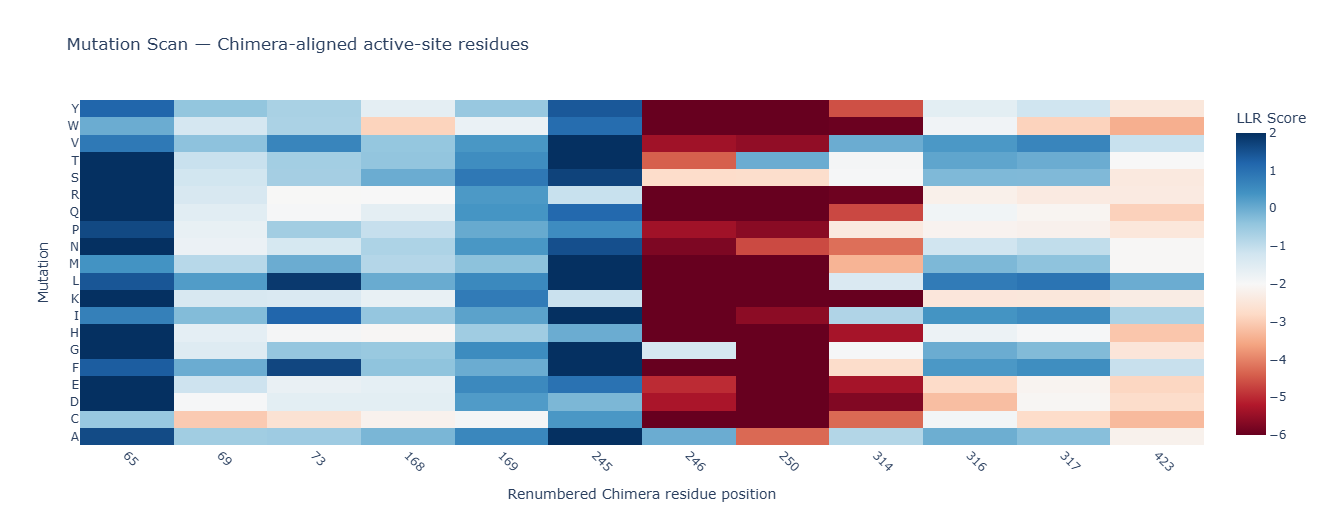
Run this notebook to generate for each position in the amino acid sequence, a “score” for what would happen to the protein if you mutated into another amino acid. It can be positive or negative for the protein. We want to identify possible mutations that are “positive”. If you run this notebook, you will see a .csv file in the sidebar. You can download it and look at it in google sheets if that’s easier.
Use the experimental data here. This dataset contains information about mutants of the L-Protein and their effect on lysis in the lab.
4. First check, does the experimental data correlate with the scores from the notebook in (b)? This should give you a clue on how well these language embeddings capture information about this protein sequence.
The experimental data seems to correlate exactly with the scores from the notebook.
5. Using information about the effect of protein mutations at these sites - both the scores and the experimental data in the drive, come up with 5 mutations for each student along with how you came up with them and why you believe they would work. 2 of the variants you submit must have mutations in the transmembrane region (refer to notes above on what amino acid positions these are) and 2 of them must be in the soluble region. Remember that you can also use the pBLAST to see which residues are conserved and not mutate them if you want to.
One easy way to generate sequence mutations could be to look for residue positions and mutations that have a positive mutational effect either in the experimental or have a positive score from step 1. And pick a combination of those mutations.
The MS2 L-protein consists of a short N-terminal soluble domain (residues 1–37) and a single transmembrane α-helix located at residues 38–60. https://www.uniprot.org/uniprotkb/P03609/entry
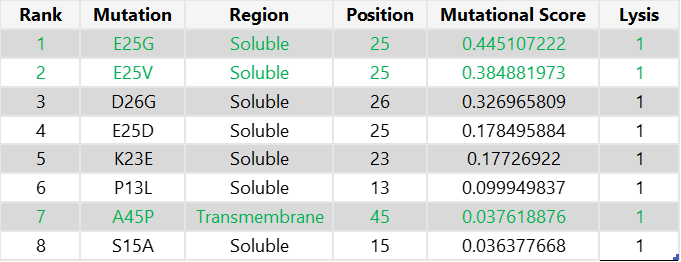
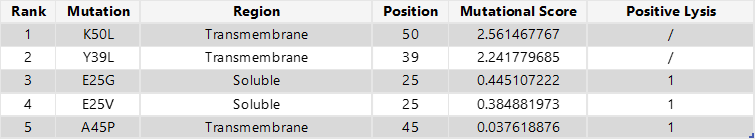
To select the final mutation set, I first focused on mutations that were positive in both the experimental dataset and the computational scoring results, thereby prioritising candidates supported by both experimental evidence and predicted mutational benefit. From this overlap, I chose the two highest-scoring mutations in the soluble region (E25G and E25V) and the only positive candidate from the transmembrane region (A45P). For the final two mutations, I selected the highest-scoring computational candidates that were not experimentally tested: K50L and Y39L; both are located in the transmembrane region.
Final 5 mutations:
E25G
E25V
A45P
K50L
Y39L
6. You can utilize Af2_Multimer to generate a Multimeric Assembly; you can do this by making your query sequence as. We want to do this because - A running hypothesis for how this protein functions is that it assembles to make a perforation in the bacterial membrane.
Figure generated using the following multimeric assembly (where each chain is separated from the other with a :):
This week we learn core molecular biology tools and techniques for processing and assembling DNA, including PCR and Gibson Assembly.
Objective:
Learn core molecular biology tools and techniques for processing and assembling DNA.
Understand PCR and Gibson Assembly.
Compare different ways of creating DNA fragments for cloning.
Explore genetic circuit design and simulation using Asimov Kernel.
Assignment: DNA Assembly
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
Answer these questions about the protocol in this week’s lab.
1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion High-Fidelity PCR Master Mix contains a high-fidelity DNA polymerase, which copies DNA with very few mistakes. It also contains dNTPs, which are the small molecules used to build new DNA strands. The mix includes a buffer to keep the chemical conditions stable and suitable for the reaction. It also has Mg2+ ions, which help the polymerase function properly.
2. What are some factors that determine primer annealing temperature during PCR?
Primer annealing temperature depends on the primer sequence, length, and GC content. The higher the GC content, the higher the temperature is for annealing. The temperature is also affected by how well the primer matches the template. Salt and buffer conditions in the reaction can change how strongly the primer binds.
3. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR uses primers and DNA polymerase to copy a specific DNA region, producing many copies. It is ideal when you need to amplify a fragment or introduce small sequence changes or overlaps. In contrast, restriction digestion uses enzymes to cut DNA at specific existing recognition sites. It is best when the DNA already contains the right cut sites and you want a simple, precise cut into fragments.
4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To make DNA suitable for Gibson cloning, the fragments need to have matching overlapping ends so they can join together correctly. The overlaps must be in the right order and orientation so the final construct assembles as planned. A crucial step is to check that the fragments are the correct size and that the DNA is clean. If there are unwanted bands or extra products, the assembly is prone to fail.
5. How does the plasmid DNA enter the E. coli cells during transformation?
Plasmid DNA enters E. coli after the cells are made competent, which means the cell membrane is prepared to allow DNA to pass through more easily. During heat shock or electroporation, the membrane becomes temporarily more permeable. This allows the plasmid DNA to move into the cell. After that, the cells recover and can begin to replicate the plasmid.
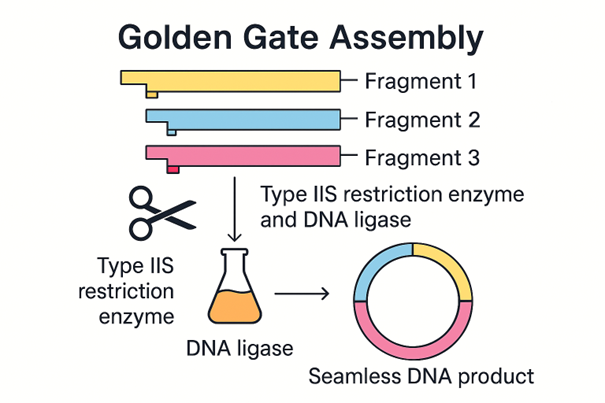
6. Describe another assembly method in detail (such as Golden Gate Assembly)
6.1 Explain the other method in 5–7 sentences plus diagrams (either handmade or online).
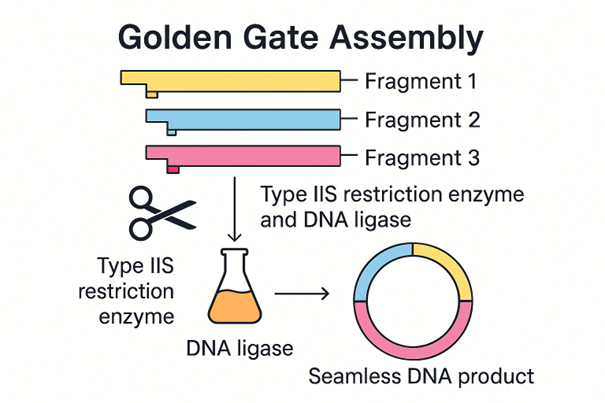
Golden Gate Assembly is a cloning method that joins DNA fragments together in a specific order. It uses Type IIS restriction enzymes, which cut outside of their recognition sequence instead of directly inside it. This creates short overhangs that can be designed to match only the correct neighbouring fragment. DNA ligase then joins the matching fragments together. One major advantage of Golden Gate Assembly is that the recognition site is usually removed during the process, so the final DNA product can be seamless. This method is very useful when multiple DNA fragments need to be assembled in one reaction.
Made using M365 Copilot.
6.2 Model this assembly method with Benchling or Asimov Kernel.
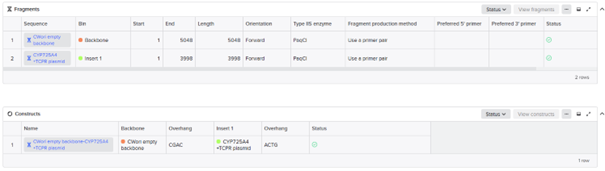
For this part of the assignment, I decided to use my final construct and designed the non-mutated cyp725a4 enzyme with induced expression, similiar to one of the articles I found (doi: 10.1016/j.pep.2017.01.008). I used Golden Gate Assembly in Benchling using the cwori backbone as the vector and the CYP725A4 + TCPR sequence as the insert. I first checked common Type IIS enzymes. BbsI, BsaI, and BsmBI were not ideal because they have internal cut sites in my construct, meaning they could cut inside the DNA instead of only at the assembly ends. I therefore chose PaqCI as a better enzyme to test because it has a longer recognition sequence and is less likely to occur internally. In a real experiment, I would add inward-facing PaqCI sites and designed overhangs to the backbone and insert using PCR primers.
Assignment: Asimov Kernel
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
Unfortunately, we never got Kernel access so I was not able to do this part as a commited listener
1. Create a Repository for your work
2. Create a blank Notebook entry to document the homework and save it to that Repository
3. Explore the devices in the Bacterial Demos Repo to understand how the parts work together by running the Simulator on various examples, following the instructions for the simulator found in the “Info” panel (click the “i” icon on the right to open the Info panel)
4. Create a blank Construct and save it to your Repository
4.1 Recreate the Repressilator in that empty Construct by using parts from the Characterized Bacterial Parts repository
4.2 Search the parts using the Search function in the right menu
4.3 Drag and drop the parts into the Construct
4.4 Confirm it works as expected by running the Simulator (“play” button) and compare your results with the Repressilator Construct found in the Bacterial Demos repository
4.5 Document all of this work in your Notebook entry - you can copy the glyph image and the simulator graphs, and paste them into your Notebook
5. Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo
5.1 Explain in the Notebook Entry how you think each of the Constructs should function
5.2 Run the simulator and share your results in the Notebook Entry
5.3 If the results don’t match your expectations, speculate on why and see if you can adjust the simulator settings to get the expected outcome
NEB’s (New England Biolabs) explanation & protocols for Gibson Assembly®
Week 7: HW genetic circuits part II
This week covers neuromorphic genetic circuits, showing how engineered gene networks can implement neural-network “perceptron”-like computation and learning.
Explore applications of neuromorphic genetic circuits.
Learn about fungal materials and possible engineering applications in fungi.
Make progress on the individual final project and first DNA design order.
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviours are Boolean functions?
Intracellular artificial neural networks (IANNs) are advantageous to Boolean genetic circuits because they can exhibit graded biological signals rather than absolute on-or-off states. In reality, cells do not have simple on and off outputs, making IANNs approximate the functionality of the cells with better accuracy. Many inputs can affect a single output and vary like a spectrum; biomolecules like miRNA, TFs, and plasmids drastically alter cellular states. As Anthony Genot explains, “Enzymes bring high nonlinearity, which we exploit to improve the sharpness of computation.” This nonlinearity is difficult to model via Boolean functions using logic gates. IANNs can also integrate many inputs with varying weights to create a more accurate representation of the biological system. This way, inhibitory and stimulatory signals are weighted rather than treated as binary. Compared with Boolean circuits, which become disproportionately complex with growing inputs, multilayer networks can approximate graded, weighted, and non-linear signals of a cell.
2. Describe a useful application for an IANN; include a detailed description of input/output behaviour, as well as any limitations an IANN might face to achieve your goal.
One potential application for IANNs could be to optimise biosynthesis pathways by controlling metabolic outputs of the microbial strain. Since IANNs allow for specific control of the cells, they could be used to mimic natural enzymatic kinetics to specify and optimise metabolic products. The IANN could help by sensing several continuous intracellular signals at the same time, such as precursor availability, intermediate accumulation, stress level, and growth state, and then using those signals to regulate key pathway enzymes in a graded way instead of a simple ON/OFF manner. For example, if harmful intermediates start to build, the system could reduce the specific pathway tied to it to relieve cellular stress and prevent cell death. Although these systems are useful, they can come with many costs. Presently, IANNs suffer from noisy intracellular signals, cell-to-cell variability, and changing cellular context, which makes it difficult to get precise and reproducible input/output behaviour. They can also suffer from cross-talk and resource competition, where different regulators interfere with each other or compete for limited cellular machinery. In biosynthesis, this is even harder because pathways need very precise balancing, and multilayer circuits respond slowly since each layer depends on transcription and translation.
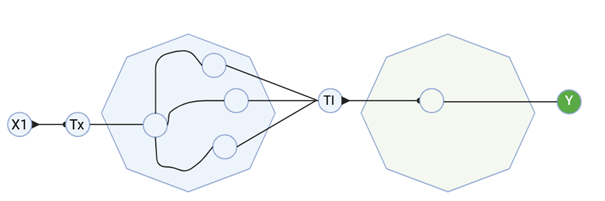
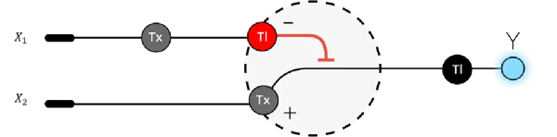
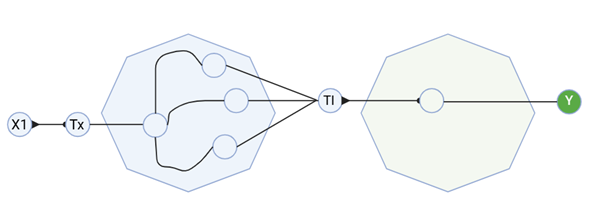
3. Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
I created a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Made using BioRender.
Assignment Part 2: Fungal Materials
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
1. What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Existing fungal materials are mostly mycelium-based materials, including packaging foams, insulation boards/panels, construction composites, and leather-like textiles made from fungal biomass grown on agricultural waste. These materials are used as substitutes for polystyrene packaging, synthetic foams, animal leather, and some lightweight building materials, because mycelium can act as a natural binder and be shaped into different forms during growth. The main advantage is that they are biobased, biodegradable, and can be produced from waste streams, which can lower environmental impact compared with plastics or animal-derived materials. Their disadvantages are that they still often have lower and more variable mechanical performance, limited water resistance and durability, and struggle to adapt to diverse environments.
2. What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
If I were to genetically engineer fungi, I would want to make them better microbial factories for complex pharmaceuticals such as paclitaxel. In that case, the goal would be to engineer fungi to express more of the paclitaxel pathway efficiently and improve precursor supply. Synthetic biology in fungi can be advantageous over bacteria for this type of problem because fungi are eukaryotes, so they are generally better at handling complex eukaryotic biosynthetic enzymes, performing post-translational modifications, secreting proteins, and supporting secondary metabolite pathways that are often harder to reconstruct in bacterial hosts. This makes fungi, especially yeasts and filamentous fungi, attractive hosts for producing plant- or fungus-derived pharmaceuticals, since many of these molecules depend on enzyme systems and intracellular organisation that are more similar to those of other eukaryotes than to bacteria. Fungi are also already widely used as industrial production organisms, which makes them a promising chassis for scaling the biosynthesis of valuable drugs. However, bacteria tend to grow faster and are usually easier to manipulate genetically, making it easier to adapt to a variety of situations and contexts.
Assignment Part 3: First DNA Twist Order
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
0. Review the Individual Final Project documentation guidelines.
I reviewed the Individual Final Project documentation guidelines.
1. Submit this Google Form with your draft Aim 1, final project summary, HTGAA industry council selections, and shared folder for DNA designs.
N/A, waiting on feedback for the final project, but I have made an explanation of my preject for Paxlitaxel on my project page
2. Review Part 3: DNA Design Challenge of the week 2 homework. Design at least 1 insert sequence and place it into the Benchling/Kernel/Other folder you shared in the Google Form above. Document the backbone vector it will be synthesized in on your website.
This week introduces synthesis of proteins using cellular machinery outside of a cell.
Homework — DUE BY START OF Apr 7 LECTURE
Homework Part A: General and Lecturer-Specific Questions
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
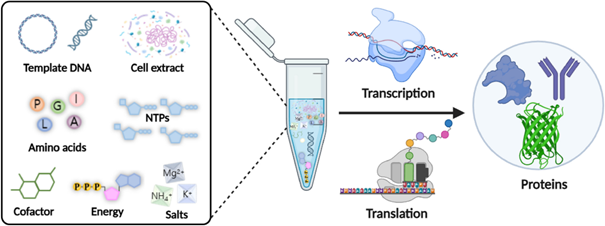
Cell-free protein synthesis is useful because proteins can be produced without needing to keep living cells alive. This gives more flexibility and control over experimental variables such as DNA concentration, energy supply, cofactors, salts, and reaction conditions.
Cell-free expression is especially beneficial for producing toxic proteins that would harm living cells, and for rapidly testing different enzymes, pathways, or genetic designs without needing to transform and grow cells each time.
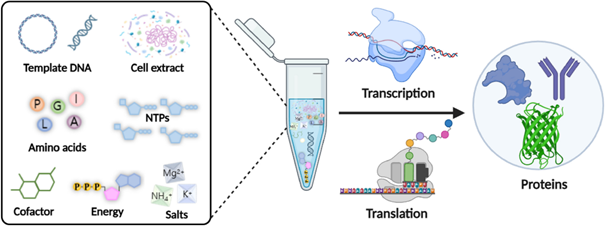
Describe the main components of a cell-free expression system and explain the role of each component.
A cell-free expression system contains a cell extract, a DNA or mRNA template, amino acids, nucleotides, an energy source, salts, and cofactors. The cell extract provides the ribosomes, polymerases, tRNAs, and enzymes needed for transcription and translation. The DNA template encodes the protein of interest. Amino acids are used to build the protein, while ATP and other energy molecules drive the reaction. Cofactors such as magnesium and potassium help maintain enzyme and ribosome activity.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy regeneration is critical because transcription and translation use ATP and other nucleotides very quickly. If ATP is depleted, ribosomes, polymerases, and other enzymes stop working, so protein synthesis decreases or stops.
One method to maintain ATP supply is to add an energy regeneration system such as phosphoenolpyruvate (PEP) with pyruvate kinase. In this setup, PEP helps regenerate ATP from ADP, allowing the reaction to keep producing protein for a longer time.
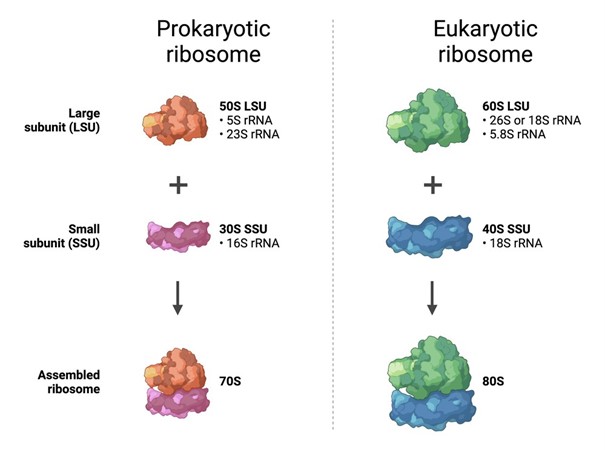
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic cell-free systems, such as E. coli extracts, are usually faster, cheaper, and easier to use. They are best for simple proteins that do not need complex folding or eukaryotic post-translational modifications.
Eukaryotic cell-free systems, such as wheat germ, insect, or mammalian extracts, are usually more expensive and slower, but they are better for proteins that need more complex folding, disulfide bonds, or eukaryotic processing.
For a prokaryotic system, I would produce a bacterial enzyme such as β-galactosidase because it is a simple bacterial protein and does not need eukaryotic modifications.
For a eukaryotic system, I would produce a human membrane receptor such as a GPCR because it requires a more complex folding environment and membrane-like conditions to function correctly.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
To optimize expression of a membrane protein, I would run a cell-free reaction with the DNA template for the target protein and include membrane-like structures such as liposomes, nanodiscs, or detergent micelles. These would give the protein a hydrophobic environment where it can insert and fold more correctly.
A major challenge is that membrane proteins can misfold or aggregate when they are produced without a membrane. I would address this by testing different membrane mimetics, changing the lipid composition, lowering the reaction temperature, and adding cofactors or chaperones if needed. I would compare the conditions by measuring both protein yield and protein activity.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Reason 1: Poor DNA template quality or incorrect DNA concentration Troubleshooting: Check the DNA quality and test a range of DNA concentrations to find the best expression level.
Reason 2: Energy supply is depleted too quickly Troubleshooting: Improve the energy regeneration system by adding components such as PEP or another ATP-regeneration substrate.
Reason 3: The protein is misfolding or aggregating Troubleshooting: Lower the reaction temperature, add chaperones, or include liposomes/nanodiscs if the protein needs a membrane-like environment.
Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
Pick a function and describe it.
a. What would your synthetic cell do? What is the input and what is the output?
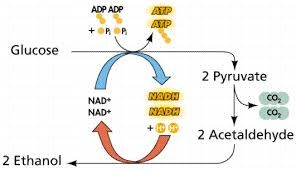
My synthetic minimal cell would produce ethanol from glucose. The input would be glucose, and the output would be ethanol. This is useful because ethanol is easy to measure and can be used as a biofuel or chemical product.
b. Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
Yes, this could be done in a bulk cell-free Tx/Tl reaction because the enzymes needed for ethanol production can work outside living cells. However, encapsulation makes the system more cell-like and allows better control over what enters and leaves the system.
c. Could this function be realized by genetically modified natural cell?
Yes, ethanol production is already commonly done using organisms such as yeast or engineered bacteria. However, a synthetic minimal cell gives more direct control over the reaction conditions and avoids some competing pathways found in living cells.
d. Describe the desired outcome of your synthetic cell operation.
The desired outcome is controlled conversion of glucose into ethanol, with predictable ethanol output and minimal side products.
Design all components that would need to be part of your synthetic cell.
a. What would be the membrane made of?
The membrane would be made from phospholipids such as POPC and POPG, with cholesterol added to improve membrane stability.
b. What would you encapsulate inside? Enzymes, small molecules.
Inside the vesicle, I would encapsulate an E. coli-based cell-free Tx/Tl system, DNA templates for the main ethanol-production enzymes, amino acids, nucleotides, ATP-regeneration components, salts, magnesium, potassium, and cofactors such as NAD⁺/NADH.
c. Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason?
A bacterial Tx/Tl system from E. coli would be sufficient because ethanol-production enzymes are simple metabolic enzymes and do not require mammalian post-translational modifications.
d. How will your synthetic cell communicate with the environment?
Glucose would enter through a membrane pore, and ethanol can diffuse out because it is small and membrane-permeable. To improve glucose entry, I would include a pore-forming protein such as α-hemolysin.
Experimental details
a. List all lipids and genes.
Lipids:
POPC — main membrane lipid
POPG — adds negative charge to the membrane
Cholesterol — improves membrane stability
Genes:
hla — encodes α-hemolysin pore for small-molecule transport
pdc from Zymomonas mobilis — converts pyruvate to acetaldehyde
adhB from Zymomonas mobilis — converts acetaldehyde to ethanol
To keep the system simpler, I would provide glucose-processing metabolites or use enzymes already present in the extract instead of encoding the entire glycolysis pathway.
b. How will you measure the function of your system?
I would measure ethanol production using an alcohol dehydrogenase-based ethanol assay, where ethanol conversion produces NADH that can be measured by absorbance at 340 nm. I could also measure glucose decrease as a second readout.
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material.
One-sentence pitch: I propose a smart textile patch containing freeze-dried cell-free biosensors that change fluorescence when exposed to sweat biomarkers linked to dehydration or heat stress.
How will the idea work? The textile would contain small dried spots of cell-free reactions embedded into a wearable patch. When sweat reaches the patch, it would rehydrate the reaction and activate protein expression or a biosensor response. If the target biomarker is present, the patch would produce a visible fluorescent signal. This could be checked with a small fluorescence viewer or phone-based imaging system.
What societal challenge or market need will this address? This could help athletes, outdoor workers, or soldiers monitor heat stress and dehydration early. It would be useful because it gives a low-cost and wearable biological readout without needing a laboratory.
How do you envision addressing the limitation of cell-free reactions? The cell-free system would be freeze-dried to improve storage stability. The patch would stay inactive until sweat provides water. Since the reaction is likely one-time use, the patch would be designed as a disposable sensor strip.
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. For this assignment, the proposal should incorporate the BioBits® cell-free protein expression system.
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. Maximum 100 words.
Long-duration space missions need simple ways to monitor microbial contamination in spacecraft water. Microbes can affect astronaut health and damage closed life-support systems. Sending samples back to Earth is slow, so astronauts need compact tools that can work directly in space. BioBits® is useful because it is freeze-dried and can produce proteins without living cells after adding water and DNA instructions. The Genes in Space toolkit also includes the P51 fluorescence viewer, which can visualize fluorescent biomolecules in small tubes. 12
Name the molecular or genetic target that you propose to study. Maximum 30 words.
A bacterial 16S rRNA gene sequence from a simulated spacecraft water sample.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. Maximum 100 words.
The 16S rRNA gene is commonly used to identify bacteria, so detecting it would show whether bacterial contamination may be present in spacecraft water. This relates directly to space biology because spacecraft are closed environments where microbial growth must be monitored carefully. A BioBits® cell-free reaction could be used as a simple biosensor that produces a fluorescent signal when the bacterial target is present. Previous ISS work showed that BioBits® cell-free reactions can function in space and produce fluorescence-based biosensor readouts. 34
Clearly state your hypothesis or research goal and explain the reasoning behind it. Maximum 150 words.
My hypothesis is that a BioBits® cell-free biosensor can detect a bacterial 16S rRNA target in a space-compatible experiment. If the target sequence is present, the biosensor should produce a fluorescent signal that can be viewed with the P51 Molecular Fluorescence Viewer. This is useful because it avoids the need to grow living cells and uses compact tools suitable for spacecraft. Since BioBits® reactions have already been tested aboard the ISS and shown to produce fluorescent outputs, a similar approach could be used for simple microbial monitoring during future missions. 13
Outline your experimental plan. Maximum 100 words.
I would test a simulated spacecraft water sample using a BioBits® cell-free reaction designed to detect a bacterial 16S rRNA target. A positive control would contain the target DNA or RNA, and a negative control would contain no target. If the target is present, the reaction should produce fluorescence. The P51 Molecular Fluorescence Viewer would be used to compare fluorescence between the sample and controls. A stronger signal in the sample or positive control would indicate successful detection of bacterial contamination. 15
Homework Part B: Individual Final Project
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
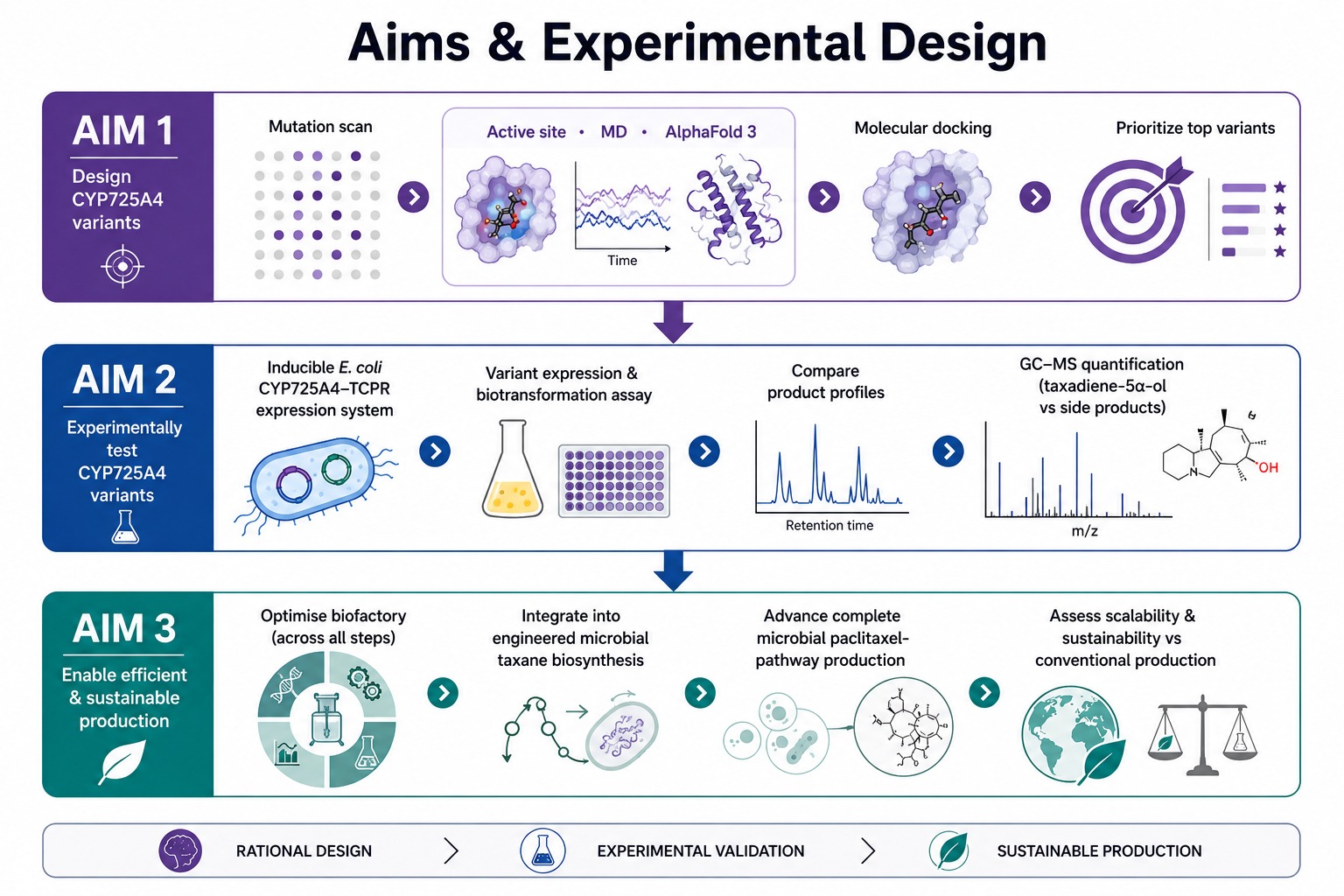
Provide Aim 1.
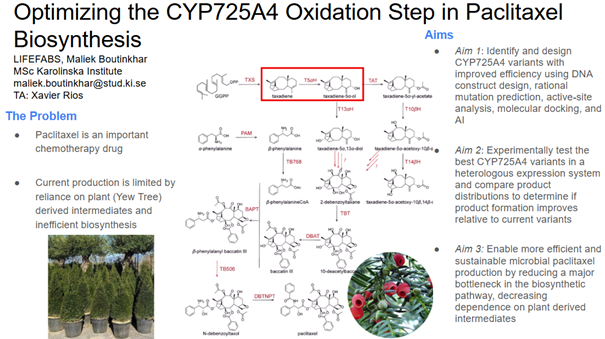
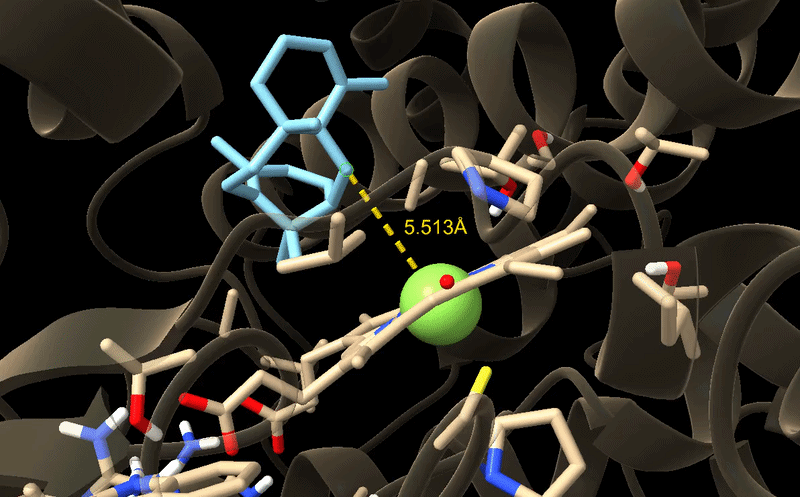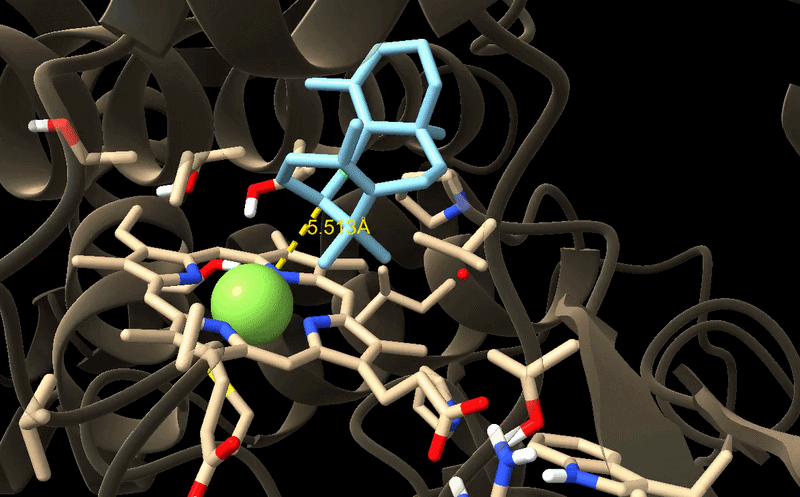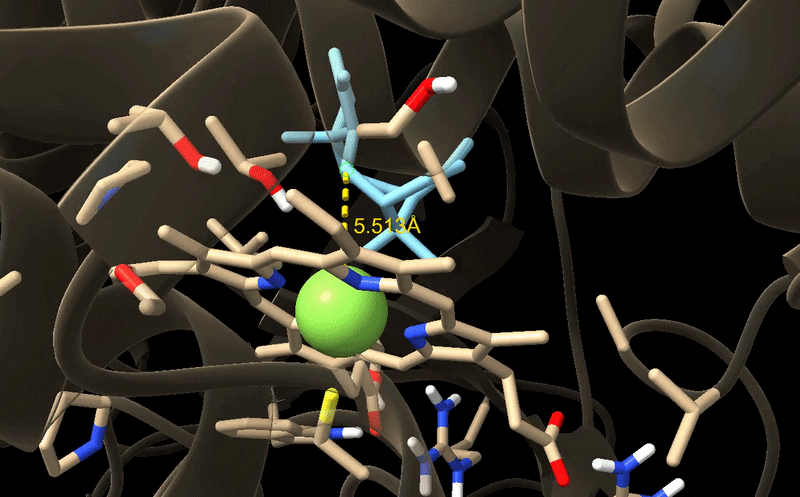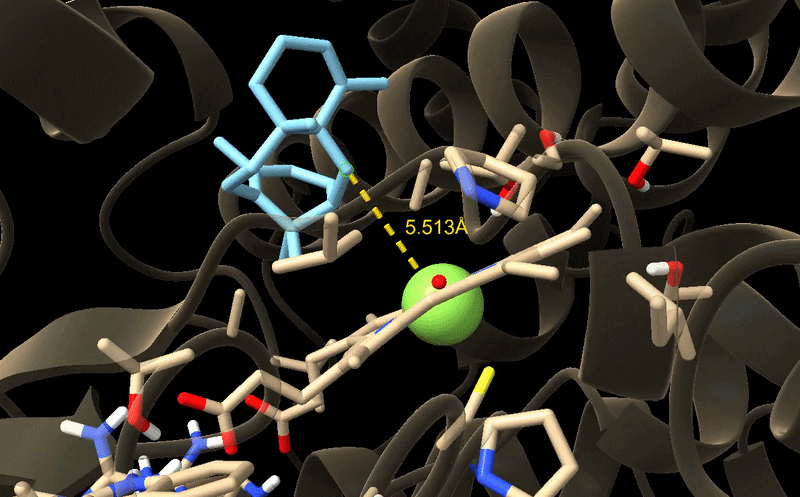
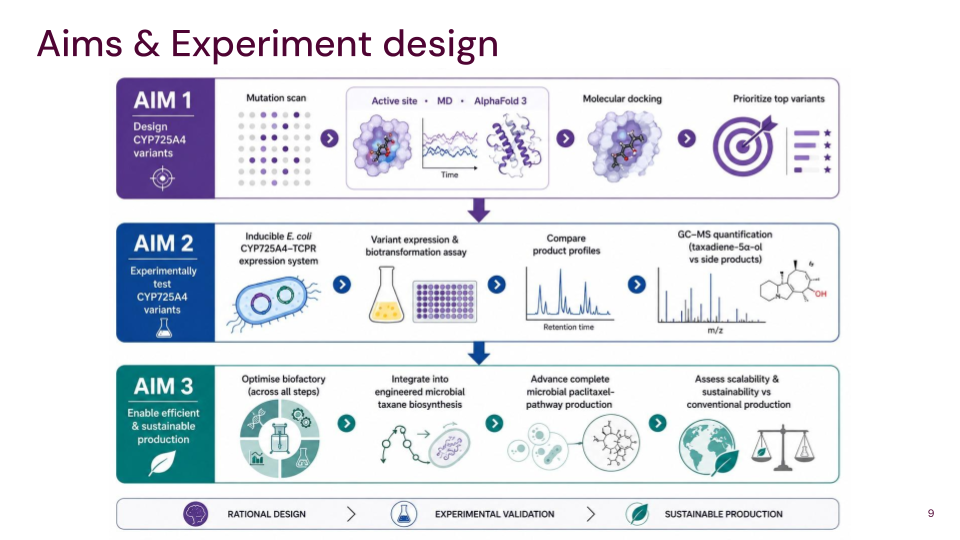
Aim 1: Identify and design CYP725A4 variants with improved efficiency using DNA construct design, rational mutation prediction, active-site analysis, molecular docking, and AI
This lecture presents a range of advanced technologies to do precision
measurement of proteins at atomic scales, characterizing chemical
composition, and detecting protein sequence and structure.
Homework is partly based on data that will be generated in the Waters Immerse Lab in Cambridge, MA. Students will characterize green fluorescent protein (eGFP, a recombinant protein standard) structure (primary, secondary/tertiary) in the lab using liquid chromatography and mass spectrometry, as well as Keyhole Limpet Hemocyanin (KLH) oligomeric states using charge detection mass spectrometry (CDMS). Data generated in the lab needed to do the homework is included both within this document and in the Appendix of the laboratory protocol.
Homework: Final Project
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
For your final project:
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
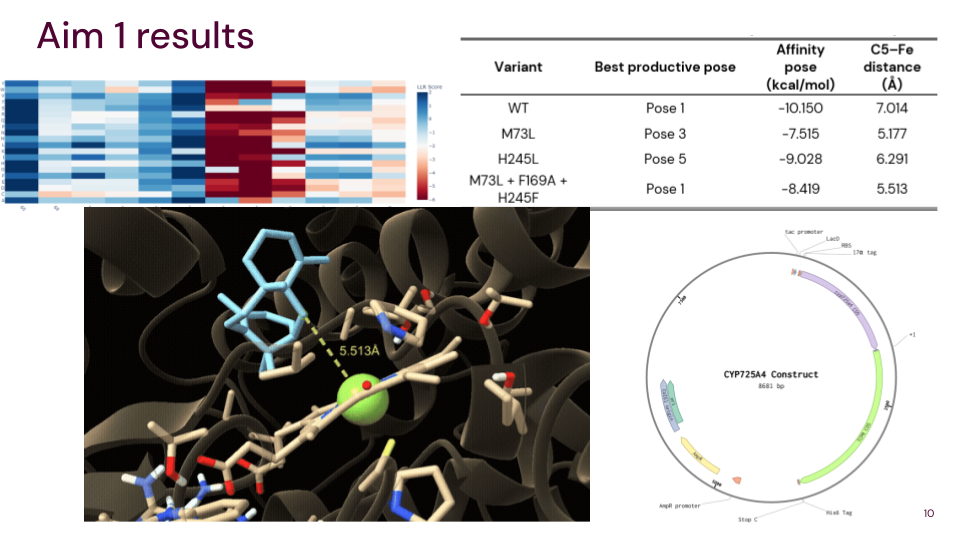
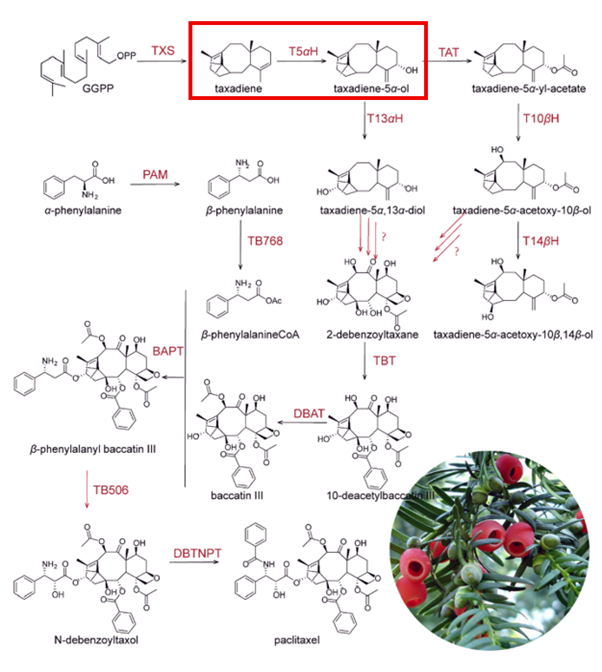
I will measure both enzyme expression and activity in the paclitaxel biosynthesis pathway, focusing on CYP725A4. The aim is to confirm that the enzyme is successfully expressed and that it is functionally active in producing the desired product.
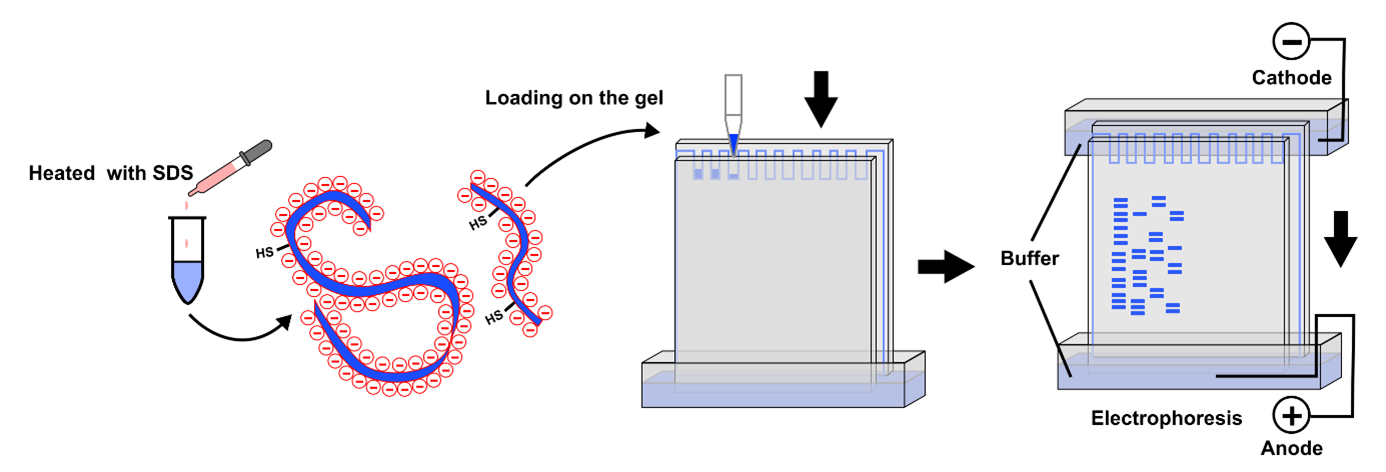
Protein expression will first be analysed using SDS‑PAGE, a simple and low-cost technique that separates proteins based on size. This allows me to confirm that a protein is present at the expected molecular weight and gives a rough indication of expression level. If more specific confirmation is required, a Western blot can be used to selectively detect the target protein using an antibody.
To evaluate enzyme activity, I will use LC‑MS to detect and quantify small molecule products such as taxadien‑5α‑ol. This method provides high sensitivity and accuracy, allowing me to confirm that the enzyme is producing the correct product.
Overall, this approach combines low-cost methods for initial validation with more precise analytical techniques for functional characterization.
Homework: Waters Part I - Molecular Weight
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
We will analyze an eGFP standard on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
eGFP Sequence: MVSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNYNSHNV YIMADKQKNG IKVNFKIRHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LSTQSALSKD PNEKRDHMVL LEFVTAAGIT LGMDELYKLE HHHHHH Note: This contains a His-purification tag (HHHHHH) and a linker (the LE before it).
The calculated molecular weight of eGFP including the His-tag is approximately
Without his tag Theoretical pI/Mw: 5.58 / 26941.48 With his tag Theoretical pI/Mw: 5.90 / 28006.60
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data and:
Determine z for each adjacent pair of peaks (n, n+1) using:
z = (m/z)_(n+1) / ((m/z)_(n+1) - (m/z)_n)
Determine the MW of the protein using the relationship between (m/z)_n, MW, and z
Calculate the accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1 using:
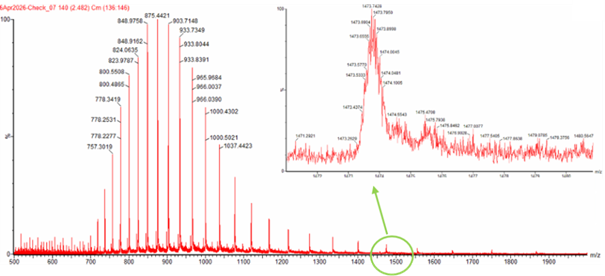
Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
The charge state cannot be clearly determined from the zoomed-in peak because the isotope peaks are not fully resolved and overlap with each other. This makes it difficult to measure the spacing between peaks, which is required to calculate the charge state accurately.
Homework: Waters Part II - Secondary/Tertiary structure
Assignees for the following sections
MIT/Harvard students
Optional but highly recommended
Committed Listeners
Optional but highly recommended
We will analyze eGFP in its native, folded state and compare it to its denatured, unfolded state on a quadrupole time-of-flight MS. We will be doing MS-only analysis (no liquid chromatography, also known as “direct infusion” experiments) on the Waters Xevo G3-QToF MS.
Based on learnings in the lab, please explain the difference between native and denatured protein conformations. For example, what happens when a protein unfolds? How is that determined with a mass spectrometer? What changes do you see in the mass spectrum between the native and denatured protein analyses?
A native protein is folded into its compact, functional structure, while a denatured protein is unfolded due to changes in solvent or pH. When the protein unfolds, more charged residues become exposed, increasing the number of charges it can carry.
In mass spectrometry, denatured proteins show a broader distribution of higher charge states, while native proteins show fewer peaks at lower charge states due to their compact structure.
Zooming into the native mass spectrum of eGFP from the Waters Xevo G3 QTof MS, can you discern the charge state of the peak at ~2800 m/z? What is the charge state? How can you tell?
From Figure 3, the zoomed inset shows isotope peaks spaced by approximately ~0.125 m/z.
Using:
z ≈ 1 / spacing
z ≈ 1 / 0.125 ≈ 8
The charge state is approximately +8.
Homework: Waters Part III - Peptide Mapping - primary structure
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
We will digest the eGFP protein standard into peptides using trypsin (an enzyme that selectively cleaves the peptide bond after Lysine (K) and Arginine (R) residues. The resulting peptides will be analyzed on the Waters BioAccord LC-MS to measure their molecular weights and fragmented to confirm the amino acid sequence within each peptide – generating a “peptide map”. This process is used to confirm the primary structure of the protein.
There are a variety of tools available online to calculate protein molecular weight and predict a list of peptides generated from a tryptic digest. We will be using tools within the online resource Expasy (the bioinformatics resource portal of the Swiss Institute of Bioinformatics (SIB)) to predict a list of tryptic peptides from eGFP.
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
Sequence used with Lysines (K) and Arginines (R) highlighted:
Copy/paste the sequence above into the input box in the PeptideMass tool to generate expected list of peptides.
Use the relevant parameters to predict peptides from eGFP.
Click “Perform the Cleavage” button in the PeptideMass tool and report the number of peptides generated when using trypsin to perform the digest.
Trypsin cleaves after lysine (K) and arginine (R) residues.
Estimated number of peptides:
(K + R) + 1 = 28
Approximately 28 peptides are expected, assuming no missed cleavages.
Based on the LC-MS data for the peptide map data generated in lab, how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
From Figure 5a, approximately 20 peaks above the threshold are observed.
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
No, the observed number of peaks is lower than the predicted number of peptides. This may be due to incomplete digestion, co-elution of peptides, or detection limits.
Identify the mass-to-charge (m/z) of the peptide shown in Figure 5b. What is the charge (z) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide ([M+H]+) based on its m/z and z.
From the spectrum: m/z ≈ 525.76 Isotope spacing ≈ 0.5 → z ≈ 2
M = (m/z × z) - z
M ≈ (525.76 × 2) - 2 ≈ 1049.5 Da
The peptide mass is approximately 1049 Da.
Predicted tryptic peptides of eGFP (Expasy PeptideMass)
Mass (Da)
Position
Peptide Sequence
4472.18
170–210
HNIEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSALSK
2566.29
217–239
DHMVLLEFVTAAGITLGMDELYK
2437.26
5–27
GEELFTGVVPILVELDGDVNGHK
2378.26
54–74
LPVPWPTLVTTLTYGVQCFSR
1973.91
142–157
LEYNYNSHNVYIMADK
1503.66
28–42
FSVSGEGEGDATYGK
1266.58
87–97
SAMPEGYVQER
1083.50
240–247
LEHHHHHH
1050.52
115–123
FEGDTLVNR
982.50
133–141
EDGNILGHK
821.39
81–86
QHDFFK
790.36
75–80
YPDHMK
769.39
47–53
FICTTGK
711.29
103–108
DDGNYK
655.38
98–102
TIFFK
602.28
211–215
DPNEK
579.31
128–132
GIDFK
507.29
164–167
VNFK
502.32
124–127
IELK
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm.
The identified peptide matches FEGDTLVNR, with a peptide mass error of approximately −970 ppm and overall protein accuracy of ~0.018%.
What is the percentage of the sequence that is confirmed by peptide mapping?
From Expasy: 90.7% coverage
From Figure 6: ~88% coverage
The peptide mapping confirms approximately 88–91% of the sequence, indicating strong agreement with the expected eGFP structure.
Homework: Waters Part IV - Oligomers
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS).
CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS:
From Figure 7: Peak at ~3.4 MDa matches the 7FU decamer Peak at ~8.33 MDa matches the 8FU didecamer Peak at ~12.67 MDa matches the 8FU 3-decamer
These peaks align closely with the expected masses of the oligomeric assemblies, indicating that the protein forms higher-order structures in solution. The presence of multiple distinct peaks suggests a mixture of oligomeric states rather than a single uniform complex. Overall, the data is consistent with known behaviour of KLH, which is known to form large, multimeric assemblies.
Homework: Waters Part V - Did I make GFP?
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
Please fill out this table with the data you acquired from the lab work done at the Waters Immerse Lab in Cambridge, or else the data screenshots in this document if you were unable to have lab work done at Waters.
Since I am a Committed Listener and did not perform the experiment myself, I used the intact LC-MS data provided in the homework document/screenshots.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST.
Make a note on your HTGAA webpages including:
what you contributed to the community bioart project,
what you liked about the project, and
what about this collaborative art experiment could be made better for next year.
I tried to make a smiley face in the bottom right quarter but I sadly had limited spots. To me I had never considered using biology and art so it was interesting to see experiments like this unfold to this scale and to have such open opportunities for all CLs. Personally, I think smaller group project could be useful, for example, if each node or small grojps in the node have their own access to a art experiment like this it could be personalised to each person and allow us to have more individual impact as its now quite difficult to have a meaningful change in the big art project.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
Referencing the cell-free protein synthesis reaction composition, provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase)
Salts/Buffer
Potassium Glutamate
HEPES-KOH pH 7.5
Magnesium Glutamate
Potassium phosphate monobasic
Potassium phosphate dibasic
Energy / Nucleotide System
Ribose
Glucose
AMP
CMP
GMP
UMP
Guanine
Translation Mix (Amino Acids)
17 Amino Acid Mix
Tyrosine
Cysteine
Additives
Nicotinamide
Backfill
Nuclease Free Water
E. coli lysate (BL21 DE3 Star, includes T7 RNA polymerase): Provides all cellular machinery (ribosomes, enzymes, tRNAs) for transcription and translation; T7 RNA polymerase transcribes DNA into mRNA.
Salts / Buffer
Potassium glutamate: Maintains intracellular-like ionic conditions needed for ribosome activity.
HEPES-KOH pH 7.5: Keeps pH stable so enzymes and proteins function properly.
Magnesium glutamate: Provides Mg²⁺ ions required for ribosome function and RNA stability.
Potassium phosphate (mono + dibasic): Acts as a buffer and supplies phosphate for metabolic reactions.
Energy / Nucleotide System
Ribose: Precursor for rebuilding nucleotides.
Glucose: Provides long-term energy via metabolic pathways.
AMP, CMP, GMP, UMP: Nucleotide building blocks that are converted into active triphosphates for transcription.
Guanine: Converted into GMP through salvage pathways to support RNA synthesis.
Translation Mix
17 amino acid mix: Supplies most amino acids needed for protein synthesis.
Tyrosine: Added separately due to solubility/stability issues.
Cysteine: Added separately because it is reactive and easily oxidized.
Additives
Nicotinamide: Supports enzyme activity and redox balance during long reactions.
Backfill
Nuclease-free water: Adjusts volume without degrading DNA or RNA.
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
The 1-hour PEP-NTP system directly supplies ATP and NTPs along with PEP for immediate energy, enabling fast but short-lived protein production. The 20-hour system uses cheaper NMPs plus ribose and glucose, relying on enzymatic recycling to gradually regenerate energy and nucleotides, allowing longer, more sustainable protein expression.
Bonus question: How can transcription occur if GMP is not included but Guanine is?
Guanine can be converted into GMP through salvage pathways in the lysate. Once GMP is regenerated, it can contribute to the nucleotide pool needed for RNA synthesis.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc. (1-2 sentences each)
sfGFP: Fast maturation and strong folding efficiency → produces fluorescence quickly.
mRFP1: Slower maturation and lower brightness → weaker signal over time.
mKO2: pH-sensitive → fluorescence decreases if reaction becomes acidic.
mTurquoise2: Very high quantum yield → strong signal even at lower expression.
mScarlet-I: High brightness but moderate acid sensitivity → may lose signal in long runs.
Electra2: Blue emission and good brightness → useful for multi-color separation but can depend on oxygen for chromophore formation.
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
For mKO2, increasing buffer strength (more HEPES or phosphate) will stabilize pH during long incubation. Since mKO2 is pH-sensitive, maintaining neutral pH should preserve fluorescence and increase total signal over 36 hours.
The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week. You can begin composing master mix compositions here.
This was the cell free master mix from RC donovans proposal:
The final phase of this lab will be analyzing the fluorescence data we collect to determine whether we can draw any conclusions about favorable reagent compositions for our fluorescent proteins.
The reaction composition for each well will be as follows:
6 μL of Lysate
10 μL of 2X Optimized Master Mix from above
2 μL of assigned fluorescent protein DNA template
2 μL of your custom reagent supplements
Total: 20 μL reaction
Part D: Build-A-Cloud-Lab | Optional Bonus Assignment
Assignees for the following sections
MIT/Harvard students
Optional
Committed Listeners
Optional
Use this simulation tool to create an interesting looking cloud lab out of the Ginkgo Reconfigurable Automation Carts.
I did not complete the optional bonus assignment to try and focus on my final project
Week 12 — Building Genomes
This week focuses on designing, synthesizing, and editing whole genomes, from minimal cells to refactored microbes and synthetic chromosomes.
Lecture (Tues, Apr 21)
Building Genomes (▶️Recording) George Church, John Glass, Jef Boeke
No additional reading or resources were listed for this week.
Week 13 — AI, SynBio, and Scaling Health Innovation (ARPA-H)
This week focused on AI, synthetic biology, and scaling health innovation through ARPA-H. The lab time was mainly dedicated to final project work and preparation.
Lecture (Tues, Apr 28)
AI, SynBio, and Scaling Health Innovation (ARPA-H) (▶️Recording) Renee Wegrzyn
The main task was to continue developing the final project and prepare for the final presentation on May 12 for MIT/Harvard students or May 13 for Committed Listeners. Individual Final Project
Reading & Resources (click to expand)
No additional reading or resources were listed for this week.
Week 14 — Bio Design & Bio Fabrication
We wrap up the term looking towards a future of Bio-Design and Bio-Fabrication.
The main task was to finish the final project and prepare for the final presentation on May 12 for MIT/Harvard students or May 13 for Committed Listeners. Individual Final Project
Reading & Resources (click to expand)
No additional reading or resources were listed for this week.
Project drafts The following three slides represent my drafts for my final project. Project one involves decaffeinating drinks using bacterial strains, project 2 and 3 are similiar in nature as both are small molecule drugs which I aim to synthesise using bacteria. Although this is ambitious I have also found that a mutual precursor such as diterpene could be made instead of the complete drug.
SECTION 1: ABSTRACT 1. Provide an abstract/summary for your project. (minimum 150 words)
This project is aimed at a critical bottleneck in the biosynthesis of paclitaxel, a widely used chemotherapy drug. Current production methods rely heavily on semi-synthesis using yew-derived intermediates such as baccatin III, which creates environmental, economic, and supply-chain limitations [1]. This creates an opportunity to develop a more sustainable and economically viable microbial production route. A major challenge in microbial paclitaxel biosynthesis is the poor catalytic efficiency and selectivity of taxadiene 5α-hydroxylase, CYP725A4. When expressed in E. coli, CYP725A4 produces taxadien-5α-ol only as a minor product and forms multiple undesired oxidised side products instead [2,3].
Subsections of Projects
Final Project Journey
Project drafts
The following three slides represent my drafts for my final project. Project one involves decaffeinating drinks using bacterial strains, project 2 and 3 are similiar in nature as both are small molecule drugs which I aim to synthesise using bacteria. Although this is ambitious I have also found that a mutual precursor such as diterpene could be made instead of the complete drug.
Optimising the oxidation reaction of Paclitaxel biosynthesis
Paclitaxel is a popular chemotherapy in several cancers such as ovarian, breast, and lung. However, the current production of it remains unsustainable from an environmental and economic perspective and can be optimised using biosynthesis. The most common way it is made on an industrial scale is via semi-synthesis by extraction of 10-deacetylbaccatin III from the European Yew (Taxus baccata) or other similar trees and eventually ending up with paclitaxel. This often is not extremely efficient (paclitaxel after extraction 10%), and contributes to environmental strain as it takes long for yew trees to mature driving up costs further
Current biosynthesis is not particularly effective either, some work has been done in generating the taxol precursors using E. coli (10.1126/science.1191652). The complete synthesis is difficult due to the inefficiency of enzymatic reactions. One of the current bottlenecks in production is the first oxidation step catalysed by taxadiene 5α-hydroxylase (CYP725A4). This enzyme converts taxadiene into taxadien‑5α‑ol but exhibits low catalytic efficiency and poor selectivity, resulting in the formation of multiple undesired side products. Heterologous expression of taxadiene‑5α‑hydroxylase (CYP725A4) results in a high side‑product to main‑product ratio and low taxadien‑5α‑ol titres due to the formation of multiple oxygenated taxane derivatives, thereby limiting metabolic flux toward paclitaxel precursors and hindering efficient microbial production (doi.org/10.1186/s12934-022-01922-1).
“The challenge for biosynthesis of paclitaxel lies on the insufficient precursor, such as taxadien-5α-ol” (Wu, QY et al. doi.org/10.1186/s40643-022-00569-5)
I want to optimise the CYP725A4‑catalysed oxidation step in paclitaxel biosynthesis, which currently exhibits low selectivity due to competing reaction pathways in its active site. This may be achieved through enzyme engineering approaches such as active site analysis, molecular docking, rational mutation prediction, or by exploring alternative enzyme variants. Improving this early biosynthetic step could increase taxadien‑5α‑ol production and enhance the overall efficiency and sustainability of microbial paclitaxel synthesis.
Final Project Slide
After some discussion with my node TAs, I settled on paclitaxel as my final project. My three goals are as follows;
Final Project IDEA Slide
Aim 1: Identify and design CYP725A4 variants with improved efficiency using DNA construct design, rational mutation prediction, active-site analysis, molecular docking, and AI
Aim 2: Experimentally test the best CYP725A4 variants in a heterologous expression system and compare product distributions to determine if product formation improves relative to current variants
Aim 3: Enable more efficient and sustainable microbial paclitaxel production by reducing a major bottleneck in the biosynthetic pathway, decreasing dependence on plant-derived intermediates
Project Journey and Raw Results
The rest of this page documents how I got to my final CYP725A4 engineering result. I included the mistakes, intermediate docking results, residue-selection process, mutation choices, raw data sequences, plasmid design, and final presentation slides.
1. Starting With the Wrong Structure: 8X3E
At the beginning, I mistakenly started with the CYP725A4 structure 8X3E. I did not realise at first that this structure already had taxadiene bound, so I visualised it in PyMOL and then ran docking in ChimeraX with AutoDock Vina. This gave me an initial result, but I later realised that this was not the correct starting point for my intended workflow because the structure was already ligand-bound.
Original wrong surface visualisation of 8X3E. This was the first structure I worked with before realising taxadiene was already bound.
Docking/visualisation result from the 8X3E structure. This was useful for learning, but it was not used as the main docking result.
Initial 8X3E Docking Output
Grid centre:
X = 78.6
Y = 70.87
Z = 33.44
Grid size:
X = 20
Y = 20
Z = 20
Best affinity:
Mode 1 = -5.934 kcal/mol
Measured C5–heme Fe distance:
12.596 Å
This mistake was useful because it showed me that taxadiene can adopt multiple conformations in the CYP725A4 active site. However, because I had started from a ligand-bound structure, I did not use this as the main result.
2. Switching to the Correct Apo Structure: 8X1W
After realising the issue with 8X3E, I switched to the CYP725A4 apo structure 8X1W. This structure did not already contain taxadiene, so it was a better starting point for testing how taxadiene docks into the active site.
Docking of taxadiene into the 8X1W apo structure. This became my correct WT baseline.
WT 8X1W Docking Result
Note: The image shown here was generated using a slightly different docking setting than the refined results reported in the table below. The table values should be treated as the final refined docking results, so the visual pose may
Best affinity:
Mode 1 = -10.15 kcal/mol
C5–heme Fe distance:
7.014 Å
I then compared the 8X1W docking result visually against the taxadiene-bound 8X3E structure. The comparison looked reasonable, which gave me confidence that the docking setup was capturing the correct binding pocket.
3. Binding-Pocket Residue Selection
I first identified 13 residues or active-site features within the broader 3.5 Å, 4 Å, and 5 Å binding-pocket scans around taxadiene: W65, PHE69, M73, SER168, F169, H245, A246, T250, V314, G316, T317, L423, and HEM440. This initial list was then narrowed by removing residues that only appeared in the 5 Å shell, such as PHE69 and SER168, and residues with mostly negative or constrained mutation profiles, such as A246, T250, V314, G316, and L423. From the remaining candidates, W65, M73, F169, and H245 were selected as first-pass mutation targets because they were close enough to influence ligand positioning while still offering useful side-chain chemical changes. Combination variants were then included to test whether multiple mutations could improve substrate positioning through epistatic effects.
Mutation scan used to narrow down candidate residues and decide which positions were reasonable to mutate.
Residues Excluded From the First-Pass Library
Residue
Reason for exclusion
PHE69
Only appeared in the 5 Å scan
SER168
Only appeared in the 5 Å scan
A246
Already small; mostly backbone/constrained
T250
Most mutations were negative
V314
Mostly negative mutation profile
G316
Glycine/backbone constrained
L423
Mostly negative; direct contact was risky
Final First-Pass Residue Positions
Position
Residue
Reason for keeping
W65
TRP
Bulky aromatic residue that may influence pocket shape
M73
MET
Hydrophobic pocket-shaping residue
F169
PHE
Aromatic residue that may affect ligand orientation
H245
HIS
Possible second-shell or local interaction effect
4. Mutation Design Logic
After selecting candidate positions, I used the mutation scan and amino acid characteristics to decide which mutations were worth testing. The goal was not just to improve binding affinity, but to improve the orientation of taxadiene relative to the heme iron, especially the C5–heme Fe distance.
Mutation
Reason tested
W65F
Reduce bulky tryptophan while keeping aromatic character
M73L
Keep hydrophobicity but change side-chain shape
M73F
Test a more aromatic substitution at the same position
F169A
Remove aromatic bulk and create more space
F169S
Reduce aromatic bulk while adding a polar side chain
H245F
Replace histidine with a hydrophobic aromatic residue
H245L
Replace histidine with a hydrophobic aliphatic residue
M73L + F169A
Test whether two pocket-shaping mutations work better together
M73L + F169A + H245F
Test whether adding H245F improves productive positioning
At this point, one difficulty was that docking produced many different conformations. This made it difficult to objectively choose the best result using affinity alone, so I focused on both docking score and C5–heme Fe distance.
5. Individual Mutation Results
M73L
M73L docking result. This mutation preserved hydrophobicity while changing the shape of the binding pocket.
Note: The image shown here was generated using a slightly different docking setting than the refined results reported in the table below. The table values should be treated as the final refined docking results, so the visual pose may
Pose
Affinity (kcal/mol)
RMSD l.b.
RMSD u.b.
C5–Fe distance (Å)
1
-8.236
0.000
0.000
7.025
2
-7.606
1.239
3.233
8.374
3
-7.515
1.698
4.996
5.177
4
-6.899
1.463
2.085
5.802
5
-6.733
1.471
4.931
7.280
M73L did not improve the top-ranked pose very much, but pose 3 gave a much shorter C5–Fe distance of 5.177 Å. This suggested that M73L could support a more productive orientation, but not consistently as the best-ranked pose.
F169A
F169A docking result. This mutation removed aromatic bulk to test whether creating more space would improve taxadiene positioning.
Note: The image shown here was generated using a slightly different docking setting than the refined results reported in the table below. The table values should be treated as the final refined docking results, so the visual pose may
Pose
Affinity (kcal/mol)
RMSD l.b.
RMSD u.b.
C5–Fe distance (Å)
1
-8.165
0.000
0.000
8.770
2
-8.021
1.297
3.518
7.070
3
-7.972
1.339
3.785
7.196
4
-7.397
1.528
4.769
9.026
5
-7.301
1.732
5.489
7.516
F169A alone did not improve the productive geometry. The best-ranked pose had a worse C5–Fe distance than WT, which showed that removing aromatic bulk alone was not enough.
M73L + F169A
M73L + F169A docking result. This tested whether combining two pocket-shaping mutations could create a more productive binding pose.
Pose
Affinity (kcal/mol)
RMSD l.b.
RMSD u.b.
C5–Fe distance (Å)
1
-7.692
0.000
0.000
8.697
2
-7.565
1.336
4.900
5.408
3
-7.482
1.212
3.068
6.960
4
-7.053
1.398
3.731
6.760
5
-6.935
1.539
2.977
5.755
The combination of M73L + F169A produced some improved poses, especially pose 2 with a C5–Fe distance of 5.408 Å. However, the best-ranked pose still had poor geometry, which made the result harder to interpret.
M73L + F169A + H245F
M73L + F169A + H245F docking result. This was the final lead mutant because the best-ranked pose also had improved productive geometry.
Pose
Affinity (kcal/mol)
RMSD l.b.
RMSD u.b.
C5–Fe distance (Å)
1
-8.419
0.000
0.000
5.513
2
-8.379
1.249
4.892
8.655
3
-7.024
1.613
4.936
6.979
4
-6.976
1.748
3.037
8.746
5
-6.714
1.372
3.529
7.210
The M73L + F169A + H245F variant gave the clearest improvement because pose 1 had both a reasonable docking score and a shorter C5–Fe distance. This made it the strongest final candidate from my tested variants.
GIF of the final best docking pose for M73L + F169A + H245F.
6. Raw Docking Results Summary
The table below shows the main raw docking results for the tested variants. I mainly used C5–heme Fe distance as the productive-geometry metric, while keeping affinity as a secondary metric.
Variant
Pose
Affinity (kcal/mol)
RMSD l.b.
RMSD u.b.
C5–Fe distance (Å)
WT
1
-10.150
0.000
0.000
7.014
WT
2
-8.937
2.083
4.767
9.032
WT
3
-8.723
1.224
3.280
8.245
WT
4
-8.675
1.498
4.784
8.458
WT
5
-8.580
1.385
2.025
9.033
W65F
1
-9.326
0.000
0.000
7.187
W65F
2
-9.232
1.408
4.824
8.165
W65F
3
-9.039
1.385
3.835
8.577
W65F
4
-8.764
1.396
3.006
7.095
W65F
5
-8.644
1.765
5.187
7.002
M73L
1
-8.236
0.000
0.000
7.025
M73L
2
-7.606
1.239
3.233
8.374
M73L
3
-7.515
1.698
4.996
5.177
M73L
4
-6.899
1.463
2.085
5.802
M73L
5
-6.733
1.471
4.931
7.280
M73F
1
-7.453
0.000
0.000
8.356
M73F
2
-6.543
1.175
4.869
5.115
M73F
3
-6.088
1.281
3.421
6.966
M73F
4
-6.084
1.513
2.171
7.570
M73F
5
-5.580
1.583
3.073
7.125
F169A
1
-8.165
0.000
0.000
8.770
F169A
2
-8.021
1.297
3.518
7.070
F169A
3
-7.972
1.339
3.785
7.196
F169A
4
-7.397
1.528
4.769
9.026
F169A
5
-7.301
1.732
5.489
7.516
F169S
1
-8.079
0.000
0.000
8.736
F169S
2
-8.029
1.336
3.810
7.138
F169S
3
-7.826
1.282
3.500
7.076
F169S
4
-7.556
1.082
1.291
7.422
F169S
5
-7.278
1.505
4.790
8.993
M73L + F169A
1
-7.692
0.000
0.000
8.697
M73L + F169A
2
-7.565
1.336
4.900
5.408
M73L + F169A
3
-7.482
1.212
3.068
6.960
M73L + F169A
4
-7.053
1.398
3.731
6.760
M73L + F169A
5
-6.935
1.539
2.977
5.755
H245F
1
-7.890
0.000
0.000
7.042
H245F
2
-6.536
1.654
2.980
6.848
H245F
3
-6.442
0.918
4.799
9.109
H245F
4
-5.912
1.316
2.627
7.595
H245L
1
-9.979
0.000
0.000
6.303
H245L
2
-9.560
1.400
3.692
9.501
H245L
3
-9.557
1.887
4.088
6.810
H245L
4
-9.275
1.522
5.013
6.754
H245L
5
-9.028
1.725
3.640
6.291
M73L + F169A + H245F
1
-8.419
0.000
0.000
5.513
M73L + F169A + H245F
2
-8.379
1.249
4.892
8.655
M73L + F169A + H245F
3
-7.024
1.613
4.936
6.979
M73L + F169A + H245F
4
-6.976
1.748
3.037
8.746
M73L + F169A + H245F
5
-6.714
1.372
3.529
7.210
7. Best Productive Poses
This table summarises the most important productive poses from the raw docking results.
Variant
Best productive pose
Affinity of that pose (kcal/mol)
C5–Fe distance (Å)
WT
Pose 1
-10.150
7.014
M73L
Pose 3
-7.515
5.177
H245L
Pose 5
-9.028
6.291
M73L + F169A + H245F
Pose 1
-8.419
5.513
The WT enzyme had the strongest predicted binding affinity, but its C5–Fe distance was 7.014 Å. The M73L + F169A + H245F variant had a shorter C5–Fe distance of 5.513 Å, suggesting improved catalytic geometry despite weaker binding affinity.
8. Variant Sequences
Below are the protein sequences used for the docking workflow.
This variant gave the best overall productive geometry because its best-ranked pose also had an improved C5–heme Fe distance.
Affinity:
-8.419 kcal/mol
C5–heme Fe distance:
5.513 Å
I was a bit surprised by the results because I expected stronger improvements across more variants. However, the data showed that many single mutations did not improve the productive geometry, and some only produced useful conformations in lower-ranked poses. This made it clear that improving CYP725A4 selectivity is difficult and that small active-site changes can create many different substrate orientations.
10. What Took the Most Time
One of the biggest challenges was how long each mutation took to make, prepare, dock, inspect, and measure. Even though I did not test a very large mutation library, each variant required several steps: generating the mutant structure, adding/transferring the heme group, preparing the receptor, running AutoDock Vina, opening the docking poses, measuring C5–heme Fe distances, and deciding which poses were meaningful.
This made the project more time-consuming than expected, especially because docking produced many conformations and not all of them were easy to interpret objectively.
11. Molecular Dynamics Attempt
Near the end of the project, I started setting up molecular dynamics simulation to test whether the best docking pose would remain stable over time. The plan was to run a 10–50 ns simulation for the lead mutant. However, I did not have enough time before the deadline because some molecular dynamics simulations can take several days to set up and run properly.
This is why molecular dynamics was moved into future work instead of being included as a completed result.
12. Plasmid Design and Benchling
As part of the DNA construct planning for future experimental validation, I designed a plasmid construct for expressing CYP725A4 variants in a heterologous E. coli system. This design connects the computational docking work to the future wet-lab testing step.
Benchling plasmid design for the CYP725A4 construct.
Below are the final presentation slides for this project. I included them here so the presentation version of the project can be viewed alongside the raw computational work and final results.
Slide 1
Slide 2
Slide 3
Final Project Report
SECTION 1: ABSTRACT
1. Provide an abstract/summary for your project. (minimum 150 words)
This project is aimed at a critical bottleneck in the biosynthesis of paclitaxel, a widely used chemotherapy drug. Current production methods rely heavily on semi-synthesis using yew-derived intermediates such as baccatin III, which creates environmental, economic, and supply-chain limitations [1]. This creates an opportunity to develop a more sustainable and economically viable microbial production route. A major challenge in microbial paclitaxel biosynthesis is the poor catalytic efficiency and selectivity of taxadiene 5α-hydroxylase, CYP725A4. When expressed in E. coli, CYP725A4 produces taxadien-5α-ol only as a minor product and forms multiple undesired oxidised side products instead [2,3].
The objective of this project is to improve the selectivity and efficiency of CYP725A4 through rational enzyme engineering. The hypothesis is that targeted mutations within the CYP725A4 active site can improve taxadiene positioning near the heme centre and reduce off-pathway reactions, thereby increasing productive C5 oxidation [4]. Specific aims include identifying key active-site residues, designing CYP725A4 mutation variants, and computationally evaluating these variants using molecular docking. Residues within 4–5 Å of the ligand were prioritised using structural analysis based on available CYP725A4 structural data, including the 8X1W apo structure [5]. Mutations were selected to test biochemical mechanisms such as hydrophobic pocket reshaping, aromatic interaction tuning, and second-shell interaction remodelling [6].
Methods include molecular docking with AutoDock Vina, structural analysis, focused mutation library design, and computational validation using binding affinity and C5–heme Fe distance as key metrics. The expected outcome is the identification of improved CYP725A4 variants that better position taxadiene for taxadien-5α-ol formation, contributing to more efficient and sustainable microbial paclitaxel biosynthesis.
SECTION 2: BACKGROUND
Provide background information and research for your final project.
Background and Literature Context
Paclitaxel is a clinically important anticancer drug, but its production remains challenging because natural extraction and semi-synthesis rely on plant-derived taxane intermediates [1]. Synthetic biology offers an alternative by engineering organisms to produce paclitaxel precursors, but the pathway remains difficult because several enzymatic steps are inefficient or poorly selective [1]. Recent reviews also describe paclitaxel production as limited by high cost, low natural abundance, and incomplete pathway optimization [7,8].
The current pathway with the exact reactin targeted marked in red
A major bottleneck is CYP725A4, a cytochrome P450 enzyme from Taxus cuspidata that catalyzes the oxidation of taxadiene toward taxadien-5α-ol (See Figure above). Sagwan-Barkdoll and Anterola showed that taxadien-5α-ol is only a minor product when CYP725A4 is expressed in E. coli, while OCT and iso-OCT are the major products [2]. This shows that CYP725A4 does not reliably direct taxadiene toward the desired oxidation pathway in a microbial host.
Rouck et al. showed that CYP725A4 can be expressed and purified in E. coli using modified constructs, including N-terminal modifications and CYP725A4-TCPR fusion strategies [3]. Their work supports the feasibility of bacterial expression, but also shows that CYP725A4 is technically difficult because it is a membrane-associated plant P450 [3].
Recent structural work has made rational engineering more realistic. Song et al. used crystallography and computational analysis to investigate CYP725A4 and showed that taxadiene oxidation can follow competing routes leading either to taxadien-5α-ol or side products such as OCT and iso-OCT [4]. The 8X1W structure provides an experimentally determined CYP725A4 apo structure from Taxus cuspidata, solved by X-ray diffraction at 2.10 Å resolution [5]. The 8X3E structure can also be implicated, as it shows a bound taxadiene, which can be used for experimental docking validation [9].
SECTION 3: VISION AND IMPACT
3a. Introduce the vision and impact of your final project. (min. 1-2 paragraphs)
Why the Project Matters
This project matters because CYP725A4 is one of the early bottlenecks in microbial paclitaxel biosynthesis. If CYP725A4 can be engineered to produce more taxadien-5α-ol and fewer side products, more pathway flux could move toward useful paclitaxel precursors [2,4].
Improving this step could make paclitaxel precursor production more sustainable by reducing dependence on slow-growing yew trees and plant extraction [1]. It could also reduce production costs and make biosynthetic manufacturing more practical. More broadly, this project contributes to synthetic biology by showing how enzyme structure, docking, and DNA construct design can be combined to improve a difficult pathway enzyme [3,4].
3b. Describe how your project is innovative (min. 3 sentences)
Novelty and Innovation
This project’s novelty lies in the ineptitude of the established pathway being economically viable. The current system has a clear inefficiency, and this oxidation step is one of the hurdles in making this pathway useful in commercial settings.
3c. Describe the bioethical considerations involved in your project. (min. 2 paragraphs)
Ethical Implications
The main ethical principles relevant to this project are beneficence, non-maleficence, responsibility, and justice. Beneficence applies because improving paclitaxel biosynthesis could support more sustainable production of an important anticancer drug. Justice is also relevant because improved biosynthetic production could eventually help lower manufacturing barriers and improve access to medicines [1,7]. However, non-maleficence is important because future experimental work would involve engineered E. coli strains expressing plant biosynthetic enzymes [3].
To ensure the project is ethical, future experiments should use non-pathogenic laboratory E. coli strains, standard biosafety containment, careful strain tracking, and responsible disposal of engineered material. An inducible expression system should be used to reduce unnecessary metabolic burden and limit uncontrolled pathway activity [3]. Potential unintended consequences include poor expression, unexpected metabolic toxicity, or environmental risk if engineered organisms were mishandled. Alternatives include testing variants first in purified enzyme assays, nanodisc systems, or cell-free systems before using living production strains.
SECTION 4: PROJECT AIMS
Outline three aims of your final project (min. 3 sentences, at least one for each aim)
Aim 1: Experimental Aim
The first aim of my final project is to identify and design CYP725A4 variants with improved productive taxadiene binding by utilising active-site analysis, rational mutation prediction, molecular docking, and DNA construct design. This aim uses CYP725A4 structural and mechanistic data, including the 8X1W apo structure and recent crystallographic/computational analysis of CYP725A4 [4,5].
Aim 2: Development Aim
The second aim is to experimentally test the best CYP725A4 variants in a heterologous E. coli expression system and compare product distributions to determine whether taxadien-5α-ol formation improves relative to wild-type CYP725A4 [2,3].
Aim 3: Visionary Aim
The third aim is to enable more efficient and sustainable microbial paclitaxel production by reducing a major enzymatic bottleneck in the biosynthetic pathway. If successful, this could reduce reliance on plant-derived intermediates and support more scalable production of paclitaxel precursors [1].
Illustrative outline of the project aims and respective methodologies
SECTION 5: EXPERIMENTAL DESIGN
Share a detailed experimental plan for your final project. Include a timeline for each part of your experimental plan (i.e., how long you would expect each step in your final project to take). (min. 15 lines/sentences—a numbered list is acceptable)
Detailed Experimental Plan
Use the CYP725A4 8X3E structure as the docking reference, and the 8X1W as the structural reference.
Dock Taxadiene on the 8X1W using Autodock Vina and identify residues within a 5 Å, 4Å, and 3.5Å shell of the taxadiene.
Exclude residues that are already small, backbone-constrained, or strongly disfavoured in mutation scans, such as A246, T250, V314, G316, and L423.
Run a mutation scan for conservation to exclude extensively nefarious mutations.
Select the most favourable mutations, in this case W65, M73, F169, and H245, as first-pass mutation targets.
Use amino acid properties to guide mutation logic; for example, M73L preserves hydrophobicity, but changes shape, while F169A removes aromatic bulk.
Design combination variants such as M73L + F169A and M73L + F169A + H245F to test epistasis.
Prepare mutant protein structures computationally.
Dock taxadiene into each model using AutoDock Vina.
Record binding affinity, RMSD values, and C5–heme Fe distance for each pose.
Compare each mutant to wild-type CYP725A4.
Select variants that improve C5–Fe distance without causing unacceptable loss of predicted binding.
Design a future E. coli expression construct using an N-terminally modified CYP725A4 fused to Taxus CPR through a flexible linker, based on previous expression work.
Add a C-terminal His6 tag for purification and use an inducible tac/trc-style promoter to reduce metabolic burden before induction.
In future wet-lab validation, express the top variants in E. coli and analyse product distribution using GC-MS.
Compare taxadien-5α-ol, OCT, and iso-OCT levels to determine whether the engineered enzyme improves product selectivity.
SECTION 6: RESULTS
Share the experimental results of your project.
Validation Chosen
I validated the computational design aspect of the project by docking taxadiene into wild-type CYP725A4 and several rationally designed CYP725A4 variants. This tested whether active-site mutations could improve productive substrate positioning, measured mainly by C5–heme Fe distance [4,5]. AlphaFold 3 was used as an orthogonal validation method to assess structural integrity. The AF3 WT model aligned closely with the experimental CYP725A4 structure (PDB 8X1W), with a backbone RMSD of 0.79 Å, confirming that AF3 accurately reproduces the native fold. The M73L + F169A + H245F mutant model showed an even lower RMSD of 0.63 Å relative to the starting structure, indicating that the introduced mutations do not disrupt the global P450 architecture. Together, these results support that the engineered active-site mutations are structurally compatible and suitable for downstream molecular dynamics simulation.
Validation Protocol
Load the CYP725A4 structural model.
Identify the taxadiene binding pocket using a 5 Å residue scan.
Refine candidate residues using ≤4 Å proximity and residue chemistry.
Generate individual and combination mutant models.
Dock taxadiene into each variant using AutoDock Vina.
Record the top docking poses and predicted binding affinities.
Measure the distance between taxadiene C5 and heme Fe using ChimeraX.
Make mutations and fold via ESMFold and transfer the heme group from the WT version.
Orthogonally validate structure with AF3 and matchmake structures
Dock taxadiene onto mutant variants.
Compare each mutant against wild-type CYP725A4.
Select variants with improved C5–Fe distance as candidates for future experimental testing.
Mutation Scan
I first identified 12 residues within the broader 3.5 Å, 4 Å, and 5 Å binding-pocket scans around taxadiene: W65, PHE69, M73, SER168, F169, H245, A246, T250, V314, G316, T317, L423, and HEM440. This initial list was then narrowed by removing residues that only appeared in the 5 Å shell, such as PHE69 and SER168, and residues with mostly negative or constrained mutation profiles, such as A246, T250, V314, G316, and L423. From the remaining candidates, W65, M73, F169, and H245 were selected as first-pass mutation targets because they were close enough to influence ligand positioning while still offering useful side-chain chemical changes. Combination variants were then included to test whether multiple mutations could improve substrate positioning through epistatic effects.
Key Docking Results
Variant
Best productive pose
Affinity of the best pose
C5–Fe distance
WT
Pose 1
-10.150 kcal/mol
7.014 Å
M73L
Pose 3
-7.515 kcal/mol
5.177 Å
H245L
Pose 5
-9.028 kcal/mol
6.291 Å
M73L + F169A + H245F
Pose 1
-8.419 kcal/mol
5.513 Å
The WT enzyme had the strongest predicted binding affinity, but its C5–Fe distance was 7.014 Å. The M73L + F169A + H245F variant had a shorter C5–Fe distance of 5.513 Å, suggesting improved catalytic geometry despite weaker binding affinity. This supports the hypothesis that productive substrate positioning may be more important than binding strength alone for improving CYP725A4 selectivity [4].
Modelled mutated protein with the best taxadiene pose
Plasmid Design and Benchling Construct
As part of the DNA construct planning for future experimental validation, I designed a plasmid construct for expressing CYP725A4 variants in a heterologous E. coli system. This design connects the computational docking work to the future wet-lab testing described in Aim 2. The plasmid design includes the CYP725A4 coding sequence, planned mutation sites, expression-control elements, and features needed for cloning and downstream validation.
I am currently trying to run an MD simulation for the binding for a substantial time of 10-50ns. However, the simulation took much longer than expected, so I have deferred this until after the project deadline.
Synthetic Biology Techniques Used
This validation used protein design, molecular docking, DNA construct planning, and database-supported structural analysis. Protein design was used to choose active-site mutations based on residue chemistry [6]. Docking was used to estimate binding poses and productive orientation. Structural databases were used because experimentally determined CYP725A4 structures now provide a stronger basis for rational engineering [5].
SECTION 7: DISCUSSION AND FUTURE WORK
7a. Discussion (2 paragraphs minimum)
One challenge is that docking does not fully capture enzyme flexibility, membrane effects, electron transfer, or true catalytic rate. This is important because CYP725A4 is a membrane-associated plant P450, and previous work showed that expression context and redox partners are important for functional testing [3].
Another limitation is that improved C5–Fe distance does not guarantee improved taxadien-5α-ol formation. CYP725A4 can form multiple products, including OCT and iso-OCT, so future validation must measure actual product distribution experimentally [2]. To overcome this, the next step should be experimental testing of the lead variants in E. coli, followed by GC-MS analysis. Molecular dynamics simulations could also be added before wet-lab testing to check whether the improved docking pose remains stable over time [4].
7b. Future Work (1 paragraph minimum)
The future plan of this project directly follows Aim 2 and Aim 3. The next step should be experimental testing of the lead CYP725A4 variants in a heterologous E. coli expression system, followed by GC-MS analysis to compare taxadien-5α-ol, OCT, and iso-OCT product distributions. This would show whether the computationally selected mutations improve product selectivity relative to wild-type CYP725A4. In the longer term, the best-performing CYP725A4 variants could be integrated into a larger microbial paclitaxel precursor pathway to support more efficient and sustainable paclitaxel biosynthesis. This connects to the visionary aim of reducing reliance on plant-derived intermediates and making microbial production of paclitaxel precursors more scalable.
SECTION 8: TECHNIQUES, TOOLS, AND TECHNOLOGY
8. We discussed and practiced various techniques related to synthetic biology throughout the semester. Place a check next to the techniques relevant to your project.
Used Techniques
Lab Safety
Bioethical Considerations
DNA Construct Design
Databases
Protein Design
Models and Notebooks
Bioproduction
Chassis Selection
Plasmid Preparation
Bacterial Culturing
Quality Control/Analysis
Bacterial Processing
Protein Purification
Primer Design or Selection
PCR Reactions
Gibson Assembly
Designing a Twist Order
Use of Benchling
Gel Electrophoresis
Not Used Techniques
Pipetting
DNA Gel Art
DNA Sequencing
DNA Editing
Restriction Enzyme Digestion
DNA Purification From Gel
Lab Automation
Creating Code for Laboratory Automation
Using Liquid Handling Robots
Creating a plan to use the Autonomous lab at Ginkgo Bioworks
Use of Boltz or PepMLM
Use of Asimov Kernel
Registry of Standard Biological Parts
Cell-Free Reactions
Freeze-Dried Cell-Free Systems
miniPCR Tools
Other Cloning Methods
CRISPR/Cas9
Designing Prime Editing gRNA
9. Expand upon two techniques you checked in the previous question by describing how you would utilize those techniques in your final project. (min. 4 sentences)
Expanded Techniques
Protein design and molecular docking:
Protein design was used to select CYP725A4 active-site mutations based on residue position, side-chain chemistry, and predicted effects on substrate orientation. Molecular docking was then used to test whether these mutations improved taxadiene positioning near the heme iron. This is appropriate because recent CYP725A4 studies show that product selectivity depends on competing catalytic pathways controlled by substrate positioning [4].
DNA construct design:
The future experimental construct is based on previous E. coli CYP725A4 expression systems. The native N-terminal membrane-anchor region would be replaced with an expression-improving peptide, CYP725A4 would be fused to Taxus CPR to support electron transfer, and a His6 tag would be added for purification [3]. An inducible promoter would be preferred to reduce metabolic stress before induction.
SECTION 9: ADDITIONAL INFORMATION
10a. List all references cited in this assignment (bullet-point list)
Tong Y, Luo YF, Gao W. Biosynthesis of paclitaxel using synthetic biology. Phytochem Rev. 2022;21(3):863-877. doi:10.1007/s11101-021-09766-0.
Sagwan-Barkdoll L, Anterola AM. Taxadiene-5α-ol is a minor product of CYP725A4 when expressed in Escherichia coli. Biotechnol Appl Biochem. 2018;65(3):294-305. doi:10.1002/bab.1606.
Rouck JE, Biggs BW, Kambalyal A, Arnold WR, De Mey M, Ajikumar PK, et al. Heterologous expression and characterization of plant taxadiene-5α-hydroxylase CYP725A4 in Escherichia coli. Protein Expr Purif. 2017;132:60-67. doi:10.1016/j.pep.2017.01.008.
Song X, Wang Q, Zhu X, Fang W, Liu X, Shi C, et al. Unraveling the catalytic mechanism of taxadiene-5α-hydroxylase from crystallography and computational analyses. ACS Catal. 2024;14(6):3912-3925. doi:10.1021/acscatal.3c05807.
RCSB Protein Data Bank. 8X1W: CYP725A4 apo structure [Internet]. RCSB PDB; 2024 [cited 2026 May 24]. Available from: https://www.rcsb.org/structure/8X1W
Mutanda I, Li J, Xu F, Wang Y. Recent advances in metabolic engineering, protein engineering, and transcriptome-guided insights toward synthetic production of Taxol. Front Bioeng Biotechnol. 2021;9:632269. doi:10.3389/fbioe.2021.632269.
Zhang S, Ye T, Liu Y, Hou G, Wang Q, Zhao F, et al. Research advances in clinical applications, anticancer mechanism, total chemical synthesis, semi-synthesis and biosynthesis of paclitaxel. Molecules. 2023;28(22):7517. doi:10.3390/molecules28227517.
RCSB Protein Data Bank. 8X3E: CYP725A4-Taxa-4,11-diene complex [Internet]. RCSB PDB; 2024 [cited 2026 May 24]. Available from: https://www.rcsb.org/structure/8X3E
10b. Create a supply list and budget for your project (bullet-point list)