Subsections of Homework
Week 1 HW: Principles and Practices
![cover image]()
![cover image]() First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
A biological engineering project I have been passionate about to bring into the world intersects art conservation, biology and design. My goal for the final project portion would be to create a conservation treatment for cultural heritage objects specifically that of ceramics and textiles utilizing synthetic biology.
The world of art conservation is a crucial field that serves to protect, and conserve the objects of our human ancestry. These objects include paintings, ceramics, textiles, baskets etc… Conservation scientists and art conservators do hands-on and theoretical work to ensure our histories are preserved for the future. They establish regulations around best practices and balance artistic intention with scientific treatment.
Conservation scientists specialize in chemistry, and though the field is inherently interdisciplinary, it wasn’t until recently that biology became a consideration in preserving cultural heritage objects. Bacteria, and fungi, are often the predators that eat away, and degrade these objects. Much of the work has come to preventing fungal growth on paintings, paper and textiles. However, in recent light of pressing issues, bacteria have come to the aid in conserving large scale monuments.
Microbiologists and art conservators have been able to team up to “train” bacteria to eat specific solvents or to grow and bring filth up to the surface of larger monuments.
I graduated with a BFA in ceramics and specialized in biofabrication. After graduation, I was fortunate to land an internship at the Walters Art Museum under the Conservation Scientist, Annette Ortiz.
I am passionate about the materiality of ceramics, as much as I am about the connective tissue it provides to our oldest relatives. Conservation science is a proactive investigation in understanding an object in all the ways it exists.
Ceramic has macro and micro cracking as issues either from the firing process, or natural degradation of the material. I propose the process of biomineralization to fill in micro and macro cracks.
Textiles is a semi-neglected sector of art conservation as the scale and nature of the work is arduous. This causes a gap in the kind of cultural heritage objects that get treated and assessed. Silk, for example, once it begins degrading, is no longer treatable as the fibers begin to split. What if there was a means to conserve silk working with protein design? In this case, fundamentals of biology become vital to conserving objects lost to time and neglect.
Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals. Below is one example framework (developed in the context of synthetic genomics) you can choose to use or adapt, or you can develop your own. The example was developed to consider policy goals of ensuring safety and security, alongside other goals, like promoting constructive uses, but you could propose other goals for example, those relating to equity or autonomy.
The provided governance/policy goals are an amalgamation of goals from the AIC code of conduct for art conservators and goals from the NSCEB report.
Respect Cultural Property and Cultural Diplomacy
- The Cultural heritage objects chosen for review will have gone through the institutional and cultural channels to properly determine necessity for treatments.
- For objects institutionalized outside of the origin culture, cultural autonomy to origin peoples and governments offer consent to scientific treatment if and when applicable.
- Assess artist intention, and cultural context to determine treatments.
Advocate for Preservation
- Active pursuit of knowledge in obtaining novel possible treatments of cultural heritage objects to ensure the conservation of such objects is prioritized.
- Consistent testing to ensure experimental treatments are safe for historical cultural heritage objects to minimize prolonged harm from biological treatment.
- All conservation methods are reversible to allow for the evolution of treatments to be applicable to all cultural heritage objects in the present and future.
Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”). Try to outline a mix of actions (e.g. a new requirement/rule, incentive, or technical strategy) pursued by different “actors” (e.g. academic researchers, companies, federal regulators, law enforcement, etc). Draw upon your existing knowledge and a little additional digging, and feel free to use analogies to other domains (e.g. 3D printing, drones, financial systems, etc.).
Implementation of Safe Scientific Examination:
- Microbiologists and Art conservators have separate codes of conducts respective to their areas of research. Various qualities of these fields demand different responses and practices.
- Due to the nature of the work, a new code of conduct decided and created by a group of art conservators and microbiologists would help in bridging these disparate practices together. This would require the merging of bio lab safety practices with conservation lab ethics in dealing with sensitive objects and materials. The implementation of Safe scientific examination would determine safe handling and transportation of both cultural heritage objects and biological materials between labs, determining various risk factors dependent on organism, object and scale, as well as institutional regulations of the participating researchers.
- It could be assumed that a code of conduct may be difficult to implement due to the wide variety of needs and resources of a given project. Failure to ensure lab safety protocols are followed within treatments could lead to contamination, exposure and further degradation of objects.
Ethical Considerations of Cultural Heritage Object Context:
<
- Conservators are trained to determine the conditions fit for treatment. This often involves the context of the objects creation and artists intent if applicable. This framework is necessary to uphold in order to maintain best practices.
- Art conservators, curators and the boards of cultural institutions will go through review to determine intention and impact of any recommended biological treatment. If biological treatment is recommended, consent from all parties including but not limited to, the institution, conservators, microbiologists, artists etc… A documented treatment plan will be developed evaluating cultural context as part of the treatment consideration. This will be shared with biologists entering the team.
- By setting ethical standards of use and execution, it can be assumed objects of consideration will be appropriately chosen for treatment. It can also be assumed that defining the cultural context of an object, and therefore designing treatment, is an ambiguous process and requires a historical understanding of who is defining/defined the ethical framework and could this contain biases that effect treatment?
Implementation of Fair Labor Contracts for Art Conservators and Interdisciplinary Collaboration:
- Unfortunately, the gap in public perception and financial support between the life sciences and humanities is quite large. This cross interdisciplinary work provides room for negotiation for equitable contracts across fields as teams involve research and cultural institutions. In my proposal, I am interested in specifying ceramics and textiles. Textile conservators do not typically hold long term positions within institutions. Object, book, and painting conservators can obtain long-term positions, while textile conservators rely on project based contracts which contributes to job insecurity for specialized professionals.
- Treatments and teams can be considered under several domains such as biotechnology, humanities and biology in which institutions can branch funding avenues to guarantee success of projects and also fair distribution of funds to contracted professionals. Cultural institutions can act as the “Principal Investigators” to projects and sub-contract microbiologists as “co-investigators” to define roles and designate appropriate funding.
- It can be assumed that project based contracts can create wider cross institutional opportunities, however, it can also maintain the same initial problem. Defining the “principal” institution is project dependent as well as defining roles. This can must be established in the suggested code of conduct intially proposed. General questions to consider: What and who is the project for? Where is funding coming from and from there, what funds get allocated to supporting labor and operational costs?
| Does the option: | Implementation of Safe Scientific Examination | Ethical Considerations of Cultural Heritage Object Context | Implementation of Fair Labor Contracts for Art Conservators and Interdisciplinary Collaboration |
|---|
| Ensure Fair Interdisciplinary Collaboration | | | |
| • By Designating Roles and Responsibilities | 2 | 2 | 1 |
| • Prioritize Project Goals | 1 | 2 | 2 |
| Foster Lab Safety | | | |
| • By preventing incident | 1 | 1 | |
| • By helping Respond | 1 | 1 | 3 |
| Preserve Cultural Heritage Objects | | | |
| • Preventive Conservation | 1 | 1 | |
| • Obliging to the Conservation Code of Conduct | 1 | 1 | 2 |
| Other considerations | | | |
| • Minimizing costs and burdens to stakeholders | | | 3 |
| • Feasibility? | 1 | 1 | 3 |
| • Not impede research | 1 | 2 | 1 |
| • Promote constructive applications | 1 | 1 | 1 |
Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties. For this, you can choose one or more relevant audiences for your recommendation, which could range from the very local (e.g. to MIT leadership or Cambridge Mayoral Office) to the national (e.g. to President Biden or the head of a Federal Agency) to the international (e.g. to the United Nations Office of the Secretary-General, or the leadership of a multinational firm or industry consortia). These could also be one of the “actor” groups in your matrix.
Reflecting on what you learned and did in class this week, outline any ethical concerns that arose, especially any that were new to you. Then propose any governance actions you think might be appropriate to address those issues. This should be included on your class page for this week.
The governance action goals all feed into each other in various aspects. However, the Implementation of Safe Scientific Examination would be one I’d prioritize as both actors of cultural and research institutions and personnel are equally involved in defining the guidelines of safe use and treatment. It requires the participation and active collaboration of microbiologists and art conservators to inform each other on best practices considering the specifications and principles of the diverse fields. It is vital that communication is established and lab safety protocols for both conservation and biological labs are decided and mutually understood.
I have become increasingly aware of how this practice would be financed and distinguishing private versus public funding. Especially when allocating funds to various institutions that operate separately and have various groups to support.
REFERENCES:
- https://edition.cnn.com/style/article/bacteria-art-restoration#:~:text=Rome%2C%20Italy%20CNN%20%E2%80%94,the%20course%20of%20two%20weeks.
- https://www.biocodexmicrobiotainstitute.com/en/bacteria-restorers-works-art-future-allies-heritage-conservation
- https://pmc.ncbi.nlm.nih.gov/articles/PMC10667932/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC10667932/
- https://www.si.edu/stories/care-victorian-silk-quilts#:~:text=The%20folds%20should%20be%20padded,the%20Smithsonian's%20Public%20Inquiry%20Services.
- https://www.k18hair.com/
- https://www.culturalheritage.org/conservation-at-work/uphold-professional-standards/code#:~:text=I,Mitigate%20Adverse%20Effects
Doctor jacobson
- Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
The error rate of polymerase is ~1:10^6. The human genome is ~3.2GBP therefore, in comparison to the error rate of the polymerase, which accounts for 3,200 mistakes in copying DNA. Biology deals with this discrepancy via the MutS repair system.
- How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
The different ways there are to code for an average human protein is 2^200 which is 1.6eX10.
All of these different codes don’t work to code for the protein of interest because of codon optimization and bias.
Dr. Le Proust:
What’s the most commonly used method for oligo synthesis currently?
From what I can assume for the slides, the most commonly used method for oligo synthesis is Phosphoramidite method by Caruthers and Electrochemical-based microarray by CombiMatrix which can be found on the chronological timeline of development.
Twist bioscience has new technology known as a silicone platform.
Why is it difficult to make oligos longer than 200nt via direct synthesis?
It is difficult to make oligos longer than 200nt via direct synthesis because of the accumulative decrease in yield and is an exponential decay.
https://www.glenresearch.com/reports/gr21-211#:~:text=Coupling%20Step,the%20concentration%20of%20phosphoramidite%20itself.
Why can’t you make a 2000bp gene via direct oligo synthesis?
You can’t make a 2000np gene via direct oligo synthesis because oligos are generally limited to 100-200bp and yields decrease significantly. https://www.lubio.ch/blog/the-challenge-of-making-long-oligos#:~:text=There%20are%20several%20challenges%20to%20synthesizing%20long,a%20fidelity%20of%20only%2099%25%20or%2010.
Dr. Church
[Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
Arginine (Arg)
Histidine (His)
Lysine (Lys)
Isoleucine (Ile)
Leucine (Leu)
Methionine (Met)
Phenylalanine (Phe)
Threonine (Thr)
Tryptophan (Try)
Valine (Val)
Lysine contingency is the concept of evolving past the production of our own Lysine and needing to obtain lysine externally. As Jurassic Park alarms, creating control factors such as a lysine contingency places a dependency of the organism we modify on researchers in an attempt to mitigate risk etc. However, like in Jurassic Park, the dinosaurs were able to obtain the lysine in their environment causing an experimental failure. This is an important ethical dilemma when designing experiments and genetically modifying organisms.
https://open.oregonstate.education/animalnutrition/chapter/proteins-structure/#:~:text=List%20of%20Essential%20Amino%20Acids%20and%20Their,(Thr)%20*%20Tryptophan%20(Try)%20*%20Valine%20(Val)
https://jurassicpark.fandom.com/wiki/Lysine_contingency
“The lysine contingency is intended to prevent the spread of the animals in case they ever got off the island. Dr. Wu inserted a gene that creates a single faulty enzyme in protein metabolism. The animals can’t manufacture the amino acid lysine. Unless they’re continually supplied with lysine by us, they’ll slip into a coma and die.”
—Ray Arnold(src)
Week 2 HW: DNA Read, Write and Edit
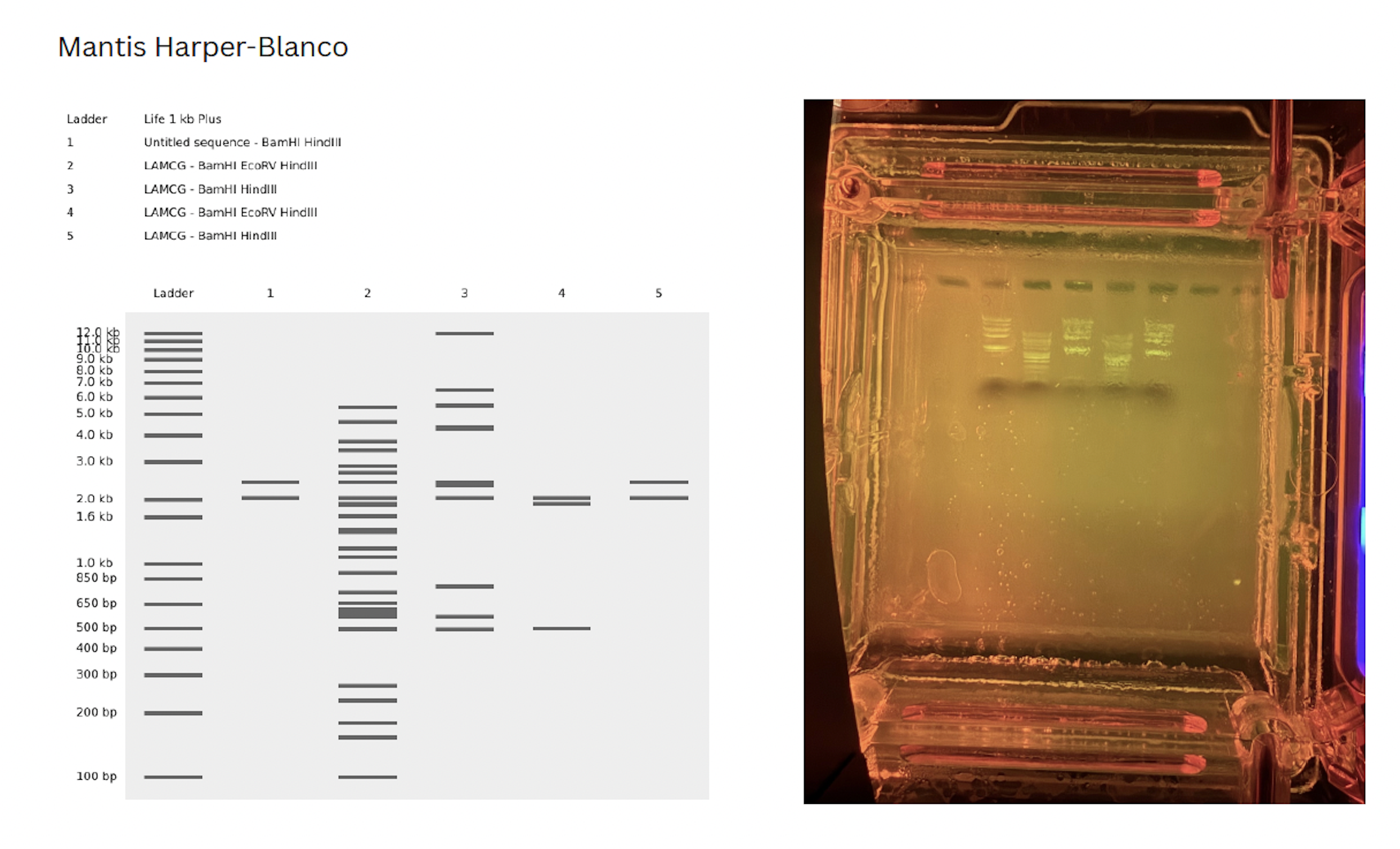
My initial confusion was trouble shooting how to cut at a specific segments of DNA. On my benchling, I copied a segment of DNA between the desired cut sites and pasted it into a “New DNA sequence” file. This was purely for artistic purposes, however, in lab, I asked our instructor how we would select for these specific sites for future gel art.
There were two options:
- Purchase or design primers to select at the specific site
OR
- Run the DNA with all of the cut sites with the selected enzymes and do a gel extraction of the desired band. Hypthetically, in the next run, only that site will appear (though the band will be faded due to less DNA content)
3.1. Choose your protein.
In recitation, we discussed that you will pick a protein for your homework that you fi nd interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose.
I chose Sericin from the organism Bombyx Mori (Silk Moth) because for my fi nal project I am interested in creating art conservation methods and protocols from synthetic biological protocols and principals to preserve cultural heritage objects and textiles. Specifi cally silk which once enters a degradation phase, is untreatable as the silk essentially powderizes. Sericin is the native gum that protects fi broin but is boiled off in the process to get the fi ne silk fi ber we use today. Sericin was considered a waste stream of the silk process but has been used in biomedical biomaterial cases and shows promise in being designed as a conservation protein glue to keep silk textiles protected.
https://www.uniprot.org/uniprotkb/P07856/entry
MRFVLCCTLIALAALSVKAFGHHPGNRDTVEVKNRKYNAASSESSYLNKDNDSISAGAHAKSVEQSQDKSKYTSGPEGVSYSGRSQNYKDSKQAYADYHSDPNGGSASAGQSRDSSLRERNVHYVSDGEAVAASSDARDENRSAQQNAQANWNADGSYGVSADRSGSASSRRRQANYYSDKDITAASKDDSRADSSRRSNAYYNRDSDGSESAGLSDRSASSSKNDNVFVYRTKDSIGGQAKSSRSSHSQESDAYYNSSPDGSYNAGTRDSSISNKKKASSTIYADKDQIRAANDRSSSKQLKQSSAQISSGPEGTSVSSKDRQYSNDKRSKSDAYVGRDGTVAYSNKDSEKTSRQSNTNYADQNSVRSDSAASDQTSKSYDRGYSDKNIVAHSSGSRGSQNQKSSSYRADKDGFSSSTNTEKSKFSSSNSVVETSDGASASRESSAEDTKSSNSNVQSDEKSASQSSSSRSSQESASYSSSSSSSTLSEDSSEVDIDLGNLGWWWNSDNKVQRAAGGATKSGASSSTQATTVSGADDSADSYTWWWNPRRSSSSSSSASSSSSGSNVGGSSQSSGSSTSGSNARGHLGTVSSTGSTSNTDSSSKSAGSRTSGGSSTYGYSSSHRGGSVSSTGSSSNTDSSTKNAGSSTSGGSSTYGYSSSHRGGSVSSTGSSSNTDSSTKSAGSSTSGGSSTYGYSSRHRGGRVSSTGSSSTTDASSNSVGSSTSGGSSTYGYSSNSRDGSVSSTGSSSNTDSNSNSAGSSTSGGSSTYGYSSNSRDGSVSSTGSSSNTDSNSNSAGSSTSGGSSTYGYSSNSRDGSVSSTGSSSNTDASTDLTGSSTSGGSSTYGYSSDSRDGSVSSTGSSSNTDASTDLAGSSTSGGSSTYGYSSDCGDGSVSSTGSSSNTDASTDLAGSSTSGGSSTYGYSSDSRDGSVSSTGSSSNTDASTDLAGSSTSGGSSTYGYSSNSRDGSVSSTGSSSNTDASTDLTGSSTSGGSSTYGYSSSNRDGSVLATGSSSNTDASTTEESTTSAGSSTEGYSSSSHDGSVTSTDGSSTSGGASSSSASTAKSDAASSEDGFWWWNRRKSGSGHKSATVQSSTTDKTSTDSASSTDSTSSTSGASTTTSGSSSTSGGSSTSDASSTSSSVSRSHHSGVNRLLHKPGQGKICLCFENIFDIPYHLRKNIGV
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
[Example: Get to the original sequence of phage MS2 L-protein from its genome
phage MS2 genome - Nucleotide - NCBI
]
To reverse translate the sericin AA sequence I used https://www.bioinformatics.org/sms2/rev_trans.html and received the DNA sequence below.
atgcgctttgtgctgtgctgcaccctgattgcgctggcggcgctgagcgtgaaagcgtttggccatcatccgggcaaccgcgataccgtggaagtgaaaaaccgcaaatataacgcggcgagcagcgaaagcagctatctgaacaaagataacgatagcattagcgcgggcgcgcatcgcgcgaaaagcgtggaacagagccaggataaaagcaaatataccagcggcccggaaggcgtgagctatagcggccgcagccagaactataaagatagcaaacaggcgtatgcggattatcatagcgatccgaacggcggcagcgcgagcgcgggccagagccgcgatagcagcctgcgcgaacgcaacgtgcattatgtgagcgatggcgaagcggtggcggcgagcagcgatgcgcgcgatgaaaaccgcagcgcgcagcagaacgcgcaggcgaactggaacgcggatggcagctatggcgtgagcgcggatcgcagcggcagcgcgagcagccgccgccgccaggcgaactattatagcgataaagatattaccgcggcgagcaaagatgatagccgcgcggatagcagccgccgcagcaacgcgtattataaccgcgatagcgatggcagcgaaagcgcgggcctgagcgatcgcagcgcgagcagcagcaaaaacgataacgtgtttgtgtatcgcaccaaagatagcattggcggccaggcgaaaagcagccgcagcagccatagccaggaaagcgatgcgtattataacagcagcccggatggcagctataacgcgggcacccgcgatagcagcattagcaacaaaaaaaaagcgagcagcaccatttatgcggataaagatcagattcgcgcggcgaacgatcgcagcagcagcaaacagctgaaacagagcagcgcgcagattagcagcggcccggaaggcaccagcgtgagcagcaaagatcgccagtatagcaacgataaacgcagcaaaagcgatgcgtatgtgggccgcgatggcaccgtggcgtatagcaacaaagatagcgaaaaaaccagccgccagagcaacaccaactatgcggatcagaacagcgtgcgcagcgatagcgcggcgagcgatcagaccagcaaaagctatgatcgcggctatagcgataaaaacattgtggcgcatagcagcggcagccgcggcagccagaaccagaaaagcagcagctatcgcgcggataaagatggctttagcagcagcaccaacaccgaaaaaagcaaatttagcagcagcaacagcgtggtggaaaccagcgatggcgcgagcgcgagccgcgaaagcagcgcggaagataccaaaagcagcaacagcaacgtgcagagcgatgaaaaaagcgcgagccagagcagcagcagccgcagcagccaggaaagcgcgagctatagcagcagcagcagcagcagcaccctgagcgaagatagcagcgaagtggatattgatctgggcaacctgggctggtggtggaacagcgataacaaagtgcagcgcgcggcgggcggcgcgaccaaaagcggcgcgagcagcagcacccaggcgaccaccgtgagcggcgcggatgatagcgcggatagctatacctggtggtggaacccgcgccgcagcagcagcagcagcagcagcgcgagcagcagcagcagcggcagcaacgtgggcggcagcagccagagcagcggcagcagcaccagcggcagcaacgcgcgcggccatctgggcaccgtgagcagcaccggcagcaccagcaacaccgatagcagcagcaaaagcgcgggcagccgcaccagcggcggcagcagcacctatggctatagcagcagccatcgcggcggcagcgtgagcagcaccggcagcagcagcaacaccgatagcagcaccaaaaacgcgggcagcagcaccagcggcggcagcagcacctatggctatagcagcagccatcgcggcggcagcgtgagcagcaccggcagcagcagcaacaccgatagcagcaccaaaagcgcgggcagcagcaccagcggcggcagcagcacctatggctatagcagccgccatcgcggcggccgcgtgagcagcaccggcagcagcagcaccaccgatgcgagcagcaacagcgtgggcagcagcaccagcggcggcagcagcacctatggctatagcagcaacagccgcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatagcaacagcaacagcgcgggcagcagcaccagcggcggcagcagcacctatggctatagcagcaacagccgcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatagcaacagcaacagcgcgggcagcagcaccagcggcggcagcagcacctatggctatagcagcaacagccgcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatgcgagcaccgatctgaccggcagcagcaccagcggcggcagcagcacctatggctatagcagcgatagccgcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatgcgagcaccgatctggcgggcagcagcaccagcggcggcagcagcacctatggctatagcagcgattgcggcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatgcgagcaccgatctggcgggcagcagcaccagcggcggcagcagcacctatggctatagcagcgatagccgcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatgcgagcaccgatctggcgggcagcagcaccagcggcggcagcagcacctatggctatagcagcaacagccgcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatgcgagcaccgatctgaccggcagcagcaccagcggcggcagcagcacctatggctatagcagcagcaaccgcgatggcagcgtgctggcgaccggcagcagcagcaacaccgatgcgagcaccaccgaagaaagcaccaccagcgcgggcagcagcaccgaaggctatagcagcagcagccatgatggcagcgtgaccagcaccgatggcagcagcaccagcggcggcgcgagcagcagcagcgcgagcaccgcgaaaagcgatgcggcgagcagcgaagatggcttttggtggtggaaccgccgcaaaagcggcagcggccataaaagcgcgaccgtgcagagcagcaccaccgataaaaccagcaccgatagcgcgagcagcaccgatagcaccagcagcaccagcggcgcgagcaccaccaccagcggcagcagcagcaccagcggcggcagcagcaccagcgatgcgagcagcaccagcagcagcgtgagccgcagccatcatagcggcgtgaaccgcctgctgcataaaccgggccagggcaaaatttgcctgtgctttgaaaacatttttgatattccgtatcatctgcgcaaaaacattggcgtg
Codon optimized for E.coli (K12) via benchling codon optimization tools with reduced hair pinning
Atgcgctttgtgctgtgctgcaccctgattgcgctggcggcgctgagcgtgaaagcgtttggccatcatccgggcaaccgcgataccgtggaagtgaaaaaccgcaaatataacgcggcgagcagcgaaagcagctatctgaacaaagataacgatagcattagcgcgggcgcgcatc
gcgcgaaaagcgtggaacagagccaggataaaagcaaatataccagcggcccggaaggcgtgagctatagcggccgcagccagaactataaagatagcaaacaggcgtatgcggattatcatagcgatccgaacggcggcagcgcgagcgcgggccagagccgcgatagcagcctgcgcgaacgcaacgtgcattatgtgagcgatggcgaagcggtggcggcgagcagcgatgcgcgcgatgaaaaccgcagcgcgcagcagaacgcgcaggcgaactggaacgcggatggcagctatggcgtgagcgcggatcgcagcggcagcgcgagcagccgccgccgccaggcgaactattatagcgataaagatattaccgcggcgagcaaagatgatagccgcgcggatagcagccgccgcagcaacgcgtattataaccgcgatagcgatggcagcgaaagcgcgggcctgagcgatcgcagcgcgagcagcagcaaaaacgataacgtgtttgtgtatcgcaccaaagatagcattggcggccaggcgaaaagcagccgcagcagccatagccaggaaagcgatgcgtattataacagcagcccggatggcagctataacgcgggcacccgcgatagcagcattagcaacaaaaaaaaagcgagcagcaccatttatgcggataaagatcagattcgcgcggcgaacgatcgcagcagcagcaaacagctgaaacagagcagcgcgcagattagcagcggcccggaaggcaccagcgtgagcagcaaagatcgccagtatagcaacgataaacgcagcaaaagcgatgcgtatgtgggccgcgatggcaccgtggcgtatagcaacaaagatagcgaaaaaaccagccgccagagcaacaccaactatgcggatcagaacagcgtgcgcagcgatagcgcggcgagcgatcagaccagcaaaagctatgatcgcggctatagcgataaaaacattgtggcgcatagcagcggcagccgcggcagccagaaccagaaaagcagcagctatcgcgcggataaagatggctttagcagcagcaccaacaccgaaaaaagcaaatttagcagcagcaacagcgtggtggaaaccagcgatggcgcgagcgcgagccgcgaaagcagcgcggaagataccaaaagcagcaacagcaacgtgcagagcgatgaaaaaagcgcgagccagagcagcagcagccgcagcagccaggaaagcgcgagctatagcagcagcagcagcagcagcaccctgagcgaagatagcagcgaagtggatattgatctgggcaacctgggctggtggtggaacagcgataacaaagtgcagcgcgcggcgggcggcgcgaccaaaagcggcgcgagcagcagcacccaggcgaccaccgtgagcggcgcggatgatagcgcggatagctatacctggtggtggaacccgcgccgcagcagcagcagcagcagcagcgcgagcagcagcagcagcggcagcaacgtgggcggcagcagccagagcagcggcagcagcaccagcggcagcaacgcgcgcggccatctgggcaccgtgagcagcaccggcagcaccagcaacaccgatagcagcagcaaaagcgcgggcagccgcaccagcggcggcagcagcacctatggctatagcagcagccatcgcggcggcagcgtgagcagcaccggcagcagcagcaacaccgatagcagcaccaaaaacgcgggcagcagcaccagcggcggcagcagcacctatggctatagcagcagccatcgcggcggcagcgtgagcagcaccggcagcagcagcaacaccgatagcagcaccaaaagcgcgggcagcagcaccagcggcggcagcagcacctatggctatagcagccgccatcgcggcggccgcgtgagcagcaccggcagcagcagcaccaccgatgcgagcagcaacagcgtgggcagcagcaccagcggcggcagcagcacctatggctatagcagcaacagccgcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatagcaacagcaacagcgcgggcagcagcaccagcggcggcagcagcacctatggctatagcagcaacagccgcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatagcaacagcaacagcgcgggcagcagcaccagcggcggcagcagcacctatggctatagcagcaacagccgcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatgcgagcaccgatctgaccggcagcagcaccagcggcggcagcagcacctatggctatagcagcgatagccgcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatgcgagcaccgatctggcgggcagcagcaccagcggcggcagcagcacctatggctatagcagcgattgcggcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatgcgagcaccgatctggcgggcagcagcaccagcggcggcagcagcacctatggctatagcagcgatagccgcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatgcgagcaccgatctggcgggcagcagcaccagcggcggcagcagcacctatggctatagcagcaacagccgcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatgcgagcaccgatctgaccggcagcagcaccagcggcggcagcagcacctatggctatagcagcagcaaccgcgatggcagcgtgctggcgaccggcagcagcagcaacaccgatgcgagcaccaccgaagaaagcaccaccagcgcgggcagcagcaccgaaggctatagcagcagcagccatgatggcagcgtgaccagcaccgatggcagcagcaccagcggcggcgcgagcagcagcagcgcgagcaccgcgaaaagcgatgcggcgagcagcgaagatggcttttggtggtggaaccgccgcaaaagcggcagcggccataaaagcgcgaccgtgcagagcagcaccaccgataaaaccagcaccgatagcgcgagcagcaccgatagcaccagcagcaccagcggcgcgagcaccaccaccagcggcagcagcagcaccagcggcggcagcagcaccagcgatgcgagcagcaccagcagcagcgtgagccgcagccatcatagcggcgtgaaccgcctgctgcataaaccgggccagggcaaaatttgcctgtgctttgaaaacatttttgatattccgtatcatctgcgcaaaaacattggcgtg
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependentor cell-freemethods,or both
.
Prior to insertion of my gene of interets, the gene cannot be inserted alone, it must exits within a plasmid ( a circular piece of DNA native to bacteria). The plasmid contains several important pieces that allows the cell to READ the DNA for transcritption and later down the line, translation then expression. These elements include a promoter (which tells the cell’s machinary where to start transcription of your gene of interest), RBS, start codon typically (ATG), CDS, His tag, stop codon and terminator. A backbone selection completes the plasmid by encoding an origin of replication, antibiotic resistance and operons etc!
To ensure I got a lot of the gene I need, I would design custom primers for amplification via PCR.
Part 4: Prepare a Twist DNA Synthesis Order
This is a practice exercise, not necessarily your real Twist order!
4.1. Create a Twist account and a Benchling account
click through for Twist signup
click through for Benchling signup
4.2. Build Your DNA Insert Sequence
For example, let’s make a sequence that will make E. coli glow fl uorescent green under UV light by constitutively (always) expressing sfGFP (a green fl uorescent protein):
- In Benchling, select New DNA/RNA sequence
- Give your insert sequence a name and select DNA with a Linear topology (this is a linear sequence that will be inserted into a circular backbone vector of our choosing).
- Go through each piece of the given DNA sequences highlighted below (Promoter, RBS, Start Codon, Coding Sequence, His Tag, Stop Codon,Terminator) and paste the sequences into the Benchling fi le one after the other (replacing the coding sequence with your codon optimized DNA sequence of interest!). Each time you add a new piece of the sequence, make sure to annotate by right clicking over the sequence and creating an annotation that describes what each piece (e.g., Promoter, RBS, etc.) is (see image below)
Promoter
(e.g.BBa_J23106): TTTACGGCTAGCTCAGTCCTAGGTATAGTGCTAGC
RBS
(e.g.BBa_B0034 with spacers for optimal expression): CATTAAAGAGGAGAAAGGTACC
Start Codon
ATG
Coding Sequence
(your codon optimized DNA for a protein of interest,sfGFP for example): AGCAAAGGAGAAGAACTTTTCACTGGAGTTGTCCCAATTCTTGTTGAATTAGATGGTGATGTTAATGGGCACAAATTTTCTGTCCGTGGAGAGGGTGAAGGTGATGCTACAAACGGAAAACTCACCCTTAAATTTATTTGCACTACTGGAAAACTACCTGTTCCGTGGCCAACACTTGTCACTACTCTGACCTATGGTGTTCAATGCTTTTCCCGTTATCCGGATCACATGAAACGGCATGACTTTTTCAAGAGTGCCATGCCCGAAGGTTATGTACAGGAACGCACTATATCTTTCAAAGATGACGGGACCTACAAGACGCGTGCTGAAGTCAAGTTTGAAGGTGATACCCTTGTTAATCGTATCGAGTTAAAGGGTATTGATTTTAAAGAAGATGGAAACATTCTTGGACACAAACTCGAGTACAACTTTAACTCACACAATGTATACATCACGGCAGACAAACAAAAGAATGGAATCAAAGCTAACTTCAAAATTCGCCACAACGTTGAAGATGGTTCCGTTCAACTAGCAGACCATTATCAACAAAATACTCCAATTGGCGATGGCCCTGTCCTTTTACCAGACAACCATTACCTGTCGACACAATCTGTCCTTTCGAAAGATCCCAACGAAAAGCGTGACCACATGGTCCTTCTTGAGTTTGTAACTGCTGCTGGGATTACACATGGCATGGATGAGCTCTACAAA
7x His Tag(Let’s add a 7×His tag at the C-terminus of the protein to enable protein purifi cation from E. coli):
CATCACCATCACCATCATCAC
Stop Codon
TAA
Terminator
(e.g.BBa_B0015):CCAGGCATCAAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTTCGTTTTATCTGTTGTTTGTCGGTGAACGCTCTCTACTAGAGTCACACTGGCTCACCTTCGGGTGGGCCTTTCTGCGTTTATA
A link to the expression cassette including all the required parts for expression in E.coli.
[https://benchling.com/s/seq-fdUK2PUX2MmY798xn0p1?m=slm-mtkTuHUVSUj7kP0YqL2I]
This insert sequence you built is commonly referred to as an expression cassette in molecular biology (a sequence you can drop into any vector and it’ll perform its function). Go ahead and download the FASTA file for the sequence you made.
4.3. On Twist, Select The “Genes” Option
4.4. Select “Clonal Genes” option
For this demonstration, we’ll choose Clonal Genes. You’ll select
clonal genesor gene fragments depending on your fi nal project. Historically, HTGAA projects using clonal genes (circular DNA) have reached experimental results 1-2 weeks quicker because they can be transformed directly into E. coli without additional assembly. Gene fragments
(linear DNA) offer greater design fl exibility but typically require an assembly or cloning step prior to transformation. An advantage is
If designed with the appropriate exonuclease protection, gene fragments can be used directly in cell-free expression.
4.5. Import your sequence
You just took an amino acid sequence of interest and converted it into DNA, codon optimized it, and built an expression cassette around it! Choose the Nucleotide Sequence option and Upload Sequence File to upload your FASTA fi le.
4.6. Choose Your Vector
Since we’re ordering aclonal gene, you will need to refer to Twist’s Vector Catalog to choose your circular backbone. You can think of this as taking your linear expression cassette for your protein of interest, and completing the rest of the circle! The backbone confers many special properties like antibiotic resistance, an origin of replication, and more. Discuss with your node to decide on appropriate antibiotic options. At MIT/Harvard, you can use Ampicillin, Chloramphenicol, or Kanamycin resistance.
Twist vectors do not contain restriction sites near the insert fragment, so make sure to flank your design with cut sites if you are intending to extract this DNA insert fragment later.
I chose the backbone pET-23(+) as shown below with my completed construct.
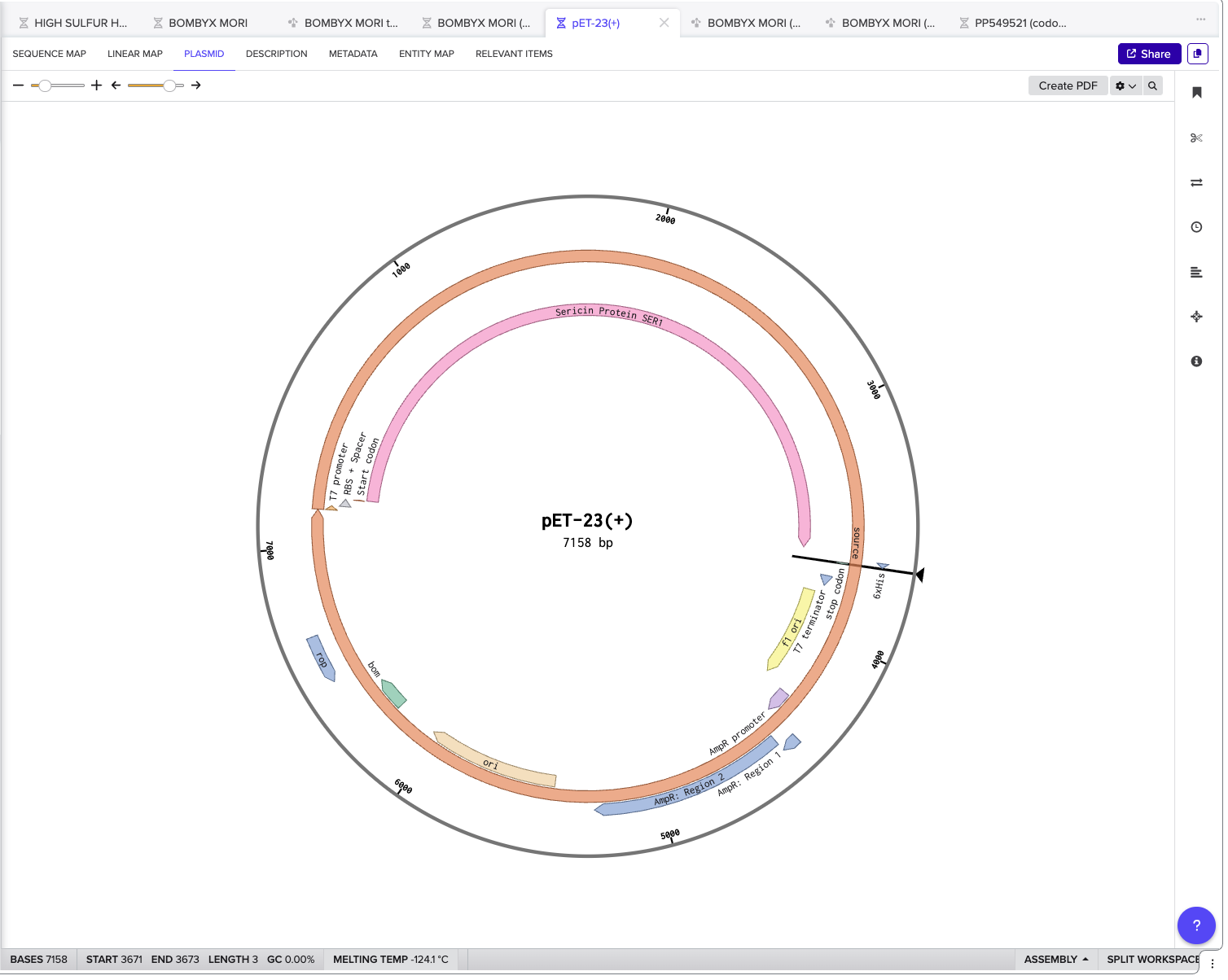
Click into your sequence and select download construct (GenBank) to get the full plasmid sequence:
Go back to your Benchling account
. Inside of a folder, click the import DNA/RNA sequence button and upload the GenBank file you just downloaded.
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why?
This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
DNA-based digital data storage technology. Source: Archives in DNA: Workshop Exploring Implications of an Emerging Bio-Digital Technology through Design Fiction - Scientifi c Figure on ResearchGate. Available from:
https://www.researchgate.net/fi gure/DNA-based-digital-data-storage-technology_fi g1_353128454
[accessed 11 Feb 2025]
In a previous workforce development program called Break into Biotech I was a part of, we chose to participate in a fi nal project to implement the skills we were learning. I chose the Microbiome group in which we would select a location, extract soil samples, extract the DNA, prep them and sequence them via the Illumna Mini Seq. Unfortunately, our experiment failed in that no data was extracted, therefore, no DNA was sequenced.
The goal was to do a microbial profi le of green wood cemetery and understand what kinds of prokaryotic life was residing in the soil.
I would love to push this research further and sequence soil surrounding natural burials to understand the vital decomposers of our bodies. New organims are constantly being discovered thanks to meta-data collection and analysis allowing us to fi nd novel traits in the most unlikely of places. I am innately curious about burial practice, death and subsequently, grief. What could these little critters tell us about the ways our bodies decompose? About our life and the decisions embodied through the fl esh inspiring the breakdown by these organisms? Perhaps even the symbiotic nature of organisms working together to break down a resource now in the soil that could release harmful chemicals? How do these organisms digest these compunds and what is excreted as a result? All of this can begin to be uncovered from sequencing.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Also answer the following questions:
- Is your method first-, second- or third-generation or other? How so?
- What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
- What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
- What is the output of your chosen sequencing technology?
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to infl ammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specifi c genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
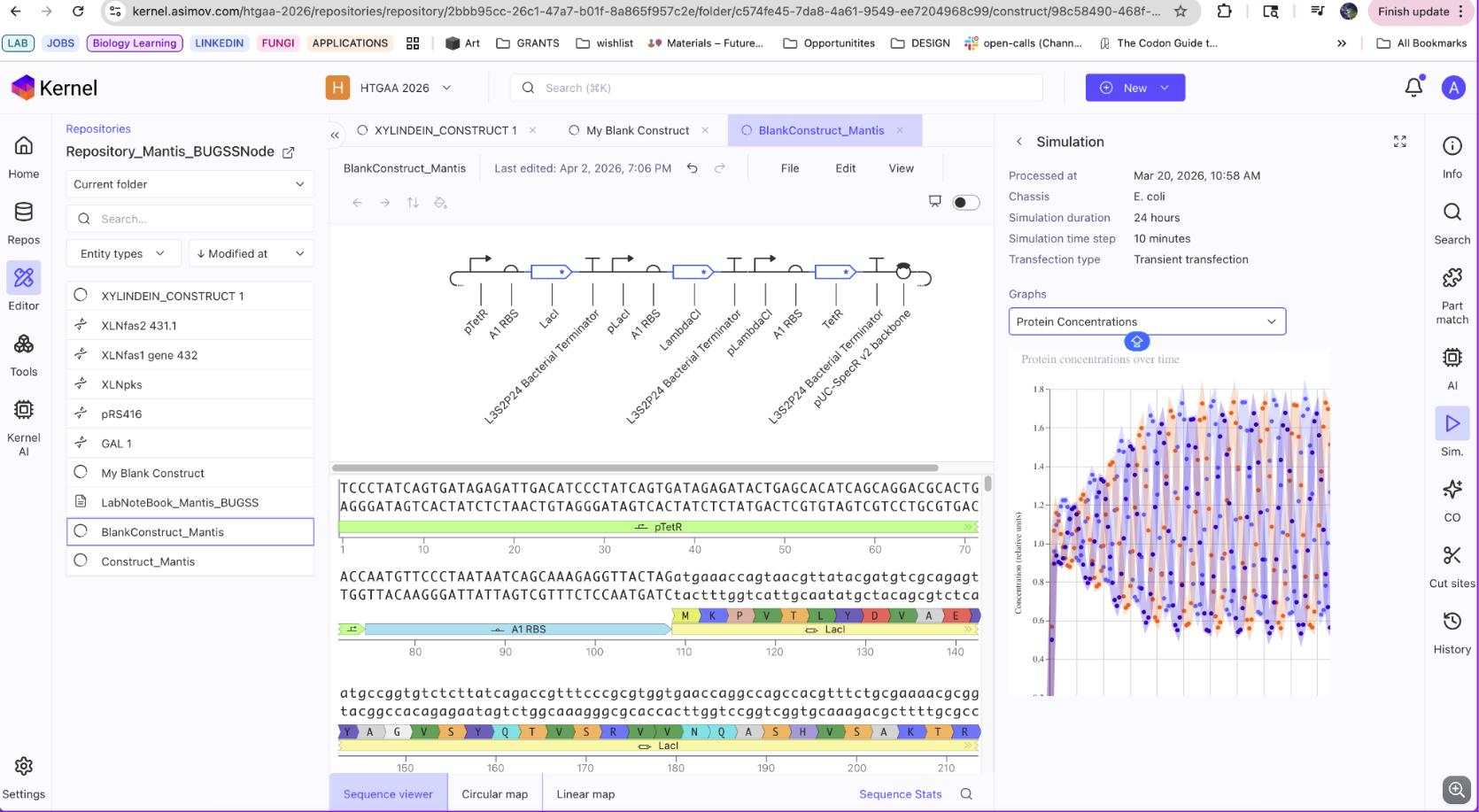
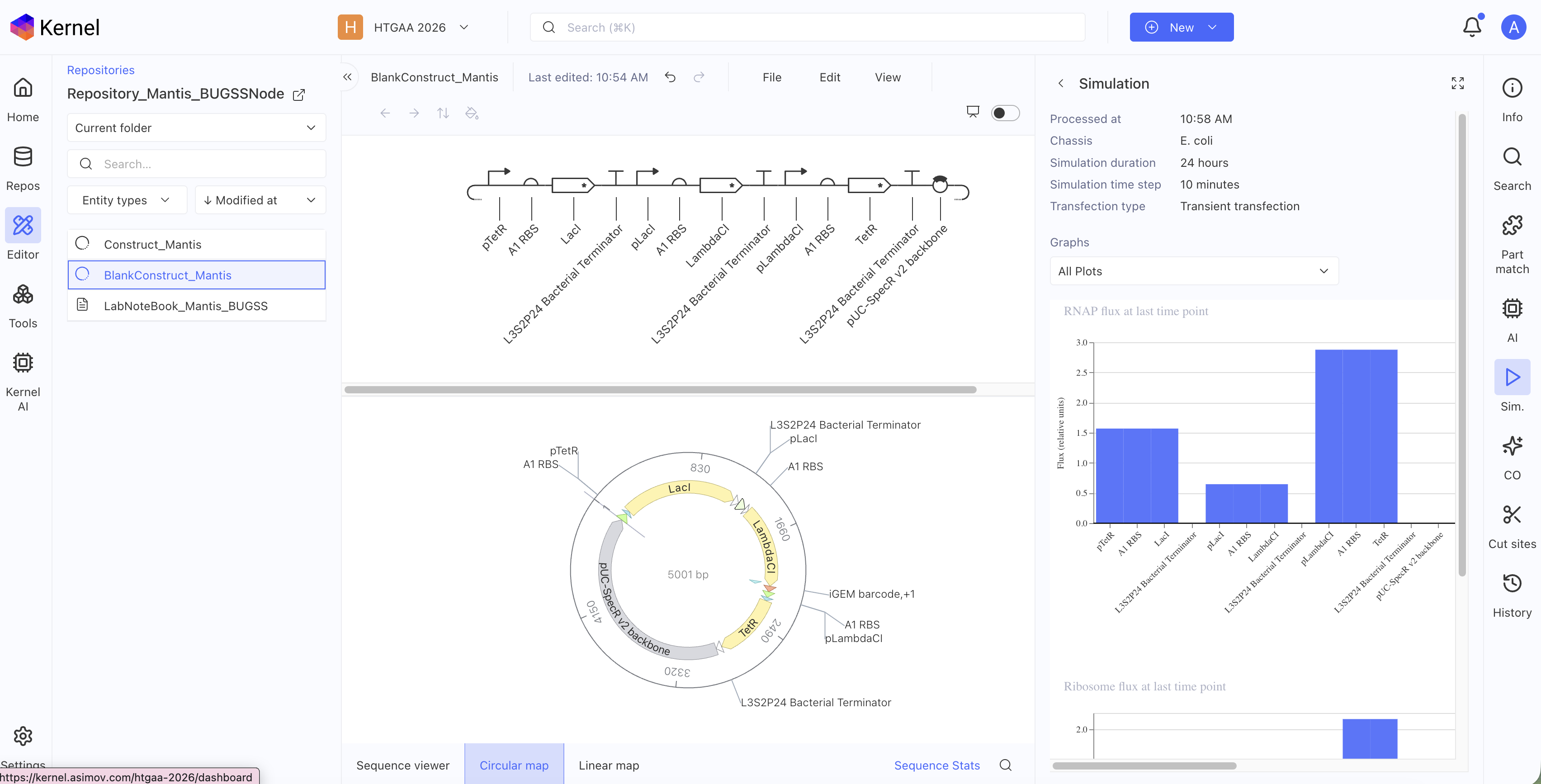
The biosynthetic gene cluster I would like to synthesize in S.cerevisiae comes from Chlorociboria Aeruginascens. The BGC is a secondary metabolite pathway that produces a pigment called Xylindein which is both beautiful and functional for its semi-conductive properties. The goal would be to produce this pigment and dye textiles for weavers!
The BGC contains a total of 7 genes!
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
I would choose phosphoramidite DNA synthesis as it is the most commonly used method for DNA synthesis. Howeverthere are complications considering my BGC is a polyketide pathway. Polyketides are some of the largest fragments, so this for of DNA synthesis is extrememly error prone and expensive due to the size of the fragments.
Also answer the following questions:
1.What are the essential steps of your chosen sequencing methods?
The following steps can be found on the HTGAA DNA synthesis slides.
a. Deprotection:
i. Acid catalyzed removal of DMT allows for subsequent base addition
b.Base Coupling:
i.A DMT protected phosphoramidite is added to the unprotected 5’ OH using a tetrazole activator
c.Capping
i.unreacted 5’ OH are acetylated to prevent further chain extension. This step helpsprevent single-base deletions at the expense of yield
d.Oxidation: Oxidation of phophite triester to phosphate using aqueous iodine.
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
The length of the chain is limited due to the error rate increasing as the chain grows. 100% coupling efficiency is near impossible. An abasic site is possible due to depurination.
https://www.twistbioscience.com/blog/science/simple-guide-phosphoramidite-chemistry-and-how-it-fi ts-twist-biosciences-commercial
https://www.compound.vc/writing/dna-synthesis-a-technical-primer
5.3 DNA Edit
(i) What DNA would you want to edit and why?
In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for fl ora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fi xation).
What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
Colossal Biosciences Inc., a biotechnology company using genetic engineering to de-extinct various historic animals such as the woolly mammoth, dodo, and dire wolf.
I would want to edit fungal DNA of fi lamentous fungi to resist contamination. There are researchers doing this work via symbiotic relationships in which the collective growth increases resiliency within a species so that even when contamination occurs, the fungi is able to resist full takeover by eating the mold itself. However, being able to grow a mycelium based product without the fear of contamination especially for regenerative engineered livign systems would be ideal. I believe this can be done by leveraging the secondary metabolic pathways found in fungi that are anti-microbial.
(ii) What technology or technologies would you use to perform these DNA edits and why?
I would employ CRISPR cas 9 technology when editing multicellular fungal polyketide pathways due to the hyper specifi city of CRISPR which could help over-express the pathway with minimal errors. In the paper listed below, the advent of CRISPR cas 9 would allow researchers more control over the expression of a secondary metabolite which is difficult to express in a host organism.
Also answer the following questions:
1. How does your technology of choice edit DNA? What are the essential steps?
a.Create gRNA(guide RNA) that binds to the specifi c target sequence. the gRNA also binds with the Cas9 enzyme. As the gRNA recognizes the targeted location the cas9 enzyme will cut the DNA for insertion. The rest is up to the hosts cell which will deploy its machinery to repair the DNA break, repairing the break with the inserted gene as though it was native. Similarly how it is done in bacteria.
2.What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
a.Choose your cas9 enzyme! Depending on your experimental needs a cas9 enzyme needs to be selected to fit the specifcs of experiment applications
b.gRNA selection: This is one of the protagonists as the insertion depends on the gRNA recognizing the target location and being repaired into the host DNA.
c. Target site selection: What are you trying to edit and why? Knowing your target site allows you
to create the experiment such as which cas9 variant, desiging of the gRNA and if there are any
PAM site close.
d. PAM and gRNA compatibility: Not all matches are made in heaven! When designing the gRNA,
it needs to have high on target specificity and needs to be close to a PAM sequence. This
compatbility includes the cas9 variant chosen to ensure correct binding to the PAM site!
3. What are the limitations of your editing methods (if any) in terms of efficiency or precision?
a. Requirement for a PAM near the site which is its own limiting factor, meaning not just ANY site
can be cut within the genome which can be inconvenient for gene therapy and polyketide
synthase edit purposes.
b. If there is another sequence that is similar to your target sequence, the gRNA could bind to an
unwanted site within the genome causing errors and incorrect placement, no integration or no
expression of the gene of interest.
c. Patenting issues arise when an organisms DNA is edited and transitions from a discovered
organism to a novel organism. What are the IP rights and how is that regulated and
distinguished? How can an organism be patented?
CRISPR-Cas for Fungal Genome Editing: A New Tool for the Management of Plant Diseases
References:
What are genome editing and CRISPR-Cas9?: MedlinePlus Genetics
What Are the Limitations of CRISPR-Cas9?
Optimizing CRISPR: Technology and Approaches for High Efficiency Gene Editing | VectorBuilder
Week 3 HW: Lab Automation
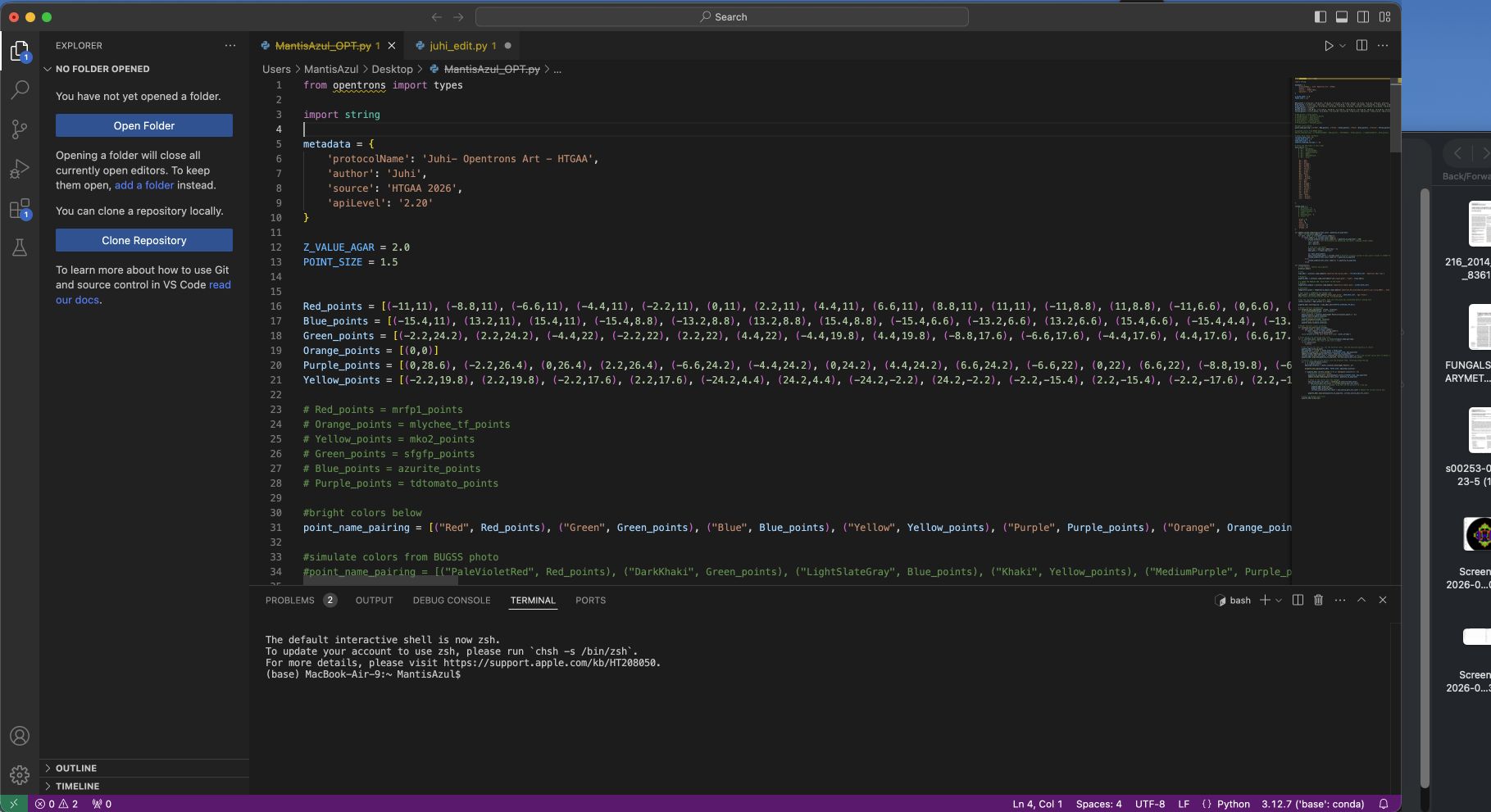

The design above was made using the platform graciously programmed by Ronan. The design is inspired by golden pieces made by Colombian craftsman. I was in Colombia at the time and had the privilege of seeing El Museo del Oro (The Gold Museum) and aimed to replicate some of the features here.
I input the coordinates from Ronan’s program into the opentrons file as seen above. There was quite a bit of trouble shooting as the equipment stated in the code was different from BUGSS supply. That caused some coordination issues within the opentrons that was eventually solved. During the lab, the majority of the time was dedicated to trouble shooting these minute bugs and I wasn’t able to run my script. However, Amanda and Joel (our instructor and TA! ((Thank you guys so much)) ran the script after lab hours which yielded this result:
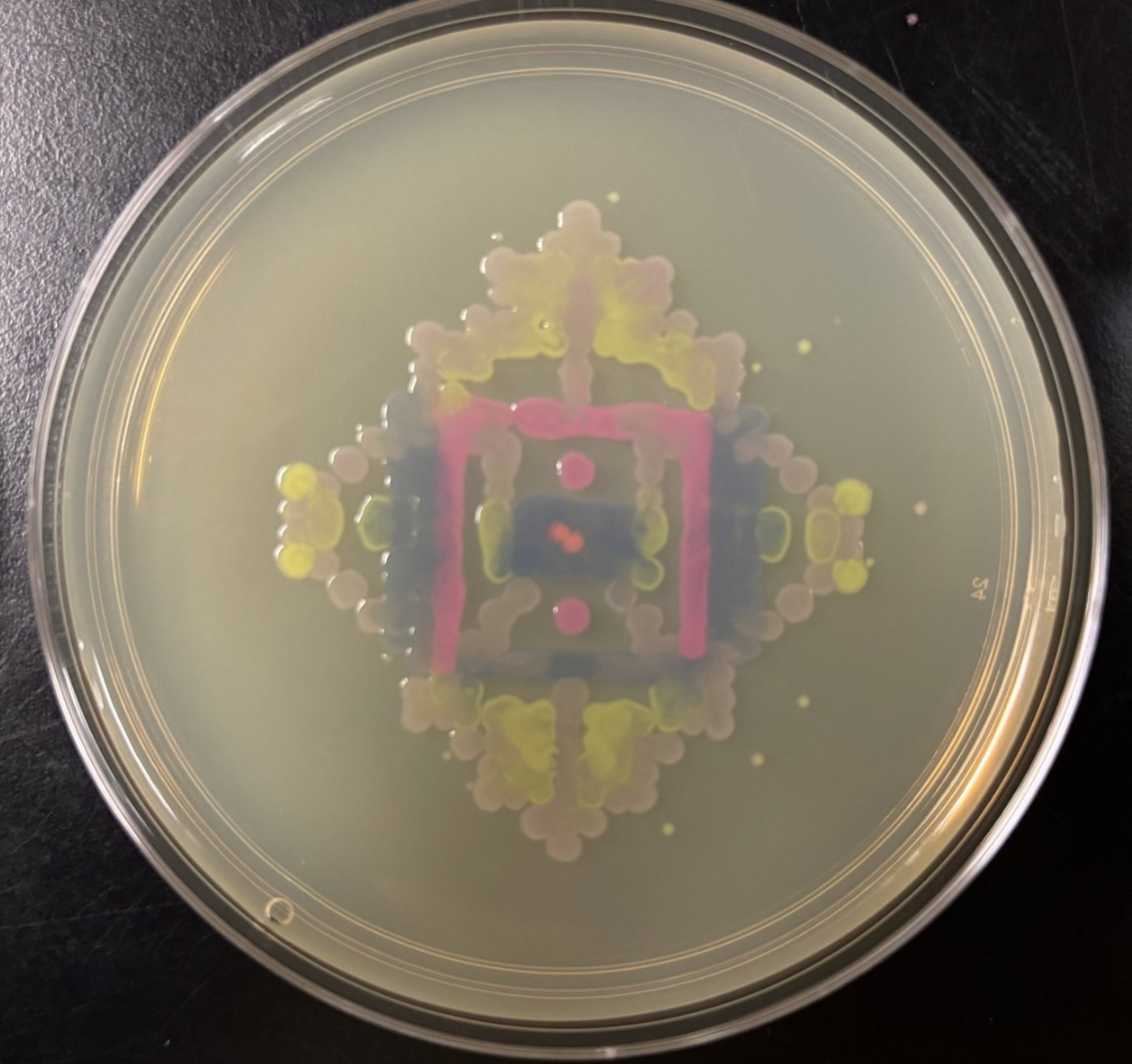
I find the bleeding of the colonies incredibly striking. There are moments where a colony will pool into another simulating layered watercolor droplets on paper.
One of the great parts about having an automated robot is being able to precisely mix, deposit, and run reactions without much intervention, and design and deploy experiments remotely.
For this week, we’d like for you to do the following:
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
https://link.springer.com/article/10.1007/s10489-025-06334-3
https://www.sciencedirect.com/science/article/pii/S2472630325000263
https://pubs.acs.org/doi/10.1021/acssuschemeng.4c05494
3D Printed Cellulose-Based Fungal Battery
Carolina Reyes, Erika Fivaz, Zsófia Sajó, Aaron Schneider, Gilberto Siqueira, Javier Ribera, Alexandre Poulin, Francis W. M. R. Schwarze, and Gustav Nyström
ACS Sustainable Chemistry & Engineering 2024 12 (43), 16001-16011
DOI: 10.1021/acssuschemeng.4c05494
3D printing technology is one of the most formally recognized and utilized automation tools in fabrication and design. Electronics are vital to our daily way of life, and have become passive pieces of hardware rarely itching at our conscience. However, e-waste is a growing issue as proper methods of disposal are difficult and inefficient. Let alone the components of electronics are not easily recyclable if at all. Researchers are beginning to investigate the world beneath our feat to find the answer to combating e-waste. Microbial fuel cells are bio-electrochemical devices that convert chemical energy to electrical energy using micro-organisms. The interest in MFCs as functional alternatives to electronics is growing and yielding some powerful results.
In the article 3D Printed Cellulose-Based Fungal Battery, the 3D printer becomes an automated wet lab tool to create a bio-degradable fungal battery in response to building more ecologically focused electronics. The 3D printer’s role is to extrude the contents of the cellulose hydrogel (mixed with carbon black and graphite flakes to conduct electricity), structural additives and yeast (or white rot mycelia). The form is a fungal ink that acts as the electrode within a microbial fuel cell sandwiched between Cathode, PEM and anode layers. The 3D printer’s design and function allows for multiple iterations of the experiment to be conducted with precision and reproducibility.
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
The aligned candidate automation tool is the OT-2 due to its liquid handling capabilities, customization (designing custom 3D printed parts and scripts) and accessibility. Since I am new to lab automation and wet lab workflows, the OT-2 is more familiar as a tool to begin solo experimental designs.
General Ideas for OT-2 Tool Implementation:
Idea one:
A final project idea is working with life to preserve cultural heritage objects. This is already being done in large scale applications, but what about small scale i.e. ceramics? My first series of tests would be testing the proper concentration of mineralization media for S.pasteurii is needed to fill macro-cracks in ceramic pieces in different clay bodies.
Make ceramic slabs and treat with various degradation methods.
- 3D print custom “seats” for ceramic samples to ensure stability in the OT-2 during operation with enough depth to contain S.pasteurii in mineralization media.
- Customize a python script to renew the mineralization media every 24 hours.
Idea Two:
I am interested in the pigment Xylindein from C.aeruginascens and would like to express this pigment in another host due to the slow growth times and poor solubility. The goal would be to heterologously express the Xylindein pathway in S.cerevisiae. Xylindein is made from a multi-fragment pathway. To ensure expression, I would start with creating constructs that individually contain one gene within the pathway. This would ensure the individual genes are capable of expression in yeast. Next, I would design two constructs each containing 3 fragments from the pathway with different selectable markers.
- OT-2: Liquid handling of the samples into a 96 well plate
- Plateloc: Seal the 96 well plate (Plate sealing would be necessary again if Xylindein pigment production is visible and can move forward to OD600 and centrifugation steps for absorbance spectroscopy)
- Inheco: Incubation at 26C
- ATC thermocycler: Perform PCR for the samples in well plates
- Xpeel: Removes the seal from the 96 well plates
Week 4 HW: Protein Design I
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
In a 500g piece of meat, there is approximately 7.8x1023 amino acids.
In researching for this question, I was unable to find further resources other than previous HTGAA pages.
Alternative question asked with minimal return:
Calculation of amino acids in meat by grams?
- Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Though this would be a quirky science fiction plot, the reason we do not become the cow when we eat the meat is because as the meat enters our bodies, it goes through a whole slew of processes that breakdown the proteins into amino acids etc. They get broken down for our bodies to use as nutrients. The proteins are not functioning in our bodies as they are broken down into building blocks.
- Why are there only 20 natural amino acids?
This question is complicated in that there aren’t any concrete answers. It is one of the great mysteries of life. There is also no truly understood theory on how these amino acids came to be on earth. However researchers are un-convinced at the “randomness” theory of the amino acid array. There are three main functions ordain the AA assemblage, hydrophobicity, charge and size. The traits gave rise to preferred amino acids that would work best to create and organize life.
https://www.chemistryworld.com/features/why-are-there-20-amino-acids/3009378.article
- Where did amino acids come from before enzymes that make them, and before life started?
In 1953, simulated a primordial soup in which various gases and elements were concocted together in a flask and emerged from this experiment were 11 amino acids. The experiment proved that amino acids could have originated from a similar environment on earth and beyond that through time and chaos further complexity was developed.
So the pathway is hypothesized to be AA by chemical synthesis, metabolic pathways from amino acid biosynthesis, and then enzyme fusion through environmental selection of evolving organisms in primordial soup.
https://www.nature.com/scitable/topicpage/an-evolutionary-perspective-on-amino-acids-14568445/
- If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
You would expect a left-handed a-helix. D-amino acids directly reflect L-amino acids, causing the “flip” of the “image”.
- Can you discover additional helices in proteins?
Yes! It is possible to discover additional helices such as π-helices that make up 15% of proteins (derived from a-helices).
- Why are most molecular helices right-handed?
Molecular helices are right handed because the L-amino acids and D-Sugars fit together in one direction via chemical chirality. I can not find a “why” and more so just the facts of their condition. WHY are helices right handed? Becuase of their structural and chemical bias. However, that is not a satisfying answer. It really only explains HOW helices are right handed.
https://www.utmb.edu/mdnews/podcast/episode/biomolecules-are-left-or-right-handed
- Why do β-sheets tend to aggregate?
Beta pleated sheets can easily interact with each other due to their side chains facing one way or another.
https://www.reddit.com/r/Mcat/comments/gql31n/why_is_it_that_the_more_beta_sheets_more/
What is the driving force for β-sheet aggregation?
Assessment from AI Overview:
The backbone of the Beta sheet contain polar C = O and N - H groups. From this they form regular hydrogen bonds with available Beta strands.
- Why do many amyloid diseases form β-sheets?
Beta sheets are low energy and thermodynamically stable and contain hydrophobic segments. They can get mis-folded more easily and disrupt cellular function.
- Can you use amyloid β-sheets as materials?
There is some research investigating amyloid beta sheets for nano materials due to their biocompatibility!
https://pmc.ncbi.nlm.nih.gov/articles/PMC8508955/
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
Briefly describe the protein you selected and why you selected it.
I selected the Fibroin protein within Bombyx Silk moths. The silk we use in silk based fibers and textiles predominantly come from the Bombyx silk moths. My intiial ideation ofor the final project is to create conservation treatments for textile pieces with biology. Understanding the key protein structures that make silk is pertinent to unraveling the problem and discovering the soltuion.
https://www.uniprot.org/uniprotkb/P21828/entry
Identify the amino acid sequence of your protein.
MKPIFLVLLVATSAYAAPSVTINQYSDNEIPRDIDDGKASSVISRAWDYVDDTDKSIAILNVQEILKDMASQGDYASQASAVAQTAGIIAHLSAGIPGDACAAANVINSYTDGVRSGNFAGFRQSLGPFFGHVGQNLNLINQLVINPGQLRYSVGPALGCAGGGRIYDFEAAWDAILASSDSSFLNEEYCIVKRLYNSRNSQSNNIAAYITAHLLPPVAQVFHQSAGSITDLLRGVGNGNDATGLVANAQRYIAQAASQVHV
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
The length of the protein is 262 amino acids. with the most common amino acid being Alanine (A), which appears 37 times.
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
By using the homology option in UNIPROT for Protein p21828, there are 14 protein sequence homologs.
Does your protein belong to any protein family?
The protein belongs to the Fibroin Light Chain Protein family according to InterPro.
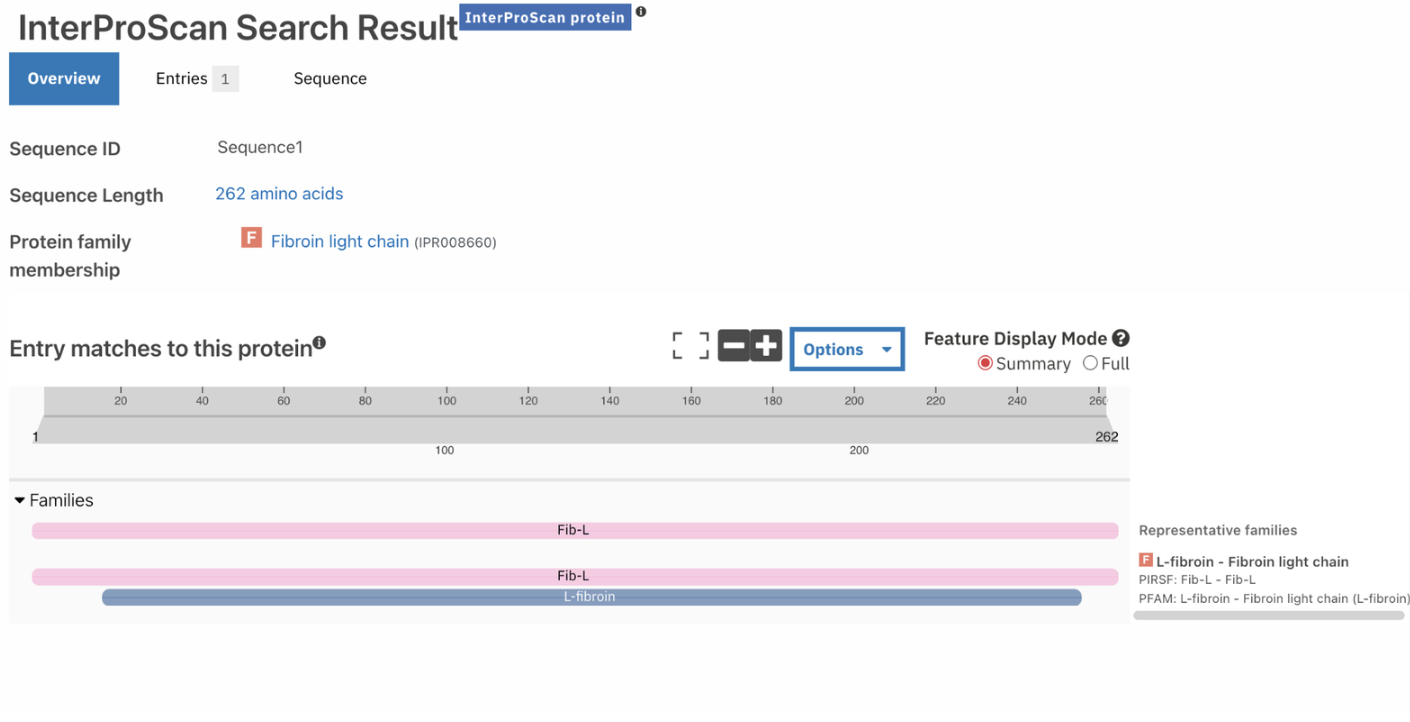
Identify the structure page of your protein in RCSB
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
According to RCSB the protein model was released in AlphaFold DB: 2021-12-09 and last Modified in AlphaFold DB: 2022-09-30
Open the structure of your protein in any 3D molecule visualization software:
CARTOON
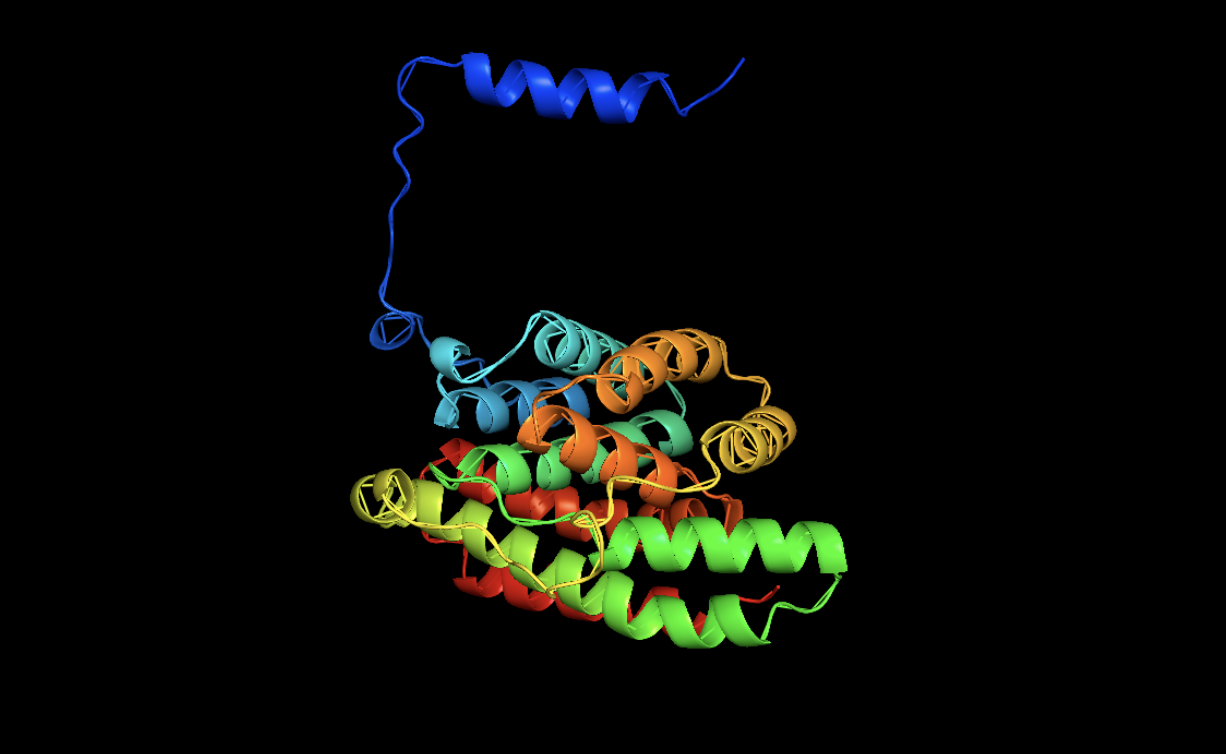
STICK
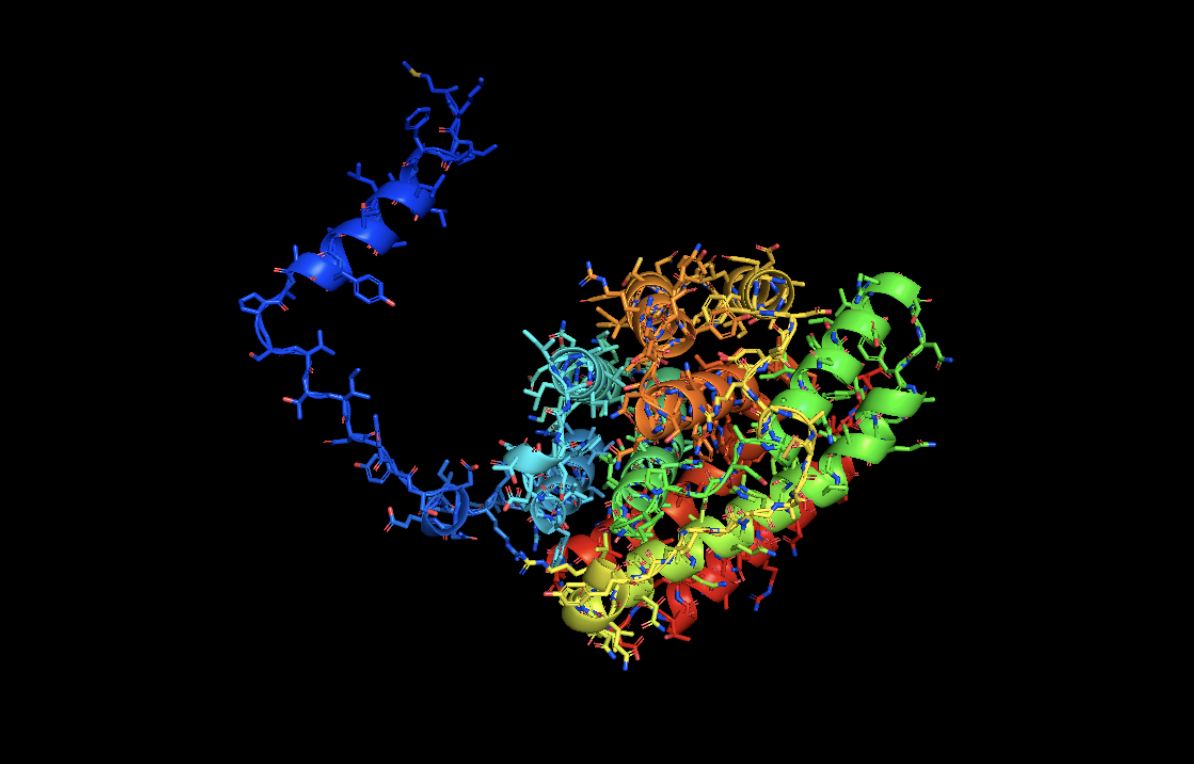
RIBBON
Color the protein by secondary structure. Does it have more helices or sheets?
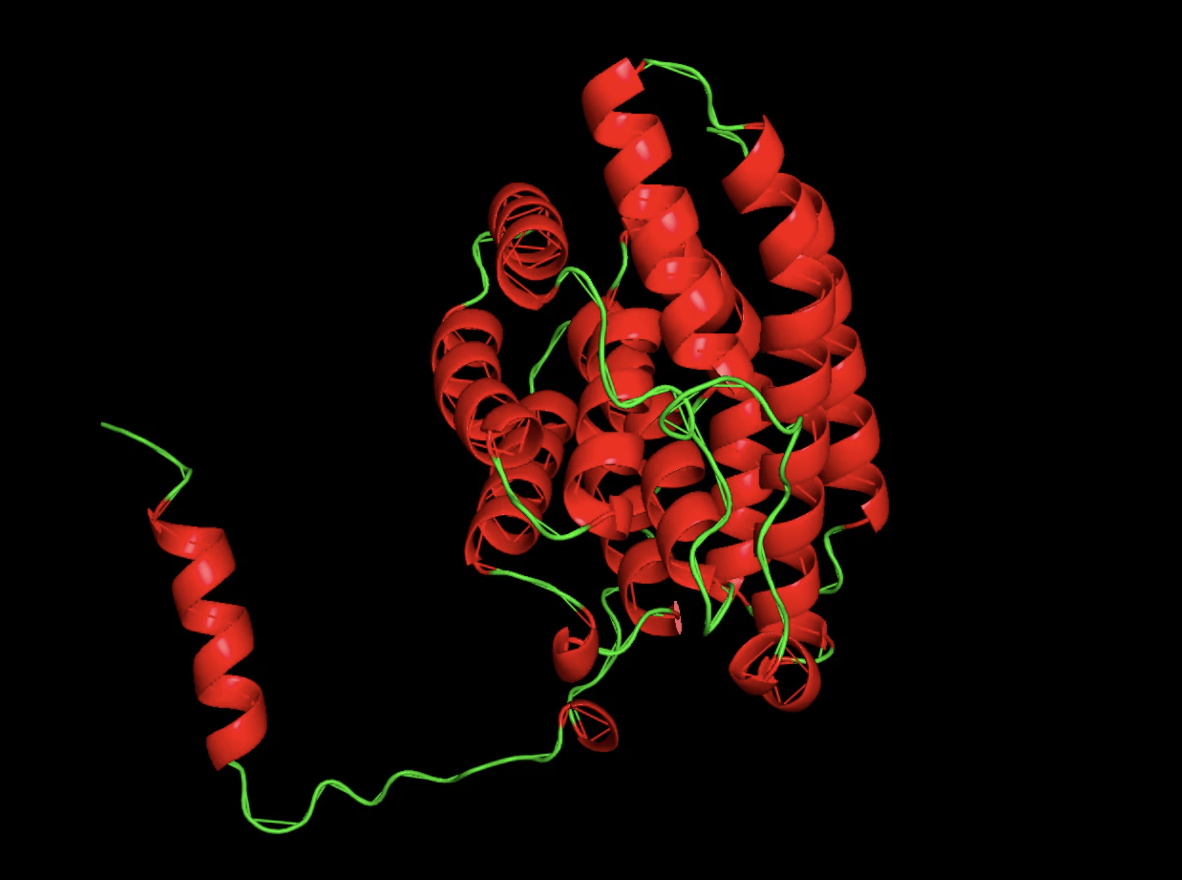
It has more helices than beta sheets!
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
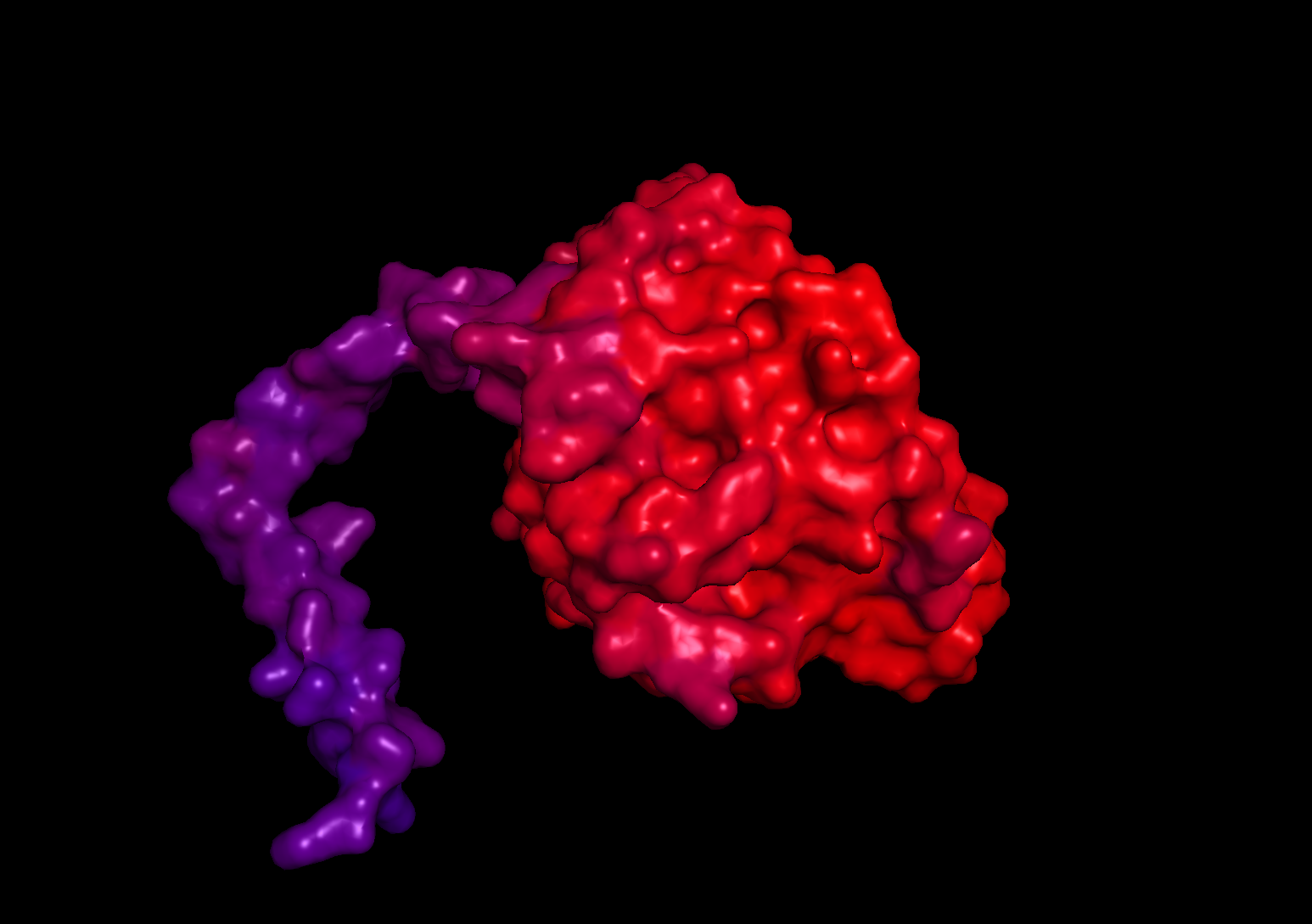
When visualizing the surface of the protein, there are no observable holes or binding pockets in the protein.
In this section, we will learn about the capabilities of modern protein AI models and test some of them in your chosen protein.
Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
Choose your favorite protein from the PDB.
We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
Deep Mutational Scans
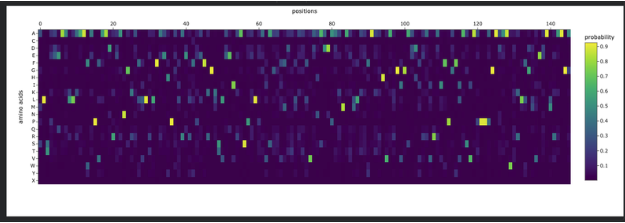
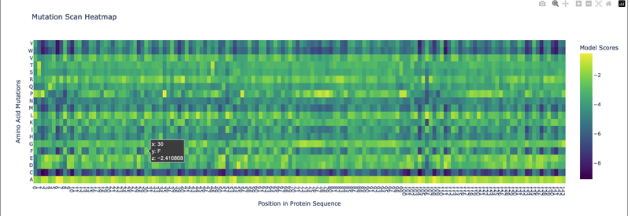
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
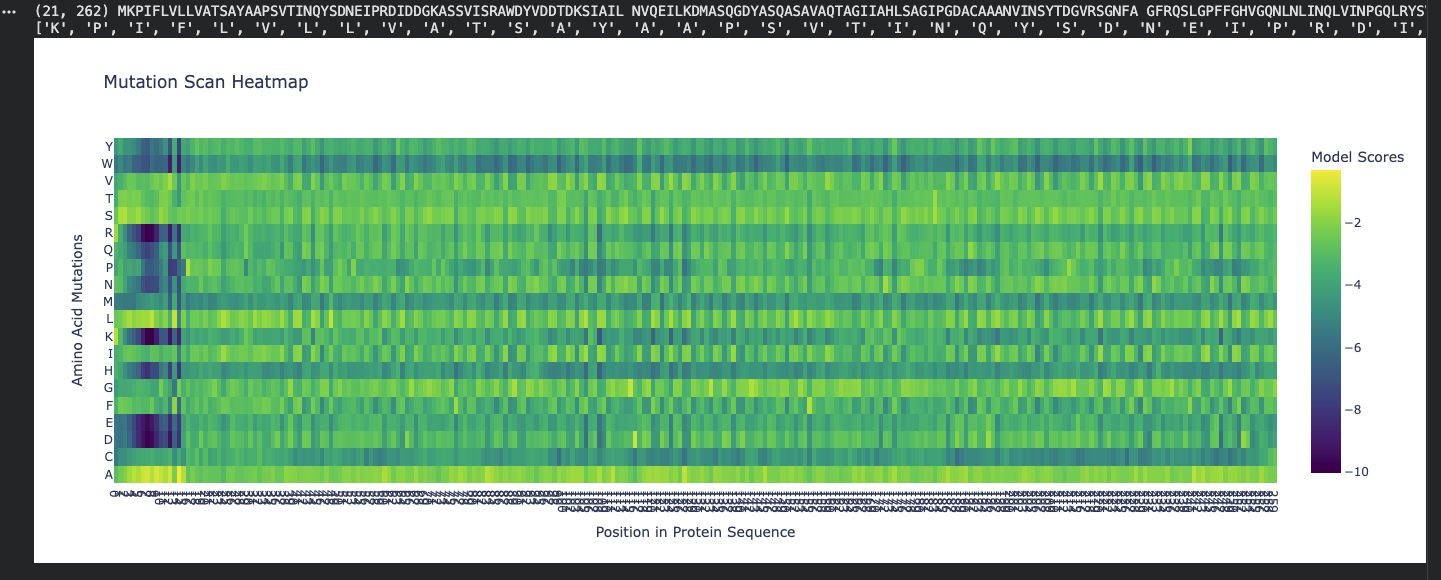
Honestly, when looking at the mutational scan, my initial thought was how visually striking the scan is. Imagine this as a weaving!
My graph being predominantly yellow to green indicates the amino acid scores are of a higher probability. Since the protein is fibroin, a well understood protein, I am not surprised by the high probability rating.
Residue: Amino Acid in the protein sequence
Position X:14 Y:A is the brightest yellow residue in the scan meaning the amino acid in the sequence has a high probabilty score than the majority. There is a general region at the beginning that is predominantly dark blue and is not reflected anywhere else in the scan. I do not understand the scan if represented spatially, however, I wonder if this is the predictive AA seqeuence that Alphafold deemed insecure notated as orange and yellow.
Latent Space Analysis
Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins?
Place your protein in the resulting map and explain its position and similarity to its neighbors.
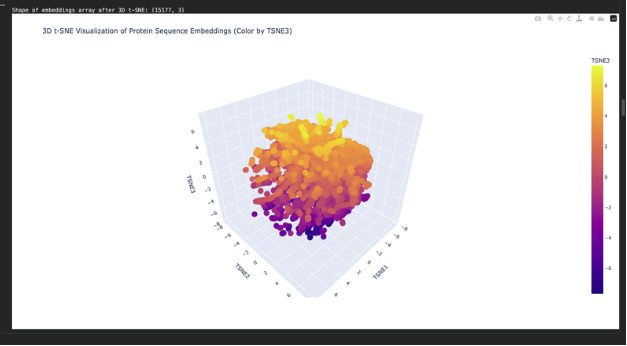
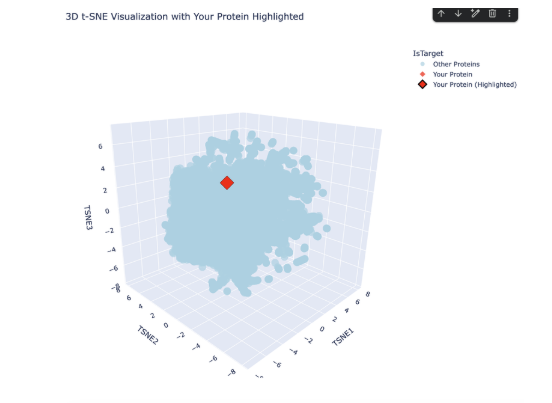
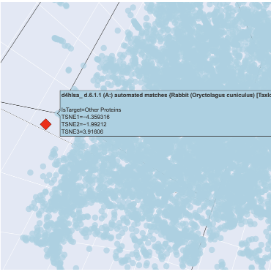
The plot map is dense and has minimal unconcentrated areas. This implies that my protein does not have distinct seprarted families. Each dot in the map is a protein sequence embedded into the model to spatially recognize proteins in relation to each other.
By my protein were proteins from rabbits, fungi and humans that contain fibroin. The family tree made a lot of sense!
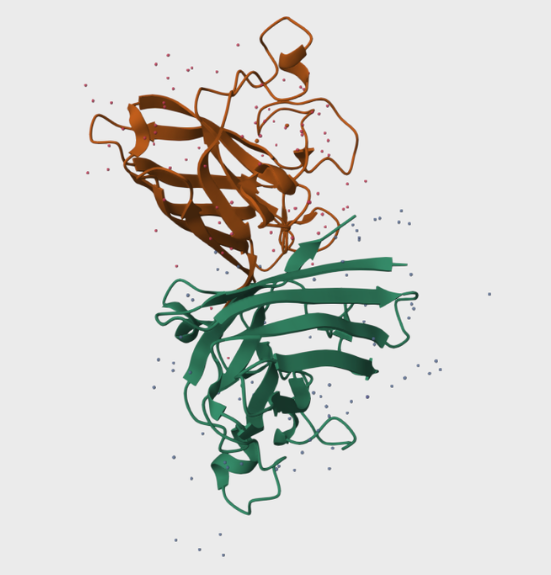
Folding a protein
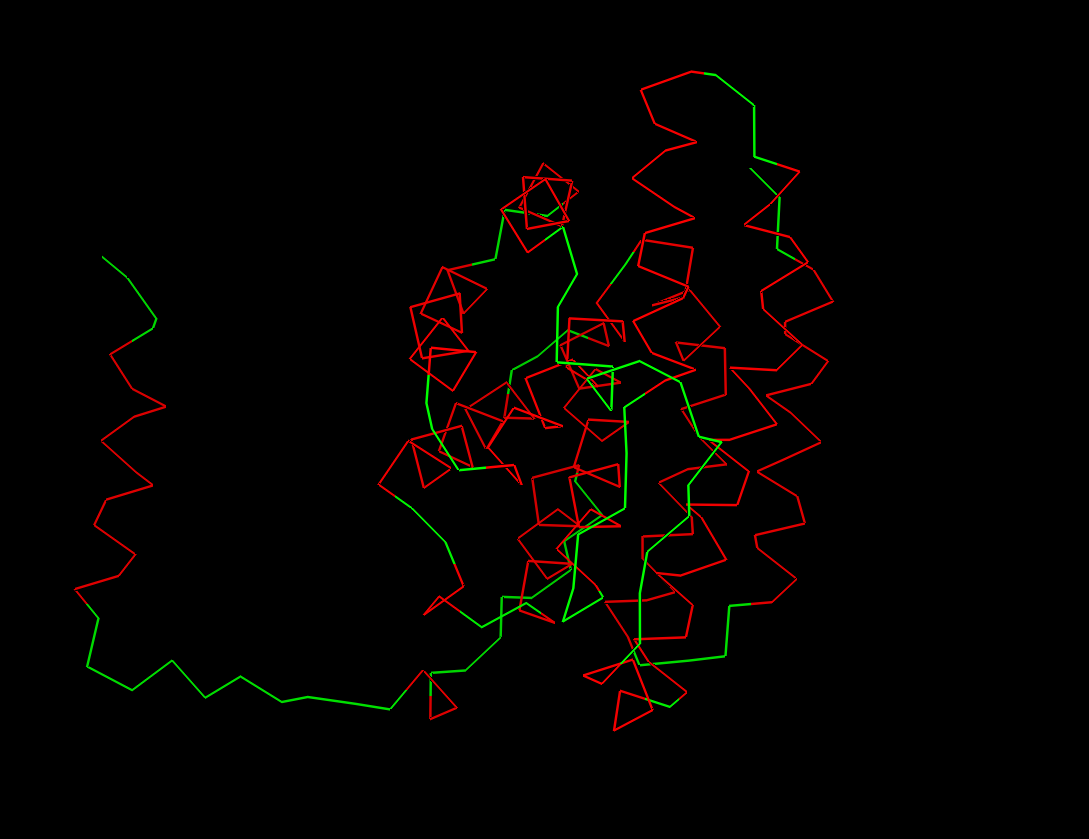
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
The predicted coordinates do not match the original structure. The ESMfold prediction is missing the arm chain extension predicted in the alpha fold simulation. The one similarity I can connect is the protein is exclusively a-helices.
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Original:
MKPIFLVLLVATSAYAAPSVTINQYSDNEIPRDIDDGKASSVISRAWDYVDDTDKSIAILNVQEILKDMASQGDYASQASAVAQTAGIIAHLSAGIPGDACAAANVINSYTDGVRSGNFAGFRQSLGPFFGHVGQNLNLINQLVINPGQLRYSVGPALGCAGGGRIYDFEAAWDAILASSDSSFLNEEYCIVKRLYNSRNSQSNNIAAYITAHLLPPVAQVFHQSAGSITDLLRGVGNGNDATGLVANAQRYIAQAASQVHV
ProteinMPNN:
ALTPEEAALLRAAWAPVAADREANGRAFMLRLFAEYPELREYFPEFKGKSLEEIAASPKLAAFSTAVFDGLERLVATADDAAAMATLLADLAKAHVAKGIGAEHVEKIRAIHPAFVASVAPPPPGADAAWDRLFGLVIAALKAAGA
Input this sequence into ESMFold and compare the predicted structure to your original.
Pretty low confidence in the scan!
Week 5 HW: Protein Design II
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Your challenge:
Design short peptides that bind mutant SOD1. Then decide which ones are worth advancing toward therapy.
You will use three models developed in our lab:
PepMLM: target sequence-conditioned peptide generation via masked language modeling
PeptiVerse: therapeutic property prediction
moPPIt: motif-specific multi-objective peptide design using Multi-Objective Guided Discrete Flow Matching (MOG-DFM)
Part 1: Generate Binders with PepMLM
Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
Amino Acid Sequence: MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
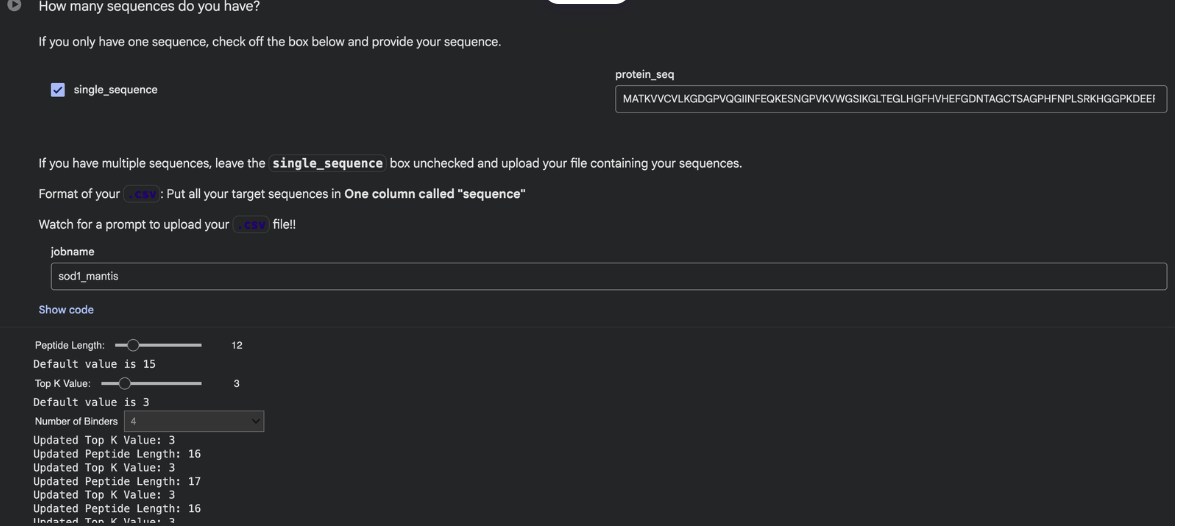
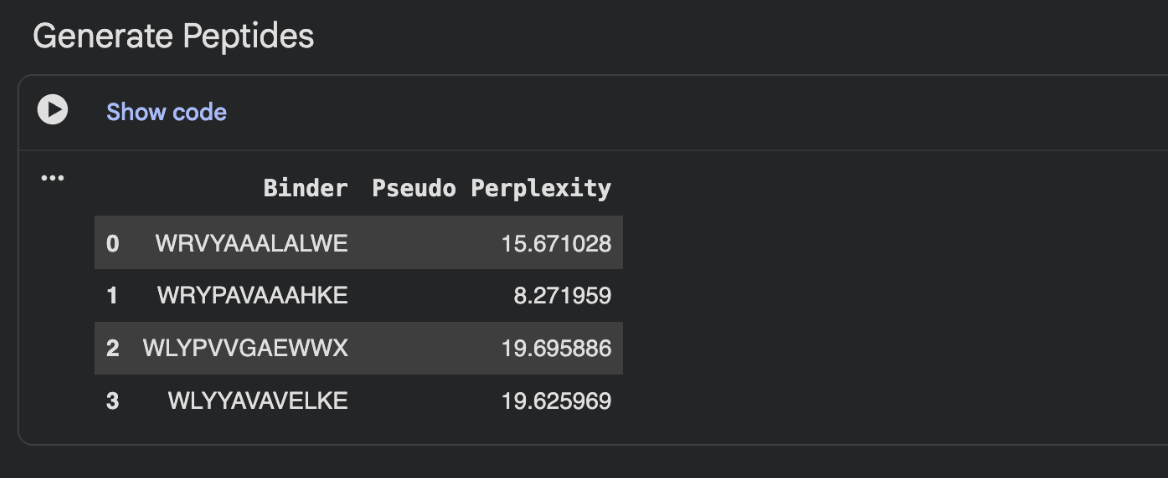
To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
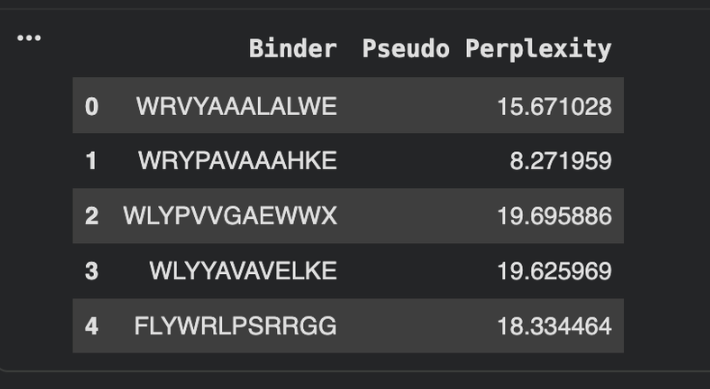
Record the perplexity scores that indicate PepMLM’s confidence in the binders.
Part 2: Evaluate Binders with AlphaFold3
Navigate to the AlphaFold Server: alphafoldserver.com
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
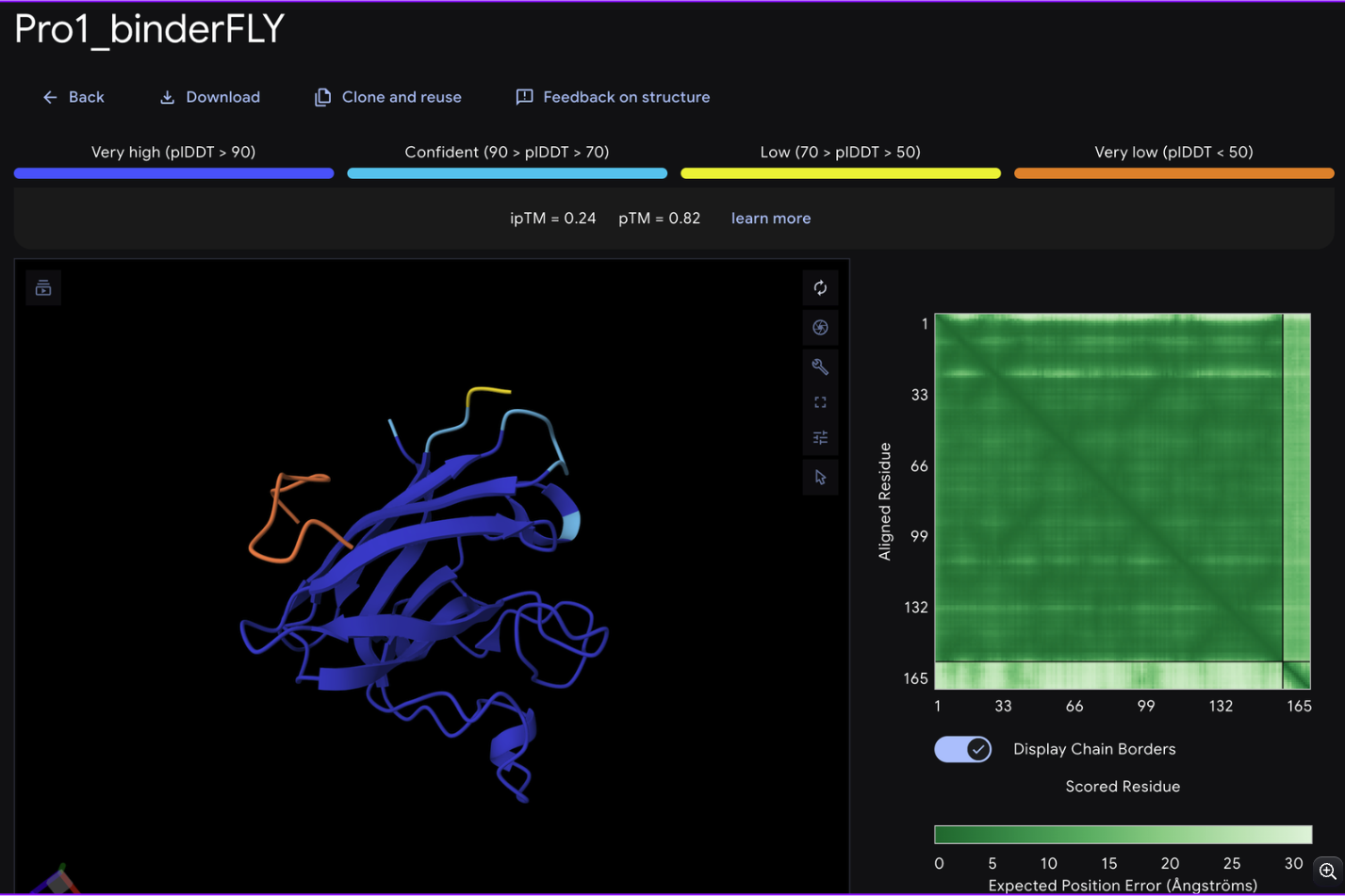
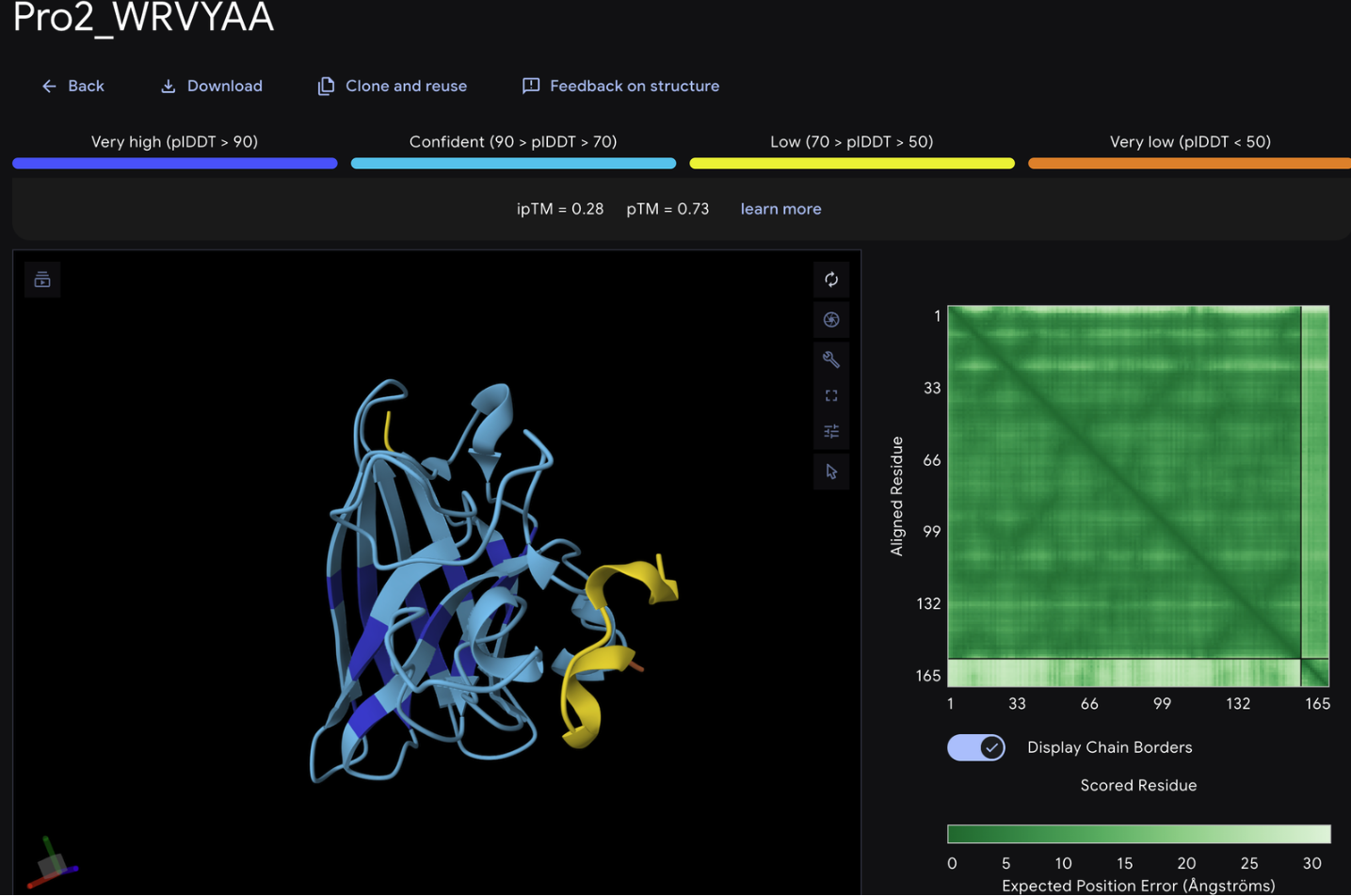
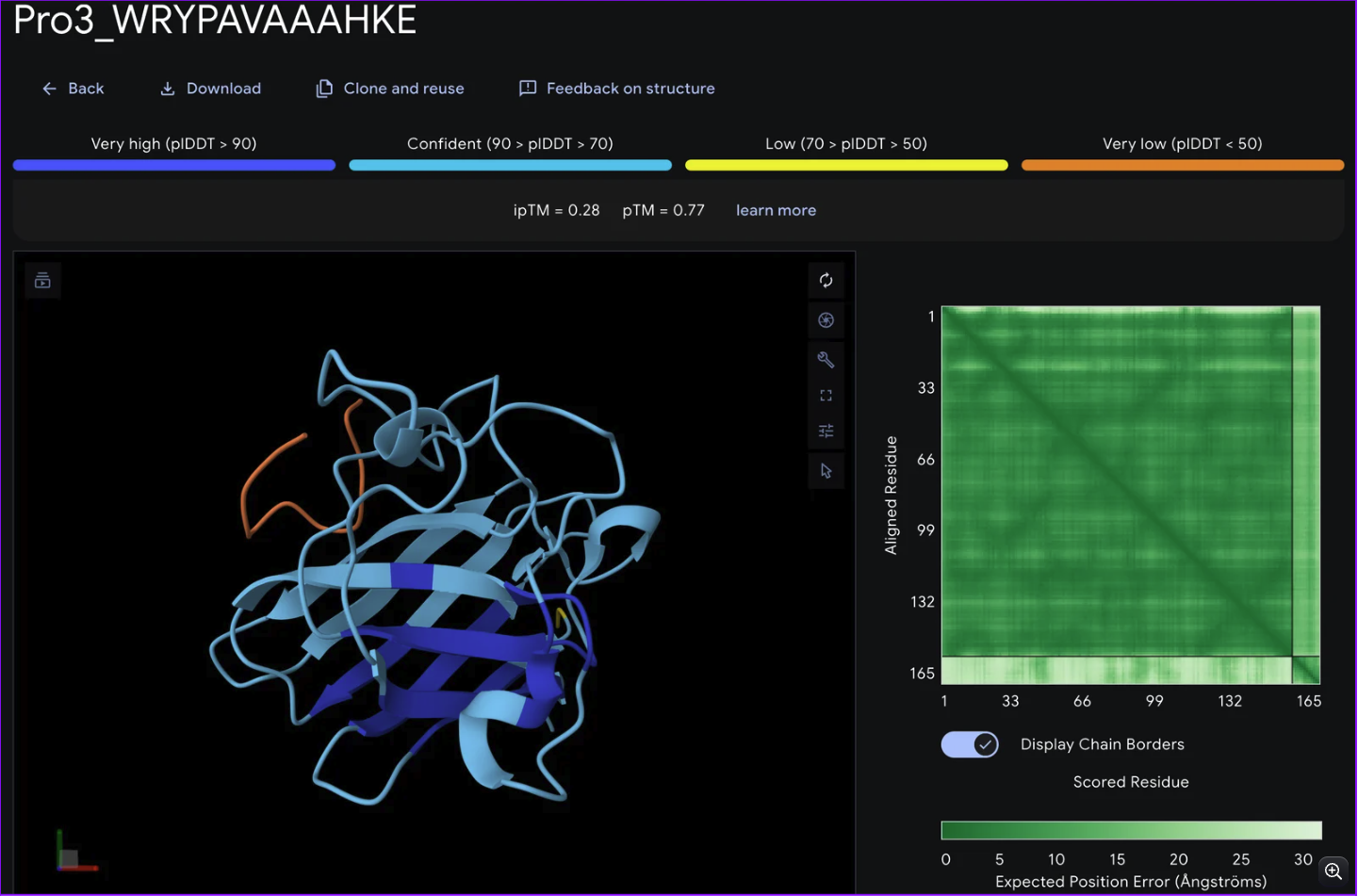
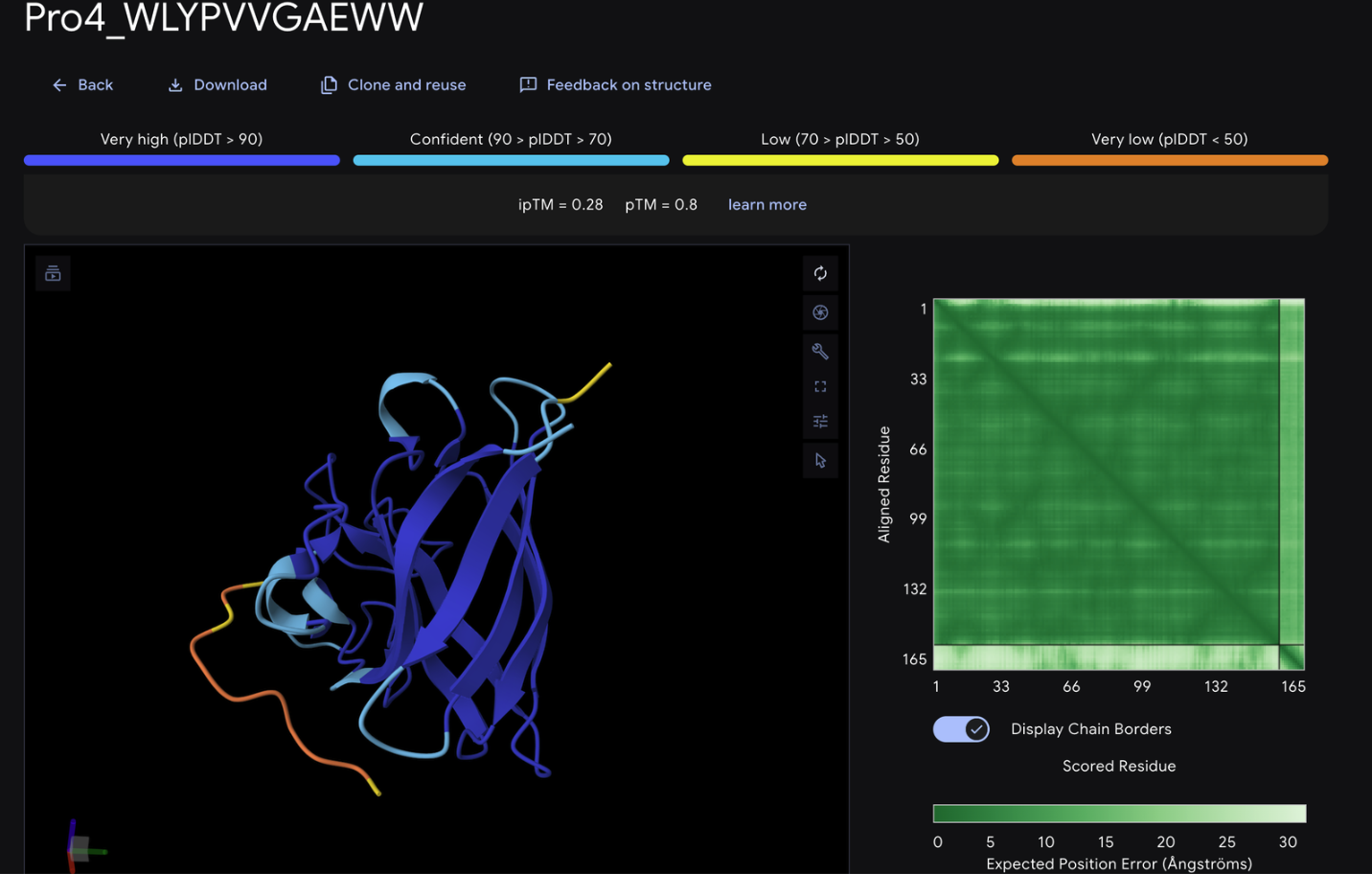
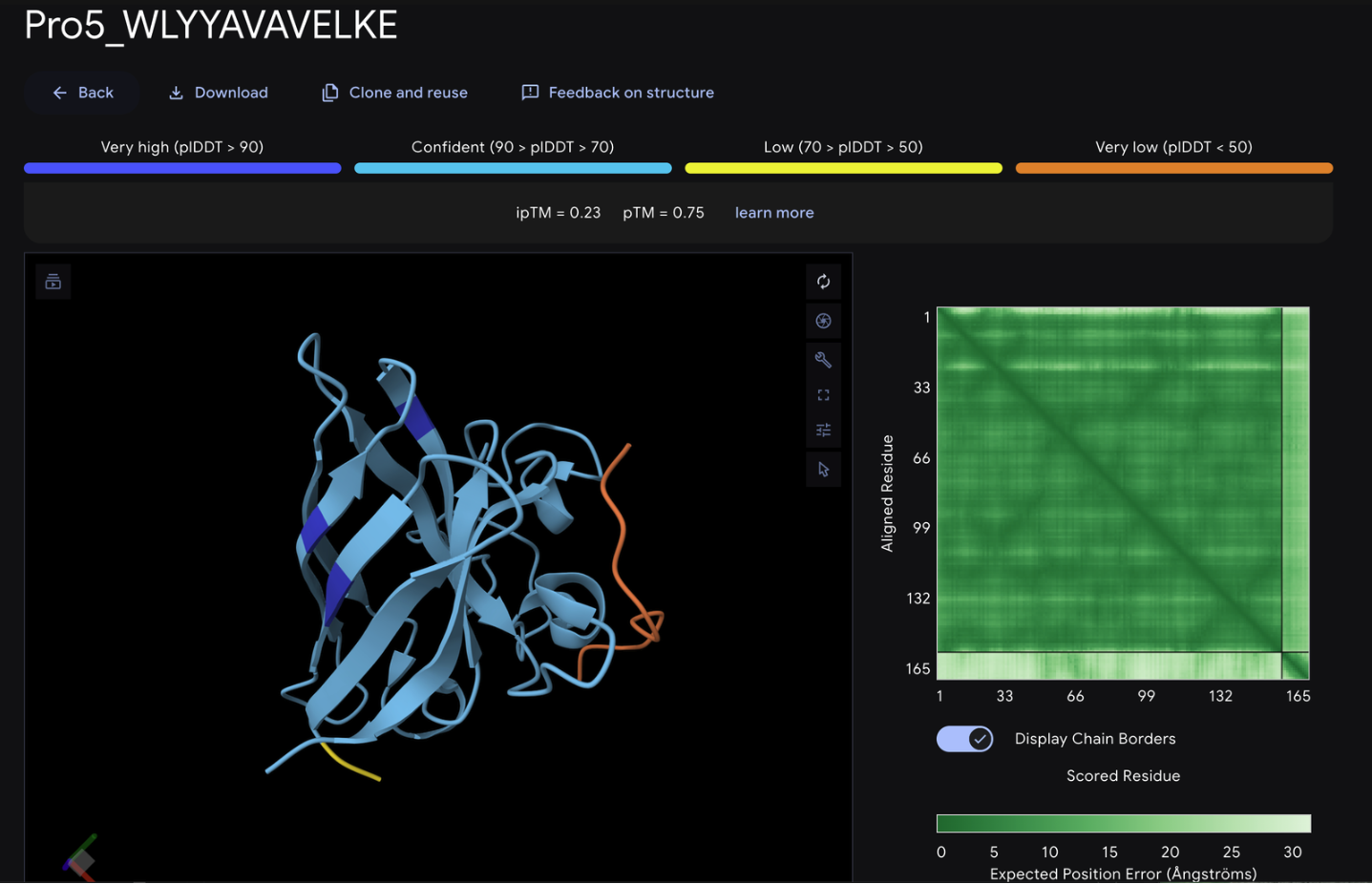
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
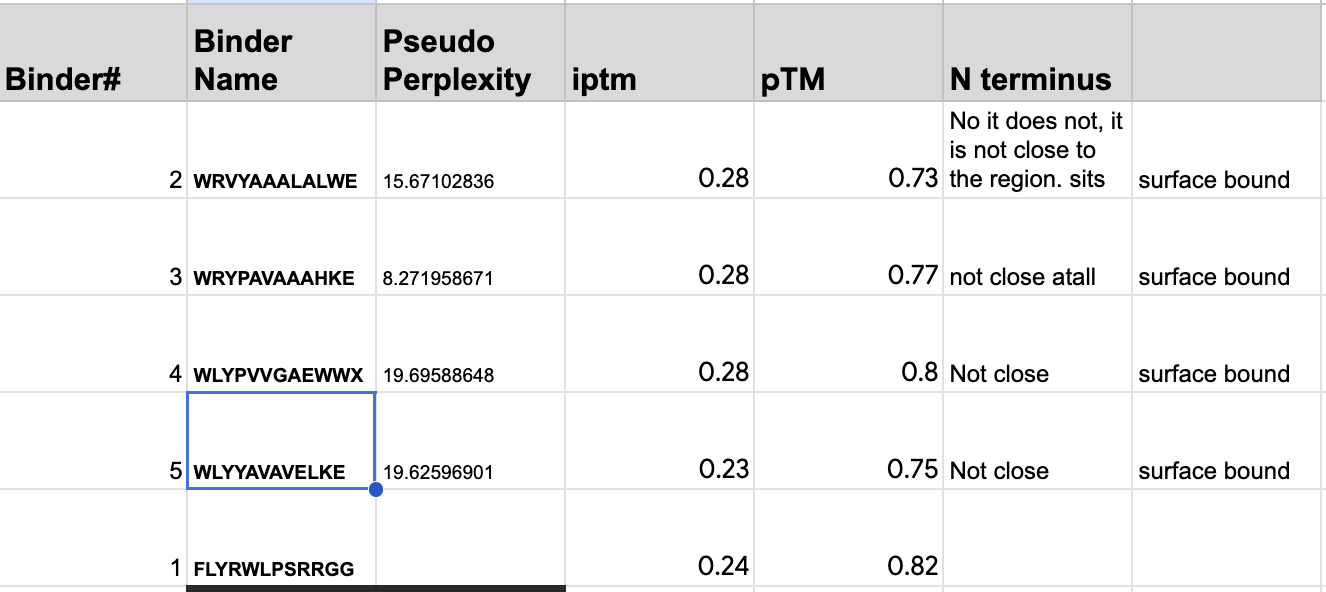
From the alphafold predictions, the peptide binders generated via pepMLM have a weak accuracy of .23-.28, meaning the probability of these binder to protein associations are unlikely to occur.
https://www.ebi.ac.uk/training/online/courses/alphafold/inputs-and-outputs/evaluating-alphafolds-predicted-structures-using-confidence-scores/confidence-scores-in-alphafold-multimer/
Vocab
Pseudo perplexity: measures the models uncertainty when predicting an amino acid sequece
iptm: accuracy of the predicted relative positions of the subunits forming the protein-protein complex.
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
The PepMLM binders do not exceed the known binder in any meaningful capacity, with alphafold giving FLYWRLPSRRGG a .24 iptm rating which is only .04 less than the generated binders. The chains via alphafold were not attached to the barrel deepening the low confidence scores of the binders.
None of the generated binder exceed the known binder of FLY
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
Paste the peptide sequence.
Paste the A4V mutant SOD1 sequence in the target field.
Check the boxes:
- Predicted binding affinity
- Solubility
- Hemolysis probability
- Net charge (pH 7)
- Molecular weight
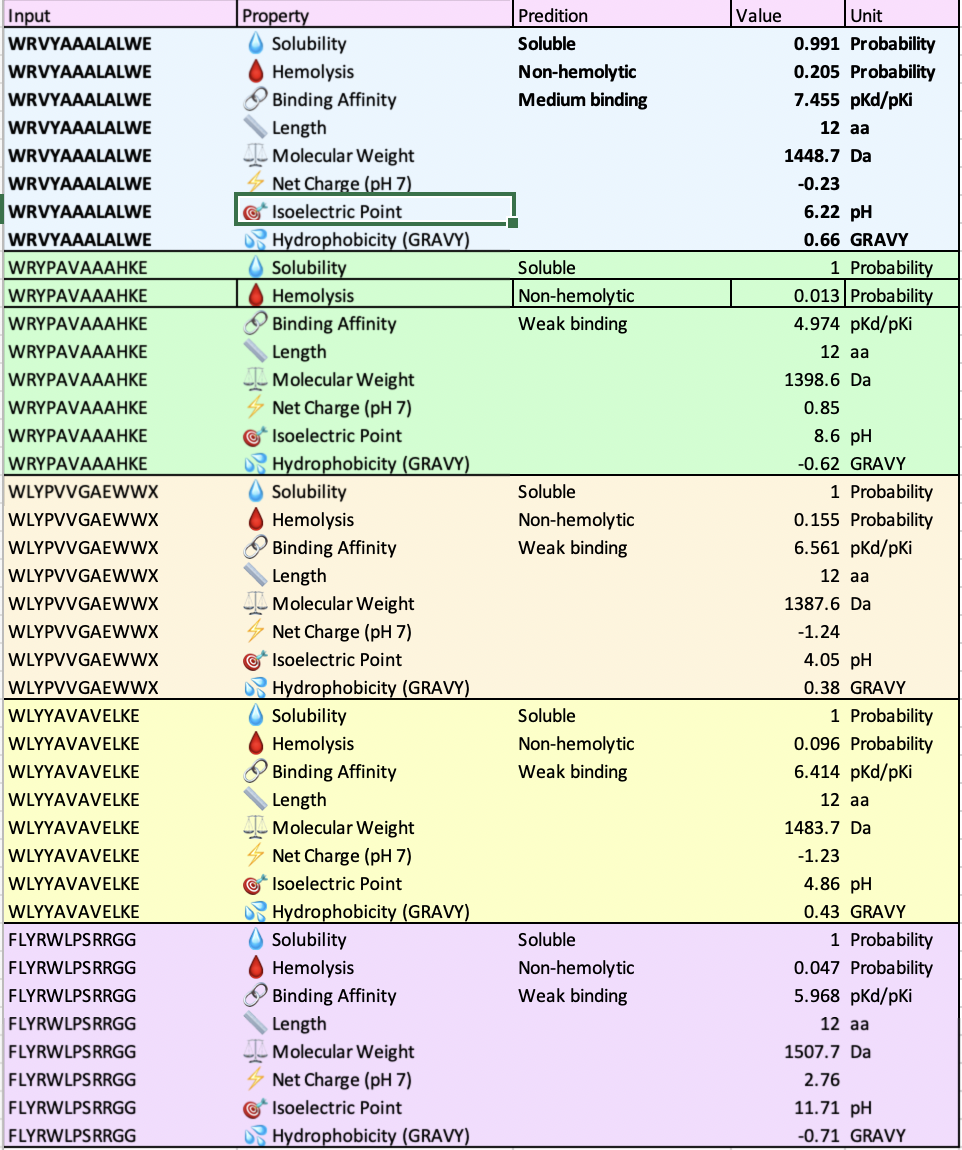
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
All of the peptides had similar molecular weights, solubility scores and four out of the five had weak binding scores which correlates to the alphafold iptm ratings of low confidence. In the alphafold 3D structure, the binders did not connect to the barrel. The pH scores range from 4.05 to 11.71.
WRVYAALALWE is the only one stated to have a medium binding affinity but has the highest hemolysis score. I am interested if this was a consistent pairing and if so what the biochemical association is.
Choose one peptide you would advance and justify your decision briefly.
WRVYAALALWE or WRYPAVAAHKE are the peptides I’d advance as candidates due to several factors.
In the alphafold #D structure, it is the only binder that details a sheet form, pseudo-proving strength in the determination of its peptide to protein binding affinity with an iptm rating of .28 (low confidence but higher compared to the lowest scores of .23 and .24.) WRVYAALALWE also has medium binding stated by peptiverse.
However, WRYPAVAAHKE also has the lowest hemolysis score of .013, solubility of 1 and an the same iptm rating as WRVYAALALWE.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
Open the moPPit Colab linked from the HuggingFace moPPIt model card
Make a copy and switch to a GPU runtime.
In the notebook:
Paste your A4V mutant SOD1 sequence.
Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
Set peptide length to 12 amino acids.
Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
Unfortunately, when I attempted to change the run type, I was met with a paywall that I am unable to bypass. I have done the experiment before and will be relying on previous runs and information gathered from my peers in the node.
When I tried running the co-lab regardless of the hardware accelerator impediment, I kept running into this issue:
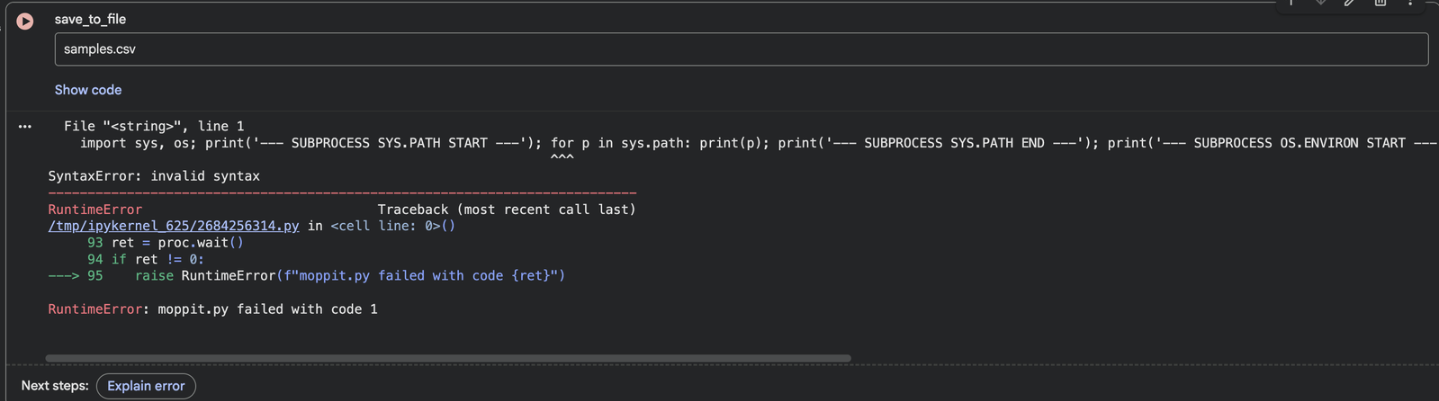
I asked gemini to evaluate the code in the case the code could be run regardless of hardware complications.
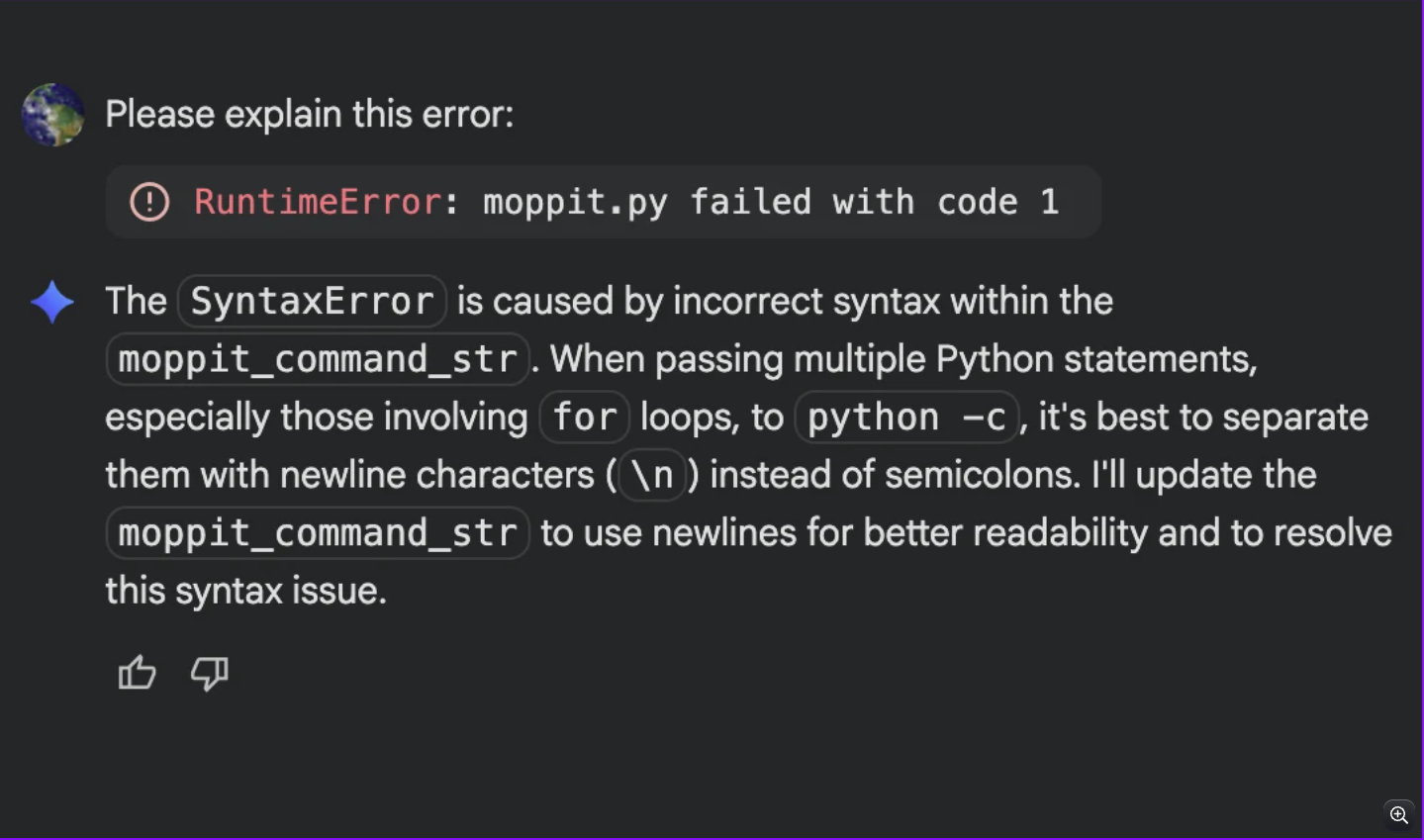
To give the experiment a fighting chance one more time, I accepted the changes gemini recommended.
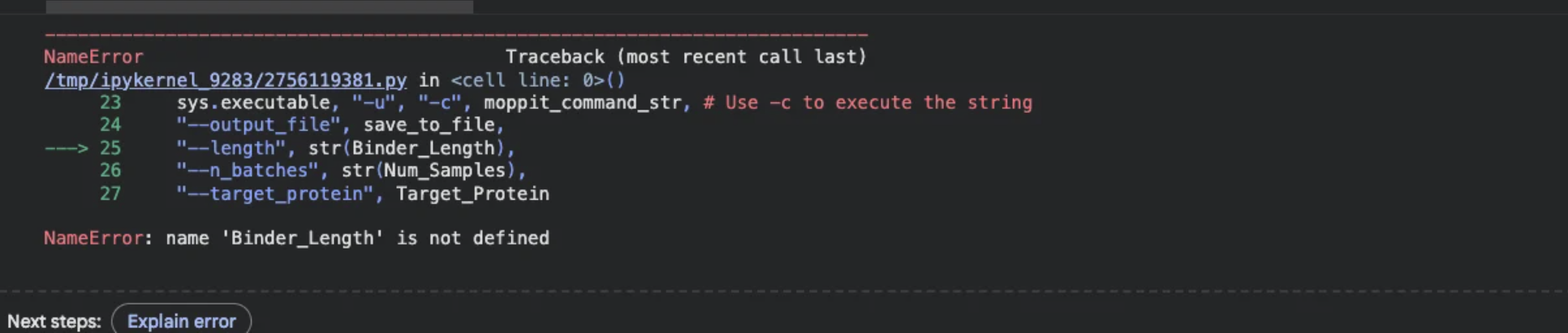
However, regardless of the edits gemini was able to administer I was already down a rabbit whole full of errors.
I pivoted to using the data Amanda Mainello, the BUGSS instructor, was able to load and save from the moPPit colab. I was not able to implement these into alpha fold for further comparisons due to9 the sequence being covered.
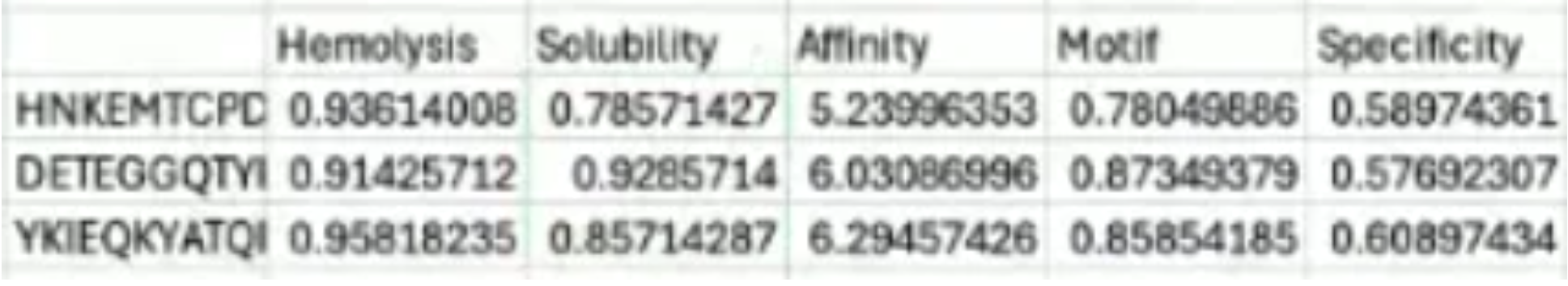
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
The moPPit scores demonstrate a higher hemolysis rate but this may be due to the different value sets of the model. The affinity scores are comparable to the ones generated by peptiverse which was between 4-7, meaning the moPPit scores would be determined as weak binding. The overall solubility scores are less than that gathered from peptiverse.
In my evaluation, I would start by understanding the value ranges and categories of the binder traits further.
From my assumptions that I can make with the information provided and with limited knowledge:
First, determine my research goals and experimental parameters that are specific to the function of the peptide to the protein.
I would evaluate the generated peptides and do a cross comparison to ensure if there are any major incongruencies in vital categories such as hemolysis, pH, specificity and binding affinity to the protein.
From doing rigorous comparisons and determining candidate peptide binders, in vivo testing in animals to study the predicted behavior to the in vivo reactions would be required prior to clinical studies.
Week 6 HW: Genetic Circuits I
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
“Phusion High-Fidelity PCR Master Mix with HF Buffer is a 2X master mix consisting of Phusion DNA Polymerase, deoxynucleotides and reaction buffer that has been optimized and includes MgCl2. All that is required is the addition of template, primers and water.” - NEB
Deoxynucelotides: dTTPs, dTCPs, dTAPs, dTGPs. These are DNA’s building blocks and having these in abundance is necessary when doing a PCR reaction so that when the primers anneal, there are synthesized nucleotides to replicate the strands/specific gene fragment. The Phusion polymerase is an “enzymatic glue” that sticks strands back together.
The reaction buffer creates the necessary environment for these reactions to occur. Often times maintaining peach and adding other ions to the solution.
https://www.neb.com/en-us/products/m0531-phusion-high-fidelity-pcr-master-mix-with-hf-buffer?srsltid=AfmBOopb-q8pqvSKuHMEB_he8yA5vAiNmyj8HZqBQVUNENKsASAHy_Ah
What are some factors that determine primer annealing temperature during PCR?
From information gathered from Integrated DNA technologies, primer annealing temperature is determined by the length and composition of the primers. To calculate the primer annealing temperature (Tm), The needed equation is the following: Ta Opt = 0.3 x (Tm of primer) + 0.7 x (Tm of product) – 14.9. The temperature is also dependent on the GC content of the DNA strands. Due to G-C pairs being bonded by three hydrogen bonds, the temperature needs to be generally higher to ensure effective annealing and specificity.
Ta: Annealing temperature
™ of the Primer: melting temperature of the less stable primer
Tm of the product: melting temp of PCR product
https://www.idtdna.com/pages/support/faqs/how-do-you-calculate-the-annealing-temperature-for-pcr#:~:text=How%20do%20you%20calculate%20the,temperature%20in%20molecular%20biology%20applications
https://www.idtdna.com/page/support-and-education/decoded-plus/annealing-temperaturefaqs#:~:text=The%20annealing%20temperature%20(Ta)%20for%20PCR%20should%20be%20selected,to%20calculate%20the%20Ta).
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Restriction enzyme digests are very site specific (sticky and blunt ends) and depend on restriction sites.
PCR is a method of amplifying segments of DNA using primers and various heat and cooling cycles in a thermocycler. You could use these two methods together by cutting at specific sites using restriction enzyme digestion, and amplify that gene OR amplify the DNA that you then will cut!
However, PCR as a separate cloning method not only amplifies regions but is generally less specific than restriction enzymes. You need less template DNA than for restriction digests.
“PCR cloning is a method in which double-stranded DNA fragments amplified by PCR are ligated directly into a vector. PCR cloning offers some advantages over traditional cloning which relies on digesting double-stranded DNA inserts with restriction enzymes to create compatible ends, purifying and isolating sufficient amounts, and ligating into a similarly treated vector of choice (see insert preparation).
With PCR amplification, this cloning technique requires much less starting template materials which include cDNA, genomic DNA, or another insert-carrying plasmid (see subcloning basics). Furthermore, PCR cloning provides a simpler workflow by circumventing the requirement of suitably-located restriction sites and their compatibility between the vector and insert. Nevertheless, there are a number of considerations related to: PCR primers and amplification conditions, the cloning method of choice and the cloning vectors used, and, finally, confirmation of successful cloning and transformation.”
https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/molecular-cloning/cloning/common-applications-strategies.html#:~:text=and%20sequencing%20method-,PCR%20cloning%20strategies,specific%20amplification%20of%20the%20template.
https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/molecular-cloning/cloning/common-applications-strategies.html#:~:text=and%20sequencing%20method-,PCR%20cloning%20strategies,specific%20amplification%20of%20the%20template.
https://www.neb.com/en-us/tools-and-resources/feature-articles/foundations-of-molecular-cloning-past-present-and-future?srsltid=AfmBOoqmgxhUA3z5QY5IMyKaWEj1T5BunNIQu05z_X0v6216QQ6_egLp
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
You want to ensure there aren’t as many consistent overhangs to minimize amplification error in which various/unpreferred regions will be amplified. As asked and implied in the previous questions, ensuring your annealing temperature is accurate and appropriate for both the primer and intended product is essential. Avoid secondary structures such as hairpinning ( which is when a strand anneals to itself!).
https://www.addgene.org/protocols/gibson-assembly/
How does the plasmid DNA enter the E. coli cells during transformation?
Plasmid DNA enters E.coli cells during transformation because E.coli, before undergoing transformation, gets treated to become “competent”. The “competency” of a cell is determined by cell wall permeability. Once introduced to heat (heat shock), pores in the cell wall open and allow foreign DNA in its surroundings to enter into the cell.
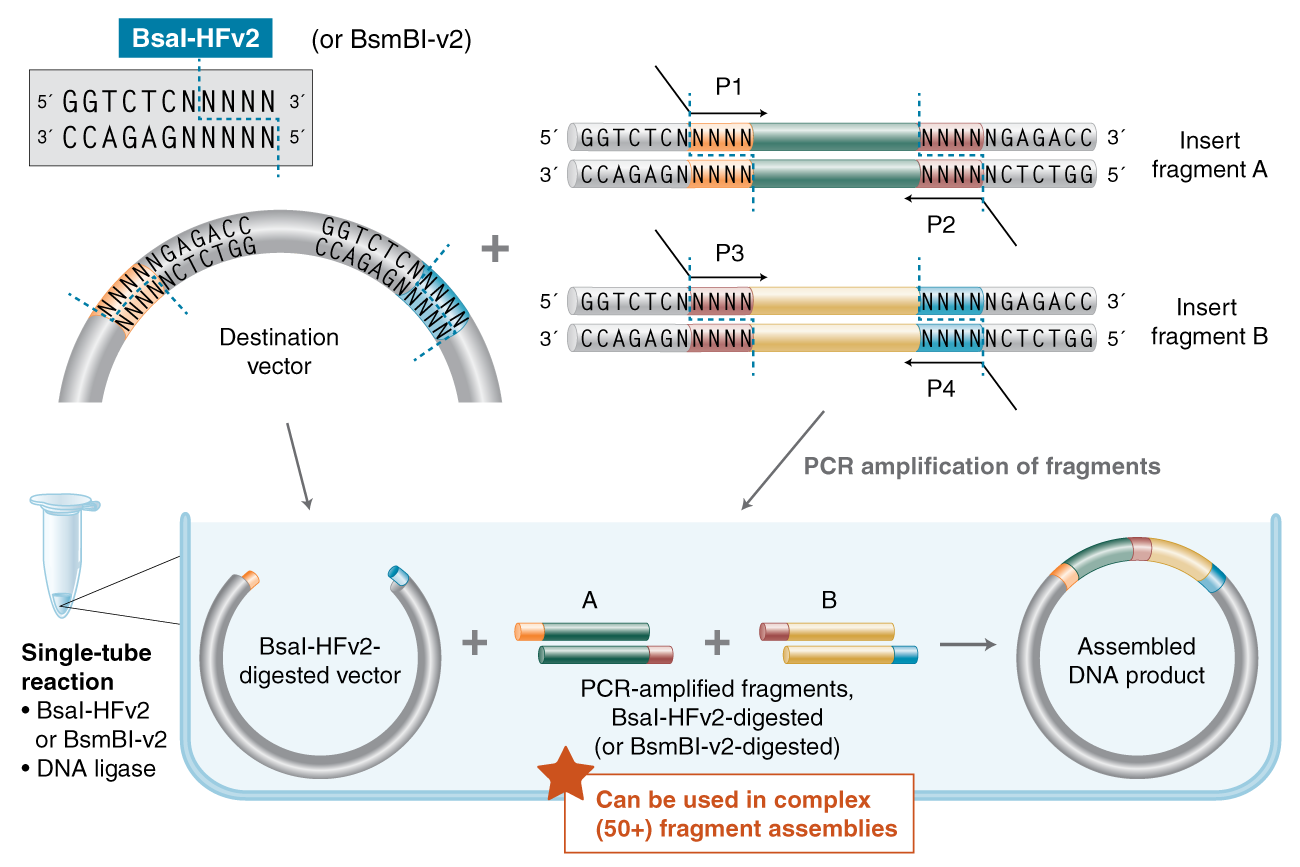
Describe another assembly method in detail (such as Golden Gate Assembly) Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online). Model this assembly method with Benchling or Asimov Kernel!
Golden Gate Assembly uses the unique features of the Type IIS restriction enzymes (example: BsaI). The type IIs restriction enzymes cleave outside of the recognition sequence. This unique ability allows the creation of custom overhangs.
This assembly method happens in a single tube! Add your backbone, enzymes and ligase the the restriction endonucleases will cleave down stream outside of the recognition sites. Not only is this assembly cloning technique efficient, but the amount of DNA fragments is increased and allows for the joining of multiple DNA fragments (50+).
Cloning Techniques Learned:
- In-Fusion cloning
- Ligation Independent Cloning
- Yeast mediated Cloning and Oligonucleotide Stitching
- TOPO cloning
https://www.addgene.org/mol-bio-reference/cloning/#:~:text=Golden%20Gate%20and%20Modular%20Cloning,assemble%20before%20you%20get%20started.
https://bitesizebio.com/26961/cloning-methods-5-different-ways-to-assemble/
https://www.snapgene.com/guides/in-fusion-cloning
https://www.youtube.com/watch?v=aBcqev1NMMo
Week 7 HW: Genetic Circuit II
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
IANNs are more “organic” in that though it is still a structured system, it does not operate on a binary code as a genetic circuit does. This makes IANNs more attuned for detecting and accounting for fluctuating systems such as varied metabolic activity and hormone changes. IANNs can also support multiple functions at once while genetic circuits, in order to scale, need be highly specific. An IANNs system is responsive while a genetic circuit is decisive is how I think of it. However, though this sounds great, IANNs is more complex to build.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
A useful application of an IANN would be the control and regulation of secondary metabolic pathways through the regulation of PPTase’s.
Input to the IANN would be environmental signals such as available sources i.e. carbon to nitrogen ratio, and chromatin reporters that sit at the BGC target sequence. This is the perfect condition to detect the cell’s readiness to initiate secondary metabolic activity.
Output from the IANN would be the PPTase expression level which sets the size of the active carrier protein pool, and a chromatin remodeling factor targeted at BGC that UNLOCKS the BGC.
The limitations of this system would be the possible off-target activation of fatty acid carrier proteins which would disrupt the secondary metabolic pathway. Also, the IANN is not designed to survive the possible flux and rapidity of shifting environmental conditions that actively determine secondary metabolism.
What are some examples of existing fungal materials and what are they used for?
Fungal materials are extremely fascinating. There are many developing fungal materials both in R&D and the market. Most notably, ECOVATIVE has mass produced fungal packaging in which a substrate is inoculated with a fungal spore and from there a mycelium network branches. The mycelium inoculates all throughout the substrate (typically sawdust) and this network stitches the sawdust particulates together to create a dense mycelial brick. Once baked, which kills the mycelium, you get a safe, biodegradable material that can replace packaging materials.
There are mycelium leathers as well that are grown on liquid. Mycelium, even in liquid, grows dense frameworks, but specifically on the liquid to air interface. It creates a floating patch that can then be treated and pressed into a fabric!
There is further research within the field of fungal materials, and the most exciting is geared towards engineered living materials in which the organism stays alive and as a result either improves the product or doesn’t affect its function. Filamentous fungi are incredibly robust in that they have no center of control. From one hyphal tip, a whole network can grow as fungi are self-regenerative. ELM’s researchers are attempting to keep fungal spores dormant with the depletion of moisture in order to keep the self regenerative properties of mycelium for damaged products.
What are their advantages and disadvantages over traditional counterparts?
Fungal materials are bio-degradable and more impressively, compostable. Meaning, you could chuck your mycelium packaging in the dirt and the natural environment can organically breakdown the waste in a non-invasive manner. Fungal materials are safe and offer an array of possibilities for non-toxic materials and pigment alternatives. You get to cultivate with life which inspires more empathy.
The main disadvantage in working with fungi to make materials is the unpredictability, and contamination issues. Fungi are living, and because they are alive, we have to feed them. This can cause contamination in the food source, or on the mycelium itself. In a BSL-1 lab, any un-predicted growth (contamination) is immediately considered a biohazard. If your work gets contaminated, there is no coming back, you must kill the mold and throw away your piece.
What might you want to genetically engineer fungi to do and why?
Fungi produce a unique class of secondary metabolites called polyketides that can be used for pharmaceuticals, materials, and design. However, secondary metabolites, as the name suggests, a secondary priority to produce because they are not required for survival. The majority of beneficial compounds are produced from SM pathways. I would engineer fungi to produce SM pathways quicker and with more consistency. My interest is in the colors that fungi make, such as xylindein in C.aeruginascens, that contain anti-microbial and semi-conductive properties. However, it takes forever to grow. The attempt to introduce pathways into unicellular organisms presents a whole slew of issues, most importantly, the native organism machinery will always work best to express a pathway of genes. If I could engineer the pathway and possibly the genes that express the tailoring enzymes in C.aeruginascens then I could strive for faster production of the pigment without the experimental pitfalls of sequential gene read in the pathway, integration into the genome etc… because it would be within the native organism already.
What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
My initial thoughts are that fungi with the added complexity are better suited at expressing multi-gene pathways while unicellular bacteria and yeast struggle to read through constructs containing more than one insert. This is both an issue for heterologous expression and homologous recombination in bacteria.
Comparing fungi and bacteria is a similar logic to comparing apples to oranges. Both are fruits or in our case, both are living systems. But besides that, their functioning similarities are minimal. The choice to synthetically engineer a fungal strain would come down to production yield, product intent and overall need.
I attempted to do the neuromorphic wizard lab multiple times but kept running into this pop up. I am unable to complete the assignment due to Anaconda failing to install into my computer consistently. I got to see my node mates complete the assignment.
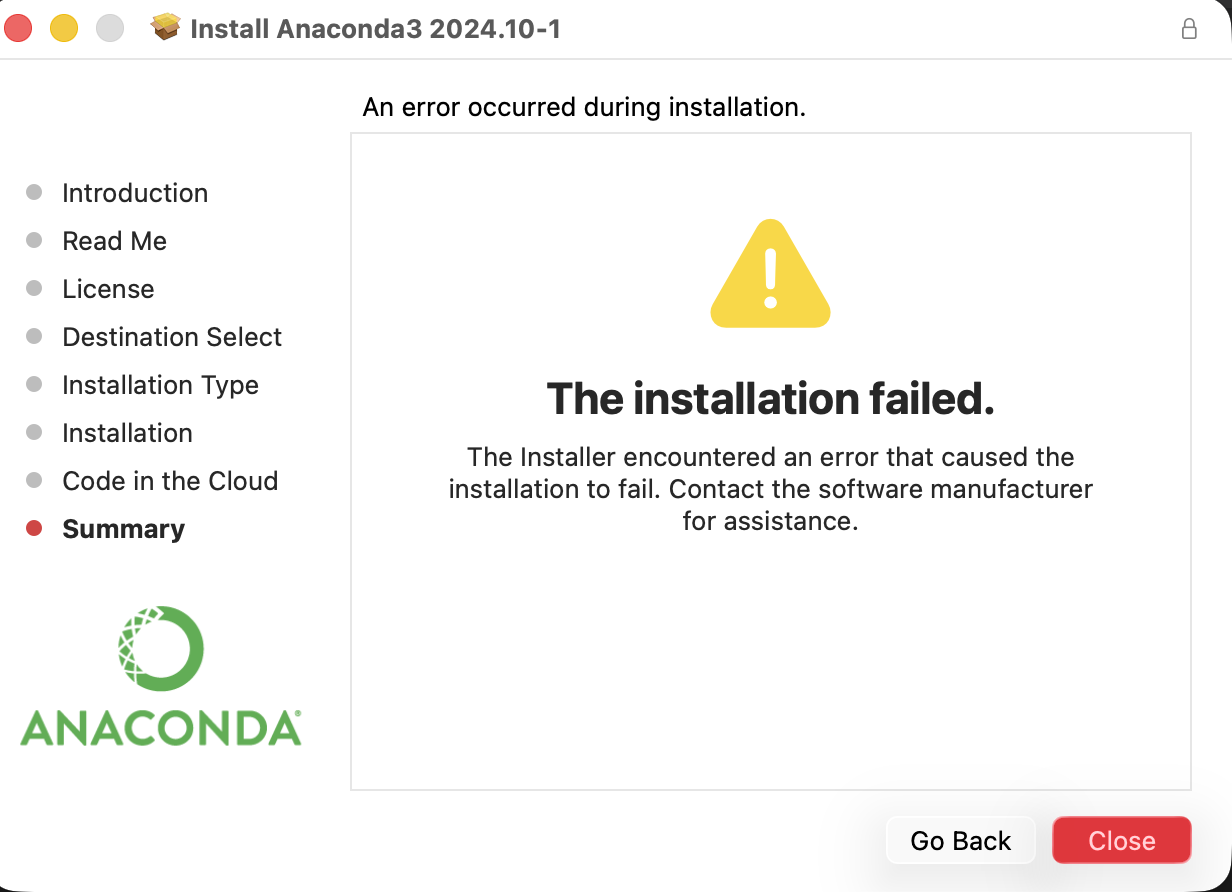
Week 9 HW: Cell Free Systems
The highlighted sections are questions I am unsure how to answer, but made the educated assumptions from the lecture and research.
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell free expression is more beneficial than cell production.
- Mutations are still a phenomenon being understood in a synthetic cell system, however, in a cell FREE system, your risk for mutational error is lessened without an entire cell incorporated.
- Therapies in the environment and on people without the elevated risk of cellular contact
- The system is overall less time intensive allowing for reactions to happen in a day rather than a multi-day to multi-week turn around time.
- So far, the cell free system is projected to be less expensive than cell dependent systems as the labor, components and reaction times are less “problematic”.
- My assumption is this also allows increases the iterative aspects to experimentation. If the system takes less time with less resources and costs, researchers can directly build upon their experiments quicker.
Describe the main components of a cell-free expression system and explain the role of each component.
Component | Role of the Component
DNA/Genes: Vital! This template specifies the protein of interest to express
Energy Regeneration/ATP: Supplies the energy for the system to do its thing! Protein synthesis is expensive and needs energy ready to be used for your desired amount of time
tRNA: tRNA works with the ribosome to form the amino acid chains (polypeptides) from the mRNA
Cell extract: ribosome, RNA polymerase: needed for transcription and translation
Buffers: Keep the environment optimal for the reaction to occur
NTPs/Amino Acids: amino acids in the system are needed to create the polypeptides that create the desire proteins - Essentially the building blocks of life
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy provision regeneration is critical because in order for the cell free system to work, it needs energy. Protein synthesis can be an energy hefty system within a cell. However, unlike the cell free system, a cell is continuously out-sourcing food, digesting and, therefore, obtaining more and more energy. A cell-free system does not have that slew of sources that a cell can access energy replenishment, so the researcher or environment has to supply energy via ATP. For continuous ATP supply, the process of phosphorylation is used.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic cell-free expression systems: Like its cellular system, it is cheaper, faster and typically yields higher results from the system compared to Eukaryotic systems. I would produce chromo-proteins in a prokaryotic cell-free system because it is typically a singular gene insert and the results can be identified through a visual indicator via its color expression.
Eukaryotic cell-free expression systems: Slower, and more expensive to produce due to the increased complexity of the system that a researcher is attempting to replicate in its broken down parts in a cell free system. However, if you are attempting a more complex pathway, a eukaryotic cell free system is a better option. I would attempt expression of Xylindein, a pigment from the organism Blue Elf Cup due to its pathway complexity.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Membrane proteins need a lipid bound environment in order to properly fold and maintain structural stability. If you attempt to express membrane proteins in a cell-free system, the proteins could aggregate and mis-fold. Membrane proteins are hydrophobic as they exist inside of the membrane, if they are in water, they precipitate to the top!
I would address these issues by adding liposomes to the mixture. The liposome will encapsulate the membrane protein within its phospholipid structure preserving its structure and function!
“Hydrophobic chemicals associate with the bilayer. This property can be utilized to load liposomes with hydrophobic and/or hydrophilic molecules, a process known as encapsulation.” - Wikipedia
Fun fact: liposomes are used for drug-delivery!
https://en.wikipedia.org/wiki/Liposome
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
If there was a low yield of my target protein I would increase the energy supply in my solution (the ATP etc) as this would extend the system’s ability to draw energy in for transcription and translation. I would tune the buffer solution to ensure the reaction is happening in a stable environment (pH, salts, trace elements etc). If my template DNA is degrading or not being expressed properly through promoter selection, I would purify my DNA to ensure a pure product and do promoter titer tests to assess the concentration or product yield of different promoters. I would mainly use inducible promoters to regulate concentration and expression without necessarily changing the construct.
1. Pick a function and describe it.
What would your synthetic cell do? What is the input and what is the output?
My synthetic cell would produce the enzyme urease: an enzyme initiating calcification.
Input: Urease enzymatic pathway.
Output: Calcification of macro and micro cracking that appears in cultural heritage objects like ceramics and stone.
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
I believe it could be realized by a cell-free tx/tl alone without encapsulation in order to get the crystallization product for cultural heritage purposes. However, if studying the origins of life and in understanding the bio-mineralization process in organisms, encapsulation is important as containment allows for specificity in the cells mechanism to crystallize.
https://pubs.acs.org/doi/10.1021/acs.chemrev.5c00659
Could this function be realized by genetically modified natural cell?
Yes and It already is via culturing S.pasteurii, however, the process is laborious and multi-step creating a feasibility complication for art conservators using this system.
Describe the desired outcome of your synthetic cell operation.
The desired outcome would be the crystallization from urease to fill micro and macro cracks in ceramic and stone cultural heritage objects without the added biohazard risk of cellular implementation. It would be a controlled system that mitigates risk and harm of art conservators and the objects themselves.
Design all components that would need to be part of your synthetic cell.
What would be the membrane made of?
The membrane would be made of phospholipids. This will mimic a cellular environment.
What would you encapsulate inside? Enzymes, small molecules.
I would encapsulate the urease enzymatic pathway (the genetic pathway) in a construct, NTPs, cytoplasm, tRNAs, ribosomes, amino acids, communication, and co factors. (This list includes the TX/TL system).
Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
My TX/TL system will come from bacterial systems as the urease pathway is being extracted from S.pastuerii.
How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
The synthetic cell will communicate (molecules moving in and out!) through ureI, a membrane protein, which creates a membrane channel for urea to pass in and out without complication.
Experimental details
List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
Lipids: Cholesterol and fatty acids
Genes: ureA, ureB, ureC, ureE.
Tx/Tl system: the bacterial cell free system from the robust species of E.coli
Output product: Bio-mineralized product via calcification
How will you measure the function of your system?
I would measure the function of my system by testing how much calcium carbonate was precipitated and calcified.
When briefly researching the direct technology or protocols to follow for assessing calcium carbonate calcification, these were the results:
- Thermogravimetric analysis
- Calcium depletion
https://pmc.ncbi.nlm.nih.gov/articles/PMC8621315/
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept.
Imagine a bandaid that contains a lyophilizied cell-free reaction where the gene is the proteins that ticks use to secrete the anesthetic when they bite. This would allow for immediate pain relief for both small and extreme topical health issues.
How will the idea work, in more detail? Write 3-4 sentences or more.
The idea starts with the immediate line of defense found in your at home first-aid kit: the bandaid. By redesigning the patch with pores that contain lyophilizied Tx/Tl system, tick anesthetic genetic pathway, and cell-free maintenance elements (energy regeneration, amino acids etc…), a topical non-invasive pain relieving system is administered. Ticks are incredibly annoying. Beyond having your blood sucked, they are incredibly difficult to notice unless you are frequently checking yourself. Ticks are literally bititng you! Which should be painful! But ticks have developed an evolutionary advantage against their host in which they release a chemical into the local area of the bite, effectively numbing any pain the host may feel for prolonged periods of time. The tickpatch leverages this capability through the cell-free system by allowing it to be shelf-stable and immediately effective when needed.
Through further investigation, tick anesthetic is NON-TOXIC to humans. Some further pluses to the system:
- Tick proteins have been tested in vivo (allergic asthma studies show HA24 safety)
- Duration of action is limited to local administration
- Lyophilized form eliminates risk of live pathogen transmission
What societal challenge or market need will this address?
The societal challenge this addresses is accessible and localized anesthetic treatments. The system would provide pain-relief available at home that are not invasive pain relievers. This could also be used by EMT professionals to treat localized incidents that do not need full anesthetic or expensive alternatives.
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
The limitation of activation would be overcome by the water content in the blood and by micro-pores imbedded in the bandaid with water to activate the cell free system if blood is not at the site. Bandaids are already a one time use application in order to mitigate contamination risks and isolate the cut.
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/ .
https://www.nasa.gov/hrp/risks/
The webpage on the above was utilized to provide background information on the issues astronauts and space personnel face when going up to space. This helped in the deciding factors of my answers below!
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
“No current ground-based study exists to test different oxygen exposure levels on humans while they’re experiencing all the stressors of spaceflight.”
Astronauts in space are dealing with a slew of issues pertaining to their health due to the extreme environmental changes the body goes through from earth to space. The risk for mild hypoxia is one of those leering issues - a condition that occurs when oxygen levels in the body become lower than usual. Mild hypoxia creates inconvenient feelings of nausea, confusion and vision impairment.
There are developing solutions to acclimate pressure to oxygen, but I propose a bio-sensor that will monitor oxygen exposure levels during space flight by using the Bio-Bits kit. The significance of this research is relevant to the health concerns that astronauts are facing. This proposal serves as a proof of concept system in identifying possible health shifts in the presence of extreme enviornmental changes.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
Hypoxia Inducible Factors (HIF proteins) are inhibited by the cellular machinery typically, however, when oxygen levels are depleted, HIF proteins are expressed and accumulate. These proteins shift the cell from an oxygen environment to an anerobic environment to maintain homeostasis during the oxygen drops.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
The genetic target of HIF proteins would diagnose possible environments in space of which hypoxia is most likely to occur through the increase of its expression. HIF proteins are only expressed when the body is expressing stress from oxygen depletion. The HIF subunits accumulate and enter the nucleus to complete the complex of the HIF proteins.
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
I hypothesize a HIF protein complex tagging system in which the higher the expression of the HIF protein complex in micro-gravity to macro-gravity environments demonstrate the increased possibility for Hypoxia to occur in astronauts during spaceflight. By doing this research in space and on the ground, researchers can begin identifying how the HIF protein complex changes in response to the extreme environments and additional cellular stressors as a result more specifically. The Bio-Bits would supply the cell-free protein expression kit to do these experiments in the active environment of which this occurs, not just the simulated environment.
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
Experimental Plan Outline:
All experiments in this set up (besides the outlined control) will be done on the ISS.
- Use miniPCR to amplify DNA encoding for the HIF Protein Complex within humans.
- Add the amplified sequence to the BioBits Cell Free System kit.
- Let the system incubate for 24 hours and induce GFP expression
- Measure Fluoresence via imageJ
- Repeat the experiment in and outside the ISS
Control:
On earth, lab personnel will conduct the same experiment in real time to compare expression of the HIF complex within earths gravitational pull.
Week 10 HW: Imaging and Measurment
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc // Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements. What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
There are many aspects of my project that will need to be measured. I will need to confirm amplification and expression of my gene(s). If pigment is yielded, I will need to measure how much pigment per cell is produced and the saturation of that pigment.
The technologies I would use to measure the aspects stated above would be polymerase chain reaction, gel electrophoresis, OD600, and Absorbance spectroscopy.
Possible technologies considering alternative methodologies and designs:
Plate reading, fluoresence Image J measurement for GFP-MAPPING strategy used in the literature.
I did a majority of homework in my lab notebook and have uploaded the images of those pages down below.
We will analyze an eGFP standard on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/eGFP Sequence:
MVSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNYNSHNV YIMADKQKNG IKVNFKIRHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LSTQSALSKD PNEKRDHMVL LEFVTAAGIT LGMDELYKLE HHHHHH
Note: This contains a His-purification tag (HHHHHH) and a linker (the LE before it).
The molecular weight of the eGFP sequence is:
Theoretical pI/Mw: 5.90 / 28006.60
Directly Shown from EXPASY:
Theoretical pI/Mw (average)
Sequence:
10 20 30 40 50 60
MVSKGEELFT GVVPILVELD GDVNGHKFSV SGEGEGDATY GKLTLKFICT TGKLPVPWPT
70 80 90 100 110 120
LVTTLTYGVQ CFSRYPDHMK QHDFFKSAMP EGYVQERTIF FKDDGNYKTR AEVKFEGDTL
130 140 150 160 170 180
VNRIELKGID FKEDGNILGH KLEYNYNSHN VYIMADKQKN GIKVNFKIRH NIEDGSVQLA
190 200 210 220 230 240
DHYQQNTPIG DGPVLLPDNH YLSTQSALSK DPNEKRDHMV LLEFVTAAGI TLGMDELYKL
EHHHHHH
Theoretical pI/Mw: 5.90 / 28006.60
Without the His-tag and LE linker:
Theoretical pI/Mw (average)
Sequence:
10 20 30 40 50 60
MVSKGEELFT GVVPILVELD GDVNGHKFSV SGEGEGDATY GKLTLKFICT TGKLPVPWPT
70 80 90 100 110 120
LVTTLTYGVQ CFSRYPDHMK QHDFFKSAMP EGYVQERTIF FKDDGNYKTR AEVKFEGDTL
130 140 150 160 170 180
VNRIELKGID FKEDGNILGH KLEYNYNSHN VYIMADKQKN GIKVNFKIRH NIEDGSVQLA
190 200 210 220 230
DHYQQNTPIG DGPVLLPDNH YLSTQSALSK DPNEKRDHMV LLEFVTAAGI TLGMDELYK
Theoretical pI/Mw: 5.58 / 26941.48
Theoretical pI/Mw (average)
Sequence:
10 20 30 40 50 60
VSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL
70 80 90 100 110 120
VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV
130 140 150 160 170 180
NRIELKGIDF KEDGNILGHK LEYNYNSHNV YIMADKQKNG IKVNFKIRHN IEDGSVQLAD
190 200 210 220 230
HYQQNTPIGD GPVLLPDNHY LSTQSALSKD PNEKRDHMVL LEFVTAAGIT LGMDELYK
Theoretical pI/Mw: 5.59 / 26810.29
PEPTIDE MASS
MVSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNYNSHNV YIMADKQKNG IKVNFKIRHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LSTQSALSKD PNEKRDHMVL LEFVTAAGIT LGMDELYKLE HHHHHH
20 K 6 R
How many peptides will be generated from tryptic digestion of eGFP?
Navigate to https://web.expasy.org/peptide_mass/
Copy/paste the sequence above into the input box in the PeptideMass tool to generate expected list of peptides.
Use Figure 4 below as a guide for the relevant parameters to predict peptides from eGFP.
Click “Perform the Cleavage” button in the PeptideMass tool and report the number of peptides generated when using trypsin to perform the digest.
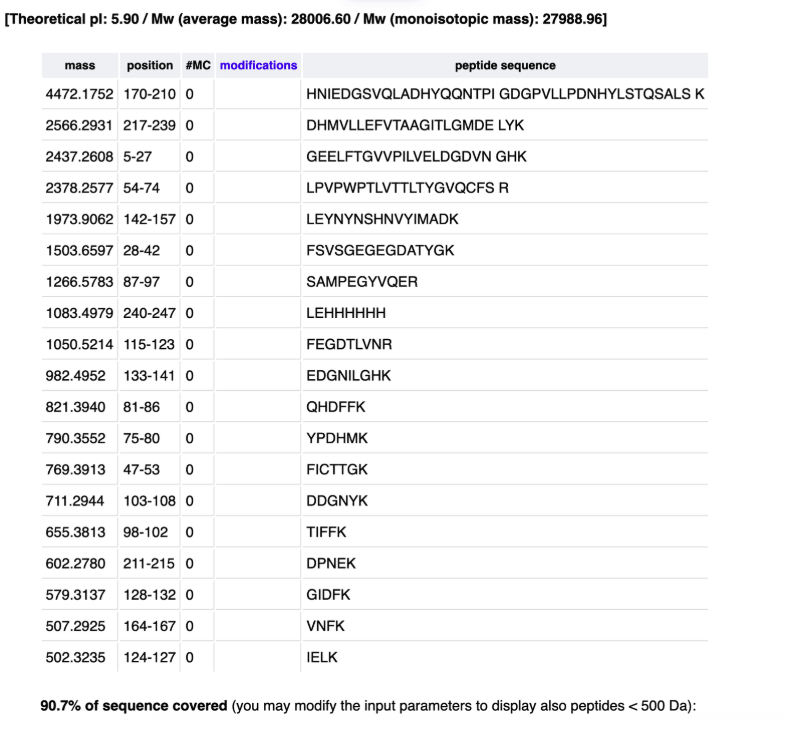
[Theoretical pI: 5.90 / Mw (average mass): 28006.60 / Mw (monoisotopic mass): 27988.96]
10 20 30 40 50 60
mvskGEELFT GVVPILVELD GDVNGHKFSV SGEGEGDATY GKltlkFICT TGKLPVPWPT
70 80 90 100 110 120
LVTTLTYGVQ CFSRYPDHMK QHDFFKSAMP EGYVQERTIF FKDDGNYKtr aevkFEGDTL
130 140 150 160 170 180
VNRIELKGID FKEDGNILGH KLEYNYNSHN VYIMADKqkn gikVNFKirH NIEDGSVQLA
190 200 210 220 230 240
DHYQQNTPIG DGPVLLPDNH YLSTQSALSK DPNEKrDHMV LLEFVTAAGI TLGMDELYKL
EHHHHHH
19 PEPTIDES
Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
21 PEAKS
More peptides in the observed, in predicted only 19 peptides
How are we classifying peaks? Perhaps 3 will not be calculated as a valid peak





Week 11-12 HW: Bioproduction
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase)
- The strain of E.coli used to lysate and extract all of the needed cellular components like polymerases for transcription and ribosomes for translation. There is reduced chromosomal degradation.
Salts/Buffer
Potassium Glutamate
- Mimics cellular environment via K+ ions
HEPES-KOH pH 7.5
- pH buffer to maintain a suitable environment for the cell free reactions.
Magnesium Glutamate
- Ribosome stability
Potassium phosphate monobasic
- pH buffer to maintain a suitable environment for the cell free reactions.
Potassium phosphate dibasic
- pH buffer to maintain a suitable environment for the cell free reactions.
Energy / Nucleotide System
Purpose:
Ribose
- Ribose is the sugar in the backbone of DNA
- Combines with nucleobases to become RNA
Glucose:
- The carbon source. Glucose drives metabolic energy regeneration. Essentially, the food to keep going.
AMP
- Adenosine monophosphate
CMP
- Cystoisine Monophosphate
GMP
- Guanosine Monophosphate
UMP
- Uridine Monophosphate
Guanine
- Nucelobase
Monophosphates:
- NMP→NDP→NTP
Supplies all components needed for energy and transcription
Translation Mix (Amino Acids)
17 Amino Acid Mix
- Standard amino acids! Made available in the solution so transcription can occur with accessible AA products
Tyrosine
- Mixed separately because unstable in a mixed solution
Cysteine
- Mixed separately because unstable in a mixed solution
- Core protein building blocks
Additives
Nicotinamide
- Extends reaction and metabolic activity
- Vitamin B3
Backfill
- Nuclease Free Water
The nuclease free water has been treated to eliminate any RNAses that could degrade the DNA and RNA in the sample. Typically, water is the solvent to get the mix to the proper concentration and acts as the body of the reaction.
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences).
The main differences between the two mixes are the energy systems, phosphate concentrations and additives. For energy, the ATPs are immediately supplied to expedite immediate growth. Over time the metabolic pathway will be exhausted and the overall system will become unstable. The addition of spermidine at 937mM boosts protein synthesis,which works for a 1 hour reaction, but will become unstable as the hours progress as synthesis uses more and more energy that is not sustained.
Bonus question: how can transcription occur if GMP is not included but Guanine is?
GMP is the guanine monophosphate and guanine can become GMP which gets converted into energy. GMP→GDP→GTP
Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems (hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each).
The amino acid sequences are shown in the HTGAA Cell-Free Benchling folder.
- sfGFP
- mRFP1
- mKO2
- mTurquoise2
- mScarlet_I
- Electra2
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
Q4-E11 Electra 2
17 AA: 4.123mM
Magnesium Glutamate: 7.402mM
Nicotinamide: 3.152mM
The goal of maximizing fluorescence over a 36 hour incubation resulted in increasing the amino acid mix, magnesium glutamate and nicotinamide.
Magnesium glutamate supports ribosomal stability which in tandem with increased amino acid support and nicotinamide (extends reaction and metabolic activity) will support long term incubation. My thought process is in increasing translational machinery and maintenance of the post-translational activity in the solution.
Post Lab Questions | Mandatory for All Students
Which genes when transferred into E. coli will induce the production of lycopene and beta-carotene, respectively?
Lycopene (pLAC-LYC): red pigment found in tomatoes. Microbes produce lycopene via a 3-enzyme pathway transferred/introduced into E.coli that can convert FPP into lycopene.
Beta-Carotene (pAC-BETA): an orange pigment altered from lycopene via a singular gene(enzyme) difference of CrtY.
The genes that will induce lycopene and beta-carotene respectively are CrtE, Crtl, and crtB with the differentiating gene creating Beta Carotene being Crt Y. CrtY is from the organism Erwinia Herbicola
Why do the plasmids that are transferred into the E. coli need to contain an antibiotic resistance gene?
Plasmids transferred into E.coli need an antibiotic resistance gene (yeast selectable markers) as a way to identify E.coli that grow with the respective gene insert. E.coli with your gene (with the resistance of the antibiotic in the media) should be the only organism to grow on the plate.
What outcomes might we expect to see when we vary the media, presence of fructose, and temperature conditions of the overnight cultures?
Fructose and 37C will lead to higher yield because 37 mimics the natural environmental temperature of E.coli. Fructose is an additional carbon source which will provide more energy for cellular growth and function.
Generally describe what “OD600” measures and how it can be interpreted in this experiment.
OD600 measures the amount of cellular growth in a substance. Bacteria, overnight, create a saturated medium that looks murky. As the cells divide during their phases of growth, the media becomes noticeably “denser”. Therefore, optical density works by sending out wavelengths of light. The reduction of light passing through the sample is used to estimate cellular growth.
The increase of bacterial growth directly increases the absorbance factor.
What are other experimental setups where we may be able to use acetone to separate cellular matter from a compound we intend to measure?
Creatively, acetone can be used to create a pigmented solution from cells by being a method that breaks up the cell via resuspension and centrifugation. You would only get the pigment within this final product.
Within the same breath, acetone can be used to precipitate proteins as acetone decreases the solubility of the proteins. If you have a gene of interest with a protein output, you can estimate the yield via centrifuging the cells into a pellet causing your desired compound from the protein to remain suspended in the acetone solution. The left supernatant then contains your protein output in the acetone solution (like carotenoids in the lab example).
OR you can use the protein precipitate in other downstream applications and analysis.
Hydrophobicity! Lipids + proteins classic!
Why might we want to engineer E. coli to produce lycopene and beta-carotene pigments when Erwinia herbicola naturally produces them?
Though Erwinia Herbicola produces lycopene and beta-carotene pigments, the bacteria does not prioritize the production of these pigments. By engineering E.coli, selecting for the production of lycopene can produce higher yields and also generate more consistent production.
Post Lab Questions | For Committed Listeners Only
You may need the following papers to answer these questions:
Gene expression pattern analysis of a recombinant Escherichia coli strain possessing high growth and lycopene production capability when using fructose as carbon source
Improvement of Biomass Yield and Recombinant Gene Expression in Escherichia coli by Using Fructose as the Primary Carbon Source
Let’s get in touch with our metabolic pathway
What are the enzymes of the carotene pathway?
The enzymes in the carotene pathway are FPP, GGPP CrtE, Crtl, and CrtB
Within this pathway, which is the rate determining step (the step that takes the longest)? Which enzyme is responsible for this step?
Farnesyl diphosphate (FPP) enzyme that makes lycopene downstream. (?)
Recitation:
Phytoene Synthase → Knockout experiments
https://pmc.ncbi.nlm.nih.gov/articles/PMC9039723/
https://www.youtube.com/watch?v=65EQViMyMR0&t=359s
https://www.youtube.com/watch?v=evWgPUc7200&t=87s
Notes for design of a DNA construct for bioproduction
The first thing to do is to decide what organism you are going to use for this (E. coli or S. cerevisiae) for production. Which would you choose and why (emphases on production differences)?
Things to initially Consider:
1. Is the intended host in the same kingdom? Prokaryote, eukaryote, Archea etc…
2. What is the protein of interest and how does that function express in the cell?
3. Rate of growth desired?
4. Length of gene insert? More post-translational difficulty and maintenance in the cell?
Can handle high copy inducible promoters!
Now choose one of the enzymes and lets outline the parts of the construct for expression
For E. coli lets create a expression vector that works as a plasmid
Origin of Replication, Promoter for antibiotic, antibiotic gene, Promoter, RBS, Start codon, gene insert, stop codon, terminator.
Now, for making a functional construct there are a variety of biological parts needed for this, like ribosome binding sites, terminators, operators and promoters. The last ones are the most important in terms of enzyme or protein production. Let’s elaborate further on this biopart.
With the links below we are going to answer a few questions and think about the correct use of promoter: (https://blog.addgene.org/plasmids-101-the-promoter-region,
https://www.addgene.org/mol-bio-reference/promoters/, https://blog.addgene.org/plasmids-101-repressible-promoters
https://blog.addgene.org/plasmids-101-inducible-promoters
What is the function of a promoter?
A promoter is a gene/DNA sequence upstream of the RBS and gene insert that initiates transcription of the sequence. It “promotes” the polymerase to read the DNA script.
What types of promoters do we have?
Promoters are specific to a class of organism. Prokaryotic and Eukaryotic organisms will use different sets of promoters as the promoter calls upon the cell machinery to initiate transcription. There are inducible and constitutive promoters.
https://byjus.com/biology/difference-between-inducible-and-constitutive-promoter/
If we wanted to turn off the transcription of a gene in response to a metabolite, what type of promoter would be most useful? What if we wanted this to increase in the presence of the metabolite?
An inducible promoter because an inducible promoter is regulated by a metabolite, pH, temperature etc! If increase of the metabolite was desired, you would use a constitutive promoter because the pathway does not receive a signal to stop under specific conditions and is thus “always on”
Now choose one of the genes of the metabolic pathway previously described (Carotene/lycopene )and choose one enzyme to make an expression construct. What promoter could you use for this? Why did you choose it?
CrtE with the constitutive promoter T7 (with T7 RNA polymerase). I chose the T7 promoter because it is widely used and drives high levels of transcription.
With the links below we are going to answer a few questions and think about the correct use of origin of rep:
https://blog.addgene.org/plasmid-101-origin-of-replication, https://blog.addgene.org/plasmids-101-plasmid-incompatibility,
https://blog.addgene.org/plasmids-101-ebook-4th-edition
What is the origin of replication?
The origin of replication is the gene/sequence that initiates replication of the plasmid in the cell. Without the origin of replication, the plasmid would not be replicated autonomously into the host cell. Though the origin of replication still relies on the host’s cell internal resources and machinery to be replicated in the cell. The ori has more A-T pairings due to bases being boned with two hydrogen bonds rather, making the sequence “easier” to denature and replicate downstream.
What types of origin of replication do we have?
Relaxed: regulated only by the DNA in the plasmid, and there is no need for host cell initiation proteins. These tend to be high copy as they are not competing with other necessities of the cell to be transcribed!
Stringent: Stringent relies on the host’s chromosome to start replication via the initiation proteins. These tend to be low copy!
Always use different origins of rep. When adding several plasmids to your host to minimize competition for host machinery and transcription.
(Extra) What are compatibility groups?
Compatibility groups in response to plasmids are the compatibility factors to ensure multiple plasmids being introduced into the host cell can be read and translated into the host. Having the same replicons on different plasmids causes the issue of competition for the cells machinery which can cause a plasmid to get lost/ignored by the cell.
In plasmids, there is the negative regulation system that distinguishes its plasmid from other plasmids in the cell. This system is based on iterons, repeated sequences in the ori that bind the initiation host machinery to begin transcription.
Now for the previously chosen promoter and gene what will be the best origin or replication?
I am choosing the pUMB1 origin in E.coli because it is a high copy origin and is only regulated from the DNA in the plasmid. With the T7 promoter and pUMB1 origin, the plasmid will be highly regulated in the host cell causing minimal complications with host machinery and high yield of the enzyme crtE.
(Mandatory for Global listeners, Optional MIT/Harvard) Elaborate further on other bioparts like RBS, terminators, operators you would use for a correct design and further bioproduction?
Ribosome Binding Site: the sequence that recruits the ribosome to bind to the RNA and begin translation. RBS typically contain a start codon prior to the gene insert to initiate direct translation of the gene and synthesis.
https://parts.igem.org/Ribosome_Binding_Sites#:~:text=A%20Ribosome%20Binding%20Site%20(RBS)%20is%20an,coding%20sequences%20begin%20with%20the%20start%20codon.
Terminators: the sequence that ends transcription. This process releases the RNA to begin translation. The Stop codon comes before the terminator.
https://blog.addgene.org/plasmids-101-terminators-and-polya-signals
Operators: sequence within the promoter that controls gene expression by acting like a switch.