Week 2 HW: DNA Read, Write and Edit
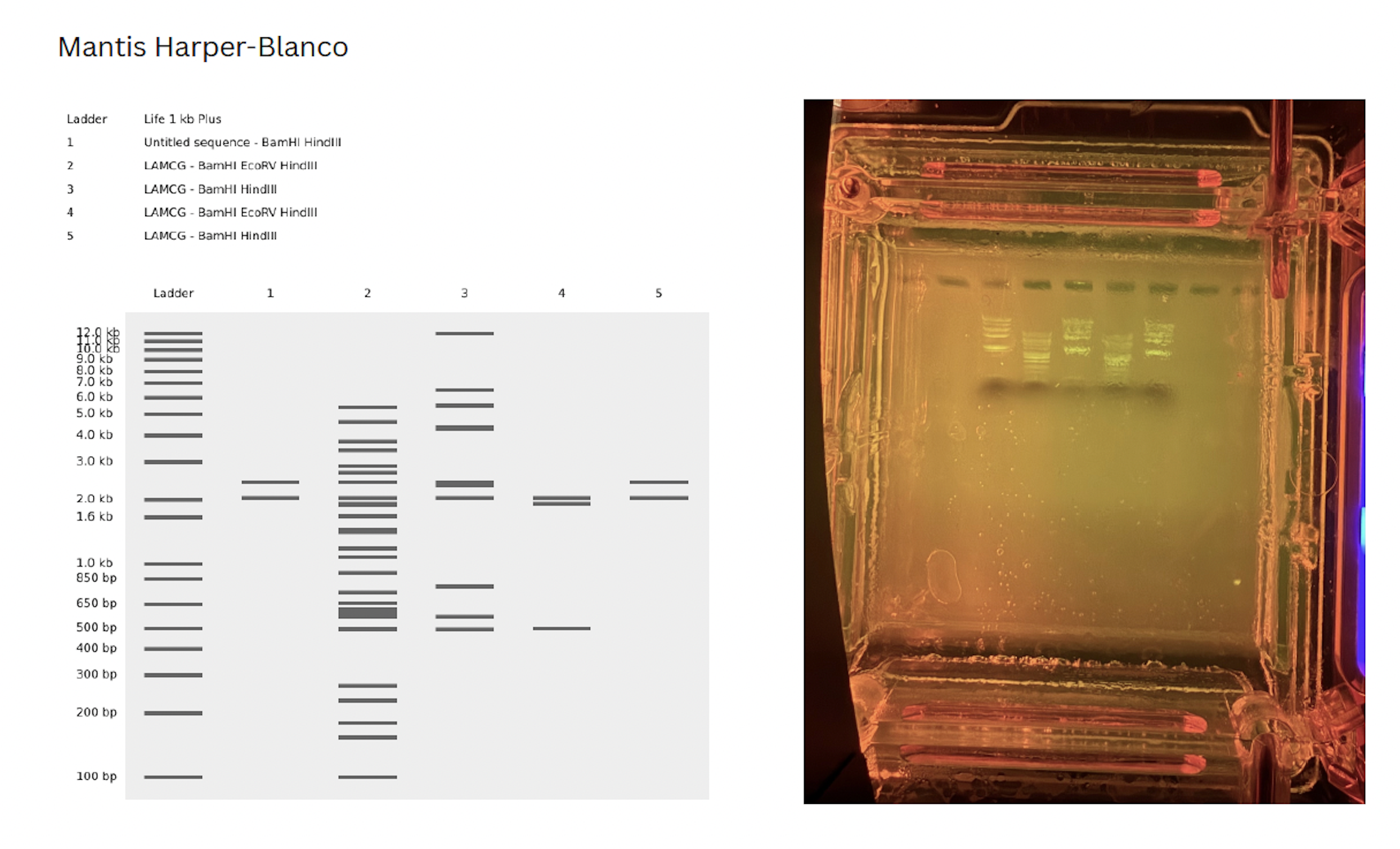
My initial confusion was trouble shooting how to cut at a specific segments of DNA. On my benchling, I copied a segment of DNA between the desired cut sites and pasted it into a “New DNA sequence” file. This was purely for artistic purposes, however, in lab, I asked our instructor how we would select for these specific sites for future gel art.
There were two options:
- Purchase or design primers to select at the specific site
OR
- Run the DNA with all of the cut sites with the selected enzymes and do a gel extraction of the desired band. Hypthetically, in the next run, only that site will appear (though the band will be faded due to less DNA content)
3.1. Choose your protein.
In recitation, we discussed that you will pick a protein for your homework that you fi nd interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose.
I chose Sericin from the organism Bombyx Mori (Silk Moth) because for my fi nal project I am interested in creating art conservation methods and protocols from synthetic biological protocols and principals to preserve cultural heritage objects and textiles. Specifi cally silk which once enters a degradation phase, is untreatable as the silk essentially powderizes. Sericin is the native gum that protects fi broin but is boiled off in the process to get the fi ne silk fi ber we use today. Sericin was considered a waste stream of the silk process but has been used in biomedical biomaterial cases and shows promise in being designed as a conservation protein glue to keep silk textiles protected.
https://www.uniprot.org/uniprotkb/P07856/entry MRFVLCCTLIALAALSVKAFGHHPGNRDTVEVKNRKYNAASSESSYLNKDNDSISAGAHAKSVEQSQDKSKYTSGPEGVSYSGRSQNYKDSKQAYADYHSDPNGGSASAGQSRDSSLRERNVHYVSDGEAVAASSDARDENRSAQQNAQANWNADGSYGVSADRSGSASSRRRQANYYSDKDITAASKDDSRADSSRRSNAYYNRDSDGSESAGLSDRSASSSKNDNVFVYRTKDSIGGQAKSSRSSHSQESDAYYNSSPDGSYNAGTRDSSISNKKKASSTIYADKDQIRAANDRSSSKQLKQSSAQISSGPEGTSVSSKDRQYSNDKRSKSDAYVGRDGTVAYSNKDSEKTSRQSNTNYADQNSVRSDSAASDQTSKSYDRGYSDKNIVAHSSGSRGSQNQKSSSYRADKDGFSSSTNTEKSKFSSSNSVVETSDGASASRESSAEDTKSSNSNVQSDEKSASQSSSSRSSQESASYSSSSSSSTLSEDSSEVDIDLGNLGWWWNSDNKVQRAAGGATKSGASSSTQATTVSGADDSADSYTWWWNPRRSSSSSSSASSSSSGSNVGGSSQSSGSSTSGSNARGHLGTVSSTGSTSNTDSSSKSAGSRTSGGSSTYGYSSSHRGGSVSSTGSSSNTDSSTKNAGSSTSGGSSTYGYSSSHRGGSVSSTGSSSNTDSSTKSAGSSTSGGSSTYGYSSRHRGGRVSSTGSSSTTDASSNSVGSSTSGGSSTYGYSSNSRDGSVSSTGSSSNTDSNSNSAGSSTSGGSSTYGYSSNSRDGSVSSTGSSSNTDSNSNSAGSSTSGGSSTYGYSSNSRDGSVSSTGSSSNTDASTDLTGSSTSGGSSTYGYSSDSRDGSVSSTGSSSNTDASTDLAGSSTSGGSSTYGYSSDCGDGSVSSTGSSSNTDASTDLAGSSTSGGSSTYGYSSDSRDGSVSSTGSSSNTDASTDLAGSSTSGGSSTYGYSSNSRDGSVSSTGSSSNTDASTDLTGSSTSGGSSTYGYSSSNRDGSVLATGSSSNTDASTTEESTTSAGSSTEGYSSSSHDGSVTSTDGSSTSGGASSSSASTAKSDAASSEDGFWWWNRRKSGSGHKSATVQSSTTDKTSTDSASSTDSTSSTSGASTTTSGSSSTSGGSSTSDASSTSSSVSRSHHSGVNRLLHKPGQGKICLCFENIFDIPYHLRKNIGV
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above. [Example: Get to the original sequence of phage MS2 L-protein from its genome phage MS2 genome - Nucleotide - NCBI ] To reverse translate the sericin AA sequence I used https://www.bioinformatics.org/sms2/rev_trans.html and received the DNA sequence below.
atgcgctttgtgctgtgctgcaccctgattgcgctggcggcgctgagcgtgaaagcgtttggccatcatccgggcaaccgcgataccgtggaagtgaaaaaccgcaaatataacgcggcgagcagcgaaagcagctatctgaacaaagataacgatagcattagcgcgggcgcgcatcgcgcgaaaagcgtggaacagagccaggataaaagcaaatataccagcggcccggaaggcgtgagctatagcggccgcagccagaactataaagatagcaaacaggcgtatgcggattatcatagcgatccgaacggcggcagcgcgagcgcgggccagagccgcgatagcagcctgcgcgaacgcaacgtgcattatgtgagcgatggcgaagcggtggcggcgagcagcgatgcgcgcgatgaaaaccgcagcgcgcagcagaacgcgcaggcgaactggaacgcggatggcagctatggcgtgagcgcggatcgcagcggcagcgcgagcagccgccgccgccaggcgaactattatagcgataaagatattaccgcggcgagcaaagatgatagccgcgcggatagcagccgccgcagcaacgcgtattataaccgcgatagcgatggcagcgaaagcgcgggcctgagcgatcgcagcgcgagcagcagcaaaaacgataacgtgtttgtgtatcgcaccaaagatagcattggcggccaggcgaaaagcagccgcagcagccatagccaggaaagcgatgcgtattataacagcagcccggatggcagctataacgcgggcacccgcgatagcagcattagcaacaaaaaaaaagcgagcagcaccatttatgcggataaagatcagattcgcgcggcgaacgatcgcagcagcagcaaacagctgaaacagagcagcgcgcagattagcagcggcccggaaggcaccagcgtgagcagcaaagatcgccagtatagcaacgataaacgcagcaaaagcgatgcgtatgtgggccgcgatggcaccgtggcgtatagcaacaaagatagcgaaaaaaccagccgccagagcaacaccaactatgcggatcagaacagcgtgcgcagcgatagcgcggcgagcgatcagaccagcaaaagctatgatcgcggctatagcgataaaaacattgtggcgcatagcagcggcagccgcggcagccagaaccagaaaagcagcagctatcgcgcggataaagatggctttagcagcagcaccaacaccgaaaaaagcaaatttagcagcagcaacagcgtggtggaaaccagcgatggcgcgagcgcgagccgcgaaagcagcgcggaagataccaaaagcagcaacagcaacgtgcagagcgatgaaaaaagcgcgagccagagcagcagcagccgcagcagccaggaaagcgcgagctatagcagcagcagcagcagcagcaccctgagcgaagatagcagcgaagtggatattgatctgggcaacctgggctggtggtggaacagcgataacaaagtgcagcgcgcggcgggcggcgcgaccaaaagcggcgcgagcagcagcacccaggcgaccaccgtgagcggcgcggatgatagcgcggatagctatacctggtggtggaacccgcgccgcagcagcagcagcagcagcagcgcgagcagcagcagcagcggcagcaacgtgggcggcagcagccagagcagcggcagcagcaccagcggcagcaacgcgcgcggccatctgggcaccgtgagcagcaccggcagcaccagcaacaccgatagcagcagcaaaagcgcgggcagccgcaccagcggcggcagcagcacctatggctatagcagcagccatcgcggcggcagcgtgagcagcaccggcagcagcagcaacaccgatagcagcaccaaaaacgcgggcagcagcaccagcggcggcagcagcacctatggctatagcagcagccatcgcggcggcagcgtgagcagcaccggcagcagcagcaacaccgatagcagcaccaaaagcgcgggcagcagcaccagcggcggcagcagcacctatggctatagcagccgccatcgcggcggccgcgtgagcagcaccggcagcagcagcaccaccgatgcgagcagcaacagcgtgggcagcagcaccagcggcggcagcagcacctatggctatagcagcaacagccgcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatagcaacagcaacagcgcgggcagcagcaccagcggcggcagcagcacctatggctatagcagcaacagccgcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatagcaacagcaacagcgcgggcagcagcaccagcggcggcagcagcacctatggctatagcagcaacagccgcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatgcgagcaccgatctgaccggcagcagcaccagcggcggcagcagcacctatggctatagcagcgatagccgcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatgcgagcaccgatctggcgggcagcagcaccagcggcggcagcagcacctatggctatagcagcgattgcggcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatgcgagcaccgatctggcgggcagcagcaccagcggcggcagcagcacctatggctatagcagcgatagccgcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatgcgagcaccgatctggcgggcagcagcaccagcggcggcagcagcacctatggctatagcagcaacagccgcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatgcgagcaccgatctgaccggcagcagcaccagcggcggcagcagcacctatggctatagcagcagcaaccgcgatggcagcgtgctggcgaccggcagcagcagcaacaccgatgcgagcaccaccgaagaaagcaccaccagcgcgggcagcagcaccgaaggctatagcagcagcagccatgatggcagcgtgaccagcaccgatggcagcagcaccagcggcggcgcgagcagcagcagcgcgagcaccgcgaaaagcgatgcggcgagcagcgaagatggcttttggtggtggaaccgccgcaaaagcggcagcggccataaaagcgcgaccgtgcagagcagcaccaccgataaaaccagcaccgatagcgcgagcagcaccgatagcaccagcagcaccagcggcgcgagcaccaccaccagcggcagcagcagcaccagcggcggcagcagcaccagcgatgcgagcagcaccagcagcagcgtgagccgcagccatcatagcggcgtgaaccgcctgctgcataaaccgggccagggcaaaatttgcctgtgctttgaaaacatttttgatattccgtatcatctgcgcaaaaacattggcgtg
Codon optimized for E.coli (K12) via benchling codon optimization tools with reduced hair pinning
Atgcgctttgtgctgtgctgcaccctgattgcgctggcggcgctgagcgtgaaagcgtttggccatcatccgggcaaccgcgataccgtggaagtgaaaaaccgcaaatataacgcggcgagcagcgaaagcagctatctgaacaaagataacgatagcattagcgcgggcgcgcatc gcgcgaaaagcgtggaacagagccaggataaaagcaaatataccagcggcccggaaggcgtgagctatagcggccgcagccagaactataaagatagcaaacaggcgtatgcggattatcatagcgatccgaacggcggcagcgcgagcgcgggccagagccgcgatagcagcctgcgcgaacgcaacgtgcattatgtgagcgatggcgaagcggtggcggcgagcagcgatgcgcgcgatgaaaaccgcagcgcgcagcagaacgcgcaggcgaactggaacgcggatggcagctatggcgtgagcgcggatcgcagcggcagcgcgagcagccgccgccgccaggcgaactattatagcgataaagatattaccgcggcgagcaaagatgatagccgcgcggatagcagccgccgcagcaacgcgtattataaccgcgatagcgatggcagcgaaagcgcgggcctgagcgatcgcagcgcgagcagcagcaaaaacgataacgtgtttgtgtatcgcaccaaagatagcattggcggccaggcgaaaagcagccgcagcagccatagccaggaaagcgatgcgtattataacagcagcccggatggcagctataacgcgggcacccgcgatagcagcattagcaacaaaaaaaaagcgagcagcaccatttatgcggataaagatcagattcgcgcggcgaacgatcgcagcagcagcaaacagctgaaacagagcagcgcgcagattagcagcggcccggaaggcaccagcgtgagcagcaaagatcgccagtatagcaacgataaacgcagcaaaagcgatgcgtatgtgggccgcgatggcaccgtggcgtatagcaacaaagatagcgaaaaaaccagccgccagagcaacaccaactatgcggatcagaacagcgtgcgcagcgatagcgcggcgagcgatcagaccagcaaaagctatgatcgcggctatagcgataaaaacattgtggcgcatagcagcggcagccgcggcagccagaaccagaaaagcagcagctatcgcgcggataaagatggctttagcagcagcaccaacaccgaaaaaagcaaatttagcagcagcaacagcgtggtggaaaccagcgatggcgcgagcgcgagccgcgaaagcagcgcggaagataccaaaagcagcaacagcaacgtgcagagcgatgaaaaaagcgcgagccagagcagcagcagccgcagcagccaggaaagcgcgagctatagcagcagcagcagcagcagcaccctgagcgaagatagcagcgaagtggatattgatctgggcaacctgggctggtggtggaacagcgataacaaagtgcagcgcgcggcgggcggcgcgaccaaaagcggcgcgagcagcagcacccaggcgaccaccgtgagcggcgcggatgatagcgcggatagctatacctggtggtggaacccgcgccgcagcagcagcagcagcagcagcgcgagcagcagcagcagcggcagcaacgtgggcggcagcagccagagcagcggcagcagcaccagcggcagcaacgcgcgcggccatctgggcaccgtgagcagcaccggcagcaccagcaacaccgatagcagcagcaaaagcgcgggcagccgcaccagcggcggcagcagcacctatggctatagcagcagccatcgcggcggcagcgtgagcagcaccggcagcagcagcaacaccgatagcagcaccaaaaacgcgggcagcagcaccagcggcggcagcagcacctatggctatagcagcagccatcgcggcggcagcgtgagcagcaccggcagcagcagcaacaccgatagcagcaccaaaagcgcgggcagcagcaccagcggcggcagcagcacctatggctatagcagccgccatcgcggcggccgcgtgagcagcaccggcagcagcagcaccaccgatgcgagcagcaacagcgtgggcagcagcaccagcggcggcagcagcacctatggctatagcagcaacagccgcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatagcaacagcaacagcgcgggcagcagcaccagcggcggcagcagcacctatggctatagcagcaacagccgcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatagcaacagcaacagcgcgggcagcagcaccagcggcggcagcagcacctatggctatagcagcaacagccgcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatgcgagcaccgatctgaccggcagcagcaccagcggcggcagcagcacctatggctatagcagcgatagccgcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatgcgagcaccgatctggcgggcagcagcaccagcggcggcagcagcacctatggctatagcagcgattgcggcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatgcgagcaccgatctggcgggcagcagcaccagcggcggcagcagcacctatggctatagcagcgatagccgcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatgcgagcaccgatctggcgggcagcagcaccagcggcggcagcagcacctatggctatagcagcaacagccgcgatggcagcgtgagcagcaccggcagcagcagcaacaccgatgcgagcaccgatctgaccggcagcagcaccagcggcggcagcagcacctatggctatagcagcagcaaccgcgatggcagcgtgctggcgaccggcagcagcagcaacaccgatgcgagcaccaccgaagaaagcaccaccagcgcgggcagcagcaccgaaggctatagcagcagcagccatgatggcagcgtgaccagcaccgatggcagcagcaccagcggcggcgcgagcagcagcagcgcgagcaccgcgaaaagcgatgcggcgagcagcgaagatggcttttggtggtggaaccgccgcaaaagcggcagcggccataaaagcgcgaccgtgcagagcagcaccaccgataaaaccagcaccgatagcgcgagcagcaccgatagcaccagcagcaccagcggcgcgagcaccaccaccagcggcagcagcagcaccagcggcggcagcagcaccagcgatgcgagcagcaccagcagcagcgtgagccgcagccatcatagcggcgtgaaccgcctgctgcataaaccgggccagggcaaaatttgcctgtgctttgaaaacatttttgatattccgtatcatctgcgcaaaaacattggcgtg
3.4. You have a sequence! Now what? What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependentor cell-freemethods,or both . Prior to insertion of my gene of interets, the gene cannot be inserted alone, it must exits within a plasmid ( a circular piece of DNA native to bacteria). The plasmid contains several important pieces that allows the cell to READ the DNA for transcritption and later down the line, translation then expression. These elements include a promoter (which tells the cell’s machinary where to start transcription of your gene of interest), RBS, start codon typically (ATG), CDS, His tag, stop codon and terminator. A backbone selection completes the plasmid by encoding an origin of replication, antibiotic resistance and operons etc! To ensure I got a lot of the gene I need, I would design custom primers for amplification via PCR.
Part 4: Prepare a Twist DNA Synthesis Order
This is a practice exercise, not necessarily your real Twist order! 4.1. Create a Twist account and a Benchling account click through for Twist signup click through for Benchling signup
4.2. Build Your DNA Insert Sequence
For example, let’s make a sequence that will make E. coli glow fl uorescent green under UV light by constitutively (always) expressing sfGFP (a green fl uorescent protein):
- In Benchling, select New DNA/RNA sequence
- Give your insert sequence a name and select DNA with a Linear topology (this is a linear sequence that will be inserted into a circular backbone vector of our choosing).
- Go through each piece of the given DNA sequences highlighted below (Promoter, RBS, Start Codon, Coding Sequence, His Tag, Stop Codon,Terminator) and paste the sequences into the Benchling fi le one after the other (replacing the coding sequence with your codon optimized DNA sequence of interest!). Each time you add a new piece of the sequence, make sure to annotate by right clicking over the sequence and creating an annotation that describes what each piece (e.g., Promoter, RBS, etc.) is (see image below)
Promoter
(e.g.BBa_J23106): TTTACGGCTAGCTCAGTCCTAGGTATAGTGCTAGC RBS
(e.g.BBa_B0034 with spacers for optimal expression): CATTAAAGAGGAGAAAGGTACC
Start Codon
ATG
Coding Sequence
(your codon optimized DNA for a protein of interest,sfGFP for example): AGCAAAGGAGAAGAACTTTTCACTGGAGTTGTCCCAATTCTTGTTGAATTAGATGGTGATGTTAATGGGCACAAATTTTCTGTCCGTGGAGAGGGTGAAGGTGATGCTACAAACGGAAAACTCACCCTTAAATTTATTTGCACTACTGGAAAACTACCTGTTCCGTGGCCAACACTTGTCACTACTCTGACCTATGGTGTTCAATGCTTTTCCCGTTATCCGGATCACATGAAACGGCATGACTTTTTCAAGAGTGCCATGCCCGAAGGTTATGTACAGGAACGCACTATATCTTTCAAAGATGACGGGACCTACAAGACGCGTGCTGAAGTCAAGTTTGAAGGTGATACCCTTGTTAATCGTATCGAGTTAAAGGGTATTGATTTTAAAGAAGATGGAAACATTCTTGGACACAAACTCGAGTACAACTTTAACTCACACAATGTATACATCACGGCAGACAAACAAAAGAATGGAATCAAAGCTAACTTCAAAATTCGCCACAACGTTGAAGATGGTTCCGTTCAACTAGCAGACCATTATCAACAAAATACTCCAATTGGCGATGGCCCTGTCCTTTTACCAGACAACCATTACCTGTCGACACAATCTGTCCTTTCGAAAGATCCCAACGAAAAGCGTGACCACATGGTCCTTCTTGAGTTTGTAACTGCTGCTGGGATTACACATGGCATGGATGAGCTCTACAAA
7x His Tag(Let’s add a 7×His tag at the C-terminus of the protein to enable protein purifi cation from E. coli):
CATCACCATCACCATCATCAC
Stop Codon
TAA
Terminator
(e.g.BBa_B0015):CCAGGCATCAAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTTCGTTTTATCTGTTGTTTGTCGGTGAACGCTCTCTACTAGAGTCACACTGGCTCACCTTCGGGTGGGCCTTTCTGCGTTTATA
A link to the expression cassette including all the required parts for expression in E.coli. [https://benchling.com/s/seq-fdUK2PUX2MmY798xn0p1?m=slm-mtkTuHUVSUj7kP0YqL2I]
This insert sequence you built is commonly referred to as an expression cassette in molecular biology (a sequence you can drop into any vector and it’ll perform its function). Go ahead and download the FASTA file for the sequence you made. 4.3. On Twist, Select The “Genes” Option
4.4. Select “Clonal Genes” option For this demonstration, we’ll choose Clonal Genes. You’ll select clonal genesor gene fragments depending on your fi nal project. Historically, HTGAA projects using clonal genes (circular DNA) have reached experimental results 1-2 weeks quicker because they can be transformed directly into E. coli without additional assembly. Gene fragments (linear DNA) offer greater design fl exibility but typically require an assembly or cloning step prior to transformation. An advantage is If designed with the appropriate exonuclease protection, gene fragments can be used directly in cell-free expression.
4.5. Import your sequence You just took an amino acid sequence of interest and converted it into DNA, codon optimized it, and built an expression cassette around it! Choose the Nucleotide Sequence option and Upload Sequence File to upload your FASTA fi le.
4.6. Choose Your Vector Since we’re ordering aclonal gene, you will need to refer to Twist’s Vector Catalog to choose your circular backbone. You can think of this as taking your linear expression cassette for your protein of interest, and completing the rest of the circle! The backbone confers many special properties like antibiotic resistance, an origin of replication, and more. Discuss with your node to decide on appropriate antibiotic options. At MIT/Harvard, you can use Ampicillin, Chloramphenicol, or Kanamycin resistance. Twist vectors do not contain restriction sites near the insert fragment, so make sure to flank your design with cut sites if you are intending to extract this DNA insert fragment later.
I chose the backbone pET-23(+) as shown below with my completed construct.
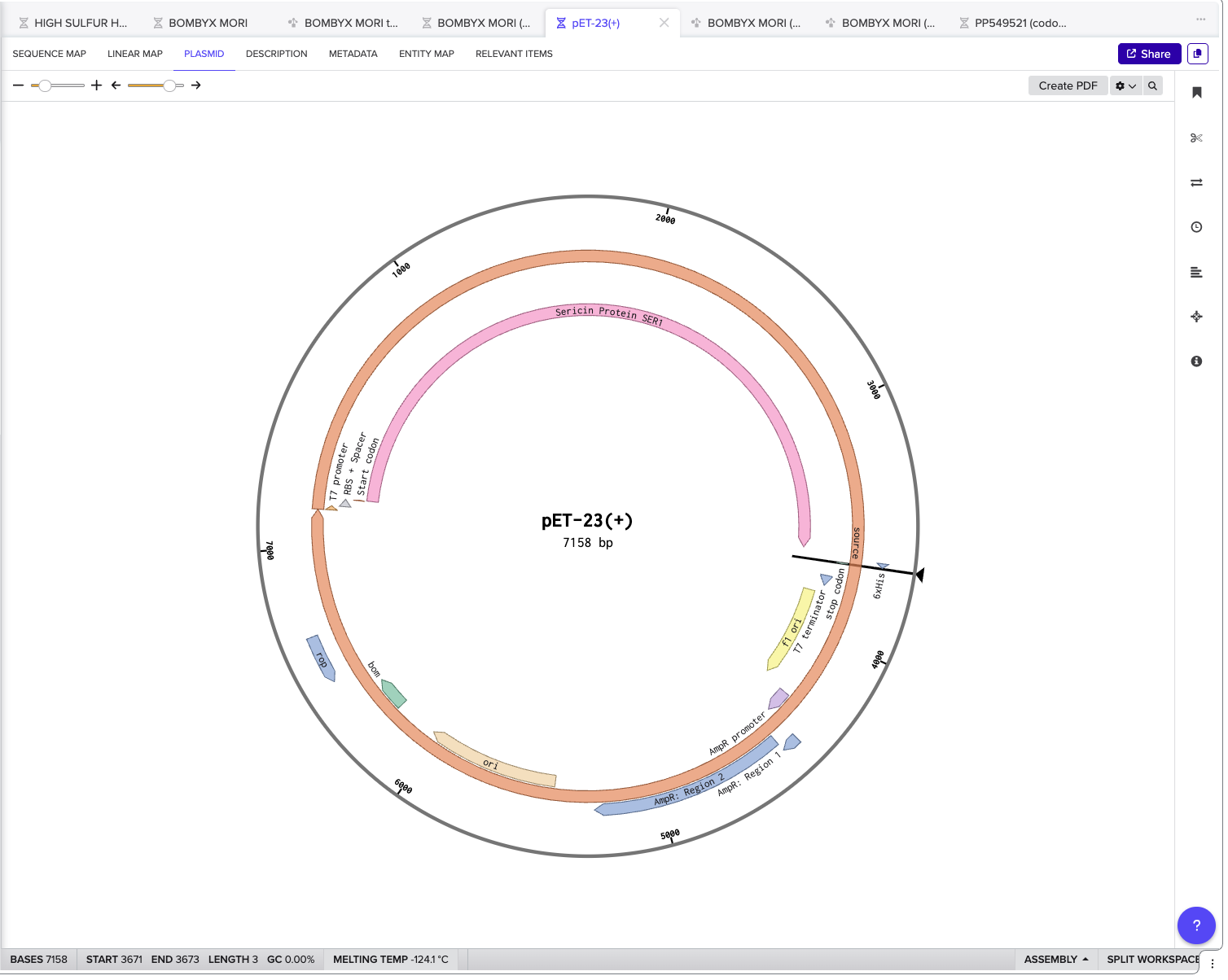
Click into your sequence and select download construct (GenBank) to get the full plasmid sequence: Go back to your Benchling account . Inside of a folder, click the import DNA/RNA sequence button and upload the GenBank file you just downloaded.
Part 5: DNA Read/Write/Edit
5.1 DNA Read (i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank). DNA-based digital data storage technology. Source: Archives in DNA: Workshop Exploring Implications of an Emerging Bio-Digital Technology through Design Fiction - Scientifi c Figure on ResearchGate. Available from: https://www.researchgate.net/fi gure/DNA-based-digital-data-storage-technology_fi g1_353128454 [accessed 11 Feb 2025]
In a previous workforce development program called Break into Biotech I was a part of, we chose to participate in a fi nal project to implement the skills we were learning. I chose the Microbiome group in which we would select a location, extract soil samples, extract the DNA, prep them and sequence them via the Illumna Mini Seq. Unfortunately, our experiment failed in that no data was extracted, therefore, no DNA was sequenced. The goal was to do a microbial profi le of green wood cemetery and understand what kinds of prokaryotic life was residing in the soil. I would love to push this research further and sequence soil surrounding natural burials to understand the vital decomposers of our bodies. New organims are constantly being discovered thanks to meta-data collection and analysis allowing us to fi nd novel traits in the most unlikely of places. I am innately curious about burial practice, death and subsequently, grief. What could these little critters tell us about the ways our bodies decompose? About our life and the decisions embodied through the fl esh inspiring the breakdown by these organisms? Perhaps even the symbiotic nature of organisms working together to break down a resource now in the soil that could release harmful chemicals? How do these organisms digest these compunds and what is excreted as a result? All of this can begin to be uncovered from sequencing.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why? Also answer the following questions:
- Is your method first-, second- or third-generation or other? How so?
- What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
- What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
- What is the output of your chosen sequencing technology?
5.2 DNA Write (i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to infl ammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specifi c genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
The biosynthetic gene cluster I would like to synthesize in S.cerevisiae comes from Chlorociboria Aeruginascens. The BGC is a secondary metabolite pathway that produces a pigment called Xylindein which is both beautiful and functional for its semi-conductive properties. The goal would be to produce this pigment and dye textiles for weavers! The BGC contains a total of 7 genes!
(ii) What technology or technologies would you use to perform this DNA synthesis and why? I would choose phosphoramidite DNA synthesis as it is the most commonly used method for DNA synthesis. Howeverthere are complications considering my BGC is a polyketide pathway. Polyketides are some of the largest fragments, so this for of DNA synthesis is extrememly error prone and expensive due to the size of the fragments.
Also answer the following questions: 1.What are the essential steps of your chosen sequencing methods? The following steps can be found on the HTGAA DNA synthesis slides. a. Deprotection: i. Acid catalyzed removal of DMT allows for subsequent base addition b.Base Coupling: i.A DMT protected phosphoramidite is added to the unprotected 5’ OH using a tetrazole activator c.Capping i.unreacted 5’ OH are acetylated to prevent further chain extension. This step helpsprevent single-base deletions at the expense of yield d.Oxidation: Oxidation of phophite triester to phosphate using aqueous iodine.
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
The length of the chain is limited due to the error rate increasing as the chain grows. 100% coupling efficiency is near impossible. An abasic site is possible due to depurination. https://www.twistbioscience.com/blog/science/simple-guide-phosphoramidite-chemistry-and-how-it-fi ts-twist-biosciences-commercial https://www.compound.vc/writing/dna-synthesis-a-technical-primer
5.3 DNA Edit (i) What DNA would you want to edit and why?
In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for fl ora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fi xation).
What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why? Colossal Biosciences Inc., a biotechnology company using genetic engineering to de-extinct various historic animals such as the woolly mammoth, dodo, and dire wolf.
I would want to edit fungal DNA of fi lamentous fungi to resist contamination. There are researchers doing this work via symbiotic relationships in which the collective growth increases resiliency within a species so that even when contamination occurs, the fungi is able to resist full takeover by eating the mold itself. However, being able to grow a mycelium based product without the fear of contamination especially for regenerative engineered livign systems would be ideal. I believe this can be done by leveraging the secondary metabolic pathways found in fungi that are anti-microbial.
(ii) What technology or technologies would you use to perform these DNA edits and why? I would employ CRISPR cas 9 technology when editing multicellular fungal polyketide pathways due to the hyper specifi city of CRISPR which could help over-express the pathway with minimal errors. In the paper listed below, the advent of CRISPR cas 9 would allow researchers more control over the expression of a secondary metabolite which is difficult to express in a host organism.
Also answer the following questions:
1. How does your technology of choice edit DNA? What are the essential steps?
a.Create gRNA(guide RNA) that binds to the specifi c target sequence. the gRNA also binds with the Cas9 enzyme. As the gRNA recognizes the targeted location the cas9 enzyme will cut the DNA for insertion. The rest is up to the hosts cell which will deploy its machinery to repair the DNA break, repairing the break with the inserted gene as though it was native. Similarly how it is done in bacteria.
2.What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
a.Choose your cas9 enzyme! Depending on your experimental needs a cas9 enzyme needs to be selected to fit the specifcs of experiment applications
b.gRNA selection: This is one of the protagonists as the insertion depends on the gRNA recognizing the target location and being repaired into the host DNA.
c. Target site selection: What are you trying to edit and why? Knowing your target site allows you to create the experiment such as which cas9 variant, desiging of the gRNA and if there are any PAM site close.
d. PAM and gRNA compatibility: Not all matches are made in heaven! When designing the gRNA, it needs to have high on target specificity and needs to be close to a PAM sequence. This compatbility includes the cas9 variant chosen to ensure correct binding to the PAM site!
3. What are the limitations of your editing methods (if any) in terms of efficiency or precision?
a. Requirement for a PAM near the site which is its own limiting factor, meaning not just ANY site can be cut within the genome which can be inconvenient for gene therapy and polyketide synthase edit purposes.
b. If there is another sequence that is similar to your target sequence, the gRNA could bind to an unwanted site within the genome causing errors and incorrect placement, no integration or no expression of the gene of interest.
c. Patenting issues arise when an organisms DNA is edited and transitions from a discovered organism to a novel organism. What are the IP rights and how is that regulated and distinguished? How can an organism be patented? CRISPR-Cas for Fungal Genome Editing: A New Tool for the Management of Plant Diseases
References: What are genome editing and CRISPR-Cas9?: MedlinePlus Genetics What Are the Limitations of CRISPR-Cas9? Optimizing CRISPR: Technology and Approaches for High Efficiency Gene Editing | VectorBuilder