Week 4 HW: Protein Design I
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
In a 500g piece of meat, there is approximately 7.8x1023 amino acids.
In researching for this question, I was unable to find further resources other than previous HTGAA pages.
Alternative question asked with minimal return:
Calculation of amino acids in meat by grams?
- Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Though this would be a quirky science fiction plot, the reason we do not become the cow when we eat the meat is because as the meat enters our bodies, it goes through a whole slew of processes that breakdown the proteins into amino acids etc. They get broken down for our bodies to use as nutrients. The proteins are not functioning in our bodies as they are broken down into building blocks.
- Why are there only 20 natural amino acids?
This question is complicated in that there aren’t any concrete answers. It is one of the great mysteries of life. There is also no truly understood theory on how these amino acids came to be on earth. However researchers are un-convinced at the “randomness” theory of the amino acid array. There are three main functions ordain the AA assemblage, hydrophobicity, charge and size. The traits gave rise to preferred amino acids that would work best to create and organize life. https://www.chemistryworld.com/features/why-are-there-20-amino-acids/3009378.article
- Where did amino acids come from before enzymes that make them, and before life started?
In 1953, simulated a primordial soup in which various gases and elements were concocted together in a flask and emerged from this experiment were 11 amino acids. The experiment proved that amino acids could have originated from a similar environment on earth and beyond that through time and chaos further complexity was developed. So the pathway is hypothesized to be AA by chemical synthesis, metabolic pathways from amino acid biosynthesis, and then enzyme fusion through environmental selection of evolving organisms in primordial soup. https://www.nature.com/scitable/topicpage/an-evolutionary-perspective-on-amino-acids-14568445/
- If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
You would expect a left-handed a-helix. D-amino acids directly reflect L-amino acids, causing the “flip” of the “image”.
- Can you discover additional helices in proteins?
Yes! It is possible to discover additional helices such as π-helices that make up 15% of proteins (derived from a-helices).
- Why are most molecular helices right-handed?
Molecular helices are right handed because the L-amino acids and D-Sugars fit together in one direction via chemical chirality. I can not find a “why” and more so just the facts of their condition. WHY are helices right handed? Becuase of their structural and chemical bias. However, that is not a satisfying answer. It really only explains HOW helices are right handed. https://www.utmb.edu/mdnews/podcast/episode/biomolecules-are-left-or-right-handed
- Why do β-sheets tend to aggregate?
Beta pleated sheets can easily interact with each other due to their side chains facing one way or another. https://www.reddit.com/r/Mcat/comments/gql31n/why_is_it_that_the_more_beta_sheets_more/ What is the driving force for β-sheet aggregation? Assessment from AI Overview: The backbone of the Beta sheet contain polar C = O and N - H groups. From this they form regular hydrogen bonds with available Beta strands.
- Why do many amyloid diseases form β-sheets?
Beta sheets are low energy and thermodynamically stable and contain hydrophobic segments. They can get mis-folded more easily and disrupt cellular function.
- Can you use amyloid β-sheets as materials?
There is some research investigating amyloid beta sheets for nano materials due to their biocompatibility! https://pmc.ncbi.nlm.nih.gov/articles/PMC8508955/
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions: Briefly describe the protein you selected and why you selected it.
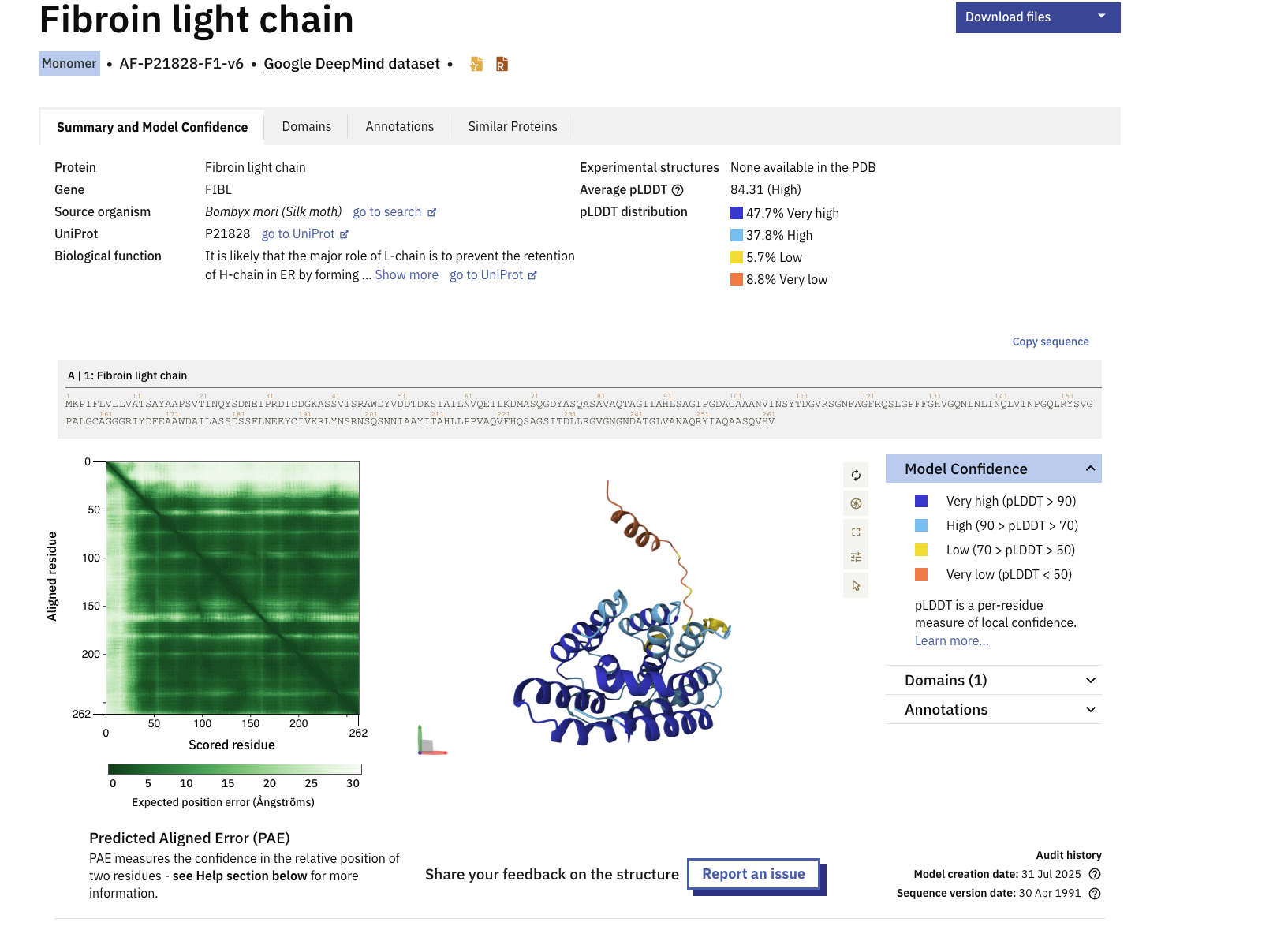
I selected the Fibroin protein within Bombyx Silk moths. The silk we use in silk based fibers and textiles predominantly come from the Bombyx silk moths. My intiial ideation ofor the final project is to create conservation treatments for textile pieces with biology. Understanding the key protein structures that make silk is pertinent to unraveling the problem and discovering the soltuion. https://www.uniprot.org/uniprotkb/P21828/entry
Identify the amino acid sequence of your protein.
MKPIFLVLLVATSAYAAPSVTINQYSDNEIPRDIDDGKASSVISRAWDYVDDTDKSIAILNVQEILKDMASQGDYASQASAVAQTAGIIAHLSAGIPGDACAAANVINSYTDGVRSGNFAGFRQSLGPFFGHVGQNLNLINQLVINPGQLRYSVGPALGCAGGGRIYDFEAAWDAILASSDSSFLNEEYCIVKRLYNSRNSQSNNIAAYITAHLLPPVAQVFHQSAGSITDLLRGVGNGNDATGLVANAQRYIAQAASQVHV
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
The length of the protein is 262 amino acids. with the most common amino acid being Alanine (A), which appears 37 times.
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs. By using the homology option in UNIPROT for Protein p21828, there are 14 protein sequence homologs.
Does your protein belong to any protein family?
The protein belongs to the Fibroin Light Chain Protein family according to InterPro.
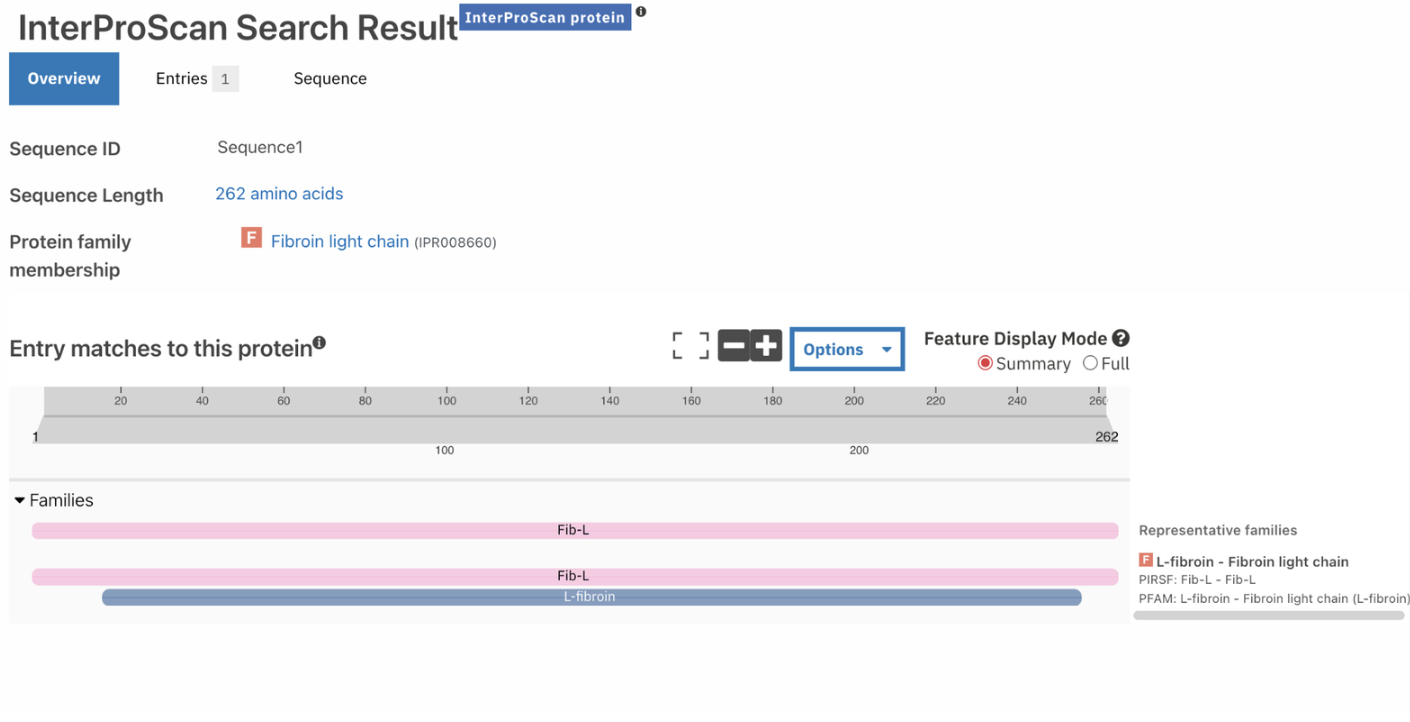
Identify the structure page of your protein in RCSB
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
According to RCSB the protein model was released in AlphaFold DB: 2021-12-09 and last Modified in AlphaFold DB: 2022-09-30
Open the structure of your protein in any 3D molecule visualization software:
CARTOON
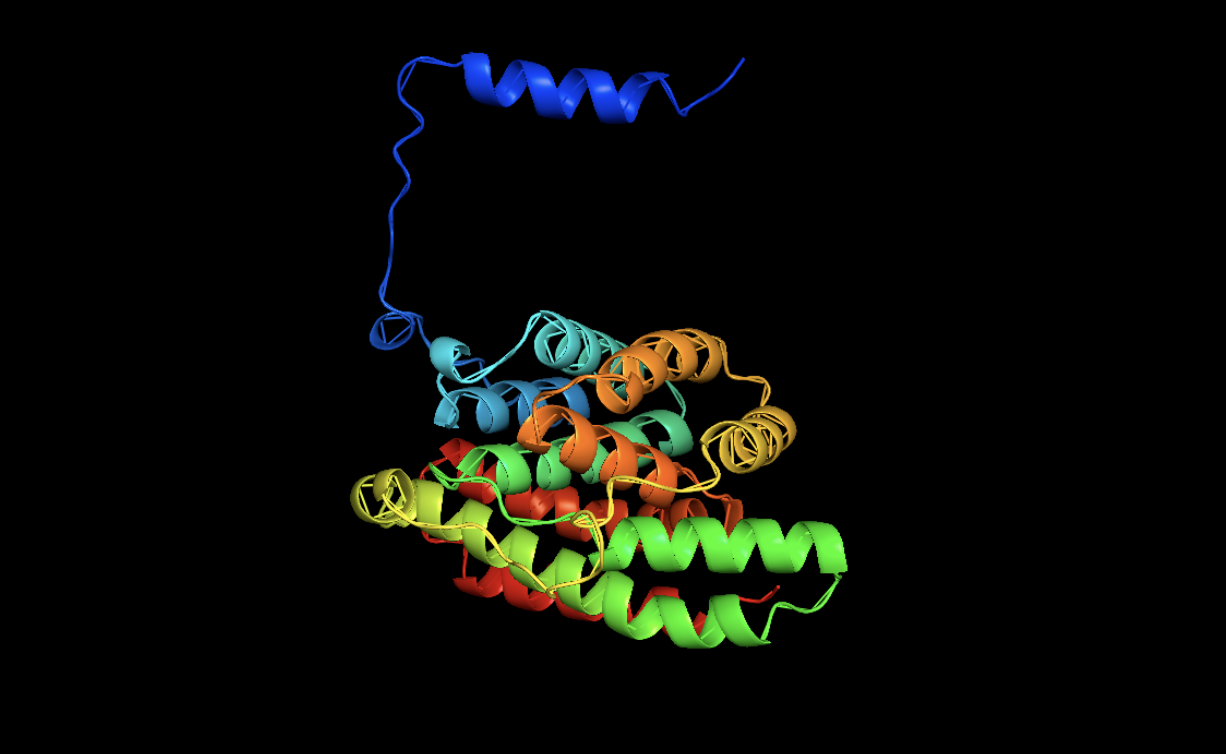
STICK
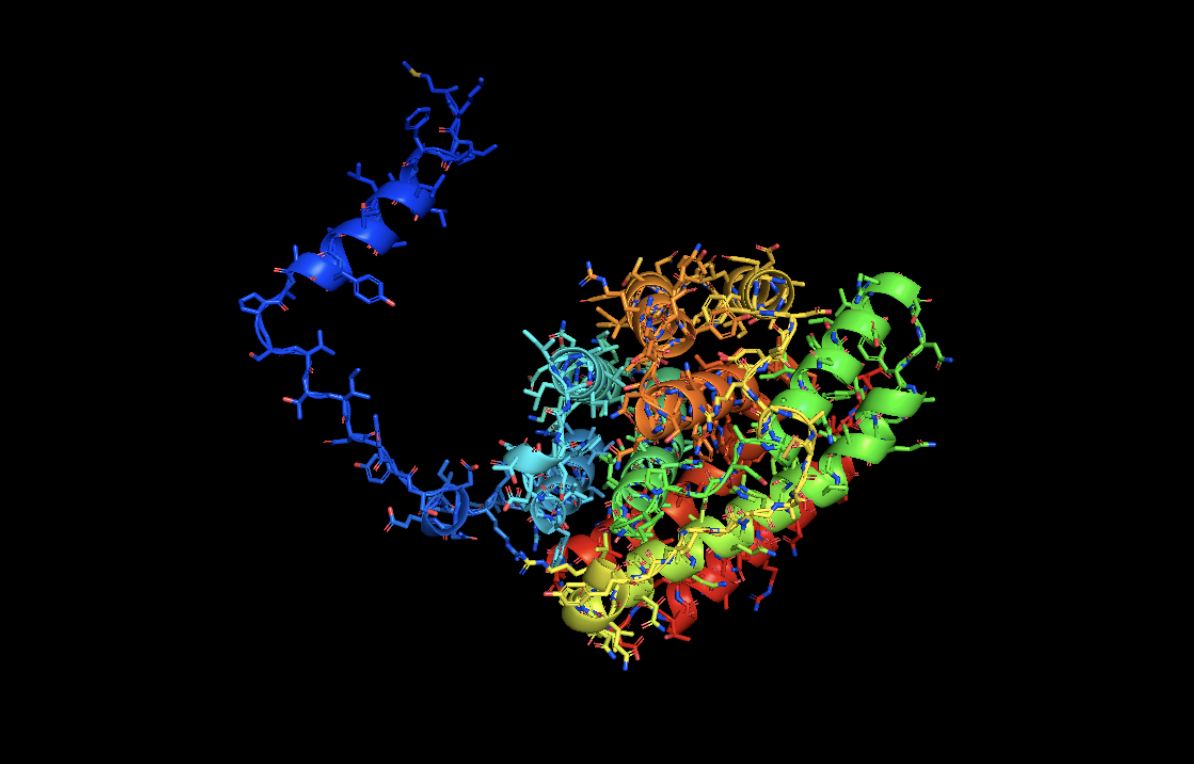
RIBBON
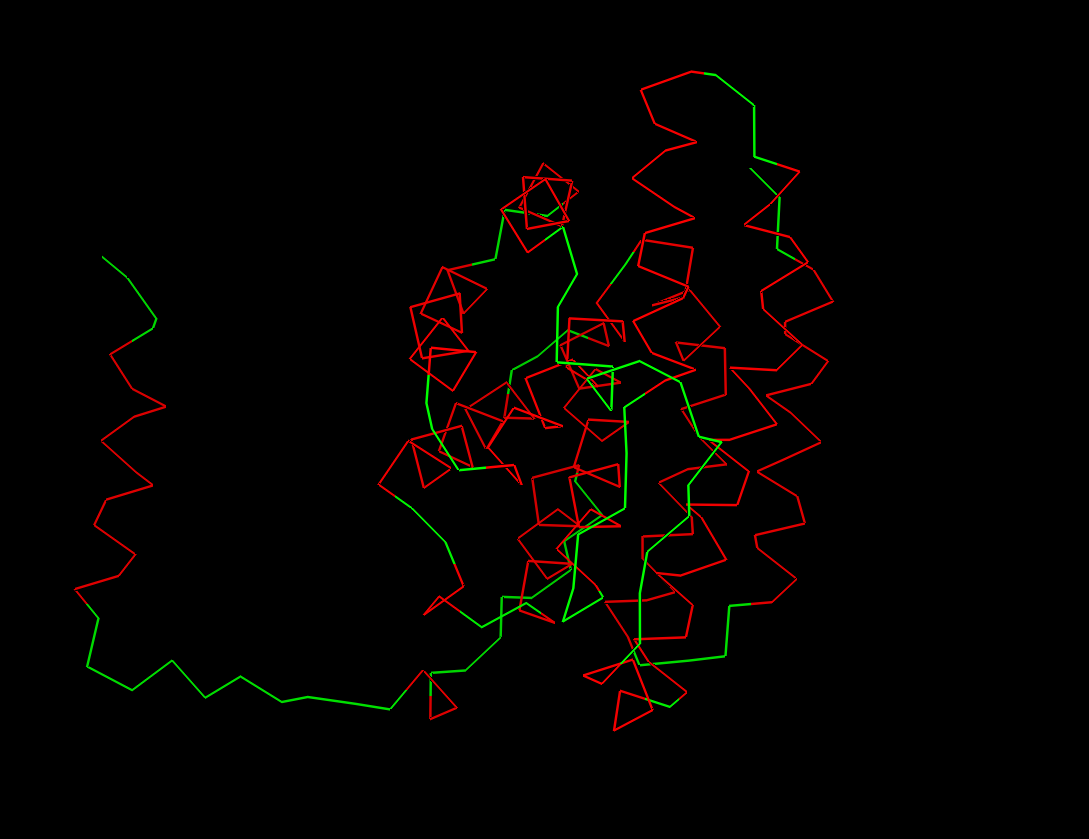
Color the protein by secondary structure. Does it have more helices or sheets?
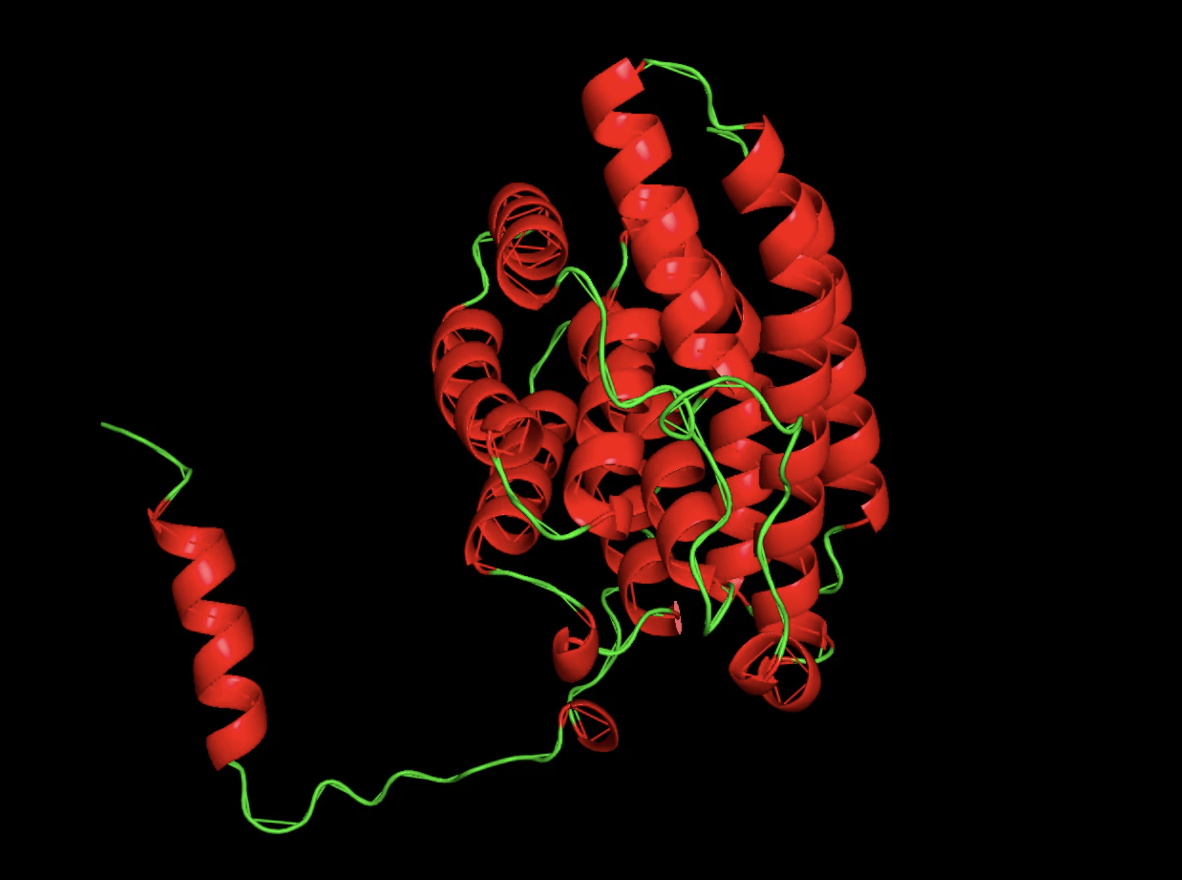
It has more helices than beta sheets!
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
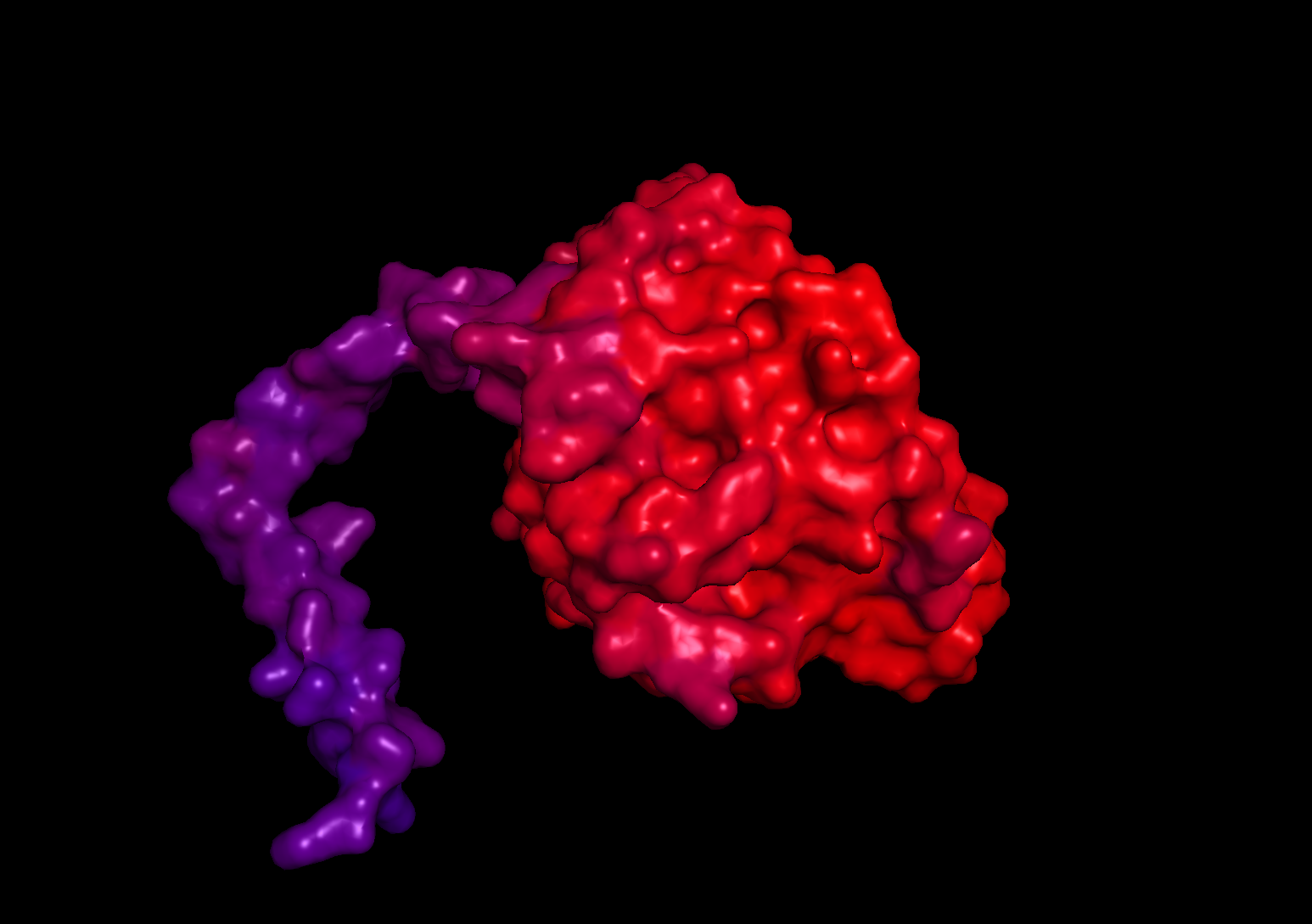
When visualizing the surface of the protein, there are no observable holes or binding pockets in the protein.
In this section, we will learn about the capabilities of modern protein AI models and test some of them in your chosen protein.
Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
Choose your favorite protein from the PDB.
We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
Deep Mutational Scans
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
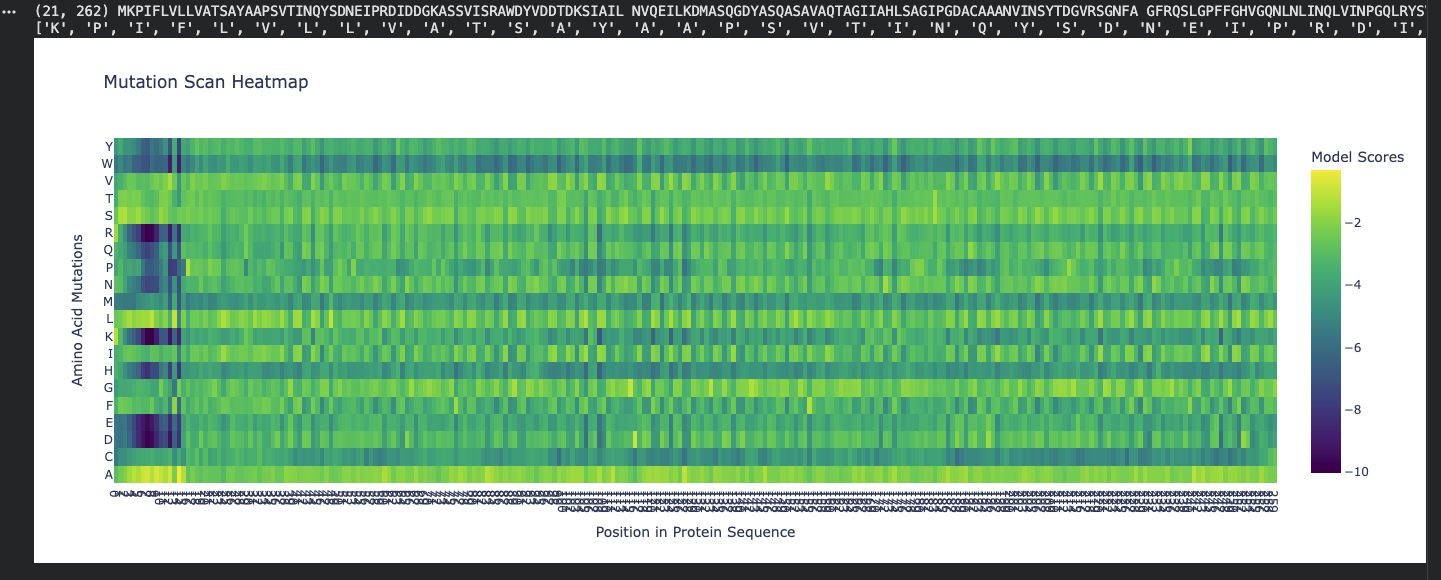
Honestly, when looking at the mutational scan, my initial thought was how visually striking the scan is. Imagine this as a weaving! My graph being predominantly yellow to green indicates the amino acid scores are of a higher probability. Since the protein is fibroin, a well understood protein, I am not surprised by the high probability rating. Residue: Amino Acid in the protein sequence Position X:14 Y:A is the brightest yellow residue in the scan meaning the amino acid in the sequence has a high probabilty score than the majority. There is a general region at the beginning that is predominantly dark blue and is not reflected anywhere else in the scan. I do not understand the scan if represented spatially, however, I wonder if this is the predictive AA seqeuence that Alphafold deemed insecure notated as orange and yellow.
Latent Space Analysis
Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins?
Place your protein in the resulting map and explain its position and similarity to its neighbors.
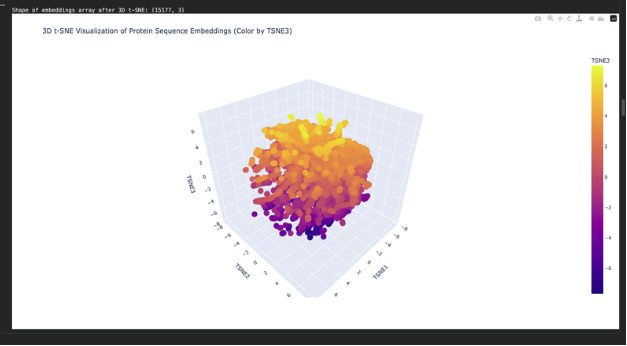
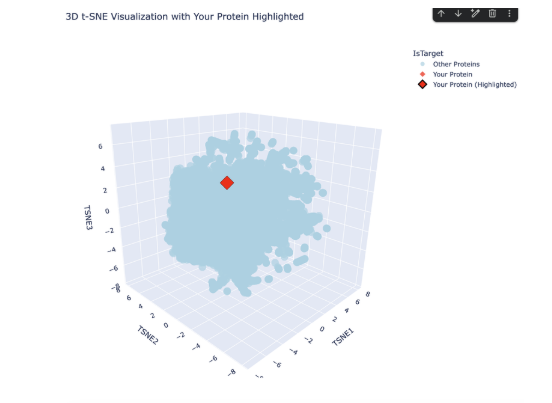
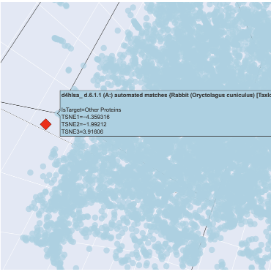
The plot map is dense and has minimal unconcentrated areas. This implies that my protein does not have distinct seprarted families. Each dot in the map is a protein sequence embedded into the model to spatially recognize proteins in relation to each other. By my protein were proteins from rabbits, fungi and humans that contain fibroin. The family tree made a lot of sense!
Folding a protein
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
The predicted coordinates do not match the original structure. The ESMfold prediction is missing the arm chain extension predicted in the alpha fold simulation. The one similarity I can connect is the protein is exclusively a-helices.
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Original:
MKPIFLVLLVATSAYAAPSVTINQYSDNEIPRDIDDGKASSVISRAWDYVDDTDKSIAILNVQEILKDMASQGDYASQASAVAQTAGIIAHLSAGIPGDACAAANVINSYTDGVRSGNFAGFRQSLGPFFGHVGQNLNLINQLVINPGQLRYSVGPALGCAGGGRIYDFEAAWDAILASSDSSFLNEEYCIVKRLYNSRNSQSNNIAAYITAHLLPPVAQVFHQSAGSITDLLRGVGNGNDATGLVANAQRYIAQAASQVHV
ProteinMPNN:
ALTPEEAALLRAAWAPVAADREANGRAFMLRLFAEYPELREYFPEFKGKSLEEIAASPKLAAFSTAVFDGLERLVATADDAAAMATLLADLAKAHVAKGIGAEHVEKIRAIHPAFVASVAPPPPGADAAWDRLFGLVIAALKAAGA
Input this sequence into ESMFold and compare the predicted structure to your original.
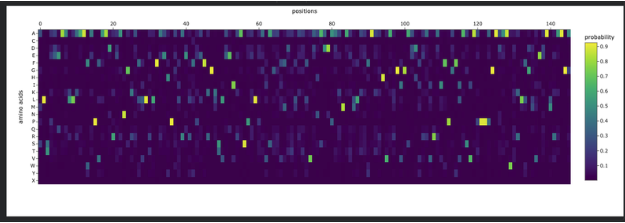
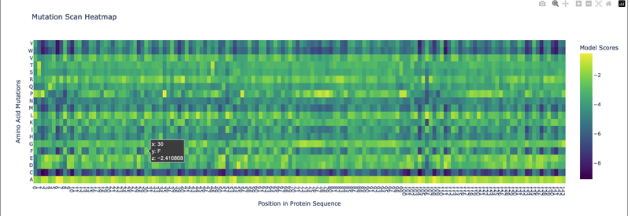
Pretty low confidence in the scan!