First, describe a biological engineering application or tool you want to develop and why After the first week of the How To Grow Almost Anything course and the projects that were presented by the panel, I decided to focus on an area that has always fascinated me: skincare and cosmetics. I have personally struggled to find products that actually worked for my skin. Some creams caused dryness, others triggered breakouts, and many were ineffective despite high prices and flashy marketing. Over time, I realized that this frustration is common—most skincare follows a “one-size-fits-all” approach, categorizing skin as oily, dry, combination, or sensitive. While these categories are a helpful starting point, they fail to capture the biological complexity and uniqueness of each person’s skin.
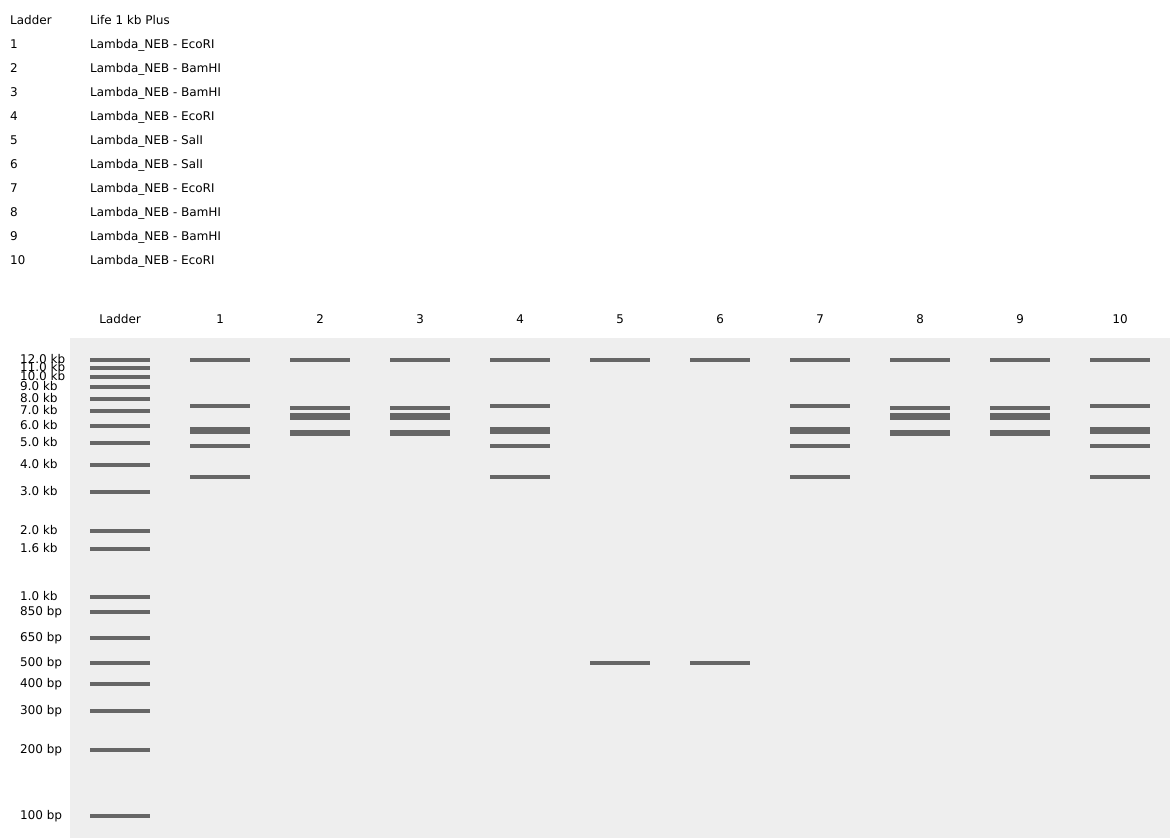
PART 1: Gel Art For the Gel Art part, I searched Roman’s Gallery and chose the little smiley face :).
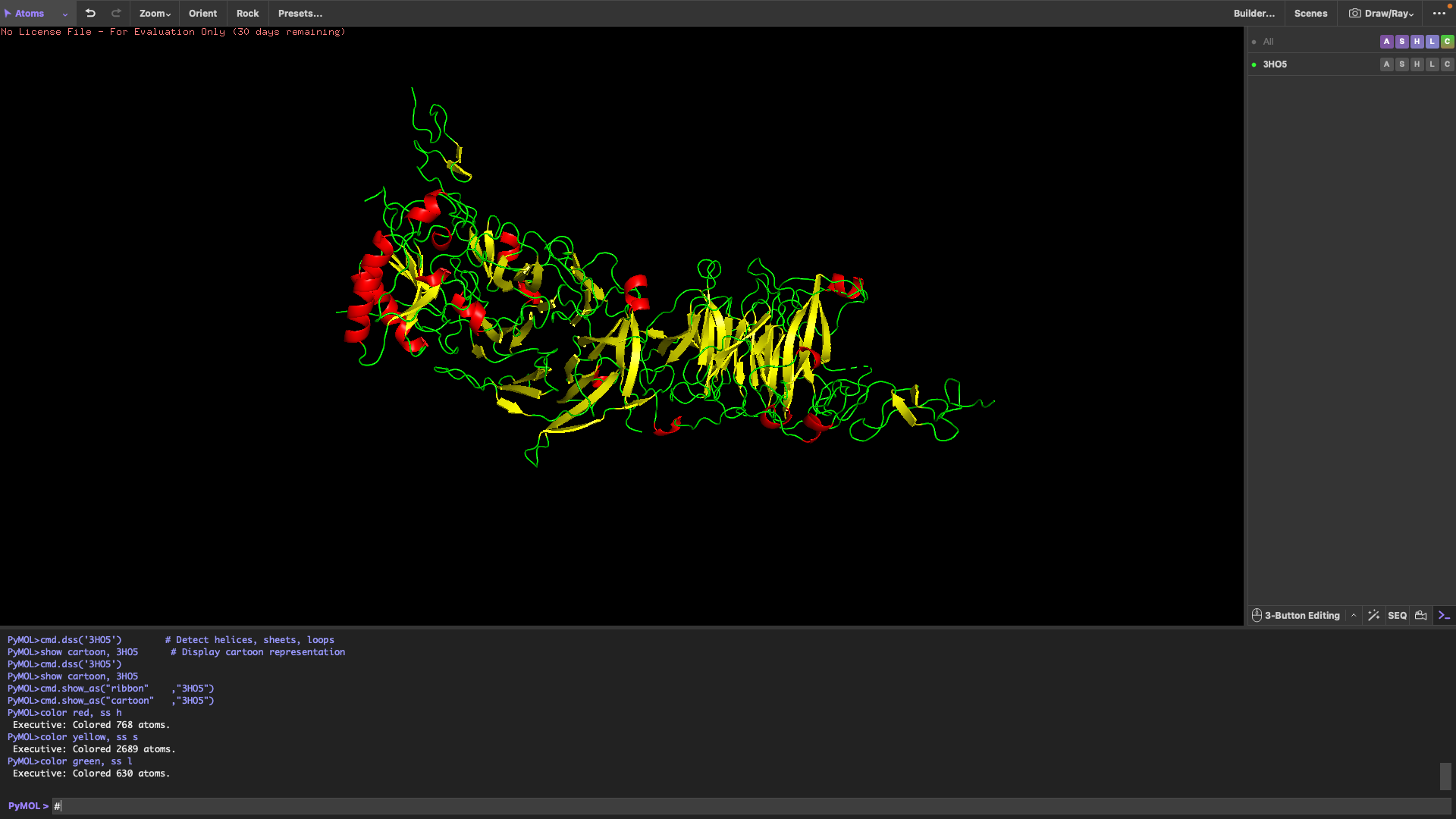
PART 3: DNA Design Challenge 3.1 Choose Your Protein The protein I have chosen for this DNA design challenge is Sonic Hedgehog (Shh), a critical signaling molecule involved in embryonic development, tissue patterning, and organ formation. Shh plays a central role in directing cell fate decisions during development, and mutations in this protein are linked to severe developmental disorders. Its importance in biology, combined with the fact that it is well-characterized and has a known amino acid sequence, makes it an ideal candidate for this exercise.
Python Script for Opentrons Artwork I used the opentrons-art.rcdonovan.com site to make a shrimp design: The script made with collab: from opentrons import types
Homework: Protein Design I Part A. Conceptual Questions 1. How many molecules of amino acids do you take with a piece of 500 grams of meat?
(on average an amino acid is ~100 Daltons)
In a 500-gram piece of meat, you are consuming approximately: 7.5275 × 10^23 amino acids
Week 5 - Protein Design Part II Part A: SOD1 Binder Peptide Design (From Pranam) Part 1: Generate Binders with PepMLM SOD1 sequence with A4V mutation:
ATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
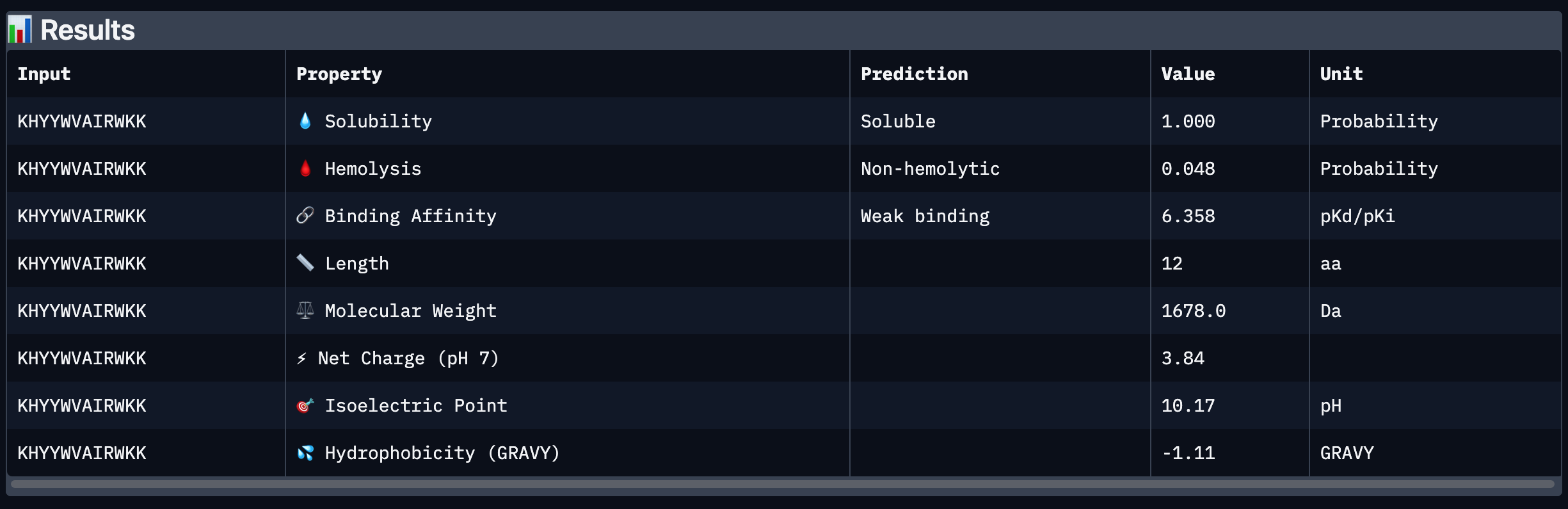
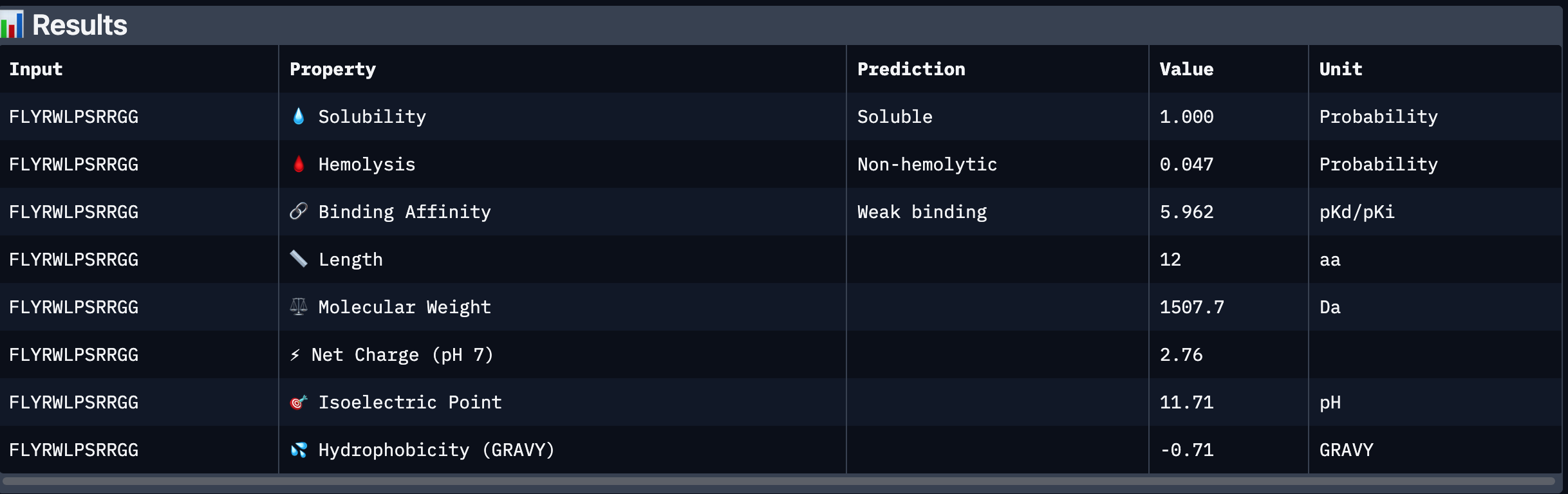
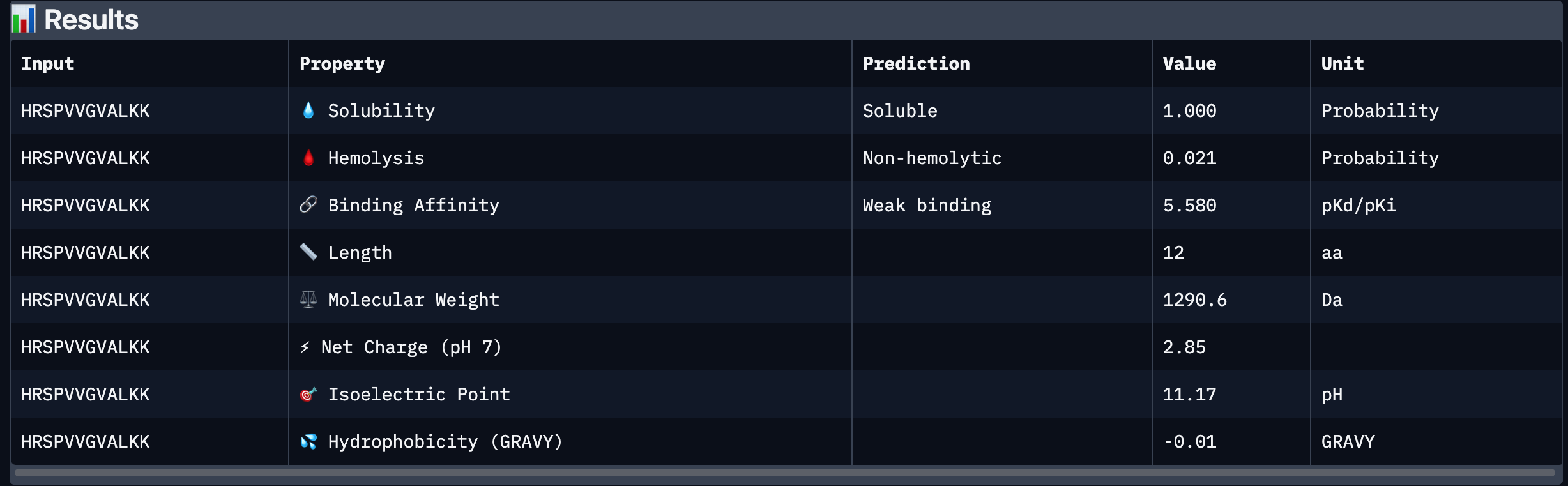
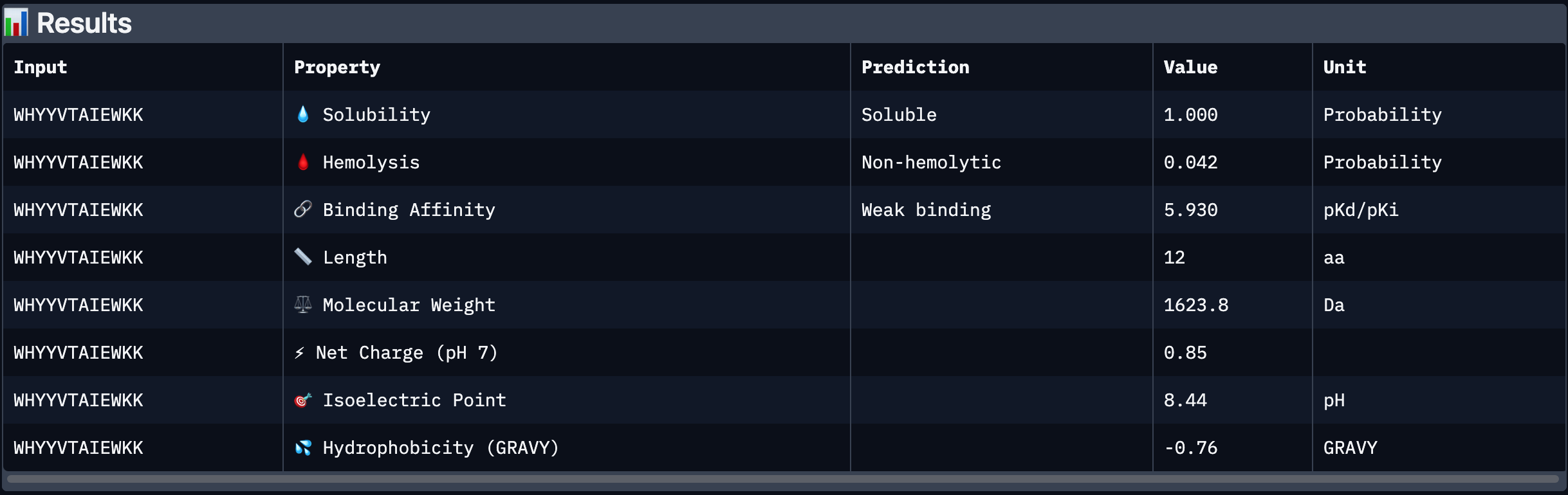
Peptide # Peptide Sequence Perplexity Score 1 WHYYVTAIEWKK 25.725023 2 KHYYWVAIRWKK 27.40476 3 HRSPVVGVALKK 12.718177 4 WRYPVAAIELKE 17.102567 5 FLYRWLPSRRGG N/A Part 2: Evaluate Binders with AlphaFold3 Peptide # Peptide Sequence ipTM 1 WHYYVTAIEWKK 0.31 2 KHYYWVAIRWKK 0.4 3 HRSPVVGVALKK 0.41 4 WRYPVAAIELKE 0.3 5 FLYRWLPSRRGG 0.28 Peptide # Peptide Sequence ipTM Binding Description 1 WHYYVTAIEWKK 0.31 Binds near the surface of the β-barrel;; mostly surface-exposed. 2 KHYYWVAIRWKK 0.40 Appears bound on the β-barrel and dimer interface, surface-bound 3 HRSPVVGVALKK 0.41 Localized near the β-barrel surface, surface-bound. 4 WRYPVAAIELKE 0.30 Surface-bound,minimal contact with N-terminal residues, approaches dimer interface. 5 FLYRWLPSRRGG 0.28 Surface-bound, does not penetrate deeply into β-barrel. The ipTM scores for the PepMLM-generated peptides range from 0.30 to 0.41, slightly higher than the known binder (0.28). Peptides 2 and 3 show the highest ipTM scores, indicating stronger predicted structural confidence in the SOD1–peptide complex.
Week 6 — Genetic Circuits Part I: Assembly Technologies Assignment: DNA Assembly 1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? Some components that are found in the Phusion High-Fidelity PCR Master Mix are:
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) 1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? -Traditional genetic circuits typically operate on Boolean logic, which processes inputs as binary states (0 or 1). IANNs offer several advantages:
-Analog Integration - unlike Boolean circuits, IANNs can process graded signals, this allows cells to respond proportionally to varying concentrations of chemicals or light.
-Scalability - to achieve complex decision-making with Boolean logic, you need a massive number of gates, which places a heavy metabolic burden on the cell, IANNs can achieve classification or pattern recognition using fewer components by adjusting binding affinities or promoter strengths.
-Non-Linear Processing - IANNs utilize activation functions, this allows them to filter noise and handle non-linear relationships between inputs.
Homework Part A: General and Lecturer-Specific Questions General homework questions 1.Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Waters Part I — Molecular Weight To calculate the theoretical molecular weight (MW) of the eGFP sequence provided, we must account for the primary amino acid sequence and the critical internal post-translational modification that creates the fluorophore.
The provided sequence contains 246 amino acids:
Subsections of Homework
Week 1 Homework: Microbiome-Tuned Skincare week
1. First, describe a biological engineering application or tool you want to develop and why
After the first week of the How To Grow Almost Anything course and the projects that were presented by the panel, I decided to focus on an area that has always fascinated me: skincare and cosmetics. I have personally struggled to find products that actually worked for my skin. Some creams caused dryness, others triggered breakouts, and many were ineffective despite high prices and flashy marketing. Over time, I realized that this frustration is common—most skincare follows a “one-size-fits-all” approach, categorizing skin as oily, dry, combination, or sensitive. While these categories are a helpful starting point, they fail to capture the biological complexity and uniqueness of each person’s skin.
This personal challenge led me to explore a potential solution: microbiome-tuned skincare. This approach involves designing cosmetic formulations that are personalized based on an individual’s skin microbial ecosystem. Every person’s skin hosts a complex community of microbes, including bacteria, fungi, and viruses, which together influence hydration, barrier function, inflammation, and susceptibility to conditions like acne, eczema, or premature aging. Normal skincare does not account for this complexity and treats all skin as if it reacts the same way. Microbiome-tuned skincare, by contrast, supports beneficial microbes while balancing or reducing harmful ones.
For example, some individuals naturally have low levels of certain beneficial bacteria, such as specific strains of Cutibacterium acnes, which help maintain the skin barrier and prevent infection. Prebiotic creams may promote the growth of beneficial microbes. On the other hand, individuals with higher levels of pro-inflammatory microbes could use formulas containing anti-inflammatory compounds that rebalance the microbial ecosystem without harming beneficial organisms. By analyzing a person’s skin microbiome, products can be customized for their unique needs rather than relying on generic formulations.
From a bioengineering perspective, this project combines synthetic biology, computational biology, and personalized healthcare. Skin samples can be collected to identify microbial composition, and machine learning algorithms can predict how different formulations will interact with these microbial communities. The result is a personalized skincare routine that improves hydration, reduces inflammation, supports anti-aging processes like collagen maintenance, and maintains overall skin health. This approach transforms skincare from a trial-and-error routine into a scientifically guided, individualized experience.
Beyond cosmetics, microbiome-tuned approaches have broader applications. Dermatologists could use similar strategies to treat skin conditions such as eczema, rosacea, or chronic acne with higher precision. Another example is wound care: microbiome-tuned wound treatments use microbial profiling and predictive analytics to personalize care, supporting beneficial microbes while inhibiting harmful ones. Monitoring the wound’s microbial environment over time can prevent infection, accelerate healing, and reduce reliance on antibiotics. Personally, the appeal of this project is both scientific and social: it addresses a real-life problem that I have experienced and has the potential to help others avoid ineffective treatments.
The skin is the largest organ of the human body and serves as a defensive barrier against pathogens while supporting sensory functions. Its microbiome is essential for health because it:
Competes with harmful microbes, preventing infections
Communicates with the immune system, regulating inflammation
Supports barrier integrity, reducing dryness and irritation
Disruption of this microbial balance, known as dysbiosis, increases the likelihood of skin issues such as acne or eczema. Dysbiosis can result from diet, environmental factors, skincare products, or antibiotics. Regular skincare often addresses only symptoms rather than the underlying microbial imbalance. Microbiome-tuned skincare aims to restore balance and promote long-term skin health.
This strategy can be implemented using three main approaches:
Microbiome Sequencing: Collect skin samples and analyze microbial DNA to assess bacterial, fungal, and viral composition.
Machine Learning Analysis: Use computational models to predict how specific ingredients will affect microbial balance and skin health.
Personalized Formulation: Develop creams, serums, or cleansers specifically designed to support beneficial microbes and reduce harmful ones.
By focusing on maintaining the health of the skin ecosystem, microbiome-tuned skincare shifts the approach from reactive to proactive care.
2. Governance/Policy Goals
Because microbiome-tuned skincare directly interacts with the body, ethical considerations are critical. The main governance goals for this project are:
Preserving Consumer Health and Safety Skincare products based on microbiomes act as biological components. Poorly designed formulas may disrupt microbial balance, leading to irritation and other skin conditions. The skin microbiome is influenced by age, genetics, ethnicity or and environmental factors, so products must be tested widely and safely. Sub-goals include:
Standardizing validation procedures for personalized formulas
Monitoring long-term effects on skin health and microbial balance
Testing across diverse populations to ensure safety and effectiveness
** Privacy and Data Security** Microbiome data is personal and could reveal information about health, lifestyle etc. Protecting this information is essential. Sub-goals include:
Anonymizing microbial data
Implementing strict consent procedures for users
Preventing misuse by third parties or commercial entities
3. Governance Actions
Action 1: Regulatory Framework for Microbiome-Based Products
Purpose: Existing cosmetic regulations are not designed for personalized microbial interventions. A framework would define safety standards, acceptable microbial strains, and validation protocols.
Design: Regulatory agencies (FDA, EMA) would require companies to submit microbial and clinical data for approval. Long-term monitoring ensures continued product efficacy.
Assumptions: Regulators accept microbiome-based evidence; companies can standardize testing across populations.
Risks of Failure & Success: Overly strict regulations could slow innovation or increase costs, while overly permissive rules may allow unsafe products to reach consumers.
Action 2: Privacy and Data Protection
Purpose: Protect sensitive microbiome data and prevent misuse.
Design: Use encryption, anonymization, secure storage, and strict consent. Independent audits and international standards would guide compliance.
Risks of Failure & Success: Data breaches or misuse could harm consumers through discrimination. Strong protections build trust and encourage adoption of microbiome-based products.
Action 3: Incentives for Broad Access and Ethical Design
Purpose: Ensure microbiome-tuned skincare benefits a wide population, not only wealthy consumers.
Design: Provide grants, subsidies, and open-source tools for small companies or academic labs.
Assumptions: Financial and technical support will broaden equitable access.
Risks of Failure & Success: Limited funding or intellectual property disputes could restrict access. Successful implementation ensures broad social impact.
4. Governance Scoring
Policy Criteria
Option 1
Option 2
Option 3
Enhance Biosecurity
Preventing incidents
1
2
3
Helping respond to incidents
1
2
3
Foster Lab Safety
Preventing incidents
1
2
3
Helping respond to incidents
2
1
3
Protect the Environment
Preventing incidents
2
3
1
Helping respond to incidents
2
3
1
Other Considerations
Minimizing costs & burden to stakeholders
3
2
1
Feasibility
2
1
2
Does not impede research
1
2
1
Promotes constructive applications
1
2
1
5. Prioritization and Recommendations
Based on the scoring, I would choose a combination of Action 1 (Regulatory Framework) and Action 2 (Privacy and Data Protection), while keeping Action 3 (Equitable Access) as a secondary focus. Regulatory oversight ensures products are safe and effective, so it is preventing disruptions to the skin microbiome. Equity initiatives are important but should follow safety and privacy considerations to ensure broad access.
Trade-offs include higher costs and slower innovation, but these are necessary to avoid harm and ensure ethical deployment. Target audiences include federal regulators (FDA, EMA), companies for privacy compliance, research institutions for open-access tools, and international bodies for cross-border standards. This combination balances safety, privacy, innovation, and societal impact, making microbiome-tuned skincare both effective and responsible.
Professor Jacobson
1. Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome, and how does biology deal with that discrepancy?
Polymerase has an error rate of roughly 1 in 10⁶ bases. The human genome contains roughly 3 billion base pairs, meaning about 3,000 mistakes could occur per replication. Biology corrects these errors with proofreading, apoptosis of damaged cells, and by allowing some variation to contribute to evolution.
2. How many different ways are there to code DNA for an average human protein? Why do all these sequences not work in practice?
Theoretically, about 10¹⁵⁷ DNA sequences could encode a 330-amino-acid protein. In practice, many sequences fail due to mRNA folding, premature translation stops, and codon bias that affects efficiency.
Dr. LeProust
1. Most commonly used method for oligo synthesis
The solid-phase phosphoramidite chemical synthesis is the most used technology today for making custom DNA oligonucleotides.
2. Why is it difficult to make oligos longer than 200 nucleotides?
In addition to side reactions and chemical limitations, it is challenging due to cumulative stepwise errors and yield drops. The maximum efficiency of each addition cycle in the synthesis of phosphoramidite is approximately 99%. The likelihood of creating an accurate product decreases to a maximum of 14% over 200 bases. The increasing length of the chain in conventional porous supports can also obstruct reagent access, which reduces efficiency even more.
3. Why can’t you make a 2000 bp gene via direct oligo synthesis?
Based on the previous answer, accumulating errors make a 2000 base pair gene (4000 nucleotides) almost impossible to synthesize. It is simply not feasible.
George Church
1. [Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
The 10 essential amino acids that animals cannot synthesize and must obtain through their diet are arginine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, and valine. The “Lysine Contingency” in Jurassic Park involved engineering dinosaurs to be unable to produce lysine so that, without lab-supplied supplements, they would die if they escaped. While this is a clever narrative device, it is biologically flawed because lysine is abundant in natural foods, meaning escaped dinosaurs could easily obtain it and survive. This highlights that while lysine is essential for all animals, relying on its absence as a control measure overlooks basic nutritional biology and ecosystem realities. A more effective contingency would need to target something unique to the lab environment rather than a common dietary requirement.
Week 2 Homework: DNA Read, Write & Edit
PART 1: Gel Art
For the Gel Art part, I searched Roman’s Gallery and chose the little smiley face :).
PART 3: DNA Design Challenge
3.1 Choose Your Protein
The protein I have chosen for this DNA design challenge is Sonic Hedgehog (Shh), a critical signaling molecule involved in embryonic development, tissue patterning, and organ formation. Shh plays a central role in directing cell fate decisions during development, and mutations in this protein are linked to severe developmental disorders. Its importance in biology, combined with the fact that it is well-characterized and has a known amino acid sequence, makes it an ideal candidate for this exercise.
Using the amino acid sequence from UniProt, I performed a reverse translation to determine the nucleotide sequence that could encode Shh. Reverse translation is the process of inferring a DNA sequence from a protein sequence, based on the codons that encode each amino acid. Because multiple codons can code for the same amino acid, this step may produce several possible DNA sequences. The resulting DNA sequence represents the gene that could be transcribed and translated to produce the Shh protein, reflecting the principles of the Central Dogma, where DNA is transcribed into RNA and RNA is translated into protein.
Once the DNA sequence is determined, I codon-optimized it for E. coli expression. Codon optimization is important because different organisms prefer certain codons over others for the same amino acid. Using preferred codons in E. coli increases translation efficiency, improves protein yield, and avoids rare codons that can slow ribosomes or create secondary structures. This ensures robust expression of Shh in bacterial systems.
3.4 You Have a Sequence! Now What?
With a codon-optimized Shh sequence, several methods can be used to produce the protein:
Cell-dependent expression: Clone the DNA into a plasmid vector and introduce it into E. coli. The bacterial machinery transcribes the DNA into mRNA and translates it into Shh protein.
Cell-free systems: Produce Shh protein directly in vitro using transcription-translation mixtures from E. coli lysates.
Both approaches demonstrate how a nucleotide sequence can be converted into a functional protein through natural molecular processes.
3.5 How Does it Work in Nature
In nature, the Shh gene is transcribed into mRNA and translated into a precursor protein, which then undergoes post-translational modifications, including cleavage and lipidation. These modifications are required for proper secretion and signaling activity. This illustrates how a single gene can produce a biologically active protein, highlighting the flow of information from DNA → RNA → protein and the importance of transcriptional and post-translational regulation.
PART 4: Prepare a Twist Synthesis Order
PART 5: DNA Read, Write, and Edit – Microbiome Skincare Applications
5.1 DNA Read – Sequencing
DNA to sequence and why: I would sequence skin microbiome DNA from individuals with different skin types or conditions, such as acne-prone, sensitive, or aged skin. This allows identification of beneficial and harmful microbes, their functional genes, and metabolites affecting skin health. Sequencing this DNA can inform development of probiotic treatments, personalized skincare, and microbial-derived bioactives.
Sequencing technology and details: I would use Oxford Nanopore long-read sequencing, a third-generation method that can read full-length microbial genomes and plasmids. Input DNA would be extracted from skin swabs, lightly sheared, and ligated with nanopore adapters. Essential steps include passing DNA through a protein nanopore, measuring ionic current changes, and decoding bases from these signal variations. The output is long-read sequences of microbial genomes and plasmids, helping resolve strain-level diversity. Limitations include lower per-read accuracy compared with short-read sequencing, but high coverage and error-correction mitigate this.
5.2 DNA Write – Synthesis
DNA to synthesize and why: I would synthesize genes encoding skin-beneficial metabolites, such as bacterial enzymes producing natural moisturizers, antioxidants, or antimicrobial peptides. For example, a sequence coding for a bacterial ceramide-synthesizing enzyme could be used to engineer probiotic strains that restore skin barrier function. This approach could create living or topical treatments that enhance skin health naturally.
Technology for synthesis and details: I would use Twist Bioscience’s array-based high-throughput DNA synthesis. Essential steps include sequential nucleotide addition via phosphoramidite chemistry, oligo cleavage and purification, and assembly into full-length gene constructs. Limitations include sequence length constraints (<200 bases per oligo), potential synthesis errors, and optimization requirements when expressing microbial genes in target probiotic strains.
5.3 DNA Edit – Editing
DNA to edit and why: I would edit the genome of commensal skin bacteria like Staphylococcus epidermidis to enhance production of anti-inflammatory molecules or UV-protective compounds. This could reduce acne, eczema flare-ups, or sun-induced skin damage while maintaining microbiome balance. Edits might include upregulating genes for antimicrobial peptides or protective pigments.
Technology for editing and details: I would use CRISPR-Cas9 coupled with plasmid-based delivery, a precise genome editing tool for bacteria. Essential steps include designing guide RNAs targeting regulatory regions, delivering Cas9 and repair templates into bacterial cells, and screening for successful edits. Inputs include plasmids carrying Cas9, guide RNAs, and donor DNA. Limitations include variable editing efficiency across bacterial strains, potential off-target mutations, and challenges in ensuring stable long-term expression of engineered traits.
Week 3 Homework: Lab Automation
Python Script for Opentrons Artwork
I used the opentrons-art.rcdonovan.com site to make a shrimp design:
##############################################################################
### PATTERNING
##############################################################################
drop_volume = 4
for color, coordinates in design_data.items():
# Pick up a new tip for each color
pipette_20ul.pick_up_tip()
source_well = location_of_color(color)
for (x, y) in coordinates:
pipette_20ul.aspirate(drop_volume, source_well)
dispense_location = center_location.move(
types.Point(x=x, y=y, z=0)
)
dispense_and_detach(
pipette_20ul,
drop_volume,
dispense_location
)
# Drop tip before switching colors
pipette_20ul.drop_tip()
Post-Lab Questions
1.Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
For this part I chose the paper: Shotgun Proteomics Sample Processing Automated by an Open‑Source Lab Robot, Han et al., Journal of Visualized Experiments, 2021. The study focuses on automating shotgun proteomics workflows using the open-source Opentrons OT‑2 liquid handling robot. Shotgun proteomics is a widely used technique for identifying and quantifying large numbers of proteins in complex samples such as tissues or cell lysates. Traditional workflows are labor-intensive, including protein extraction, concentration measurement, chemical reduction and alkylation, enzymatic digestion, and peptide clean-up. Manual handling introduces variability and errors, which can affect reproducibility and data quality. Han et al. addressed these issues by developing a semi-automated workflow on the OT‑2, reducing manual labor, improving consistency, and making proteomics automation more accessible for academic labs.
The OT‑2 robot provides a platform for automated liquid handling. Han et al. configured the OT‑2 with single-channel electronic pipettes, a magnetic module, a temperature module, and specialized tube racks and deep-well plates. Their automated workflow included three main steps:
Protein reduction, alkylation, and digestion: proteins were prepared for enzymatic cleavage into peptides. Peptide clean-up using SP3 paramagnetic beads: proteins in common detergents can interfere with downstream analysis. LC-MS/MS analysis: After automation, peptides were dried, reconstituted, and analyzed by LC-MS/MS
Three Python scripts supported the automation: NoSP3_digestion.py, SP3_peptide_cleanup.py, SP3_digestion.py .
Han et al., 2021, demonstrate that low-cost open-source robots like the OT‑2 can automate complex proteomics workflows effectively. Their study highlights the benefits of automation: consistent sample preparation, reduced manual labor, flexibility for different sample types, and affordable access for academic or resource-limited laboratories.
2.Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
Final Project Ideas
Idea I: A Programmable Probiotic Platform for Microbiome-Modulated Skincare
My first idea is a project that explores how engineered living systems can be used to modulate the skin microbiome. Instead of relying on chemical antimicrobials, this project proposes a programmable probiotic designed to selectively respond to acne-associated microenvironments. Acne is often linked to dysbiosis and inflammatory strains of Cutibacterium acnes, rather than the mere presence of bacteria. The system uses a skin-safe bacterial chassis engineered with a pH-sensitive genetic circuit. When exposed to the slightly acidic conditions characteristic of inflamed acne lesions, the circuit activates expression of a targeted antimicrobial peptide.
My second idea is a synthetic biology project that creates living flowers that glow in response to environmental triggers, such as allergens, pollutants, or changes in pH. Instead of passive decoration, these flowers act as living biosensors, showing you what’s happening around them. The flower’s cells are engineered with a genetic circuit that detects a specific trigger, like pollen proteins or chemical pollutants. When the trigger is present, the circuit switches on and activates bioluminescent genes, making the flower glow. The glow stops when the trigger is gone.
My third idea is a living, edible fungal patch designed to produce essential vitamins such as B12, B9 and K2. Fungi are grown in small hydrogel scaffolds containing optimized nutrients, which support both growth and vitamin synthesis. As the patch develops over several days,it can be observed the living system in action, with growth and optional color indicators reflecting vitamin production.
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) In a 500-gram piece of meat, you are consuming approximately: 7.5275 × 10^23 amino acids
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish? Humans don’t become the animals they eat because digestion is a process of proteolysis, where enzymes break down foreign proteins into their universal amino acid subunits.
3. Why are there only 20 natural amino acids? There are only 20 natural amino acids because they get the job done in the exact number they are. They cover all the basics, so evolution probably just found a “sweet spot” with these 20 and stuck with it. Making more would require the body to build brand-new ways of using them, and since the 20 we have can make millions of different proteins, that would be very complicated.
4. Can you make other non-natural amino acids? Design some new amino acids. You can certainly trick the system and build amino acids that don’t exist in nature. We can maybe design one called “Magnetite,” it has a tiny magnetic cluster on its side chain so we could move proteins around using magnets. They are basically like custom-made Lego bricks with special powers like built-in magnets that the standard ones in the nature sets don’t have.
5. Where did amino acids come from before enzymes that make them, and before life started? Before life and enzymes existed, amino acids formed through geochemical processes. The Miller-Urey experiment has shown that discharging energy into a primitive atmosphere of methane, ammonia, and hydrogen produces organic compounds.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect? While natural L-amino acids form right-handed alpha-helices, a polymer made of D-amino acids would adopt a left-handed conformation.
7. Can you discover additional helices in proteins? Yes, they have been discovered as rare but stable variants. The 3-helix is more tightly wound and elongated, appearing at the termini of regular helices. The pi-helix is wider and shorter, it is found near functional sites in enzymes.
8. Why are most molecular helices right-handed? Most molecular helices are right-handed because that direction keeps the amino acid side chains from crashing into the backbone.
9. Why do β-sheets tend to aggregate? Beta-sheets aggregate because of the hydrophobic effect, where water molecules force non-polar side chains together to hide them from the aqueous environment, making the sheets into stacks. Once they meet, unsatisfied hydrogen bonds along the edges act like a chemical glue, locking the strands into stable fibers called amyloids.
Part B. Protein Analysis & Visualisation
1. Protein choice: I selected Sonic Hedgehog (SHH) because it is one of the most famous and critically important proteins in developmental biology. It acts as a “morphogen,” meaning it provides a chemical map that tells an embryo where to put its limbs, fingers, and even the different parts of the brain. Also, because I did it in a previous assignment.
2.
The length of the protein is: 462 amino acids
The most common amino acid is: A, which appears 56 times
Sonic Hedgehog (SHH) belongs to a very specific and small family of signaling proteins appropriately named the Hedgehog (Hh) family.
It has more hydrophilic residues than hydrophobic residues.
I can’t really tell.
Part C. Using ML-Protein Design Tools
C1: Protein choice I chose the p53 protein.
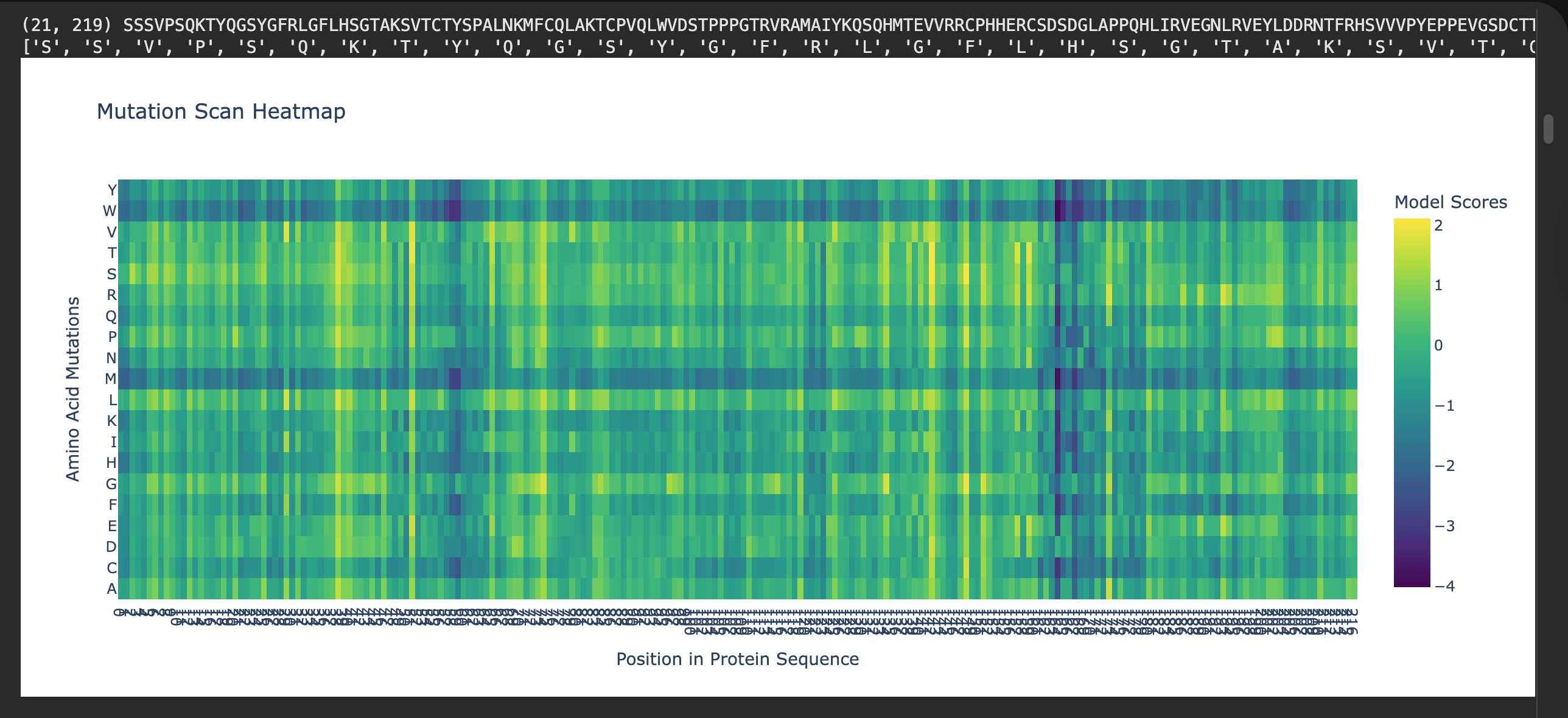
1. Deep mutational scans
I choose residues 102–292 as a “dark zone” because any mutation here breaks the protein’s ability to grip DNA.
Residue: Arginine 248 (R248)
Mutation: R248W
2. Latent Space Analysis
C2: Protein Folding
C3:Protein Generation
Part D:
Project Proposal: Engineering a Minimal MS2-L Lysis Engine
Primary Goal:
Our group aims to increase the functional stability of the MS2 lysis (L) protein. We will achieve this by eliminating the N-terminal domain (residues 1–36). This truncation removes the “regulatory brake” that normally makes lysis dependent on the host chaperone DnaJ, resulting in a more potent, autonomous lysis protein, thus lysis will be achieved a lot faster, beacuse MS2-L will be functional from the moment of translation which gives less time for the proteases to degrade it before it attached to the cellular membrane.
Tools & Approaches
We propose a computational pipeline to validate and optimize this truncated variant:
AlphaFold3 / AlphaFold-Multimer: We will model the truncated L protein in a cramped lipid bilayer environment to predict how the remaining transmembrane helix (TMH) inserts itself. We will also use it to confirm the loss of binding affinity to E. coli DnaJ.
Protein Language Models (ESM-2 / ESM-1v): We will use these models to perform in silico mutagenesis on the remaining C-terminal sequence. Our goal is to identify “stabilizing” mutations that strengthen the alpha-helical propensity of the membrane-spanning region.
Molecular Dynamics (MD) Simulations (OpenMM or Gromacs): Since lysis involves membrane distortion, we will simulate the truncated protein within a model bacterial membrane to observe its ability to disrupt lipid packing.
Potential Pitfalls
Membrane Toxicity in in silico models: Most protein design tools are trained on soluble proteins. Modeling a protein whose entire job is to destroy the membrane (lysis) may lead to unstable simulations or “unphysical” results.
Expression Levels: In a real-world lab setting, a highly toxic protein might kill the host cells so quickly that we cannot produce high enough titers of the phage for study.
Binds near the surface of the β-barrel;; mostly surface-exposed.
2
KHYYWVAIRWKK
0.40
Appears bound on the β-barrel and dimer interface, surface-bound
3
HRSPVVGVALKK
0.41
Localized near the β-barrel surface, surface-bound.
4
WRYPVAAIELKE
0.30
Surface-bound,minimal contact with N-terminal residues, approaches dimer interface.
5
FLYRWLPSRRGG
0.28
Surface-bound, does not penetrate deeply into β-barrel.
The ipTM scores for the PepMLM-generated peptides range from 0.30 to 0.41, slightly higher than the known binder (0.28). Peptides 2 and 3 show the highest ipTM scores, indicating stronger predicted structural confidence in the SOD1–peptide complex.
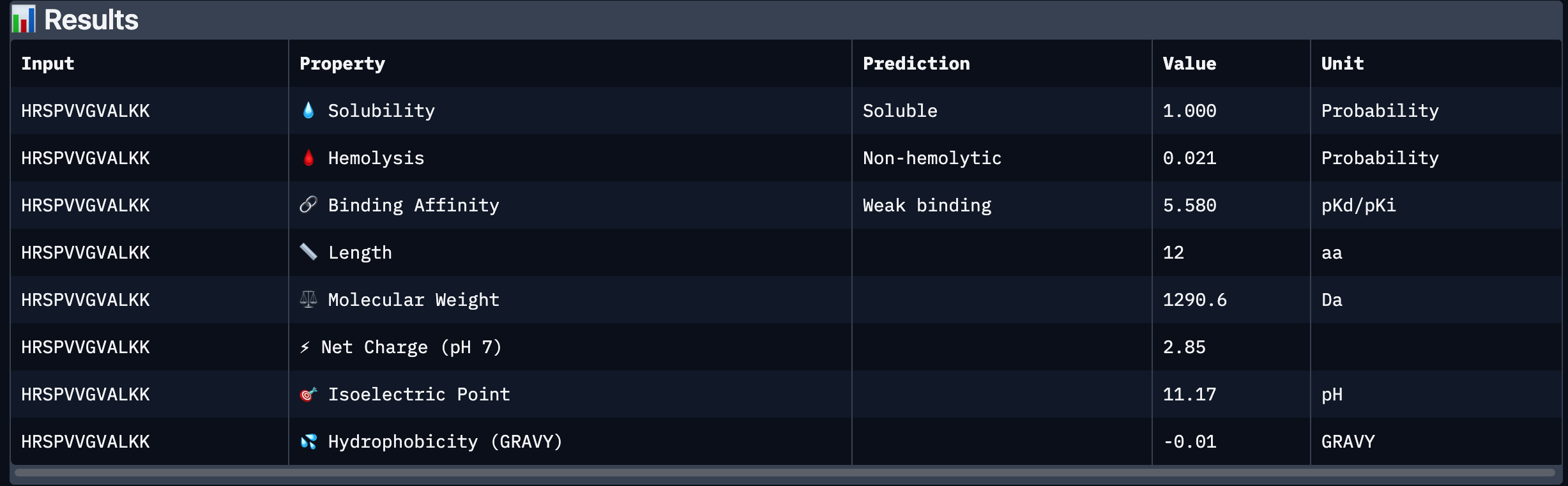
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
After analysing all the peptides and their predicted binding and therapeutic properties I concluded that their affinity binding does not rise with the ipTM index, all of my peptides are soluble( constant index of 1) and are non-hemolytic.
I chose peptide 2 (HRSPVVGVALKK), because it has one of the highest ipTM (0.40)and the highest binding affinity(6.35).
Part 4: Generate Optimized Peptides with moPPIt
Week 6 Homework: Genetic Circuits Part I
Week 6 — Genetic Circuits Part I: Assembly Technologies
Assignment: DNA Assembly
1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Some components that are found in the Phusion High-Fidelity PCR Master Mix are:
Phusion DNA Polymerase: a enzyme that assembles the new DNA strand and proofreads for errors.
Nucleotide bases (A, T, C, G) used as building blocks for the new DNA.
Optimized reaction buffer including MgCl2 :maintains the optimal pH and ionic strength to keep the enzyme stable during thermal cycling.
2. What are some factors that determine primer annealing temperature during PCR?
Some factors That determine the primer annealing temperature during PCR are:
Base Composition: higher G-C content increases the melting temperature
Primer Length: longer primers generally have higher Tm values.
Primer Concentration: higher concentrations can shift the kinetics of annealing.
3. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR
Restriction Enzyme Digest
Protocol
Thermal Cycling: You move through specific temperatures (98°C, ~60°C, 72°C) to unzip, prime, and build DNA.
Isothermal Incubation: You hold the sample at a steady temperature (usually 37°C) to let the enzymes “chew” the DNA.
Input DNA
Needs very little “template” DNA; it creates millions of copies from almost nothing.
Needs a high concentration of DNA because you are only cutting what is already there.
Customization
High: You can add “overhangs” or mutations to the ends of your DNA.
Low: You can only cut where a specific “recognition site” (like GAATTC) already exists.
You can use PCR when you need to copy-paste a gene out of a genome or prepare fragments for Gibson Assembly by adding specific overlapping ends.
You can use Digest for diagnostic purposes (to check if your plasmid is correct) or for traditional cloning if the gene already has the right cut sides at the ends.
4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To ensure that the DNA sequences are appropriate:
each fragment must share a 20–40 bp sequence with the fragment next to it
purity: must “clean" your DNA (using a kit or gel extraction)
If there are leftover Polymerase or Restriction Enzymes from the previous steps, they will destroy the Gibson reaction
If you use a digest, ensure the restriction site isn’t leaving behind DNA that will ruin the protein’s code
5. How does the plasmid DNA enter the E. coli cells during transformation?
Plasmid DNA enters E. coli cells during transformation through temporary pores formed in the cell membrane. These pores are created artificially using methods like: heat shock and electroporation
6. Describe another assembly method in detail (such as Golden Gate Assembly)
a.Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
b.Model this assembly method with Benchling or Asimov Kernel!
Golden Gate Assembly (GGA) allows you to snap multiple pieces of DNA together in a specific order, all in one go. Most restriction enzymes (like EcoRI) cut DNA right inside their recognition sequence. Golden Gate uses Type IIS restriction enzymes (like BsaI or BbsI), which are unique because they bind to a specific DNA sequence but cut the DNA a few base pairs away from that site. Because the enzyme cuts outside its designated sequence, you can design the sticky ends to be whatever 4 letters you want.By designing the end of Fragment A to match the beginning of Fragment B, you ensure they can only stick to each other.This allows for directional assembly, meaning the parts will never move into the wrong position.In GGA, you put your DNA fragments, the Type IIS enzyme, and DNA Ligase into a single tube. You then cycle the temperature:
cooler (~37°C): the enzyme cuts the DNA
warmer (16°C or 25°C): the ligase pastes the matching ends together
In GGA the recognition sites are designed to be “thrown away.” When the enzyme cuts the fragment, the recognition site is on the piece of DNA that gets discarded. Once the fragments are ligated together into the final circular plasmid, the recognition site is gone.
Week 7 Homework: Genetic Circuits Part II: Neuromorphic Circuits
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
-Traditional genetic circuits typically operate on Boolean logic, which processes inputs as binary states (0 or 1). IANNs offer several advantages: -Analog Integration - unlike Boolean circuits, IANNs can process graded signals, this allows cells to respond proportionally to varying concentrations of chemicals or light. -Scalability - to achieve complex decision-making with Boolean logic, you need a massive number of gates, which places a heavy metabolic burden on the cell, IANNs can achieve classification or pattern recognition using fewer components by adjusting binding affinities or promoter strengths. -Non-Linear Processing - IANNs utilize activation functions, this allows them to filter noise and handle non-linear relationships between inputs.
2. Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
Application: A cell designed to detect a specific cancer profile by sensing multiple microRNA (miRNA) biomarkers. Input: The inputs ) are various miRNA sequences overexpressed in a specific tumor type. These inputs act as inhibitors or activators for the IANN. Output: The output is the expression of a pro-apoptotic gene (causing cell death) or a secreted cytokine to alert the immune system. The output triggers only if the weighted sum of inputs exceeds a specific threshold
Limitations: -Metabolic Load: High-level computation consumes cellular resources (ATP, ribosomes), potentially slowing cell growth. -Signal Decay: Biological components degrade over time, which can shift the weights and lead to false positives.
3.Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Assignment Part 2: Fungal Materials
1.What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Fungal materials primarily utilize mycelium, the root-like structure of fungi.
Material
Use
Adv
Disadv
Mycelium Packaging
Biodegradable alternative to Styrofoam
Fully compostable; low energy to produce
Lower impact resistance than some plastics
Fungal Leather
Sustainable textile for fashion
No animal cruelty; uses 90% less water than bovine leather
Lower structural load-bearing capacity than concrete
2. What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
Engineering fungi to biomineralize (deposit calcium carbonate) or secrete anti-microbial peptides. This could create “self-healing” buildings where cracks in fungal bricks are filled by the fungi themselves, or packaging that prevents food spoilage.
Advantages of Fungi vs. Bacteria: -Fungi are eukaryotes, meaning they can perform complex post-translational modifications (like glycosylation) that bacteria (prokaryotes) cannot. This is essential for producing human-like proteins. -The filamentous growth of mycelium provides a natural 3D scaffold. Bacteria usually form disorganized biofilms, whereas fungi create structural networks. -Fungi are professional secretors. They are evolved to pump out massive amounts of enzymes to break down wood and organic matter, making them superior cell factories for harvesting products. -Fungi can often thrive in more acidic or low-moisture environments where many industrial bacteria would perish.
Assignment Part 3: First DNA Twist Order
Week 9 HW: Cell-Free Systems
Homework Part A: General and Lecturer-Specific Questions
General homework questions
1.Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis (CFPS) offers superior flexibility and control compared to in vivo methods. You can directly add DNA templates, tweak reaction conditions like temperature or pH on the fly, and incorporate non-natural amino acids without genetic engineering, which is impossible in living cells due to their complex regulation. It also allows real-time monitoring and manipulation of variables like cofactor concentrations.
CFPS is better in two cases: (1) toxic proteins, like bacterial toxins that kill host cells during expression; (2) membrane proteins, where cell-free systems use nanodiscs or liposomes to mimic membranes without cellular toxicity or folding issues.
2.Describe the main components of a cell-free expression system and explain the role of each component.
A typical CFPS systemhas:
-DNA template: Encodes the target protein; often under a strong promoter like T7.
-Ribosomes: Translate mRNA into protein; sourced from crude cell lysates.
-RNA polymerase (T7 RNA pol), NTPs for mRNA synthesis.
-Other proteins you need
-Translation factors: Aminoacyl-tRNA synthetases, elongation/initiation factors to charge tRNAs and assemble proteins.
-Energy sources: ATP, GTP, creatine phosphate.
-Amino acids and cofactors: Building blocks and Mg²⁺/K⁺ ions for stability.
-Lysate: Crude extract providing most components.
3.Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy regeneration is critical because CFPS rapidly consumes ATP/GTP, halting reactions within minutes without replenishment, unlike cells with metabolic pathways.
A phosphoenolpyruvate (PEP)/pyruvate kinase system: PEP donates phosphate to ADP via kinase, continuously regenerating ATP. Add 20-50 mM PEP and 0.1 mg/mL pyruvate kinase to your reaction mix.
4.Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Feature
Prokaryotic
Eukaryotic ( wheat germ or HeLa)
Speed
Fast (hours), high yield for simple proteins
Slower, lower yield but better folding
Post-translation
Minimal glycosylation
Full glycosylation, chaperones
Cost/Complexity
Cheap, simple
Expensive, needs eukaryotic factors
I would produce GFP in prokaryotic CFPS, it’s prokaryotic-optimized, high-yield, no glycosylation needed. I would produce a glycoprotein like erythropoietin in eukaryotic CFPS for proper N-glycosylation essential for activity.
5.How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Challenges: Membrane proteins aggregate without lipids, misfold, or insert poorly.
Design:Use a lipid-detergent mix (1% Brij-58 + POPC liposomes) or nanodiscs (MSP1D1 belt proteins + lipids).Co-express with chaperones (Skp) from a polycistronic template.Optimize Mg²⁺ (10-15 mM) and temperature (25°C) for insertion.Test via fluorescence (GFP) or functional assays ( transport activity).This solubilizes proteins during synthesis, boosting yield 10-fold.
6.Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Reason 1: Inhibitory DNA impurities =>Strategy: Purify plasmid with column kits and test dilutions.
Reason 2: Suboptimal energy depletion =>Strategy: Add fresh PEP/GTP mix and monitor ATP via luciferase assay.
Reason 3: Protease degradation => Strategy: Use protease-deficient lysates and add inhibitors like PMSF.
Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
1.Pick a function and describe it.
a.What would your synthetic cell do? What is the input and what is the output?
Function: Detect arsenic in water and signal via fluorescence.
Input: arsenite . Output: fluorescent resorufin.
b.Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
No, it needs encapsulation for controlled release and membrane signaling.
c.Could this function be realized by genetically modified natural cell?
Yes (E. coli with ars operon), but synthetic version allows modularity for sensors without genomic integration.
d.Describe the desired outcome of your synthetic cell operation.
Synthetic cells glow pink near toxic arsenic levels, enabling visual water safety checks.
2.Design all components that would need to be part of your synthetic cell.
-Lipids: POPC, cholesterol.
-Genes: arsR (repressor, UniProt P0A1C7), aHL (hlyA from S. aureus, UniProt P0DQI1), luciferase (luxA from Vibrio fischeri).
-Biological cells: None
b.How will you measure the function of your system?
Fluorescence plate reader (560 nm excitation) for resorufin, dose-response curve vs. arsenite.
Homework question from Peter Nguyen
1.Write a one-sentence summary pitch sentence describing your concept.
Smart fabric-embedded freeze-dried cell-free sensors that change color in response to sweat pH, alerting wearers to dehydration.
2.How will the idea work, in more detail? Write 3-4 sentences or more.
Freeze-dry E. coli CFPS with pH-sensitive GFP plasmid and pH indicator dyes into cotton fibers via lyophilization-spray coating. Water from sweat rehydrates the mix, triggering protein expression, acidic pH shifts GFP fluorescence from green to yellow, visible under UV or as embedded dye release.
3.What societal challenge or market need will this address?
Addresses dehydration and heat stress for athletes, outdoor workers, and other people exposed to Romania’s increasingly frequent heatwaves.
4.How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
Stability via trehalose cryoprotectant (extends shelf-life 1+ year) or one-time use mitigated by modular reloading or multi-compartment fabrics; water activation natural from body moisture.
Homework question from Ally Huang
1.Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting.
Microgravity impairs plant growth on long missions, reducing food and oxygen because of the weakened roots and altered gene expression (EXPANSIN genes). Vital for Mars missions needing sustainable hydroponics, scientifically intriguing as it reveals gravity’s role in auxin signaling and cell wall remodeling,BioBits CFPS enables rapid, resource-light testing in space.
2.Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches.
EXP1 expansin gene from Arabidopsis thaliana.
3.Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses.
EXP1 loosens cell walls for root elongation; microgravity downregulates it, stunting growth. Studying EXP1 expression in CFPS mimics space conditions), linking to pathways disrupted off-Earth, and informing genetic fixes for space crops.
4.learly state your hypothesis or research goal and explain the reasoning behind it.
Overexpression of EXP1 in BioBits CFPS will restore simulated microgravity-impaired root growth signals, as measured by downstream fluorescent reporters.
Reasoning: Earth-based studies show EXP1 compensates gravity loss in clinostats; CFPS bypasses live plant limits in space, allowing direct gene dosing. Hypothesis tests if EXP1 alone boosts growth factors without full cellular contex
5.Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc
Week 10 HW: Measurement Technology
Waters Part I — Molecular Weight
To calculate the theoretical molecular weight (MW) of the eGFP sequence provided, we must account for the primary amino acid sequence and the critical internal post-translational modification that creates the fluorophore.
In eGFP, a spontaneous cyclization and oxidation occurs within the tripeptide sequence Thr65–Tyr66–Gly67 . During this maturation process, the protein loses:
One water molecule (H₂O =18.01 Da)
Two hydrogen atoms (H₂ =2.01 Da)
Total mass loss = 20.02 Da.Using standard average isotopic masses for the 246 residues:
Raw sequence MW ≈ 27,828.1 Da
Minus maturation loss ≈ −20.0 Da
Predicted MW: 27,808.1 Da
2
The relationship between mass-to-charge ratio and molecular weight in ESI-MS is:
m/z = (MW + zH⁺) / z
Rearranged: MW = z(m/z) − z(1.008)
Using a the formula: z = (m/zₙ₊₁) / [(m/zₙ) − (m/zₙ₊₁)]
If z ≈ 31 and m/z ≈ 898.05:
MW = 31 × (898.05) − 31 × (1.008)
MW ≈ 27,808.3 Da
This value matches the corrected theoretical mass.
Accuracy is calculated using:
Accuracy (ppm) = |MWₑₓₚ − MWₜₕₑₒ| / MWₜₕₑₒ × 10⁶
Accuracy = |27,808.3 − 27,808.1| / 27,808.1 × 10⁶
Accuracy ≈ 7.2 ppm
3.
Determining the charge state from a single zoomed-in peak of an intact protein like eGFP is not possible on a system with 30,000 resolution because the isotopic spacing is too narrow to be resolved. For a 28 kDa protein, the individual isotopic peaks merge into a single, smooth isotopic envelope due to the natural peak width and the instrument’s resolution limits. Consequently, you observe a hump representing the average mass rather than distinct isotopic lines, requiring the use of the adjacent peak method across the full mass spectrum to mathematically derive the charge.
Waters Part III — Peptide Mapping - primary structure
Here’s your section cleaned up, standardized, and ready to paste into Google Docs:
1.
To analyze the eGFP peptide map, we apply principles of enzymatic digestion and high-resolution mass spectrometry.
This indicates very high mass accuracy, consistent with high-resolution MS systems.
7.
Sequence coverage refers to the percentage of amino acids identified through MS/MS fragmentation relative to the full protein sequence.
Typical eGFP coverage: 85%–98%
Missing regions typically include:
Very small, hydrophilic peptides that do not retain on the LC column;Large or poorly ionizing peptides
Homework: Waters Part IV — Oligomers
Oligomeric Species
Calculation
Theoretical Mass (MDa)
Observed Peak (MDa)
7FU Decamer
340 kDa × 10
3.40
3.4
8FU Didecamer
400 kDa × 20
8.00
8.33
8FU 3-Decamer
400 kDa × 30
12.00
12.67
8FU 4-Decamer
400 kDa × 40
16.00
~17.0
7FU Decamer (3.4 MDa):
The peak at 3.4 MDa corresponds to a decamer composed of 10 subunits of the 7FU polypeptide.
8FU Didecamer (8.33 MDa):
This is the most intense peak in the spectrum. Although the theoretical mass is 8.0 MDa, the observed value of 8.33 MDa suggests an effective subunit mass closer to ~415 kDa under experimental conditions.
8FU 3-Decamer (12.67 MDa):
The peak at 12.67 MDa represents a structure composed of 30 subunits .
8FU 4-Decamer (17.0 MDa):
The cluster of peaks around 17.0 MDa corresponds to a 40-subunit assembly.
The clear resolution of these very large complexes is enabled by Charge Detection Mass Spectrometry (CDMS).Unlike conventional mass spectrometry, which measures the mass-to-charge ratio (m/z) of ion populations, CDMS directly measures both the charge (z) and m/z of individual ions. This allows accurate mass determination even for megadalton-scale assemblies.
The spectrum indicates that the protein strongly favors formation of didecamers (8.33 MDa). The presence of 3-decamer and 4-decamer species further demonstrates the ability of the complex to stack into larger cylindrical assemblies.