Week 4 Homework: Protein Design I
Homework: Protein Design I
Part A. Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat?
(on average an amino acid is ~100 Daltons)
In a 500-gram piece of meat, you are consuming approximately: 7.5275 × 10^23 amino acids
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Humans don’t become the animals they eat because digestion is a process of proteolysis, where enzymes break down foreign proteins into their universal amino acid subunits.
3. Why are there only 20 natural amino acids?
There are only 20 natural amino acids because they get the job done in the exact number they are. They cover all the basics, so evolution probably just found a “sweet spot” with these 20 and stuck with it. Making more would require the body to build brand-new ways of using them, and since the 20 we have can make millions of different proteins, that would be very complicated.
4. Can you make other non-natural amino acids? Design some new amino acids.
You can certainly trick the system and build amino acids that don’t exist in nature. We can maybe design one called “Magnetite,” it has a tiny magnetic cluster on its side chain so we could move proteins around using magnets. They are basically like custom-made Lego bricks with special powers like built-in magnets that the standard ones in the nature sets don’t have.
5. Where did amino acids come from before enzymes that make them, and before life started?
Before life and enzymes existed, amino acids formed through geochemical processes. The Miller-Urey experiment has shown that discharging energy into a primitive atmosphere of methane, ammonia, and hydrogen produces organic compounds.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
While natural L-amino acids form right-handed alpha-helices, a polymer made of D-amino acids would adopt a left-handed conformation.
7. Can you discover additional helices in proteins?
Yes, they have been discovered as rare but stable variants. The 3-helix is more tightly wound and elongated, appearing at the termini of regular helices. The pi-helix is wider and shorter, it is found near functional sites in enzymes.
8. Why are most molecular helices right-handed?
Most molecular helices are right-handed because that direction keeps the amino acid side chains from crashing into the backbone.
9. Why do β-sheets tend to aggregate?
Beta-sheets aggregate because of the hydrophobic effect, where water molecules force non-polar side chains together to hide them from the aqueous environment, making the sheets into stacks. Once they meet, unsatisfied hydrogen bonds along the edges act like a chemical glue, locking the strands into stable fibers called amyloids.
Part B. Protein Analysis & Visualisation
1. Protein choice:
I selected Sonic Hedgehog (SHH) because it is one of the most famous and critically important proteins in developmental biology. It acts as a “morphogen,” meaning it provides a chemical map that tells an embryo where to put its limbs, fingers, and even the different parts of the brain. Also, because I did it in a previous assignment.
2.
- The length of the protein is: 462 amino acids
- The most common amino acid is: A, which appears 56 times
- Sonic Hedgehog (SHH) belongs to a very specific and small family of signaling proteins appropriately named the Hedgehog (Hh) family.
- 250 homologous proteins in other species
3.
- PDB ID: 3HO5
- Description: RCSB 3HO5
- Resolution: 3.01 Å = moderate quality
- Solved Date: 2009
- Non-protein molecules: Zinc (Zn²⁺), Calcium (Ca²⁺), water, buffer components
- Classification: Hedgehog protein family
4.
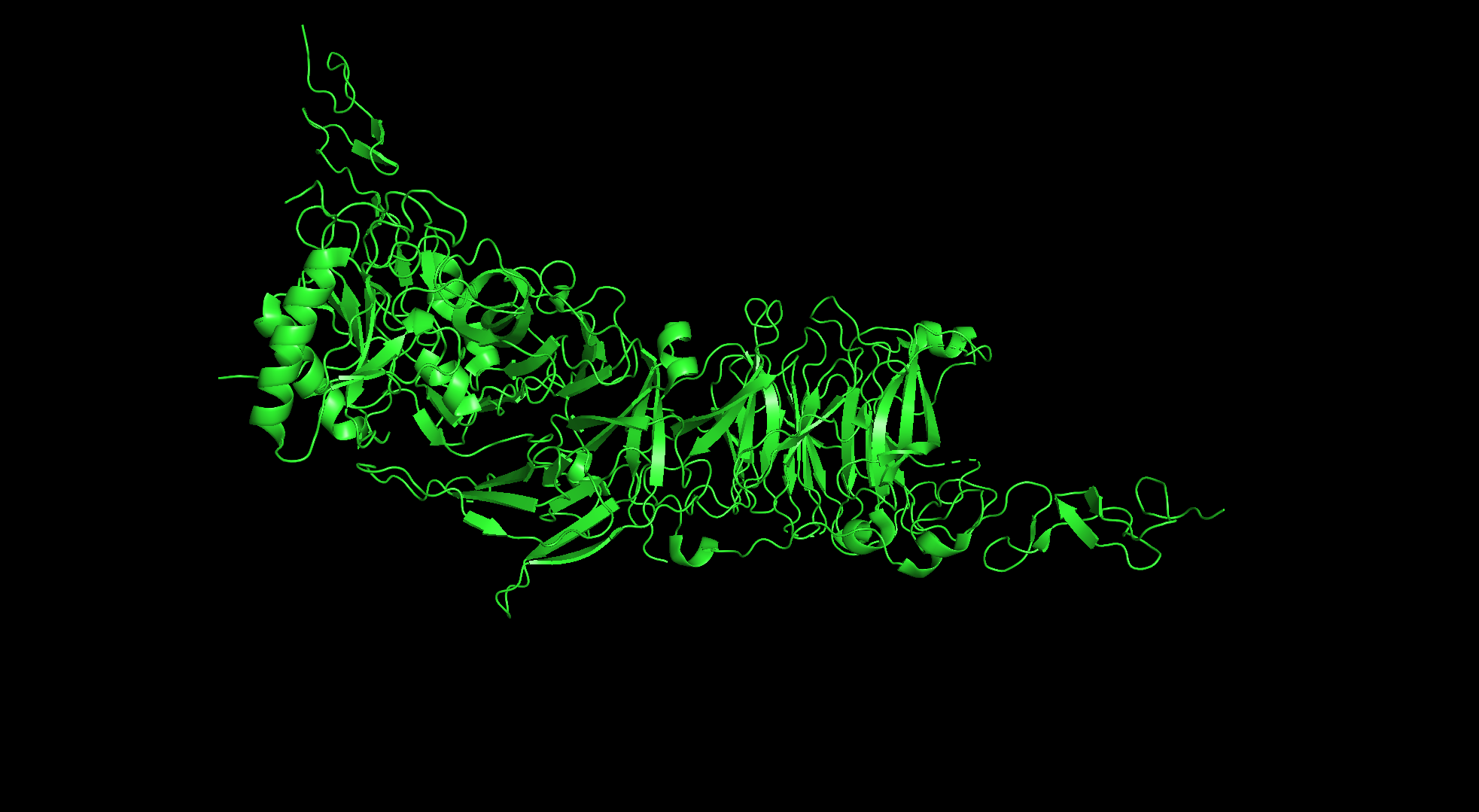
Ribbon
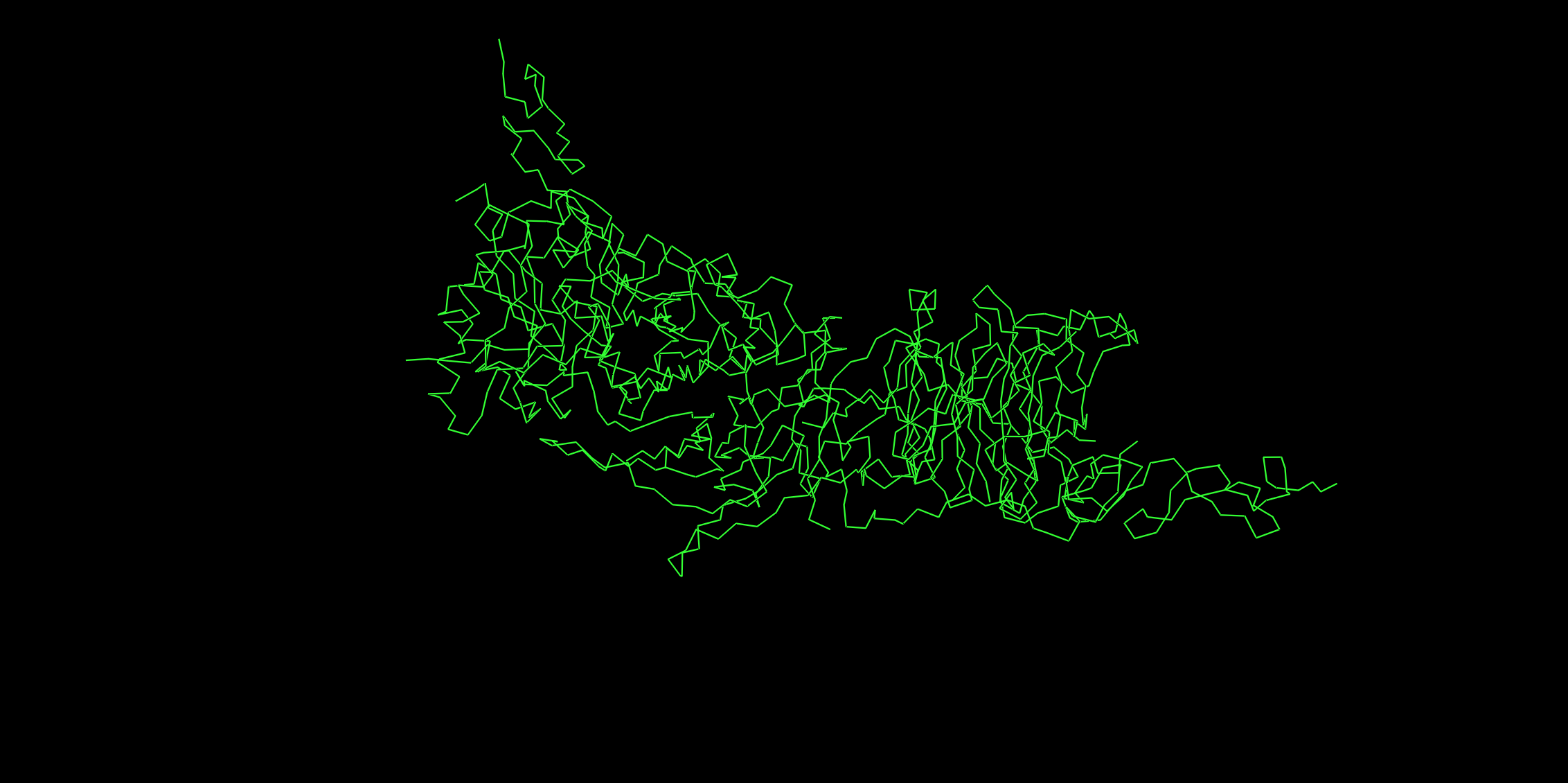
Cartoon
Sticks

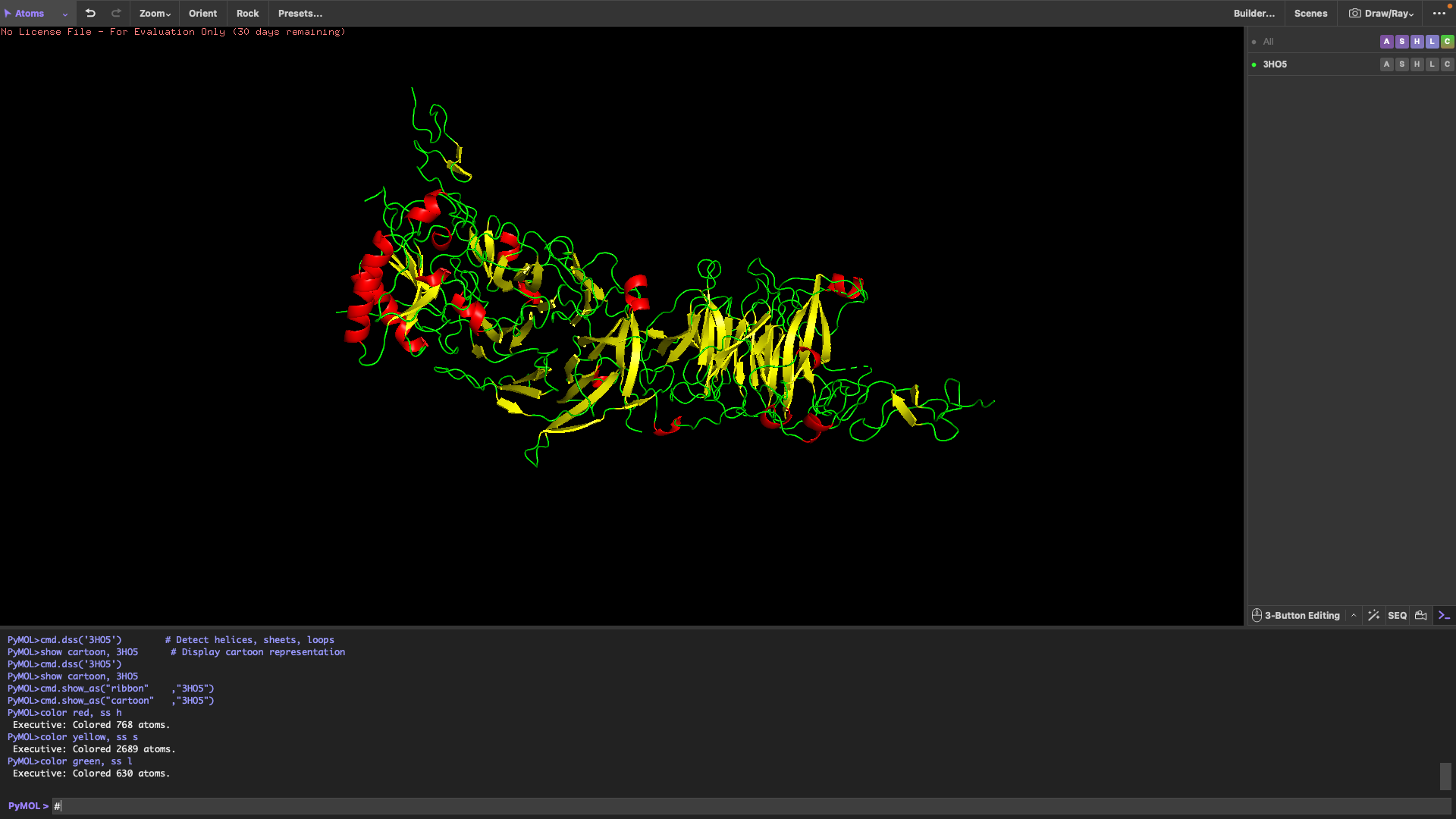
It has more sheets than helicases.
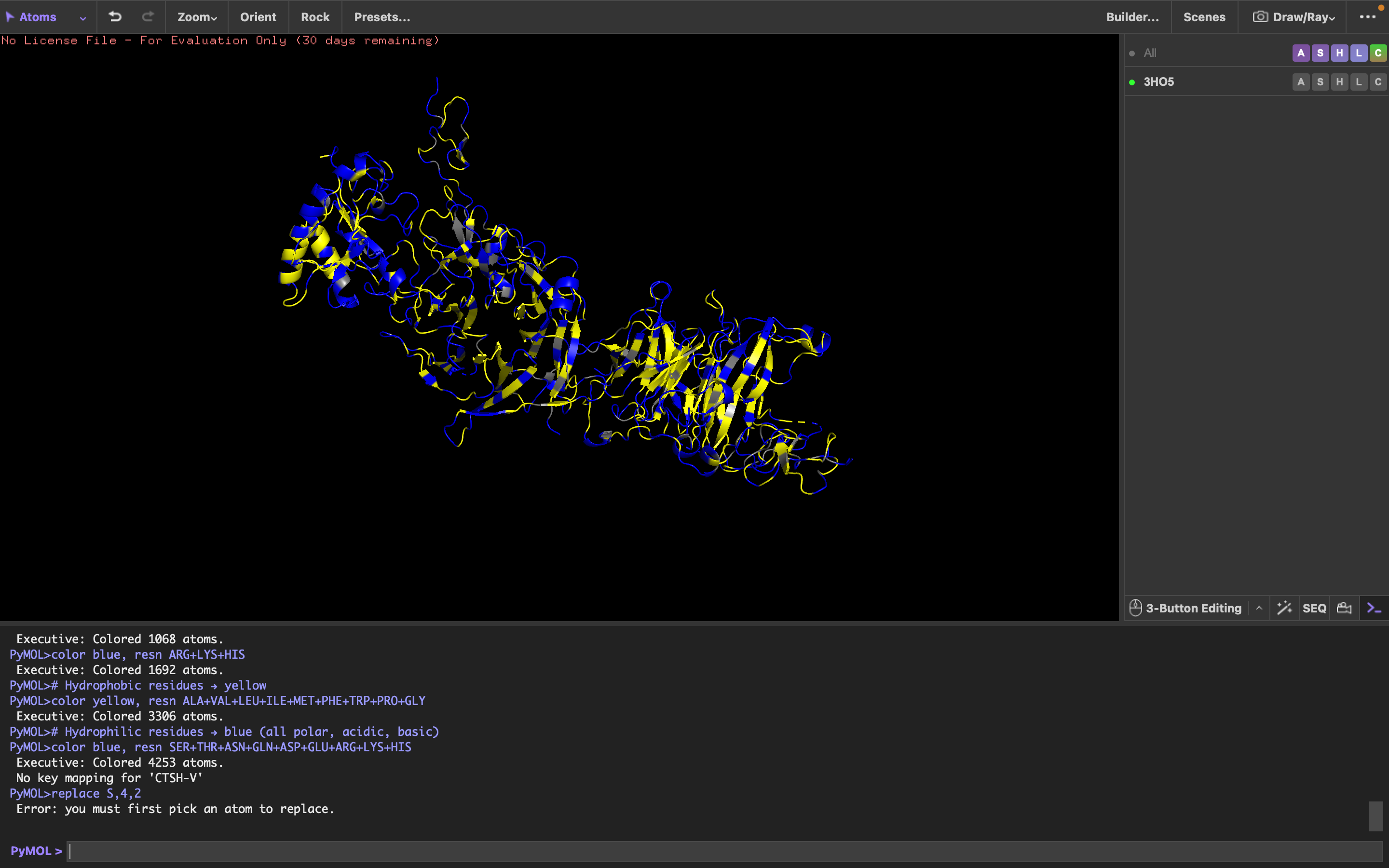
It has more hydrophilic residues than hydrophobic residues.
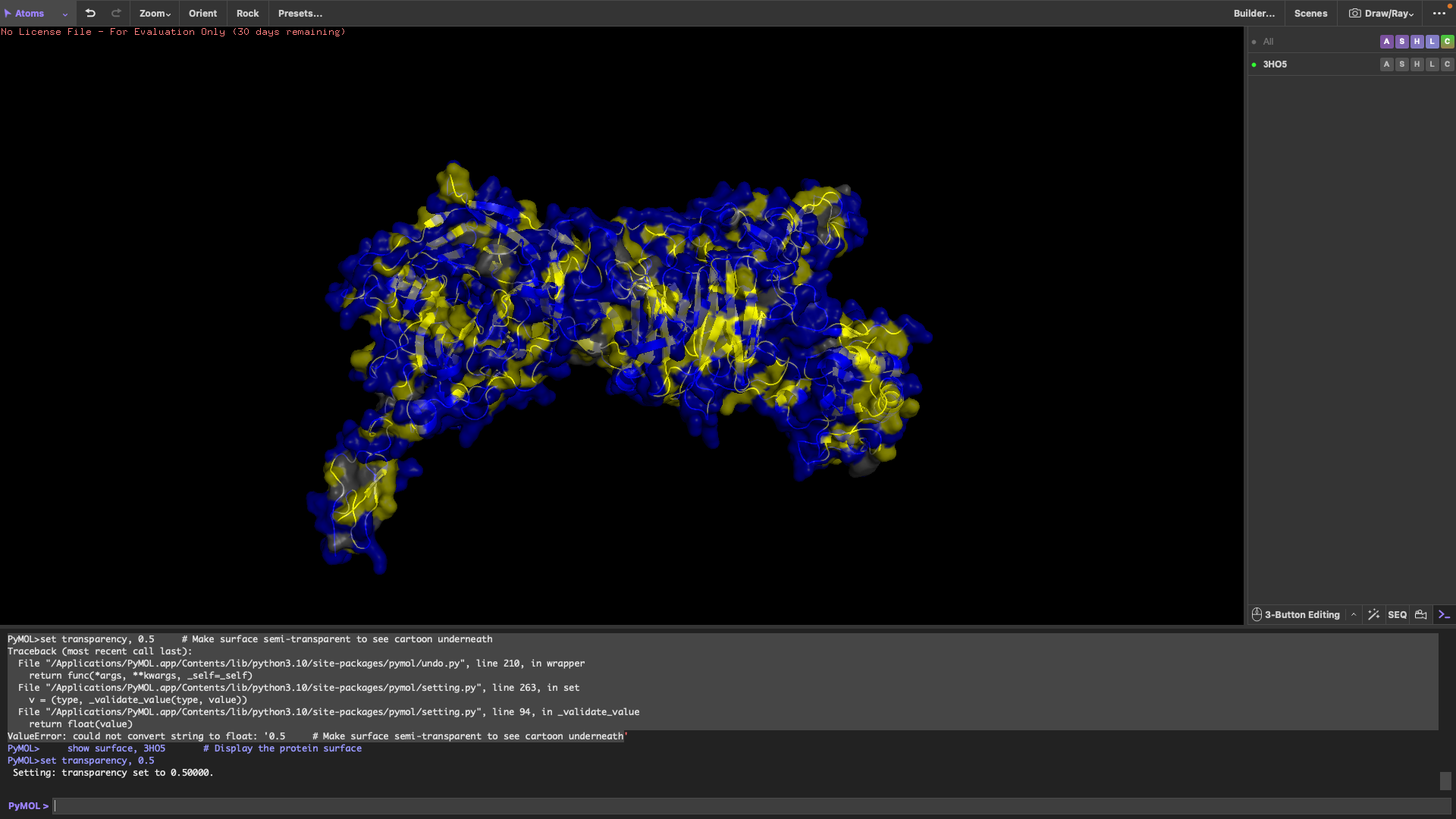
I can’t really tell.
Part C. Using ML-Protein Design Tools
C1: Protein choice
I chose the p53 protein.
1. Deep mutational scans
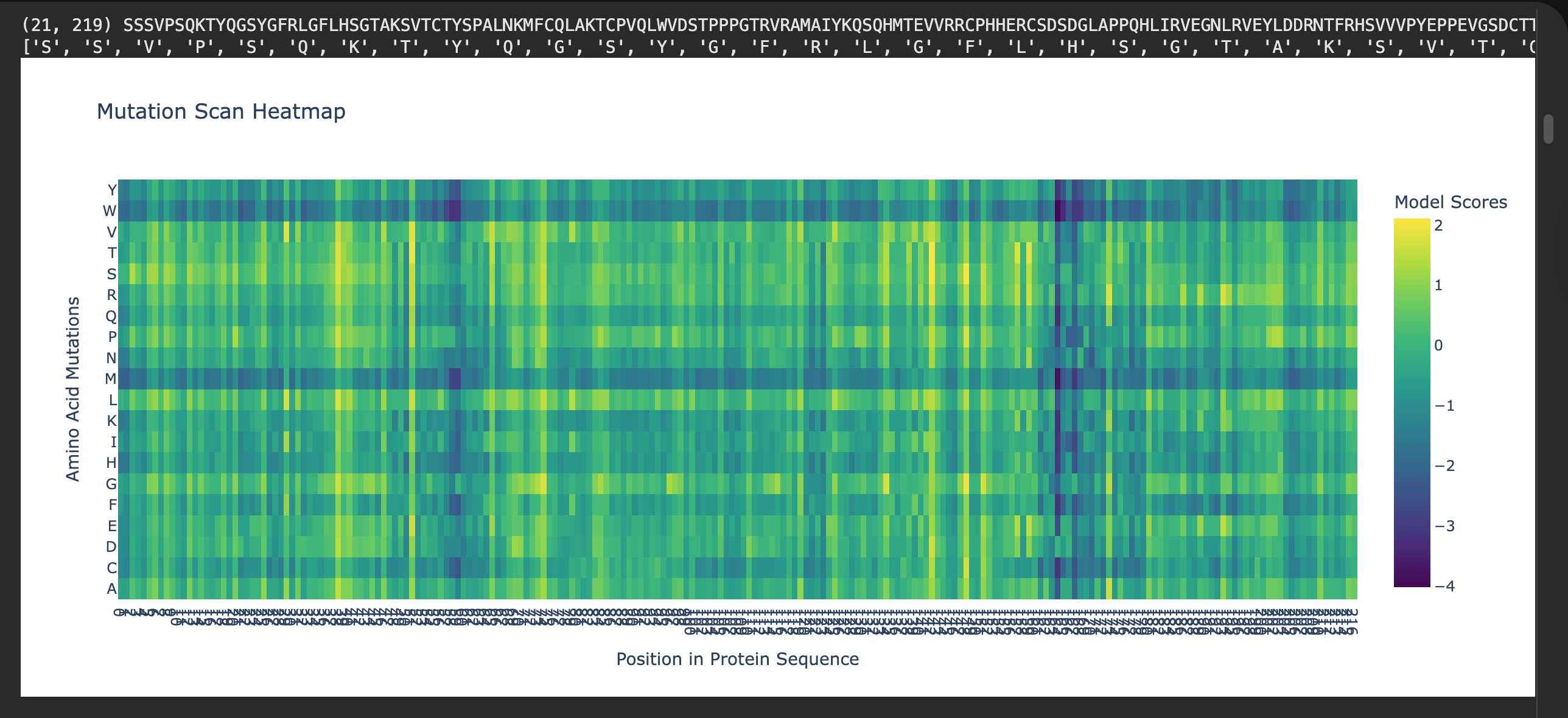
I choose residues 102–292 as a “dark zone” because any mutation here breaks the protein’s ability to grip DNA.
- Residue: Arginine 248 (R248)
- Mutation: R248W
2. Latent Space Analysis
C2: Protein Folding
C3:Protein Generation
Part D:
Project Proposal: Engineering a Minimal MS2-L Lysis Engine
Primary Goal: Our group aims to increase the functional stability of the MS2 lysis (L) protein. We will achieve this by eliminating the N-terminal domain (residues 1–36). This truncation removes the “regulatory brake” that normally makes lysis dependent on the host chaperone DnaJ, resulting in a more potent, autonomous lysis protein, thus lysis will be achieved a lot faster, beacuse MS2-L will be functional from the moment of translation which gives less time for the proteases to degrade it before it attached to the cellular membrane.
Tools & Approaches We propose a computational pipeline to validate and optimize this truncated variant:
AlphaFold3 / AlphaFold-Multimer: We will model the truncated L protein in a cramped lipid bilayer environment to predict how the remaining transmembrane helix (TMH) inserts itself. We will also use it to confirm the loss of binding affinity to E. coli DnaJ.
Protein Language Models (ESM-2 / ESM-1v): We will use these models to perform in silico mutagenesis on the remaining C-terminal sequence. Our goal is to identify “stabilizing” mutations that strengthen the alpha-helical propensity of the membrane-spanning region.
Molecular Dynamics (MD) Simulations (OpenMM or Gromacs): Since lysis involves membrane distortion, we will simulate the truncated protein within a model bacterial membrane to observe its ability to disrupt lipid packing.
- Potential Pitfalls Membrane Toxicity in in silico models: Most protein design tools are trained on soluble proteins. Modeling a protein whose entire job is to destroy the membrane (lysis) may lead to unstable simulations or “unphysical” results.
Expression Levels: In a real-world lab setting, a highly toxic protein might kill the host cells so quickly that we cannot produce high enough titers of the phage for study.
- Pipeline Schematic Design: Truncate N-terminus (1-36).
Optimize: Run ESM-1v to find high-probability stabilizing mutations.
Predict: Fold top candidates in AlphaFold3 to ensure TMH orientation.
Simulate: Insert into a virtual membrane to verify disruptive “toxicity.”