1. Biological engineering application or tool to develop and why
Idea
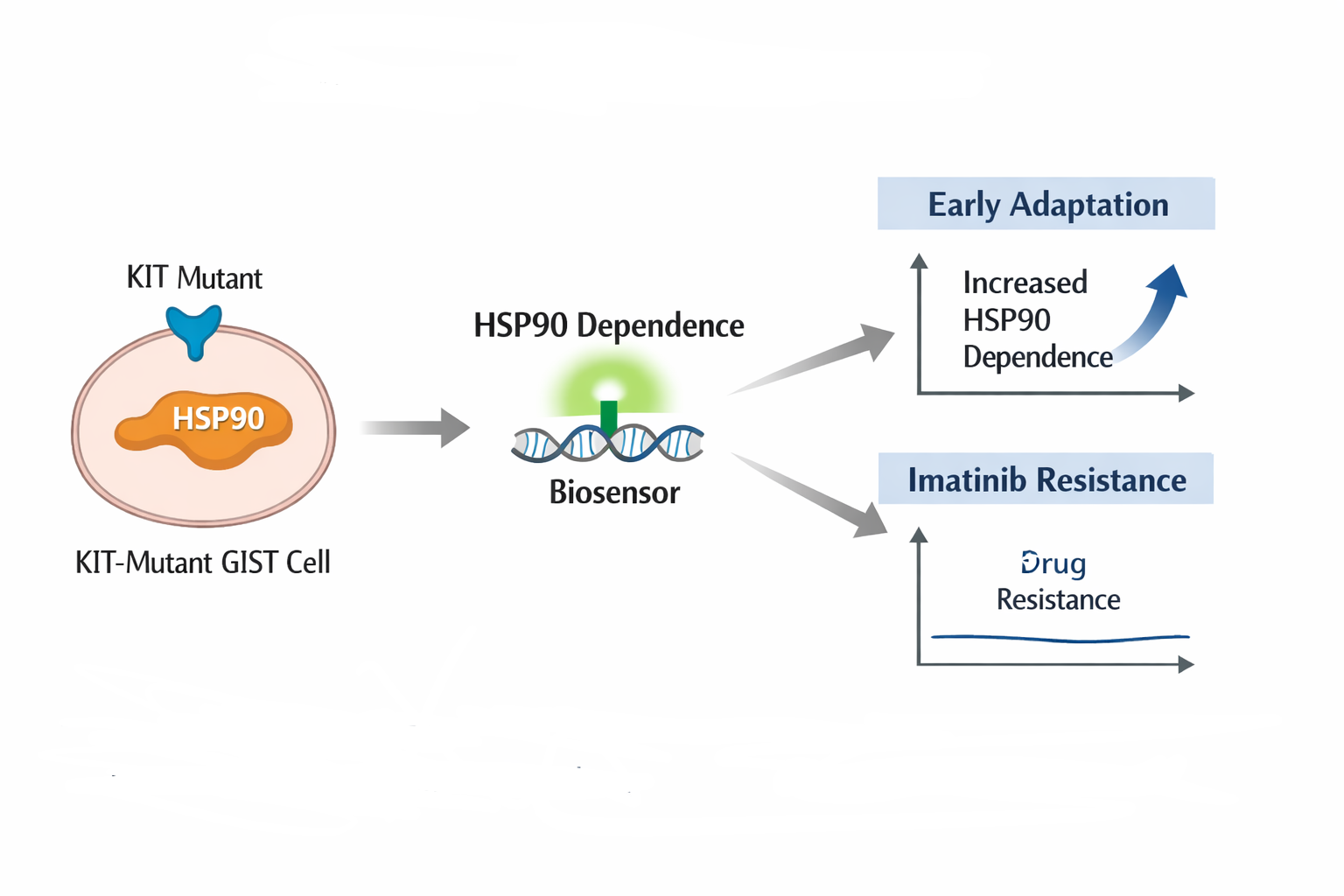
Mi idea its a biological biosensor designed to detect early changes in dependence on the molecular chaperone HSP90 in KIT mutant gastrointestinal stromal tumors (GIST), as functional indicators of cellular adaptation preceding the development of resistance to imatinib.
Figure 1. Conceptual schematic of the proposed biological biosensor.
Image generated using AI for graphical representation purposes only.
Gastrointestinal Stromal Tumors (GIST)
Gastrointestinal stromal tumors (GIST) are rare mesenchymal neoplasms but represent the most common subtype of sarcoma of the gastrointestinal tract. The majority of GISTs harbor activating mutations in the receptor tyrosine kinases KIT or PDGFRA, which has enabled the development of targeted therapies such as imatinib. Despite these advances, progression to metastatic disease remains a major clinical challenge, even among patients who share similar mutational profiles (Hemming et al., 2018a; Antonescu et al., s. f.).
Imatinib and KIT Mutations
Imatinib mesylate is a selective tyrosine kinase inhibitor that binds to the ATP binding site of receptor tyrosine kinases, thereby blocking their phosphorylation and downstream proliferative and anti apoptotic signaling cascades. In GIST, its primary therapeutic targets are KIT and, in specific subgroups, PDGFRA, resulting in inhibition of constitutive oncogenic signaling and induction of cell cycle arrest and apoptosis (Fletcher & Rubin, 2007). This approach represented one of the earliest successes of molecularly targeted therapy in oncology, leading to substantial improvements in survival in patients with advanced or metastatic disease (Fletcher & Rubin, 2007).
Approximately 70–85% of GISTs carry activating mutations in KIT, most frequently in exon 11, followed by exons 9, 13, and 17. These mutational subtypes strongly influence tumor biology and therapeutic response to imatinib. Tumors harboring exon 11 mutations exhibit the highest response rates, whereas other mutational subtypes often require dose escalation or develop secondary resistance through additional kinase domain mutations, ultimately leading to clinical progression after an initial response (Fletcher & Rubin, 2007; Killock, 2022).
Figure 2. Molecular structure of imatinib, highlighting its role as a tyrosine kinase inhibitor in KIT-driven tumors and the biological context in which resistance mechanisms may emerge.
Role of HSP90 in GIST and Therapeutic Resistance
Heat shock protein 90 (HSP90) is a molecular chaperone essential for the stability and functional activity of multiple oncogenic proteins. In GIST, KIT both wild type and mutant critically depends on HSP90 to maintain its active conformation. Inhibition of HSP90 results in destabilization and proteasomal degradation of KIT, including variants harboring mutations associated with imatinib resistance, positioning HSP90 as an alternative or complementary therapeutic target to conventional tyrosine kinase inhibitors (NIHMS212509; Frontiers in Immunology, 2024).
In addition, HSP90 is involved in biological processes linked to tumor progression and therapeutic resistance, which has driven interest in combination strategies for patients with advanced or refractory GIST (Frontiers in Immunology, 2024).
Acquired Resistance to Imatinib: A Central Clinical Problem
Despite the initial efficacy of imatinib in most patients with GIST, a substantial proportion eventually develops acquired resistance, leading to tumor progression and limited therapeutic options. This resistance is driven by heterogeneous molecular mechanisms that cannot always be explained solely by the emergence of new KIT or PDGFRA mutations, complicating both prediction and timely clinical monitoring.
Biosensor Design Concept
The proposed biosensor is conceived as a functional molecular sensor designed to detect cellular states associated with acquired resistance to imatinib in GIST, rather than relying solely on static mutational information. While current clinical stratification primarily focuses on the presence or absence of mutations in KIT or PDGFRA, resistance to imatinib frequently arises through dynamic molecular adaptations that are not fully captured by genomic profiling alone (Fletcher & Rubin, 2007; Killock, 2022). This biosensor aims to bridge that gap by translating complex intracellular signaling states into a measurable and interpretable output, reflecting functional tumor behavior beyond mutational status.
Conceptual Design
At a conceptual level, the biosensor would operate by coupling a biological recognition element sensitive to KIT associated signaling activity or HSP90 dependent protein stability to a reporter output that reflects the functional response of the cell to imatinib exposure. Rather than detecting specific mutations, the sensor would respond to changes in intracellular conditions indicative of loss of KIT dependency, activation of alternative survival pathways, or increased reliance on molecular chaperones such as HSP90, all of which have been implicated in imatinib resistance in GIST (Fletcher & Rubin, 2007; NIHMS212509; Frontiers in Immunology, 2024).
Need for New Functional Analytical Tools
In this context, there is an unmet need for tools capable of capturing the functional state of tumor cells and their dependence on KIT-associated oncogenic pathways, beyond static mutational characterization. A biosensor designed to detect dynamic changes in molecular activity related to imatinib sensitivity or resistance could complement current genomic approaches and provide a pathway toward earlier identification of therapeutic resistance.
2. Governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm).
Clinical safety and non-malfeasance
How to ensure the biosensor is used strictly to support cancer treatment and does not cause harm?
-How to prevent its use as a standalone diagnostic tool, and ensure that clinical decisions are not made solely based on biosensor readouts without proper medical interpretation?
Responsible clinical integration
How should the biosensor be integrated into clinical workflows so that it provides additional biological information rather than replacing clinician judgment?
Who is authorized to interpret the results, and under what conditions should the data influence treatment decisions?
Equitable access
Who will have access to this technology?
How can barriers related to cost, infrastructure, or healthcare disparities be minimized so that the benefits of early adaptation detection are available to a broader patient population and not only to a privileged few?
Research integrity and prevention of misuse
How can the biosensor be governed to ensure it is used to better understand KIT-mutant tumor biology and mechanisms of drug adaptation, rather than being applied in ways that could intentionally or unintentionally cause harm?
3. Different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”)
Governance Action 1: Clinical use restriction policy
Purpose: Currently, emerging biosensors may be interpreted as direct indicators of treatment response or resistance. This action proposes restricting the use of the biosensor strictly as a supportive clinical and research tool, not as a standalone diagnostic or decision making instrument.
Design: This would require hospitals and clinical research institutions to implement internal guidelines stating that biosensor outputs must be interpreted by trained oncologists or molecular pathologists. Ethics committees and hospital review boards would approve its use, and researchers would include clear disclaimers in publications and clinical protocols.
Assumptions: This action assumes that clinicians will follow institutional guidelines and that proper training will be available. It also assumes that misuse mainly occurs at the interpretation stage rather than during data generation.
Risks of Failure & “Success”: If the policy fails, clinicians might still rely too heavily on biosensor readouts. If it succeeds too well, overly strict rules could slow down clinical adoption or discourage innovation, delaying potential patient benefits.
Governance Action 2: Mandatory validation and transparency requirements (technical strategy)
Purpose: At present, early biomarkers may be applied before their limitations are fully understood. This action proposes requiring rigorous validation and transparency regarding uncertainty, sensitivity, and limitations of the biosensor.
Design: Academic researchers and biotech developers would be required to publish validation data, uncertainty ranges, and known limitations alongside biosensor results. Regulatory agencies or funding bodies could mandate this as a condition for approval or funding.
Assumptions: This assumes that validation metrics can adequately capture real-world biological variability and that transparency will lead to more responsible interpretation rather than confusion.
Risks of Failure & “Success”: Failure could occur if validation data are incomplete or misleading. If successful, excessive emphasis on uncertainty might reduce clinician confidence or limit the biosensor’s perceived usefulness.
Governance Action 3: Equity oriented access and deployment incentives
Purpose: Advanced molecular tools often benefit only well-resourced institutions. This action proposes promoting equitable access so the biosensor does not widen existing healthcare disparities.
Design: Public research institutions, funding agencies, or non-profit organizations could subsidize deployment in public hospitals or low-resource settings. Open research protocols or cost-reduction strategies could be encouraged at early development stages.
Assumptions: This assumes that cost and infrastructure are the main barriers to access, and that equitable deployment is feasible once the technology is validated.
Risks of Failure & “Success”: If unsuccessful, the technology may remain inaccessible to most patients. If overly successful, rapid deployment without adequate training or infrastructure could increase misinterpretation or misuse
4. Score each of the governance actions against the rubric of policy goals.
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
1
1
3
• By helping respond
2
1
n/a
Foster Lab Safety
• By preventing incident
2
1
n/a
• By helping respond
3
1
n/a
Protect the environment
• By preventing incidents
n/a
n/a
n/a
• By helping respond
n/a
n/a
n/a
Other considerations
• Minimizing costs and burdens to stakeholders
1
2
3
• Feasibility?
1
1
2
• Not impede research
1
2
2
• Promote constructive applications
2
1
1
5. Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties.
Based on the scoring, I would prioritize a combination of Option 1 (clinical-use restriction policy) and Option 2 (mandatory validation and transparency requirements), as these most directly support non-malfeasance by preventing harmful clinical misuse and reducing misinterpretation of biosensor outputs. While Option 3 (equitable access incentives) is ethically important, prioritizing it too early could increase risks if the technology is deployed before sufficient validation. This approach assumes that clinicians and researchers will follow institutional guidelines and that validation data will be communicated clearly. A key trade-off is that stricter rules and validation requirements may slow adoption and increase administrative burden, but this is justified to protect patient safety. This recommendation is primarily directed toward academic research institutions, clinical research hospitals, and institutional review boards (IRBs), which play a central role in approving, overseeing, and guiding the ethical use of emerging biomedical technologies. This exercise also highlighted the ethical risk of over-relying on early biological signals, even when technologies are developed with good intentions, as well as the tension between promoting access and ensuring safety. As a result, governance actions that clearly define scope of use, emphasize transparency, and support phased deployment are essential to prevent unintended harm.
Questions from lecture’s 2
Homework Questions from Professor Jacobson: [Lecture 2 slides]
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
R/:DNA polymerase is the enzyme that copies DNA in cells. It is very accurate, but not perfect. It usually makes one mistake every 100 million to 10 billion bases (DNA letters). The human genome is about 3 billion bases long, so if there were no corrections, we could get a few errors each time the genome is copied. But biology has special systems that fix those mistakes. These systems include proofreading (checking as the DNA is copied) and DNA repair mechanisms. Thanks to these, most errors are fixed, and the final copy is very accurate.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
R/: There are many different ways to write the DNA code for a human protein. That’s because the genetic code is redundant most amino acids can be coded by more than one set of three DNA letters (called codons). For example, the amino acid leucine has six different codons. So, a protein with 300 amino acids can have many thousands of possible DNA sequences that all make the same protein. But in practice, not all of these versions work well. Some codons are rare and slow down protein production. Some DNA sequences can fold into weird shapes that block the process. Also, each organism has codon preferences, meaning it uses some codons more than others. That’s why scientists choose special codons when they want the protein to be made properly in a specific cell.
Homework Questions from Dr. LeProust: [Lecture 2 slides]
What’s the most commonly used method for oligo synthesis currently?
R/: The most common way to make short pieces of DNA (called oligos) is by using a chemical method that adds one DNA letter at a time. This method is called phosphoramidite synthesis. It’s like building a word by adding one letter at a time, very carefully. Scientists do this on a small surface in the lab. It works well for making short DNA pieces, which are used in many experiments.
Why is it difficult to make oligos longer than 200nt via direct synthesis?
R/: It’s hard to make DNA oligos longer than 200 bases because the process isn’t perfect. Each time a new DNA letter is added, there’s a small chance it goes wrong or doesn’t stick. If you’re only adding a few letters, most of the strands are correct. But if you try to add 200 letters, those small mistakes add up and many of the strands end up incomplete or with errors. That’s why longer DNA pieces are usually made by putting together shorter, more accurate pieces.
Why can’t you make a 2000bp gene via direct oligo synthesis?
R/: You can’t make a 2000bp gene directly with oligo synthesis because the process isn’t accurate enough for something that long. When scientists make DNA, they add one letter at a time, and each step has a small chance of making a mistake. For short pieces of DNA (like 100–200 bases), the process works well. But if you try to make something as long as 2000 bases all at once, too many mistakes happen, and most of the DNA ends up broken or wrong. So instead, scientists make shorter pieces and then join them together to build the full gene.
Homework Question from Professor George Church: [Lecture 2 slides]
Choose ONE of the following three questions to answer; and please cite AI prompts or paper citations used, if any.
[Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
R/: The 10 essential amino acids in all animals are: histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, valine, and arginine (which is considered essential in some conditions, like during growth or illness). These amino acids are called “essential” because animals cannot make them on their own and must obtain them from food sources.
The concept of the “Lysine Contingency,” as described in Professor George Church’s lecture (slide #4), is a biosafety strategy in synthetic biology. It involves engineering organisms to be dependent on lysine, meaning they cannot survive without an external supply of it. This acts as a safety switch if the organism escapes into the environment, where lysine is not available, it cannot grow or survive.
Understanding that lysine is essential and not naturally produced by the organism itself makes this strategy seem very effective and logical. It adds a layer of control to prevent genetically modified organisms from spreading outside the lab, which is an important concern in biotechnology research.
Sources:
Prof. George Church – Lecture 2, Slide #4: “The Genetic Code”