Week 5 HW: Protein design part II
week-05-hw-protein-design-part-ii
Part A: SOD1 Binder Peptide Design (From Pranam) Part 1: Generate Binders with PepMLM
- Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation. original sequence:
sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn]
OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
A4V mutation:
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card: https://colab.research.google.com/drive/1tOhdz-ZO91A0u--wk65WY69CSMcL0e70?usp=sharing
Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
Record the perplexity scores that indicate PepMLM’s confidence in the binders.
Binder Pseudo Perplexity
0 WHYPPVGAEHKE 19.688582
1 WRYPATAARWGX 7.422996
2 WRYPVVAAELWX 12.680934
3 WHYYVVGVAWKX 17.178792
FLYRWLPSRRGG
Part 2: Evaluate Binders with AlphaFold3 https://alphafoldserver.com/fold/79f46573a7da07d6
- Navigate to the AlphaFold Server: alphafoldserver.com
- For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
- Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
- In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
https://alphafoldserver.com/fold/79f46573a7da07d6
For WHYPPVGAEHKE peptide:
- ipTM = 0.28
- pTM = 0.83
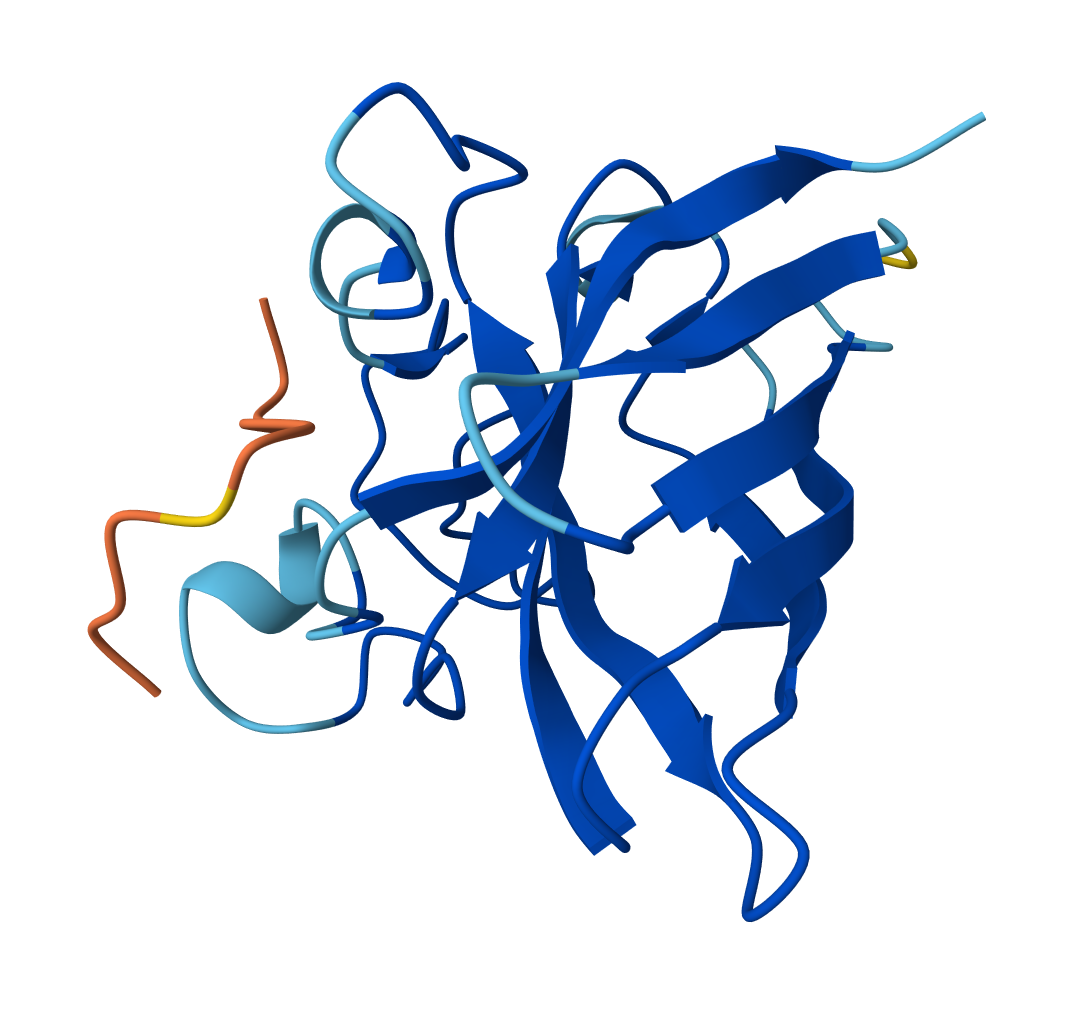
For WRYPATAARWGX peptide:
- ipTM = 0.42
- pTM = 0.87
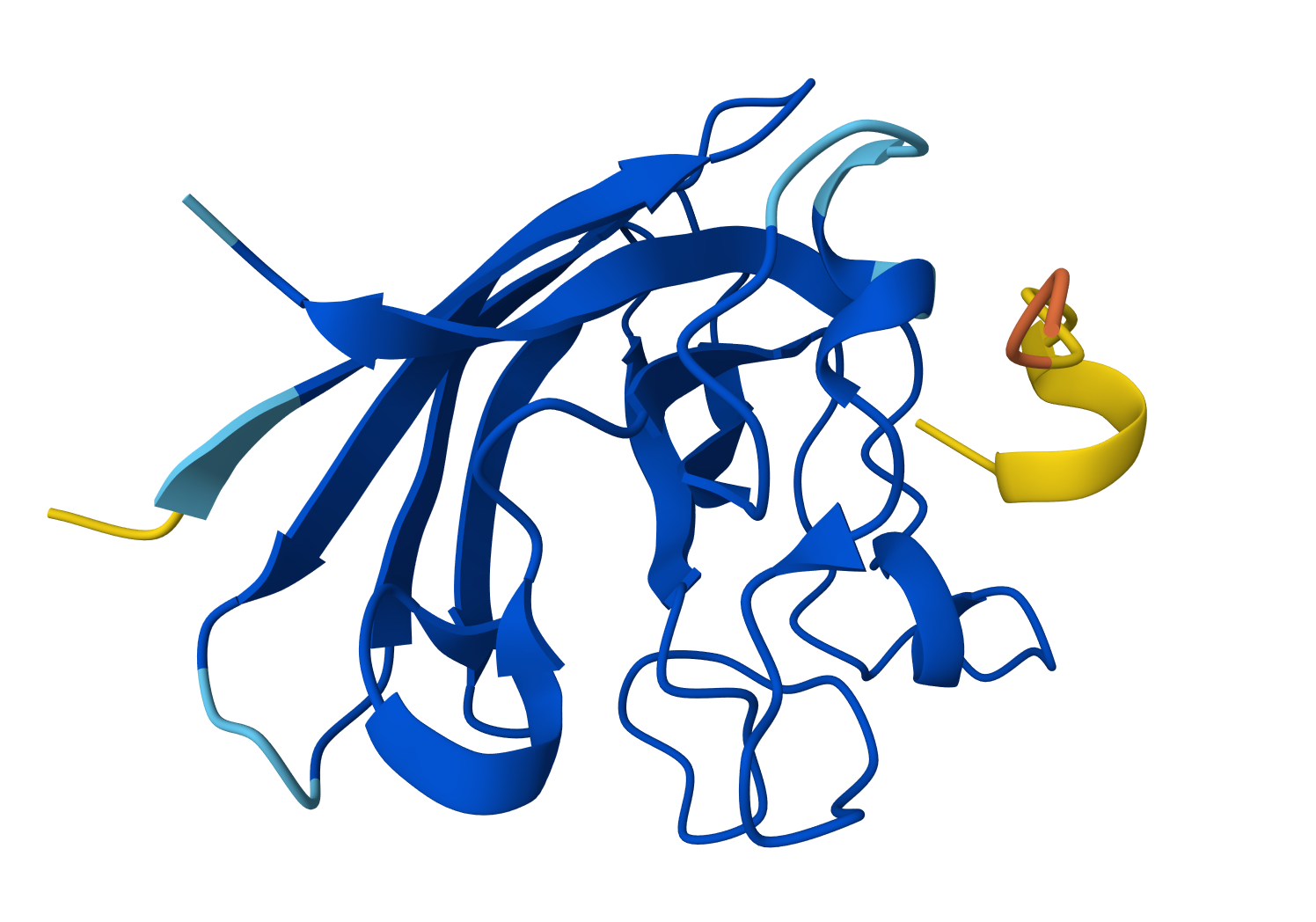
For WRYPVVAAELWX peptide:
- ipTM = 0.3
- pTM = 0.83
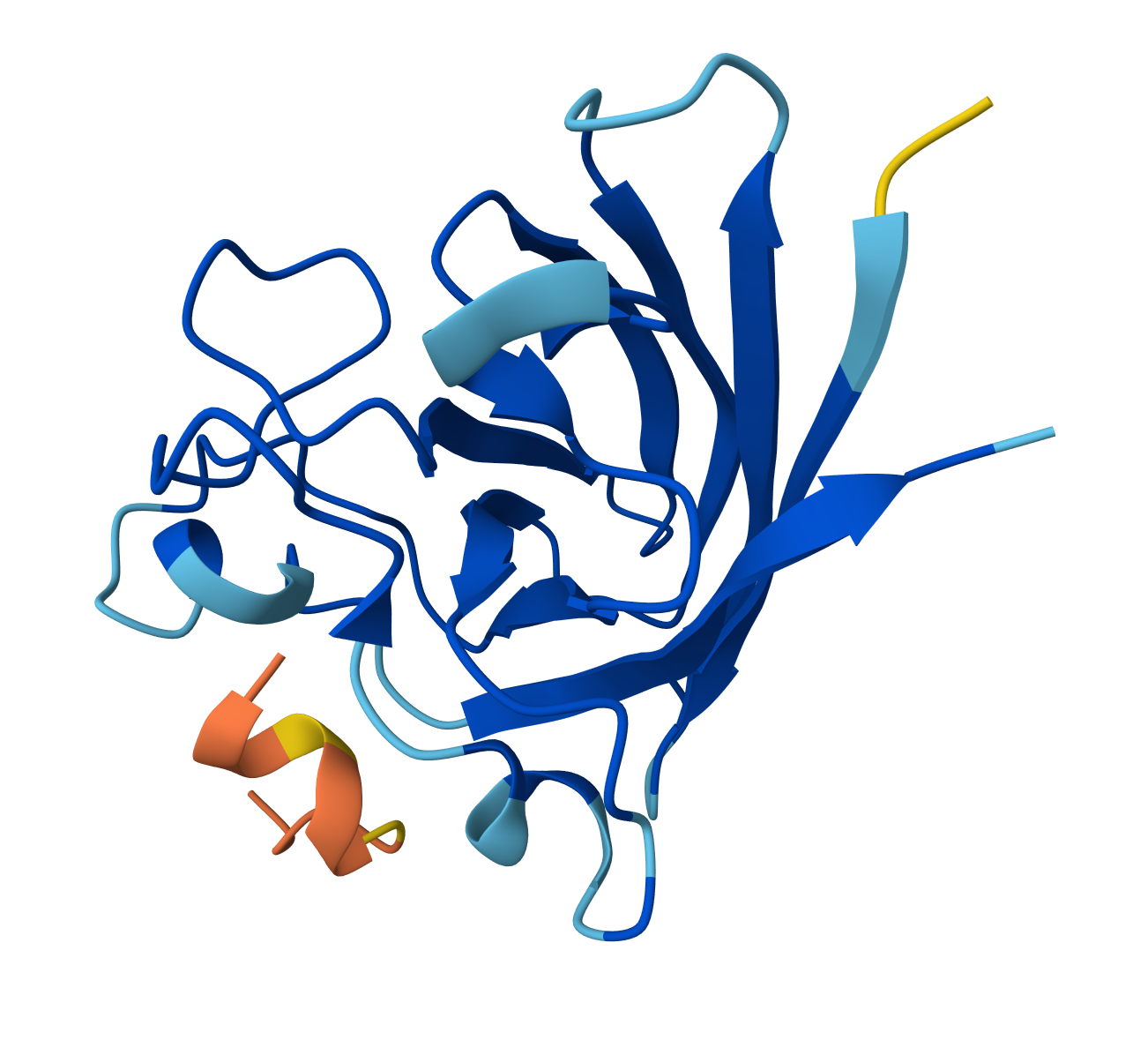
For WHYYVVGVAWKX peptide:
- ipTM = 0.42
- pTM = 0.81
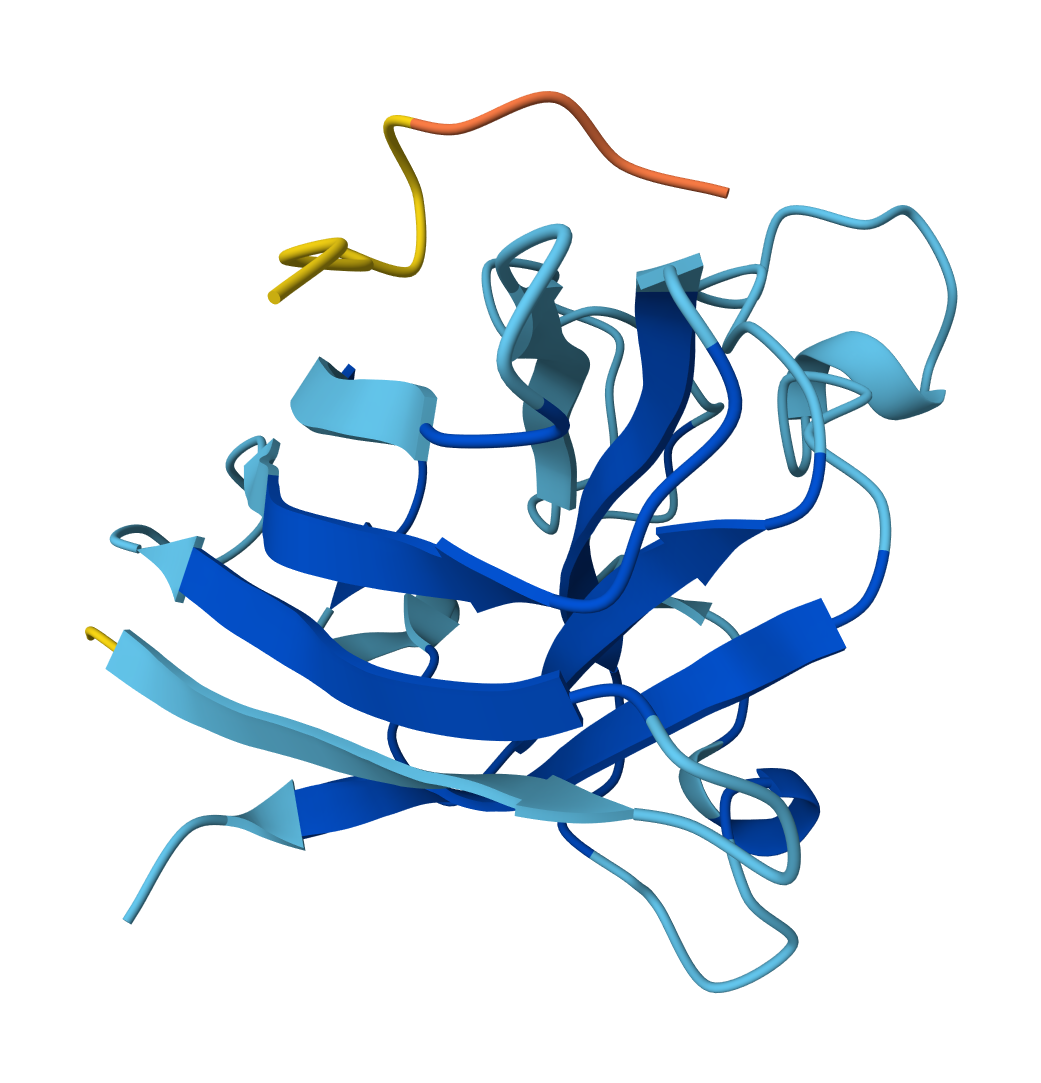
For FLYRWLPSRRGG peptide:
- ipTM = 0.31
- pTM = 0.83
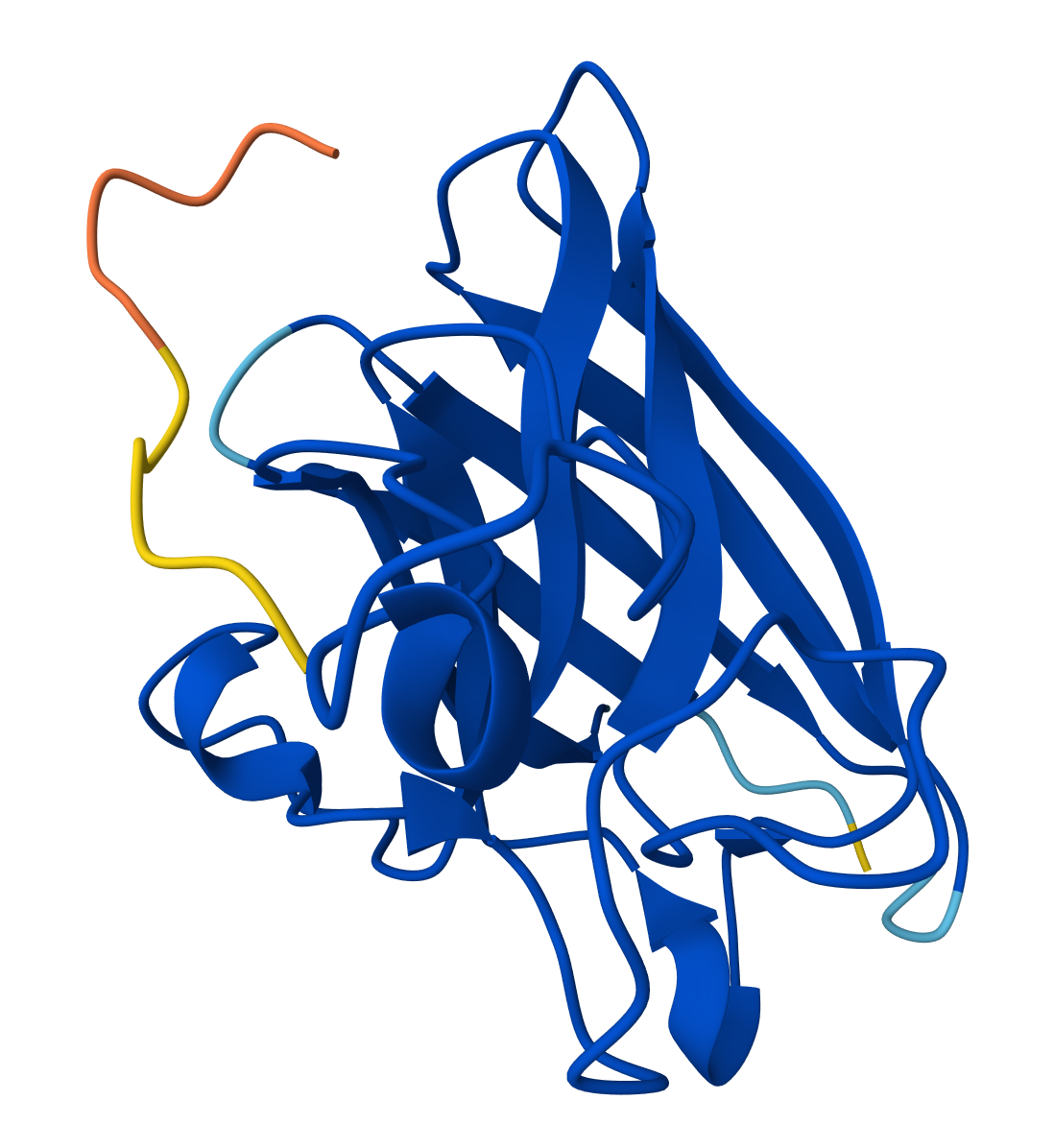
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse https://huggingface.co/spaces/ChatterjeeLab/PeptiVerse For each PepMLM-generated peptide:
- Paste the peptide sequence.
- Paste the A4V mutant SOD1 sequence in the target field.
- Check the boxes
- Predicted binding affinity
- Solubility
- Hemolysis probability
- Net charge (pH 7)
- Molecular weight
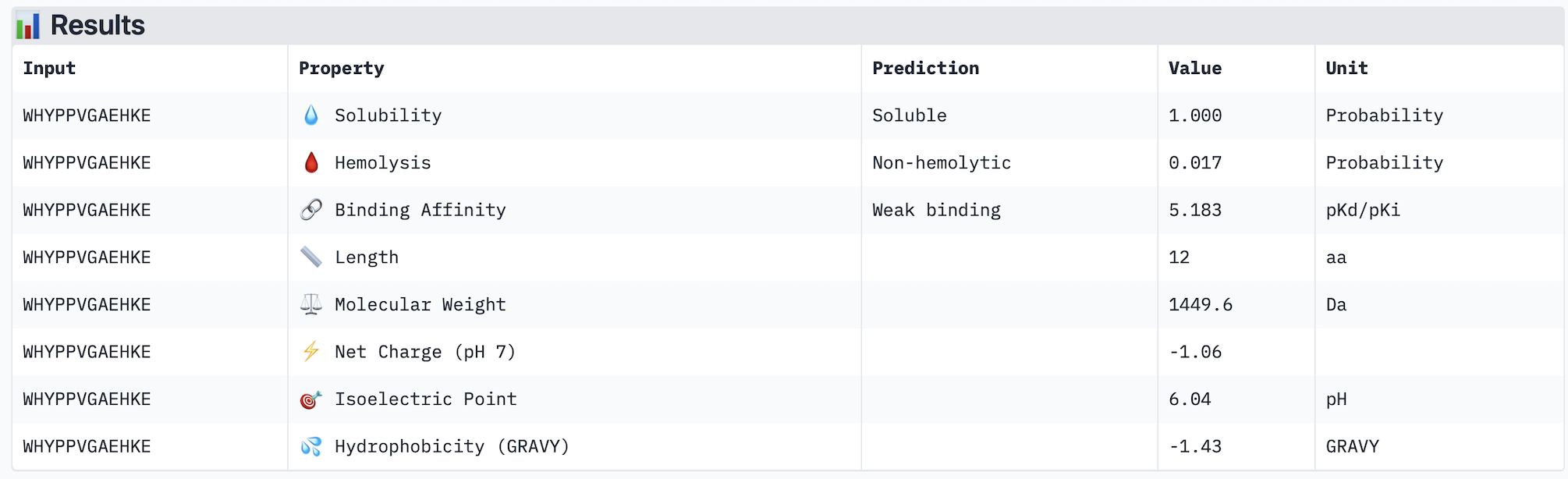
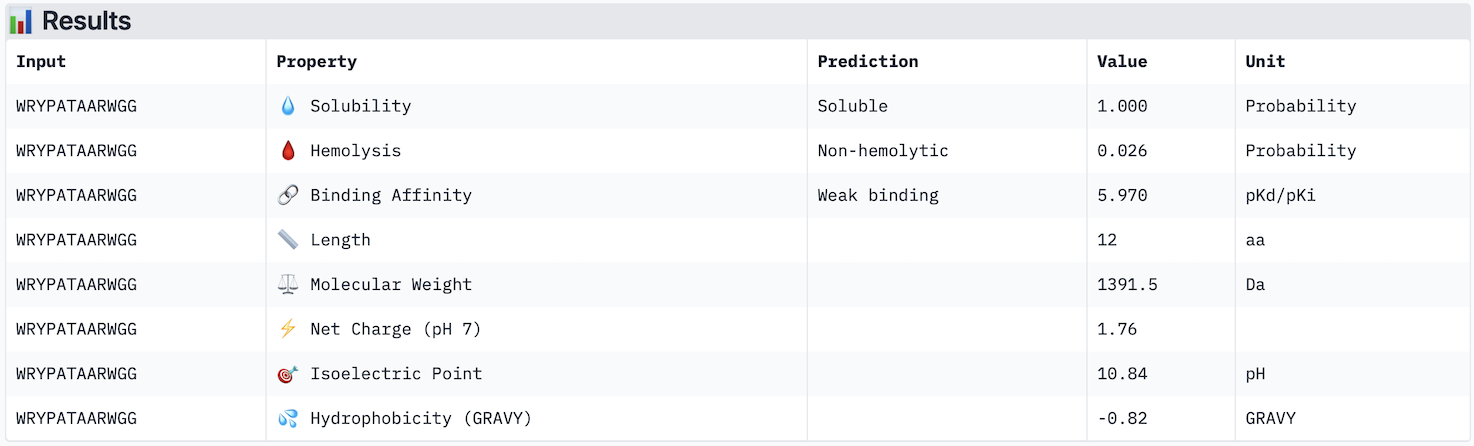
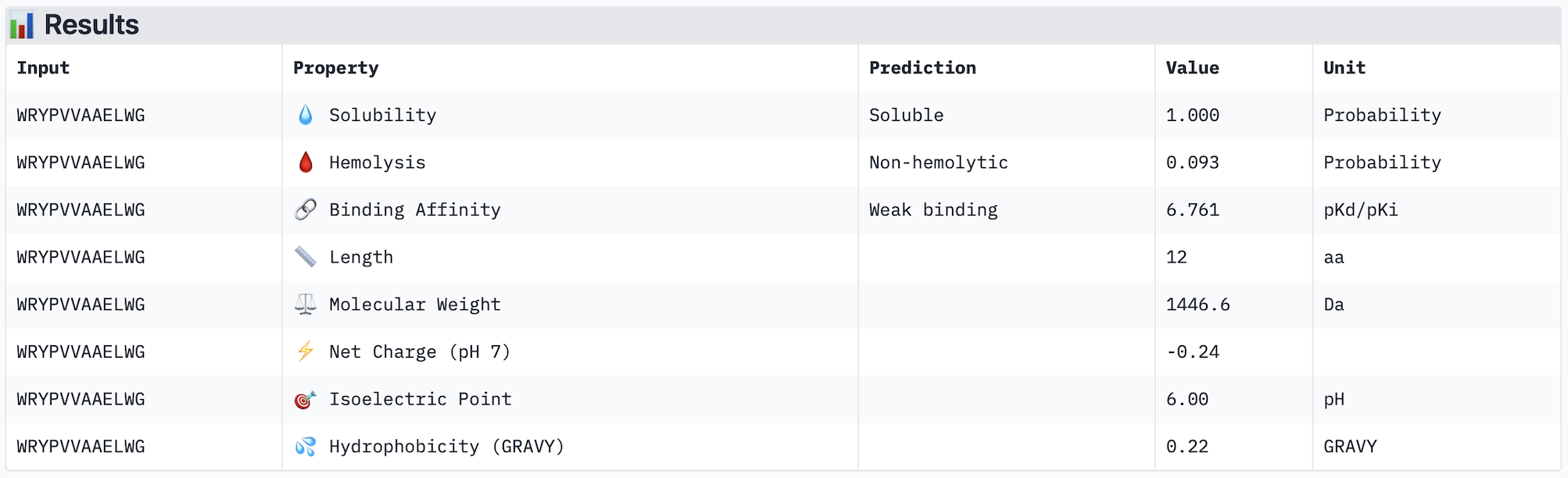
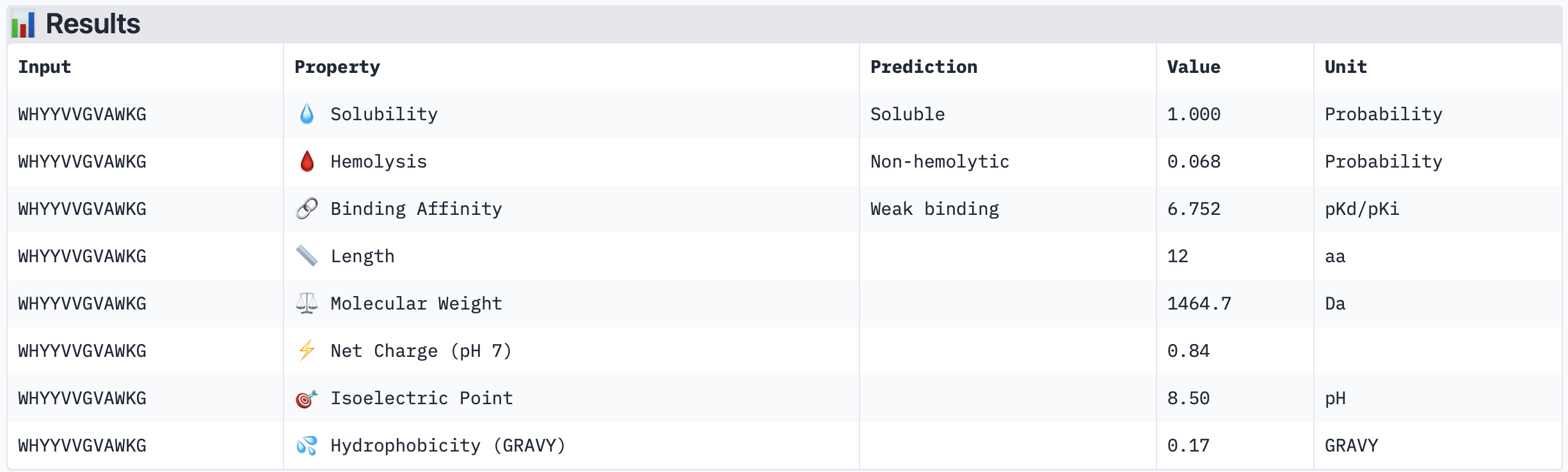
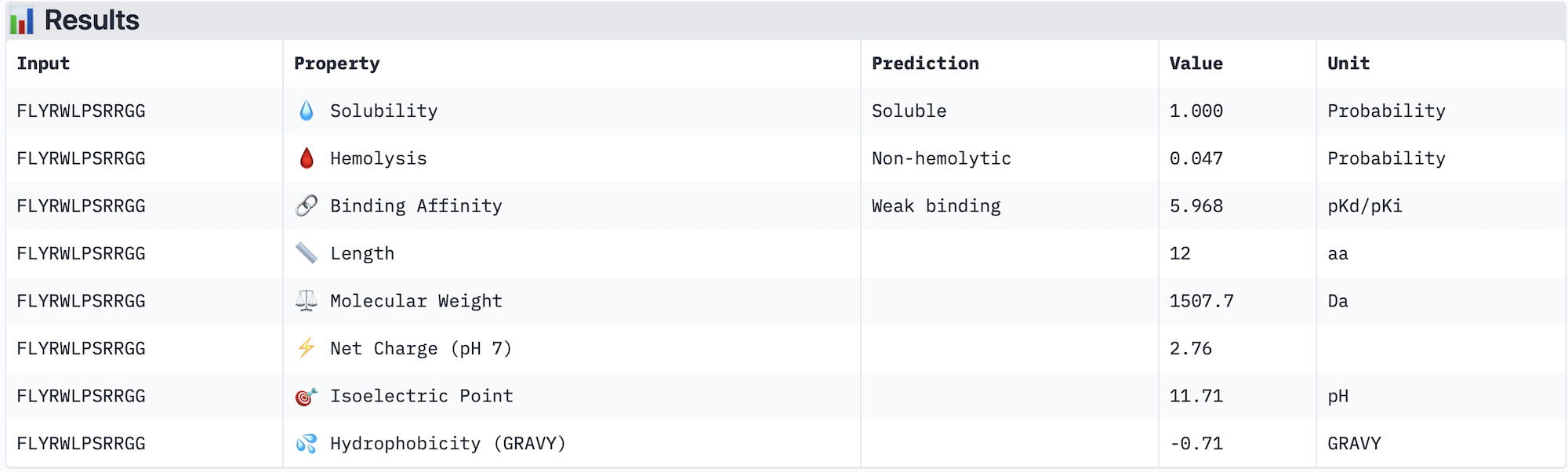
Across the five peptides evaluated, ipTM scores ranged from 0.28 to 0.42, all below the 0.5 threshold typically considered indicative of confident interface prediction, suggesting that none of the peptides form a highly stable predicted complex with A4V mutant SOD1. Interestingly, the two peptides with the highest ipTM scores (WRYPATAARWGG and WHYYVVGVAWKG, both 0.42) did not show the strongest predicted binding affinity in PeptiVerse — instead, WRYPVVAAELWG had the highest affinity (pKd 6.761) despite a lower ipTM of 0.30, indicating that structural confidence and predicted affinity do not perfectly correlate in this case. Notably, none of the peptides were predicted to be hemolytic or poorly soluble, making all of them therapeutically safe from a preliminary standpoint. The known binder FLYRWLPSRRGG performed modestly, with an ipTM of 0.31 and affinity of 5.968, and was outperformed by several PepMLM-generated candidates. The peptide that best balances structural confidence and therapeutic properties is WHYYVVGVAWKG, with the joint-highest ipTM (0.42), strong predicted affinity (6.752), excellent solubility, and very low hemolysis probability (0.068). I would advance WHYYVVGVAWKG for further development based on this combination of structural and therapeutic predicted properties.
Part 4: Generate Optimized Peptides with moPPIt
Due to GPU memory constraints with the available T4 runtime (moPPIt requires an A100 or L4 GPU), the notebook encountered errors during execution. However, based on the moPPIt framework, the key difference between moPPIt generated peptides and PepMLM generated peptides is the level of design control. While PepMLM samples peptides conditioned only on the target sequence, moPPIt uses Multi Objective Guided Discrete Flow Matching (MOG-DFM) to simultaneously optimize binding affinity, solubility, and hemolysis while steering the peptide toward specific residues in this case residues 1-7 near the A4V mutation site. This means moPPIt peptides would be expected to show more consistent engagement with the N-terminal region of SOD1, potentially with better therapeutic profiles overall.
Before advancing any moPPIt-generated peptide to clinical studies, I would evaluate them through the following steps: (1) run PeptiVerse to assess binding affinity, solubility, and hemolysis as done for PepMLM peptides; (2) submit to AlphaFold3 to confirm structural engagement with residues 1-7; (3) perform in vitro binding assays such as surface plasmon resonance (SPR) to measure actual Kd values; (4) test cytotoxicity in neuronal cell lines; and (5) assess proteolytic stability since short peptides are rapidly degraded in vivo
Part C: Final Project: L-Protein Mutants
Stage 1: Engineer novel L-protein mutants using protein design tools Stage 2: Synthesize the L-protein mutant gene via Twist Stage 3: Clone the L-protein mutant gene into a plasmid using Gibson Assembly Stage 4: Test the L-protein mutant’s structural integrity using the Nuclera system Stage 5: Test the L-protein in E. coli with plaque assays