Week 5 HW: Protein Design Part II
Part 1: Generate Binders with PepMLM
1. Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
Superoxide Dismutase 1 (SOD1) is a human enzyme that plays a critical role in protecting cells from oxidative stress by catalyzing the conversion of superoxide radicals into oxygen and hydrogen peroxide. It is a small, 154-amino-acid protein that typically forms a stable homodimer and contains a β-βarrel core structure with metal cofactors, copper and zinc, essential for its catalytic activity. Mutations in SOD1, such as the A4V variant, are associated with familial amyotrophic lateral sclerosis (ALS), a neurodegenerative disorder. SOD1 is widely expressed in the cytoplasm and is a key model protein for studying protein folding, aggregation, and targeted protein degradation strategies.
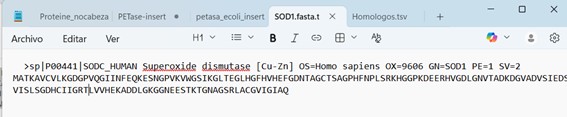
Figure 1. Superoxide Dismutase 1 (SOD1) sequence, fasta file.
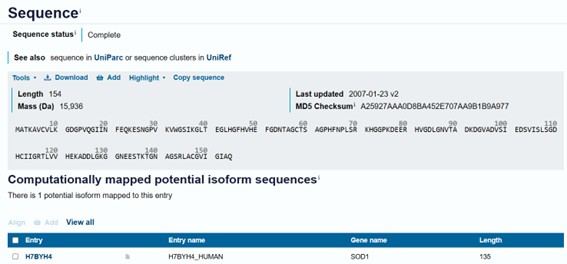
Figure 2. Superoxide Dismutase 1 (SOD1) sequence.
Mutation A4V was inserted:
Original:
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQMutated:
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
2. Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
Figure 3. PepMLM Colab.
3. Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
As the original script generated four peptides containing some invalid characters, I improved the script to produce only valid peptides without those characters. You can access the script cell here:
Additionally, I included a scritp to calculate the pseudo-perplexity of a known peptide.
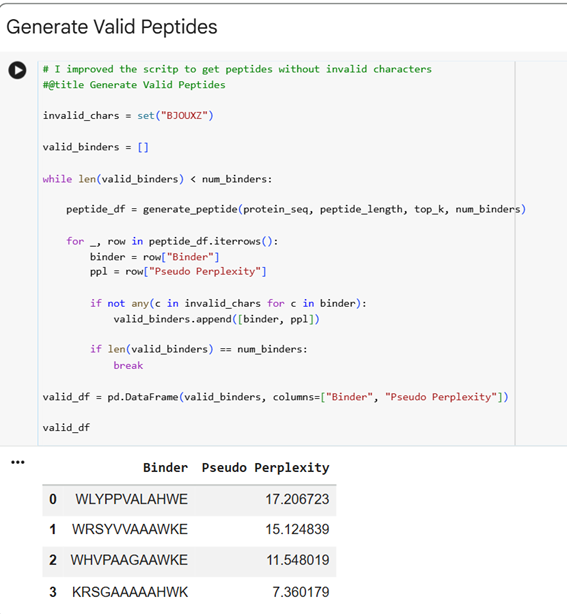
The .cvs file gives the peptides and the Pseudo Perplexity values:
Pseudo-perplexity measures the model’s confidence in a peptide sequence given the target protein context. Lower values indicate that the peptide is more consistent with patterns of protein–peptide interactions learned during training, while higher values suggest lower confidence. In the table, WRSGAAGAAWWK has the lowest pseudo-perplexity (7.12), indicating the model is most confident in this generated peptide, whereas the known binder FLYRWLPSRRGG has the highest value (20.64), reflecting comparatively lower model confidence. The other generated peptides fall in between, showing moderate confidence according to the model.
4. To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
- I open the test.csv file in excel, added a line and insert the SOD1-binding peptide FLYRWLPSRRGG. Record the perplexity scores that indicate PepMLM’s confidence in the binders.
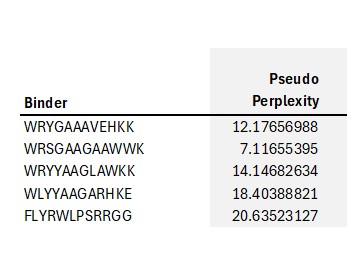
Figure 6
PepMLM assigns lower pseudo-perplexity scores to several generated peptides compared with the known SOD1-binding peptide FLYRWLPSRRGG. The peptide WRSGAAGAAWWK shows the lowest perplexity (7.12), indicating that the model predicts it as the most probable binder among the candidates.
Part 2: Evaluate Binders with AlphaFold3
1. Navigate to the AlphaFold Server: alphafoldserver.com
2. For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
Figure 7
Figure 8
3. Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
The predicted binders 1–4 localize near residues 96–102 of the protein, a region that forms one of the parallel β-strands contributing to the β-sheet structure. In contrast, the known binder (binder 5) appears near residues 11–15. None of the binders are located close to the N-terminus or C-terminus of the protein. Additionally, the peptides do not appear tightly attached to the protein surface or inserted into a binding pocket, suggesting that they remain relatively exposed rather than strongly surface-bound or buried.
The predicted interaction confidence, measured by ipTM, is relatively low for all binders, with values ranging from 0.25 to 0.44. Binder 2 shows the highest interaction score (ipTM = 0.44), followed by binder 4 (0.39) and binder 1 (0.34), while binders 3 (0.28) and 5 (0.25) show lower confidence. These values indicate moderate to low confidence in the predicted peptide–protein interactions, suggesting that the binding poses may be weak or uncertain.
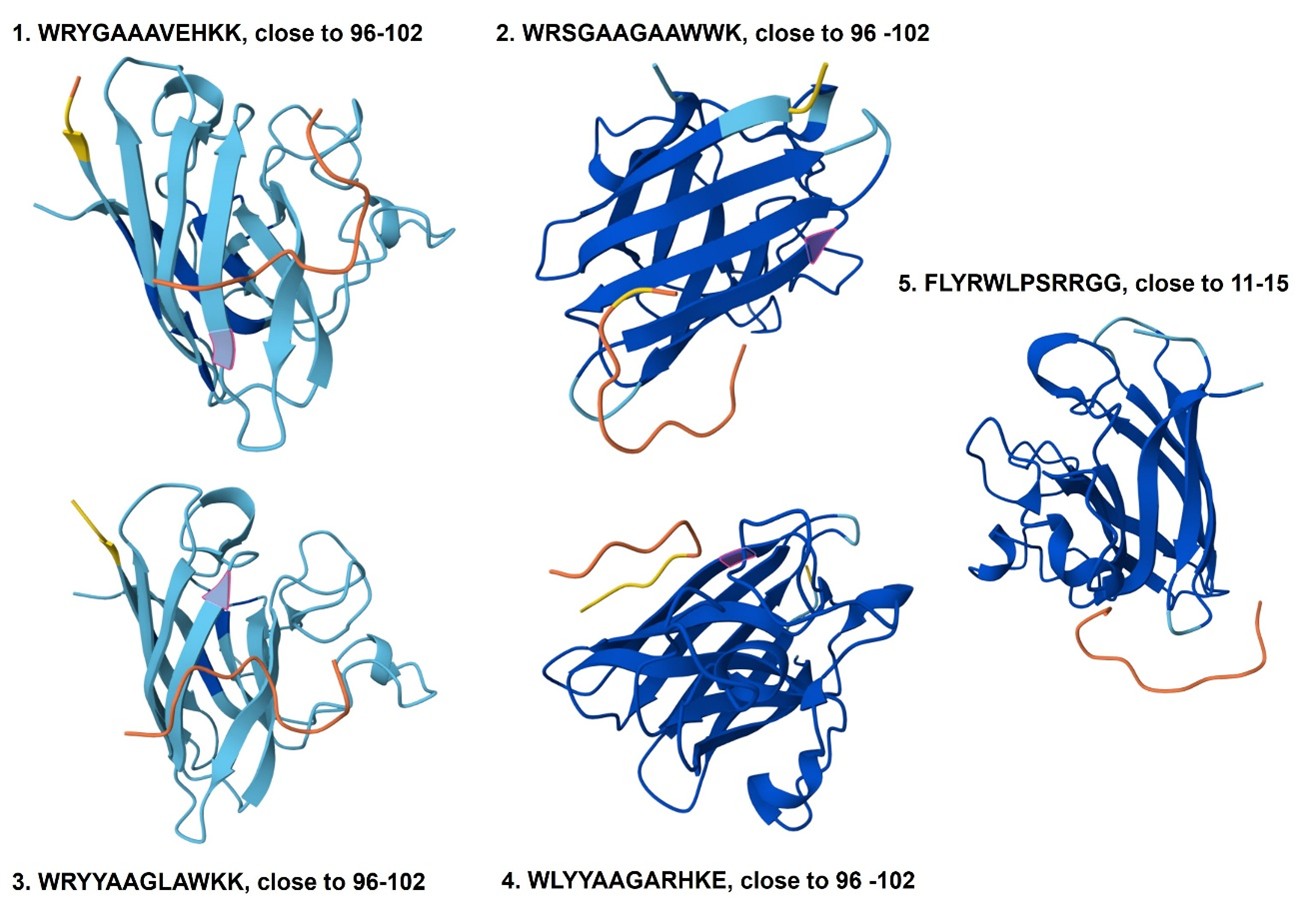
Figure 9
4. In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
The predicted complexes show low ipTM values (0.25–0.44), indicating weak confidence in protein–peptide interactions. The PepMLM-generated peptide WRSGAAGAAWWK has the highest ipTM score (0.44), exceeding that of the known binder FLYRWLPSRRGG (0.25), but none of the peptides appear to form a stable interaction with SOD1 in the predicted models.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse**
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
1. Paste the peptide sequence.
2. Paste the A4V mutant SOD1 sequence in the target field.
- Check the boxes:
- Predicted binding affinity
- Solubility
- Hemolysis probability
- Net charge (pH 7)
- Molecular weight
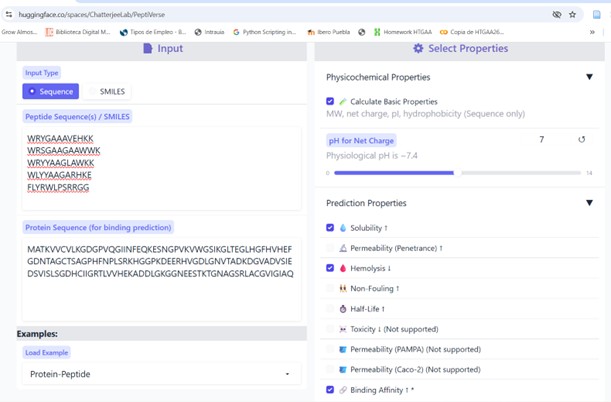
Figure 10
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Choose one peptide you would advance and justify your decision briefly.
The predicted binding affinities range from pKd/pKi values of ~5.4 to 6.9, indicating weak to moderate binding overall. The peptide WRSGAAGAAWWK shows both the highest predicted affinity (pKd 6.94) and the highest ipTM value (0.44), suggesting some agreement between the structural prediction from AlphaFold3 and the affinity prediction from Peptiverse. However, most peptides did not appear to form stable interactions with SOD1 in the structural models, remaining mostly surface-proximal rather than clearly bound. All peptides show good predicted solubility (score = 1) and very low hemolysis probabilities (<0.05), indicating favorable therapeutic properties. Among the candidates, WRSGAAGAAWWK (2) best balances predicted binding strength, structural confidence, and safety properties.
See all values from alphaFold web y pepTiVerse per peptide:
| Binder | Pseudo Perplexity | ipTM | pTM | Solubility | Hemolysis | Binding Affinity (pKd/pKi) | Length (aa) | Molecular Weight (Da) | Net Charge (pH 7) | Isoelectric Point (pH) | Hydrophobicity (GRAVY) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| WRYGAAAVEHKK | 12.17656988 | 0.34 | 0.75 | 1 | 0.021 | 5.436 | 12 | 1415.6 | 1.85 | 9.7 | -1 |
| WRSGAAGAAWWK | 7.11655395 | 0.44 | 0.84 | 1 | 0.032 | 6.939 | 12 | 1346.5 | 1.76 | 11 | -0.46 |
| WRYYAAGLAWKK | 14.14682634 | 0.28 | 0.73 | 1 | 0.024 | 6.735 | 12 | 1512.8 | 2.76 | 10 | -0.66 |
| WLYYAAGARHKE | 18.40388821 | 0.39 | 0.87 | 1 | 0.029 | 5.887 | 12 | 1464.6 | 0.85 | 8.5 | -0.82 |
| FLYRWLPSRRGG | 20.63523127 | 0.25 | 0.83 | 1 | 0.047 | 5.968 | 12 | 1507.7 | 2.76 | 11.71 | -0.71 |
Notes:
- ipTM score estimates the confidence of protein–peptide interactions. Values above 0.7 indicate a reliable interaction, values between 0.5 and 0.7 suggest a possible interaction, and values below 0.5 indicate that an interaction is unlikely.
- Solubility is predicted as a binary value where 0 indicates not soluble and 1 indicates soluble.
- Hemolysis probability predicts whether a peptide may damage red blood cells; 0 indicates non-hemolytic and 1 indicates hemolytic.
- Binding affinity (pKd/pKi) reflects binding strength (−log10 of Kd or Ki), where higher values indicate stronger binding. Values <5 indicate very weak binding (>100 µM), 5–6 weak (~100 µM), 6–7 moderate (~10 µM), 7–8 good (~1 µM), and >8 strong binding (<100 nM).
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
1. Open the moPPit Colab linked from the HuggingFace moPPIt model card.
2. Make a copy and switch to a GPU runtime.
Figure 11
3. In the notebook:
- Paste your A4V mutant SOD1 sequence.
- Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
- Set peptide length to 12 amino acids.
- Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
4. After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
moPPIt Generated Peptides
| Binder | Hemolysis | Solubility | Affinity | Motif |
|---|---|---|---|---|
| GGGRQKCFTLNM | 0.9349 | 0.75 | 6.5764 | 0.5096 |
| SKKQKTITCELC | 0.9677 | 0.8333 | 7.2126 | 0.6561 |
| KTCKKSFEKKQN | 0.9739 | 0.9167 | 6.2008 | 0.6707 |
| GTTCIQGKKKDE | 0.9785 | 0.9167 | 6.3731 | 0.5156 |
The peptides generated with moPPIt differ from the PepMLM peptides because their generation was guided by multiple objectives defined in the notebook, including affinity, motif targeting, solubility, hemolysis, and specificity. In contrast, PepMLM generates peptides based mainly on sequence likelihood conditioned on the target protein sequence. As a result, the moPPIt peptides show moderate predicted affinities (pKd ≈ 6.2–7.2), relatively high motif scores (≈0.51–0.67), and good solubility predictions (0.75–0.92). The PepMLM peptides also show moderate affinities but were not optimized simultaneously for these therapeutic objectives during generation.
However, when one of the generated peptides was modeled with the SOD1 structure using AlphaFold, the peptide did not appear to form a stable interaction with the protein surface. This suggests that although the predicted affinity and motif scores are moderate, the structural predictions do not clearly support strong binding.
Comparison of PepMLM and moPPIt Generated Peptides
| Method | Binder | Hemolysis | Solubility | Affinity |
|---|---|---|---|---|
| PepMLM | WRYGAAAVEHKK | 0.021 | Yes | 5.436 |
| PepMLM | WRSGAAGAAWWK | 0.032 | Yes | 6.939 |
| PepMLM | WRYYAAGLAWKK | 0.024 | Yes | 6.735 |
| PepMLM | WLYYAAGARHKE | 0.029 | Yes | 5.887 |
| PepMLM | FLYRWLPSRRGG | 0.047 | Yes | 5.968 |
| moPPIt | GGGRQKCFTLNM | 0.9349 | 0.75 | 6.5764 |
| moPPIt | SKKQKTITCELC | 0.9677 | 0.8333 | 7.2126 |
| moPPIt | KTCKKSFEKKQN | 0.9739 | 0.9167 | 6.2008 |
| moPPIt | GTTCIQGKKKDE | 0.9785 | 0.9167 | 6.3731 |
Before advancing any peptide toward clinical studies, further validation would be necessary. Additional structural modeling and docking analyses should confirm stable binding to the intended region of SOD1. Promising candidates should then be synthesized and tested experimentally through in vitro binding assays, hemolysis and cytotoxicity tests, and stability analyses to verify their predicted properties. Only peptides showing clear binding and favorable safety profiles would be considered for further development.
Part C: Final Project: L-Protein Mutants
High level summary: The objective of this assignment is to improve the stability and auto-folding of the lysis protein of a MS2-phage. This mechanism is key to the understanding of how phages can potentially solve antibiotic-resistance.
HTGAA 2026 – Protein Design II: Phage homework
1. Background Bacteriophages are viruses that infect bacteria. In this homework, I am working with MS2, a small RNA phage that infects Escherichia coli. My goal is to design mutants of the MS2 L-protein that can overcome a bacterial resistance mechanism.
2. The L-protein
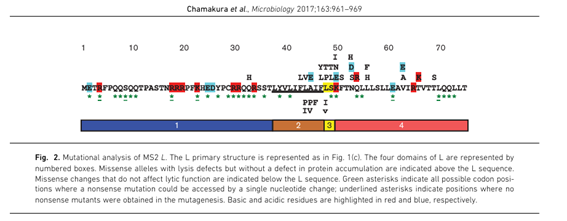
Figure 12. From Chamakura et al. 2017.
3. The DnaJ problem The L-protein needs DnaJ to work. DnaJ (UniProt P08622) is a bacterial chaperone from E. coli that physically interacts with the soluble domain of L-protein to activate lysis. Bacteria can mutate DnaJ to become resistant to the phage — the best-characterized resistance mutation is P330Q in the C-terminal domain of DnaJ. When this mutation is present, the interaction with L-protein is disrupted and the phage cannot lyse the cell.
To fight this resistance, I designed L-protein mutants — especially in the soluble domain — that might interact with DnaJ differently, making it harder for the bacterium to escape. To evaluate this, I used AF2-Multimer (ColabFold) to co-fold each L-protein mutant with two versions of DnaJ:
- DnaJ wild-type: to check whether the mutation changes the natural interaction
- DnaJ P330Q: to check whether the mutant can still interact with the resistant form of DnaJ
4. Reading the experimental data The paper by Chamakura et al. (2017) provides a mutational analysis of the L-protein. For each position, missense mutations were tested and scored for two things:
- Lysis: does the mutant still lyse the cell? (1 = yes, 0 = no)
- Protein levels: is the mutant protein stable and detectable? (1 = yes, 0 = no) A good candidate mutation must have Lysis = 1 and Protein = 1. This means the protein is stable and still functional. Mutations with Lysis = 0 are defective and not useful.
5. MS2 L-protein candidate mutations (Lysis = 1, Protein = 1) from L-protein mutants - excel To filter the candidates, I used the Excel file provided in the homework materials: L-protein Mutants. This file contains the experimental results from Chamakura et al. (2017), with each mutation scored for Lysis and Protein levels. From the full dataset of 35 functional mutations, I filtered for those with both Lysis = 1 and Protein = 1. This gave me 10 strong candidates to choose from.
Soluble domain (pos 1–40)
| Position | AA change | bp change | Lysis | Protein | Note |
|---|---|---|---|---|---|
| 13 | P → L | C→T (pos 38) | 1 | 1 | |
| 15 | S → A | T→G (pos 43) | 1 | 1 | |
| 18 | R → G | A→G (pos 52) | 1 | 1 | RRR cluster (DnaJ binding site) |
| 18 | R → I | G→T (pos 53) | 1 | 1 | RRR cluster (DnaJ binding site) |
| 30 | R → Q | G→A (pos 89) | 1 | 1 | |
| 30 | R → L | G→T (pos 89) | 1 | 1 | |
| 31 | R → I | G→T (pos 92) | 1 | 1 |
Transmembrane domain (pos 41–75)
| Position | AA change | bp change | Lysis | Protein | Note |
|---|---|---|---|---|---|
| 44 | L → P | T→C (pos 131) | 1 | 1 | TM start |
| 45 | A → P | G→C (pos 133) | 1 | 1 | TM start |
| 46 | I → F | A→T (pos 136) | 1 | 1 | TM start |
6. Conservation analysis with ClustalOmega To make sure our candidate mutations are well-chosen, I used BLAST to find similar L-protein sequences from other MS2 phage strains. I then aligned all the sequences using ClustalOmega to see which positions are conserved across evolution.
In the alignment, conserved positions are marked with * — these are amino acids that never change across strains, meaning they are probably essential for the protein to work. Variable positions have no symbol, meaning mutations there are better tolerated.
The results confirmed:
- The RRR cluster (positions 18–20) shows variation across strains. One natural MS2 strain already has isoleucine (I) at position 18 instead of arginine — exactly our mutation R18 → I. This makes our choice even more justified: if nature already “tried” this mutation and the phage survived, it is a safe and reasonable design.
- The transmembrane domain (positions 44–46) is conserved, with
*symbols in the alignment. This confirms these positions are structurally important. However, the experimental data from Chamakura et al. (2017) shows that mutations L44 → P and A45 → P maintain Lysis = 1 and Protein = 1, so I can proceed with confidence. - Position 30 (R) is also well conserved, suggesting it plays an important role in the soluble domain.
Overall, the conservation analysis validates our 5 final mutations: they target functionally important sites, but the experimental data confirms they are compatible with lysis activity.
The alignment was generated using ClustalOmega with BLAST results from the homework materials.
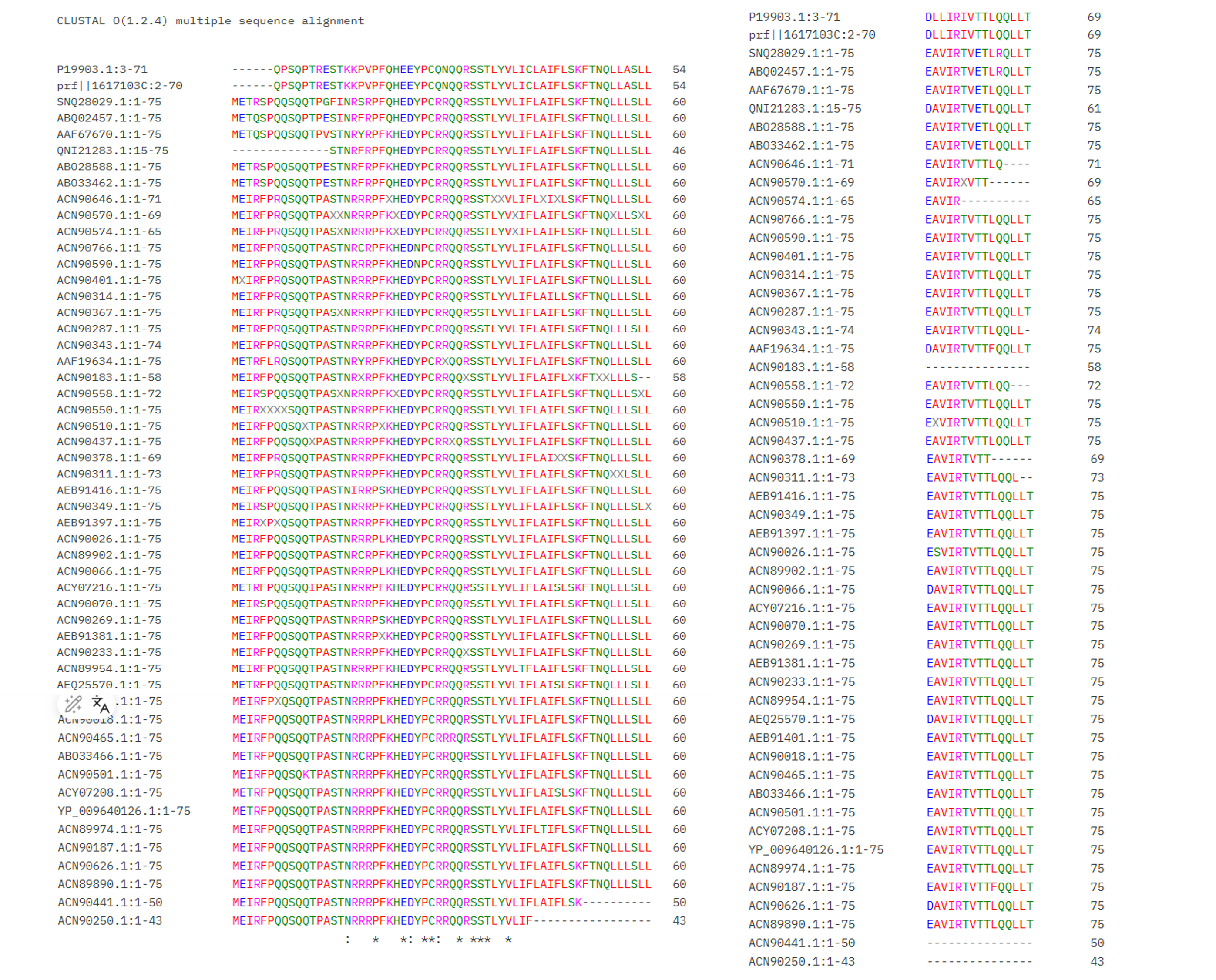 Figure: Multiple sequence alignment of MS2 L-protein homologs. Asterisks () indicate fully conserved positions.*
Figure: Multiple sequence alignment of MS2 L-protein homologs. Asterisks () indicate fully conserved positions.*
Download alignment file (.aln)
7. The 5 final mutants From the 10 candidates, I selected 5 mutations based on their biological relevance and experimental validation. The central question guiding my design is: can we engineer an L-protein that still lyses bacteria even when DnaJ carries the resistance mutation P330Q?
DnaJ normally interacts with the soluble domain of L-protein to activate lysis. When DnaJ is mutated at P330, this interaction is disrupted. Our hypothesis is that mutations in the L-protein — especially in the region that contacts DnaJ — could change the interaction interface, allowing the phage to lyse resistant bacteria.
For the soluble domain, I chose two mutations at position 18, both in the RRR cluster (positions 18–20). This cluster is the main contact point between the L-protein and DnaJ. Replacing the positively charged arginine with a neutral amino acid may shift how DnaJ recognizes this region, potentially allowing interaction through a different surface that the P330Q mutation does not affect.
For the transmembrane domain, I chose positions 44 and 45, at the start of the TM region. Replacing leucine or alanine with proline breaks alpha-helices, changing the geometry of the TM region and potentially affecting membrane insertion independently of DnaJ.
For the free choice, I selected R30 → Q, also in the soluble domain. Position 30 is another arginine outside the RRR cluster. Mutating it tests whether the broader charged region — not just positions 18–20 — contributes to DnaJ recognition.
All 5 mutations have Lysis = 1 and Protein = 1 in the experimental data.
| # | Mutation | Domain | Strategy |
|---|---|---|---|
| 1 | R18 → G | Soluble | Alter DnaJ contact surface |
| 2 | R18 → I | Soluble | Alter DnaJ contact surface |
| 3 | L44 → P | Transmembrane | Change TM geometry |
| 4 | A45 → P | Transmembrane | Change TM geometry |
| 5 | R30 → Q | Soluble (free choice) | Alter DnaJ contact surface |
8. Genomic overlap analysis The MS2 genome is very small and compact. The L-protein gene overlaps with two other genes: the coat protein gene and the replicase gene (rep). This means that a mutation in the L-protein gene can also change an amino acid in one of these other proteins, because they share the same nucleotides but are read in a different reading frame.
This genomic structure is shown in the Benchling snapshot from Leverkus et al. (2023):
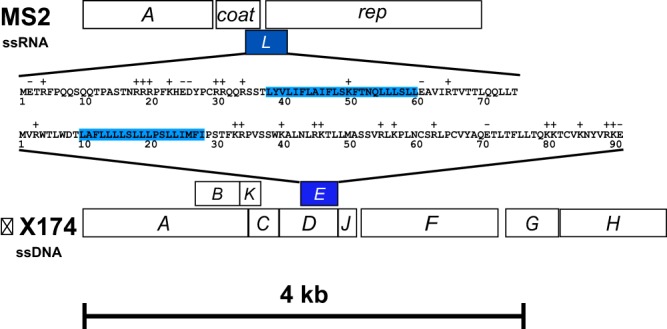
Figure: The L-protein gene (lys gene, green) overlaps with the coat protein gene (cp gene, orange) and the replicase gene (rep gene, pink). Each row shows the amino acids decoded in that reading frame from the same nucleotide sequence. Source: Leverkus et al. (2023), PMC5446614.
To check whether our 5 mutations are safe for the other genes, I wrote a Python script (with AI assistance) that reads the MS2 genome (NC_001417.2), locates each mutation in the genome, and checks what happens in the overlapping reading frames. The notebook is available here: MS2 Overlap Analysis
| Mutation | Overlapping gene | Codon # | Original | Mutated | Effect |
|---|---|---|---|---|---|
| R18 → G | none | — | — | — | No overlap ✓ |
| R18 → I | none | — | — | — | No overlap ✓ |
| R30 → Q | rep | 2 | TCG (S) | TCA (S) | Synonymous ✓ |
| L44 → P | rep | 16 | CCT (P) | CCC (P) | Synonymous ✓ |
| A45 → P | rep | 17 | CGC (R) | CCC (P) | Missense ⚠️ |
Four of the five mutations are safe for the overlapping genes. R18 → G and R18 → I fall in a region with no overlap at all. R30 → Q and L44 → P overlap with the rep gene but produce synonymous mutations — the amino acid in the replicase does not change. The only concern is A45 → P, which changes an arginine (R) to a proline (P) in the replicase (rep codon 17). This is a known limitation and would need to be tested experimentally.
Note: A synonymous mutation means the DNA sequence changes but the amino acid stays the same, because multiple codons can encode the same amino acid (e.g. CCT and CCC both code for Proline).
9. Python script The analysis was done in three cells in the MS2 Overlap Analysis Google Colab notebook
- Cell 1 — Load genome and verify L-protein translation
- Cell 2 — Analyze genomic overlap for the 5 selected mutations
- Cell 3 — Generate mutant sequences and DnaJ P330Q for AlphaFold
10. PepMLM results — pseudo perplexity analysis
I used PepMLM-650M to generate peptide binders for each of the 5 mutant L-protein sequences. PepMLM is a protein language model that generates short peptides and scores them using pseudo perplexity (PPL) — a measure of how “natural” or plausible a peptide is given the target protein. A lower PPL means the peptide fits the protein surface better.
For each mutant, I generated 8 candidate binders (15 amino acids long) and selected the one with the lowest PPL. As a biological reference, I used the fragment TNRRRPFKHEDYPCR (positions 14–28 of the original L-protein), which contains the RRR cluster — the known DnaJ binding site identified by Chamakura et al. (2017). Since this is a real functional fragment, we expect it to have a low PPL, and we use it as a baseline.
| Mutant | Best binder | Pseudo Perplexity |
|---|---|---|
| Reference (RRR cluster) | TNRRRPFKHEDYPCR | 2.988 |
| R30Q | STLGLLLADLLAKLL | 8.471 |
| R18I | STEGQQLADDLAFIL | 9.492 |
| R18G | TTEEQLQLDDEGFIF | 12.102 |
| L44P | TTWELQLLYDEGLLL | 15.185 |
| A45P | TTFGQLESRDGLLIL | 17.985 |
The reference fragment has the lowest PPL (2.988), consistent with it being a real, functional sequence. All mutant binders score higher, which is expected — the mutations alter the protein surface compared to the wild-type. Among the mutants, R30Q and R18I produce the most plausible binders (lowest PPL), suggesting their protein surface remains well-structured despite the mutation. A45P scores highest, consistent with it being the most structurally disruptive mutation — it also introduces a missense change in the rep gene (see Section 8).
Note: Pseudo perplexity is a computational score, not an experimental measurement. These results are used to rank and prioritize candidates, and need to be validated with structural prediction (AlphaFold) and experimental testing.
📓 PepMLM Pseudo-Perplexity Notebook
Script 📓 PepMLM Pseudo-Perplexity Notebook
The original PepMLM notebook was adapted to handle multiple protein sequences at once, reading them from a .csv file (one column called sequence) and generating binders for all 5 mutants in a single run. The key modification is a loop over all input sequences in the “Generate Valid Peptides” cell.
- Cell 1 — Install packages
- Cell 2 — Inputs and parameters
The original notebook assumes a single sequence. We set single_sequence = False and upload the all_sequences.csv file generated in the overlap analysis notebook (Section 9). Parameters: peptide_length = 15, top_k = 3, num_binders = 8.
- Cell 3 — Load model
- Cell 4 — Generate valid peptides (adapted for multiple sequences)
This is the key cell that was modified. The original notebook generates binders for a single sequence. Here we loop over all 5 mutant sequences and filter out any peptides containing ambiguous amino acid characters (B, J, O, U, X, Z).
- Cell 5 — Add mutant names and sort by PPL
- Cell 6 — Compare with reference binder and download results
11. Structural predictions with AF2-Multimer
We used ColabFold to run AF2-Multimer predictions for all 5 L-protein mutants co-folded with DnaJ. We ran two sets of jobs to answer two questions:
- Set A: Does each mutant still interact with normal DnaJ (wild-type)?
- Set B: Does any mutant interact better with the resistant DnaJ (P330Q)?
In ColabFold, both sequences are entered in the query_sequence field separated by :, which
tells AF2-Multimer to model them as two chains of a complex. We used the full DnaJ sequence
(376 aa, UniProt P08622) as chain B, and
each L-protein variant (75 aa) as chain A.
The key confidence score for our analysis is ipTM (interface predicted TM-score, 0–1): it measures how confidently AlphaFold predicts the interface between the two proteins. Values below 0.5 indicate low interface confidence; values above 0.8 are considered reliable. We also report pTM (overall complex confidence) and pLDDT (per-residue confidence). See figure below:
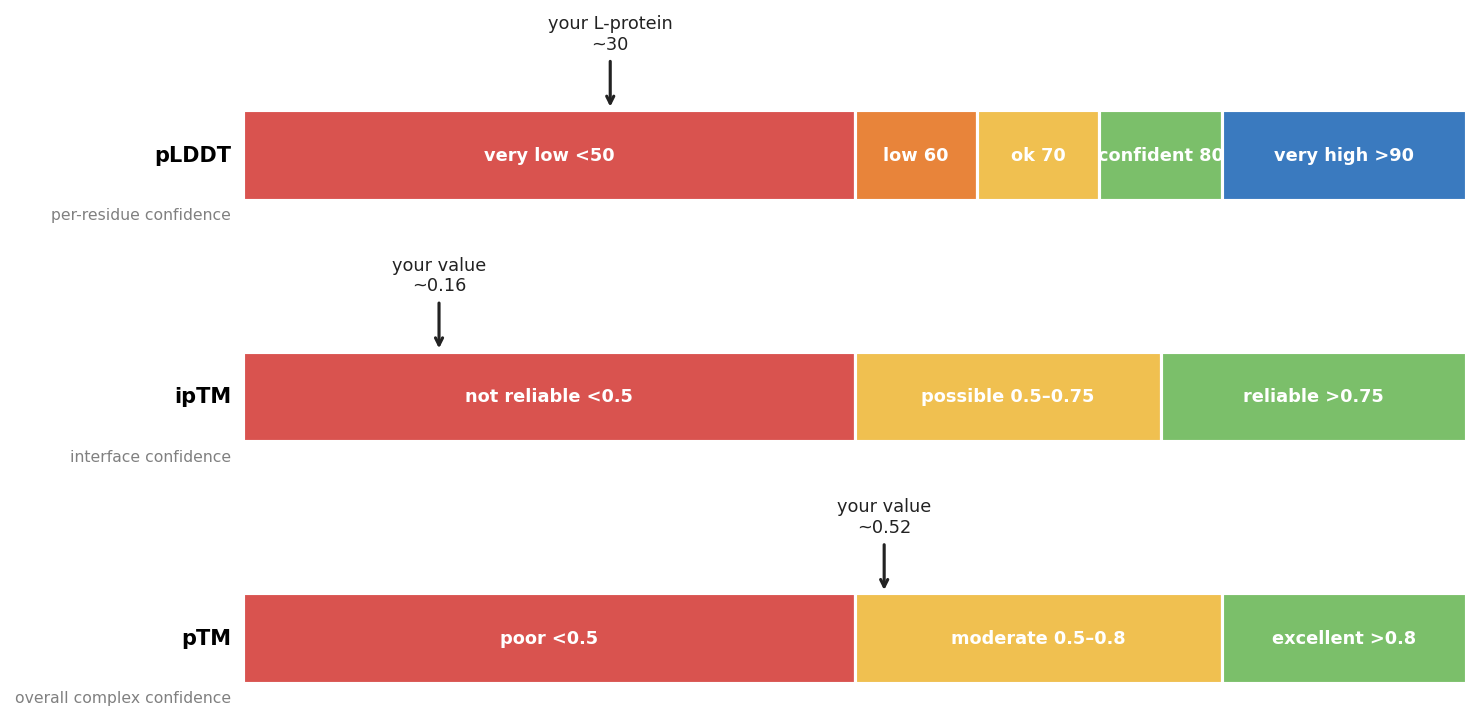
Important limitation: AF2-Multimer is not well-suited for membrane proteins. The L-protein transmembrane domain (positions 41–75) is predicted with low confidence (pLDDT < 50), which explains the very low ipTM values across all jobs. We use the wild-type result as a baseline and compare mutants relative to it, rather than interpreting the absolute values.
- Set A — L-protein mutants vs DnaJ wild-type
Question: Do our mutations preserve the ability to interact with normal DnaJ?
| Mutant | Best ipTM | Avg ipTM | pTM (avg) | pLDDT (avg) |
|---|---|---|---|---|
| Wild-type (control) | 0.161 | 0.134 | 0.523 | 75.6 |
| R18G | 0.160 | 0.137 | 0.523 | 75.7 |
| R18I | 0.160 | 0.138 | 0.523 | 75.7 |
| R30Q | 0.160 | 0.134 | 0.524 | 75.6 |
| L44P | 0.162 | 0.135 | 0.526 | 75.5 |
| A45P | 0.167 | 0.137 | 0.526 | 75.5 |
Answer: Yes. All mutants show ipTM scores essentially identical to the wild-type control. None of the mutations dramatically disrupt the predicted complex with DnaJ WT.
Download results — Set A (L-protein mutants vs DnaJ wild-type): WT · R18G · R18I · R30Q · L44P · A45P
- Set B — L-protein mutants vs DnaJ P330Q (resistant)
Question: Does any mutant recover interaction with the resistant DnaJ?
| Mutant | Best ipTM | Avg ipTM | vs WT baseline |
|---|---|---|---|
| Wild-type (baseline) | 0.160 | 0.138 | — |
| R18G | 0.161 | 0.143 | ↑ +0.001 |
| R18I | 0.161 | 0.141 | ↑ +0.001 |
| R30Q | 0.158 | 0.136 | ↓ −0.002 |
| L44P | 0.168 | 0.140 | ↑ +0.008 |
| A45P | 0.167 | 0.140 | ↑ +0.007 |
Answer: L44P and A45P show the highest ipTM scores against DnaJ P330Q — both above the wild-type baseline and consistent across all 5 predicted models. R18G and R18I show marginal improvement, and R30Q scores slightly below baseline.
To identify which residues are responsible for the predicted interaction, we used PyMOL to select all L-protein atoms within 5 Å of DnaJ. The iterate command loops over each selected atom and prints its residue number, residue name, and chain — giving us the exact list of contact residues at the interface.
- L44P contact residues: His24, Asp26 (soluble domain) and Leu56 (TM domain)
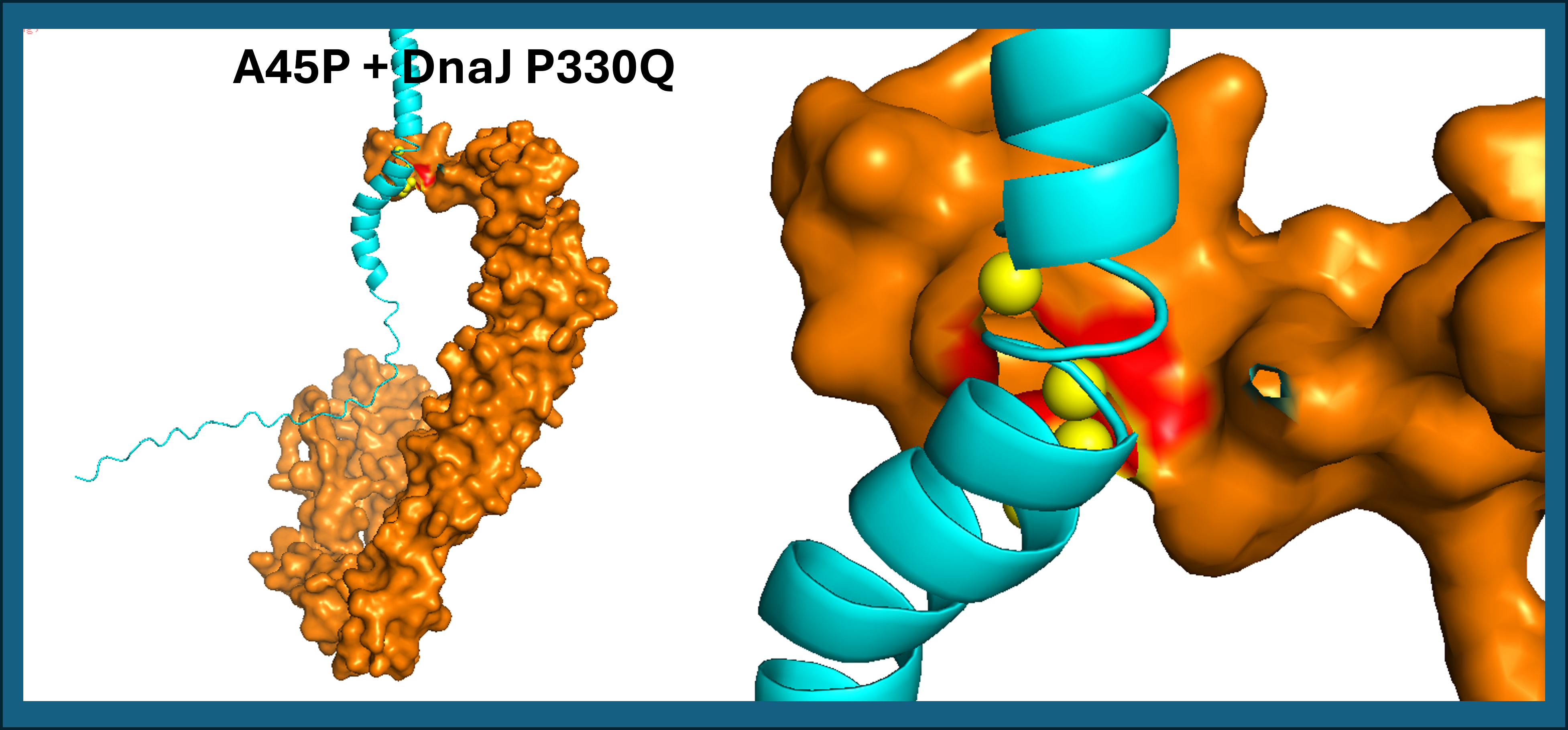
- A45P contact residues: Leu48, Thr52, Leu56 (TM domain only)
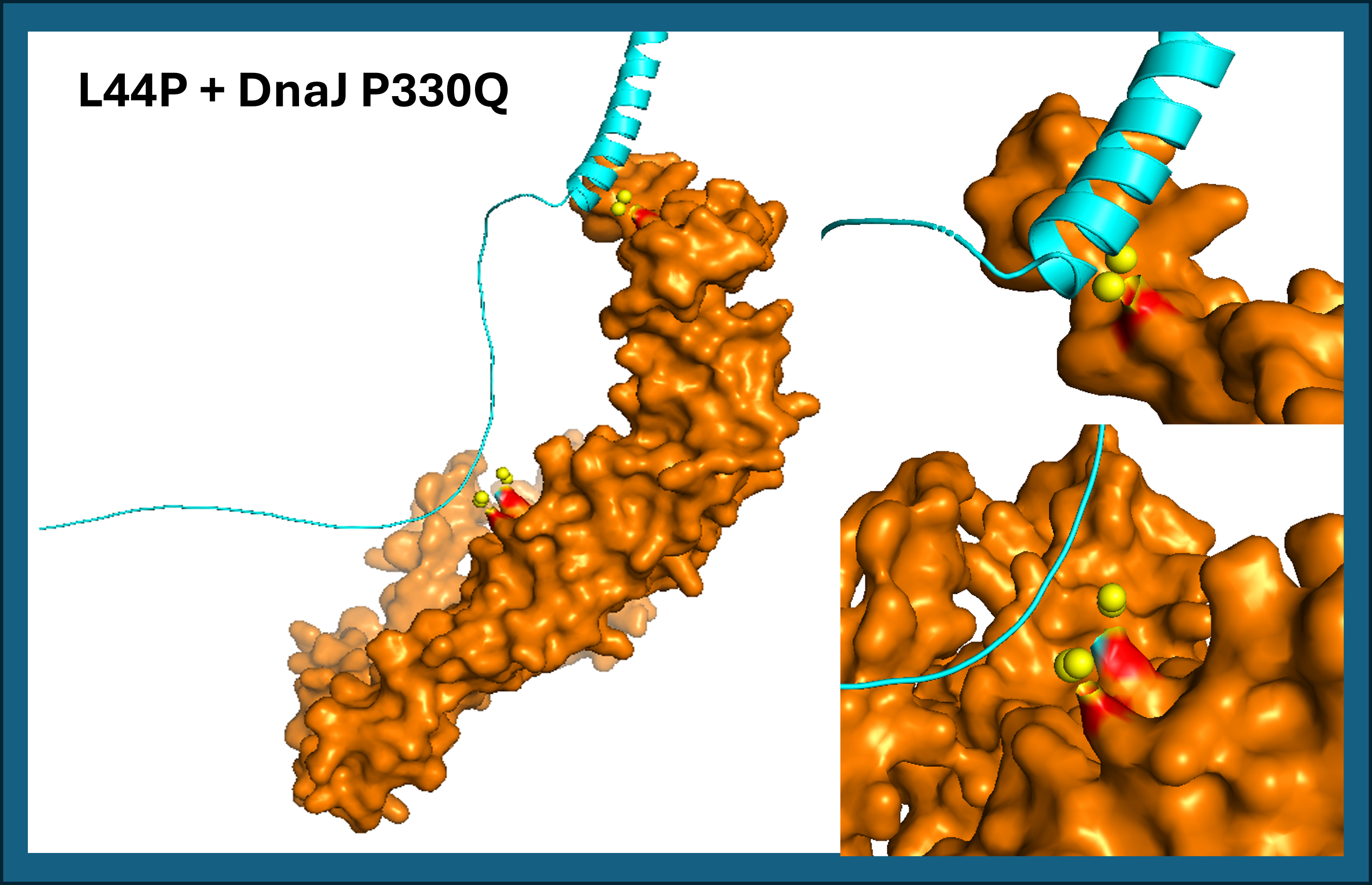
L44P shows two predicted contact zones with DnaJ P330Q: one in the soluble domain (His24, Asp26) and one in the transmembrane domain (Leu56). See Figure 16.
A45P contacts DnaJ exclusively through the transmembrane domain (Leu48, Thr52, Leu56), with no involvement of the soluble region. Both results suggest that proline mutations at positions 44 and 45 reposition the TM helix against DnaJ P330Q (See Figure 17). L44P additionally engages the soluble domain, potentially creating a more stable interaction surface.
The following PyMOL script was used to visualize the interface and list the contact residues mentioned above:
Download results — Set B (L-protein mutants vs DnaJ P330Q): WT · R18G · R18I · R30Q · L44P · A45P
12. Open Question: How do we define a “good” mutant?
A good mutant is one that can still kill bacteria — including resistant ones. Computationally, we use ipTM (predicted interface confidence with DnaJ) and pseudo-perplexity (how natural the sequence looks) as proxies. A mutant scores better if it has higher ipTM against resistant DnaJ P330Q than the wild-type, and lower pseudo-perplexity. By these criteria, L44P and A45P are our best candidates.
Conclusions
All five mutants maintain structural integrity when co-folded with wild-type DnaJ (Set A), confirming that none of the designed mutations disrupt the overall complex. This is a prerequisite for any mutant to be functional.
Against the resistant DnaJ P330Q (Set B), L44P and A45P stand out as the top candidates. Both show consistently higher ipTM scores across all 5 predicted models compared to the wild-type L-protein baseline. Notably, both mutations are located in the transmembrane domain (positions 44–45), suggesting that TM helix geometry may play a role in adapting to the P330Q change in DnaJ. PyMOL interface analysis confirms this: L44P contacts DnaJ P330Q at two zones (His24/Asp26 in the soluble domain and Leu56 in the TM domain), while A45P contacts exclusively through the TM domain (Leu48, Thr52, Leu56). By contrast, R30Q scores slightly below baseline, and R18G and R18I show only marginal improvement.
These are computational predictions with known limitations for membrane proteins. Experimental validation — such as phage lysis assays with resistant bacteria — would be required to confirm whether L44P or A45P actually overcome DnaJ P330Q resistance in vivo.
References
Chamakura KR, Tran JS, Young R. (2017). MS2 Lysis of Escherichia coli Depends on Host Chaperone DnaJ. Journal of Bacteriology, 199(12). https://doi.org/10.1128/JB.00058-17
Chamakura KR, Sham LT, Reed CA, Coleman S, Bernhardt TG, Young R. (2017). Mutational analysis of the MS2 lysis protein L. Microbiology, PMC5775895. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5775895/
Mezhyrova J, Martin J, Börnsen C, Dötsch V, Frangakis AS, Morgner N, Bernhard F. (2023). In vitro characterization of the phage lysis protein MS2-L. Microbiome Research Reports, 2:28. https://doi.org/10.20517/mrr.2023.28
Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M. (2022). ColabFold: making protein folding accessible to all. Nature Methods, 19, 679–682. https://doi.org/10.1038/s41592-022-01488-1
Chatterjee et al. PepMLM-650M — peptide binder generation using masked language models. GitHub: https://github.com/programmablebio/pepmlm
This homework was completed with the assistance of Claude AI (Anthropic).