Week 2 HW: DNA Read, Write, & Edit
Week 2 Lecture Prep
Homework Questions from Professor Jacobson
Questions:
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy? How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Answers: DNA polymerase, when it includes proofreading activity, has an error rate of approximately 1 mistake per 10⁶ nucleotides incorporated (10⁻⁶). This proofreading function allows the enzyme to detect and correct mismatched bases during DNA replication, significantly improving accuracy.
The human genome is approximately 3.2 × 10⁹ base pairs long. If DNA replication occurred with an error rate of 10⁻⁶, this would result in roughly: ~3,200 errors per full genome replication At first glance, this seems extremely high and potentially harmful for a complex organism like humans.
Biology addresses this problem through multiple layers of error correction, not just the polymerase itself:
- Proofreading by DNA polymerase (3’→5’ exonuclease activity) corrects errors as replication occurs.
- Post-replication DNA repair systems, such as mismatch repair, detect and fix remaining errors after replication.
- Additional repair pathways (base excision repair, nucleotide excision repair) correct DNA damage caused by environmental factors.
Together, these systems reduce the final mutation rate to approximately 1 error per 10⁹–10¹⁰ nucleotides, meaning that fewer than one error typically remains per cell division. This remarkable fidelity allows large genomes to remain stable over time.
An average human protein contains roughly 300–400 amino acids, corresponding to about 1,000–1,200 base pairs of coding DNA. Because the genetic code is degenerate (most amino acids are encoded by multiple codons), there are astronomically many possible DNA sequences that could encode the exact same protein sequence. In theory, the number of valid coding sequences increases exponentially with protein length.
Although many DNA sequences can theoretically encode the same protein, not all work biologically due to several constraints:
- Codon bias - Cells prefer certain codons based on available tRNAs, affecting translation efficiency.
- mRNA secondary structure - Certain sequences form strong hairpins or structures that block ribosome binding or movement.
- Translation speed and protein folding - Codon choice influences how fast a protein is translated, which affects proper folding.
- Regulatory signals - Some sequences may unintentionally introduce stop signals, splice sites, or degradation signals.
- Host compatibility - A sequence optimized for humans may not express well in bacteria, yeast, or other systems used in synthetic biology.
DNA is not just a code for proteins—it is a finely tuned system optimized for accuracy, efficiency, and regulation. This is why in biological engineering and synthetic biology, designing DNA requires much more than simply translating amino acid sequences into codons.
Homework Questions from Dr. LeProust
Questions: What’s the most commonly used method for oligo synthesis currently?, Why is it difficult to make oligos longer than 200nt via direct synthesis? Why can’t you make a 2000bp gene via direct oligo synthesis?
Answers: The most commonly used method for oligonucleotide synthesis today is solid-phase phosphoramidite chemical synthesis. In this method, DNA is synthesized one nucleotide at a time on a solid support (such as controlled pore glass or silicon). Each synthesis cycle consists of four main steps: coupling, capping, oxidation, and deprotection, and this cycle is repeated until the desired sequence length is reached. This approach is highly automated and is the foundation of both traditional column-based synthesizers and modern high-throughput platforms like silicon-based microarrays.
It is difficult to synthesize oligos longer than ~200 nucleotides because errors accumulate with each synthesis cycle. Each nucleotide addition has a small but non-zero failure rate (incomplete coupling, side reactions, or deletions). As the oligo gets longer:
- The probability of producing a full-length, error-free molecule drops exponentially
- Truncated sequences accumulate
- Overall yield of correct full-length product becomes very low
Additionally, long sequences are more prone to secondary structure formation, GC bias, and depurination, which further reduce synthesis efficiency and quality.
You cannot make a 2000 bp gene via direct oligo synthesis because the combined error rate and yield loss make it impractical and unreliable. At that length: - The chance that every single coupling step succeeds is extremely low - The vast majority of molecules would contain errors or truncations - Purifying a correct full-length 2000 bp product from the error background would be infeasible Instead, long genes are made by assembling many shorter oligos (typically 150–200 nt) using enzymatic assembly methods such as PCR-based assembly or ligation, followed by sequencing verification to ensure accuracy.
Homework Question from George Church
Question: What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
Answer: The 10 essential amino acids in animals are those that cannot be synthesized de novo and must be obtained from the diet. These are:
- Histidine
- Isoleucine
- Leucine
- Lysine
- Methionine
- Phenylalanine
- Threonine
- Tryptophan
- Valine
- Arginine (essential in growing animals and conditionally essential in adults)
Lysine is particularly important because it is often the most limiting amino acid in plant-based diets and many staple.
Conclusion (personal reflection)
Working through these topics reshaped how I think about biological engineering. I was struck by how biology embraces imperfection—DNA polymerase makes errors, oligo synthesis accumulates mistakes, and metabolic pathways have gaps—yet life remains remarkably stable through layered correction, repair, and interdependence. Rather than aiming for flawless systems, biology succeeds by managing error and constraint. I also gained a deeper appreciation for why synthetic biology relies on modular design and assembly instead of direct construction. The chemical limits of DNA synthesis and the evolutionary reality of essential amino acids show that engineering decisions are always bounded by fundamental biological rules. Overall, this reinforced for me that responsible bioengineering is not about overpowering biology, but about understanding its limits and designing within them. These constraints are not obstacles—they are guideposts for safer, more ethical, and more resilient innovation.
Part 1: Benchling & In-silico Gel Art "
Following the instructions described in HTGAA 2026 week 02
Make a free account at Benchling https://www.benchling.com/
Import the Lambda DNA https://www.neb.com/en/tools-and-resources/interactive-tools/dna-sequences-and-maps-tool

- Sequence file FASTA format: lambdafsa.txt
- Sequences file GenBank format: lambdagbk.txt
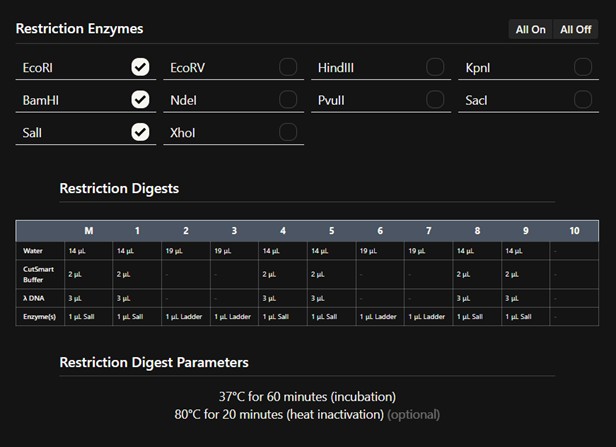
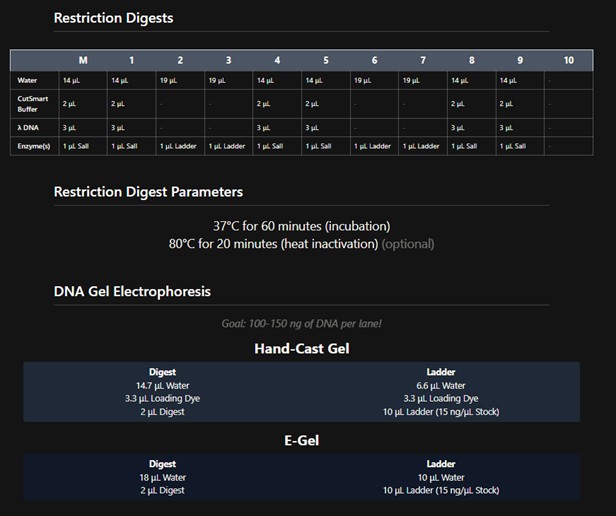
Simulate Restriction Enzyme Digestion with EcoRI

Create a pattern/image in the style of Paul Vanouse’s Using DNA Gel Art Interface To obtain this pattern:
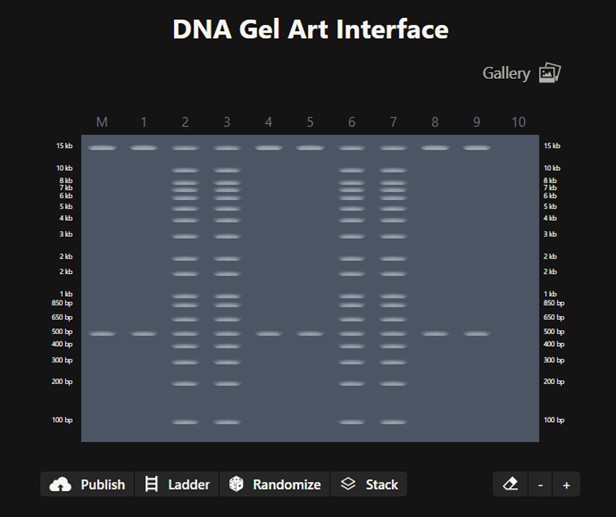
This is the protocol obtained
Part 2: NA to Committed Listeners
Part 3: DNA Design Challenge
- Choose your protein:
- Name: Bombyx mori 6-pyruvoyl tetrahydrobiopterin synthase purple (purple), NCBI Reference Sequence: NM_001046638.1
- Sequence:
- Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence
Following the instructions described in HTGAA 2026 week 02
Using Reverse translate tool ProteinIQ
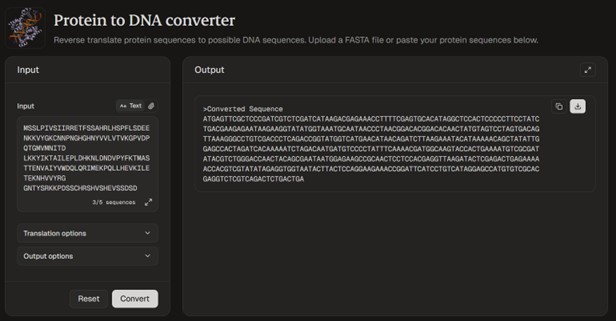 Converted sequence obtained:
Converted sequence obtained:
- Codon optimization
Codon optimization is a useful tool when expressing genes heterologously (in a different host organism), if problems occur when cloning a gene, or when optimizing gene expression level. VectorBuilder’s Codon Optimization tool is in-built into the Design Studio and can be used.
codon optimization vectorbuilder
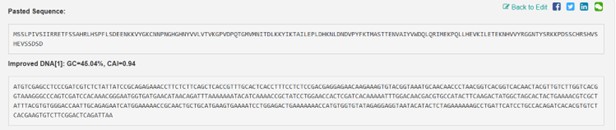
- Improved DNA: GC:45.04%, CAI=0.94
- You have a sequence! Now what?
To produce the Bombyx mori 6-pyruvoyl tetrahydrobiopterin synthase (purple) protein from its DNA sequence, the gene must first be expressed in a biological system capable of transcription and translation. This can be achieved using either cell-dependent or cell-free technologies.
One common approach is heterologous expression in living cells, such as E. coli. In this case, the DNA sequence encoding the purple protein would be inserted into an expression vector containing the necessary regulatory elements, including a promoter (such as T7), a ribosome binding site (RBS), and a transcription terminator.
Once the plasmid is introduced into bacterial cells through transformation, the host’s cellular machinery carries out transcription and translation. The promoter recruits RNA polymerase, which transcribes the DNA into messenger RNA (mRNA). The ribosome then binds to the mRNA at the RBS and translates the nucleotide sequence into a polypeptide chain, assembling amino acids according to the genetic code. The result is the recombinant purple protein produced inside the cell.
After expression, the protein can be purified, often using affinity tags such as a His-tag if included in the construct. Alternatively, eukaryotic systems such as yeast, insect cells, or silkworm-derived systems could be used if post-translational modifications or proper folding require a more complex cellular environment.
- How does it work in nature/biological systems?
Part 4: Prepare a Twist DNA Synthesis Order
Something is wrong with my Twist account, and I haven’t received a response. I couldn’t use this tool, but I did use Vectorbuilder
 Vectorbuilder design
Following the instructions of the Online Vector Design tool:
Vectorbuilder design
Following the instructions of the Online Vector Design tool:
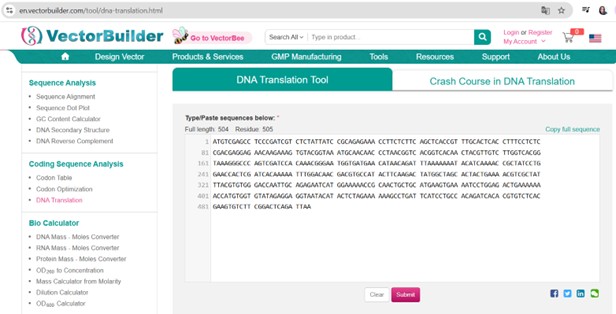
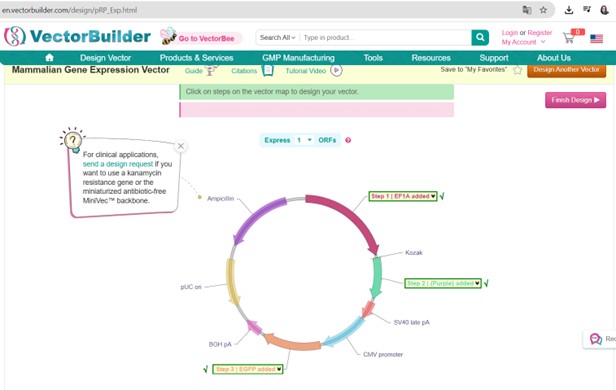
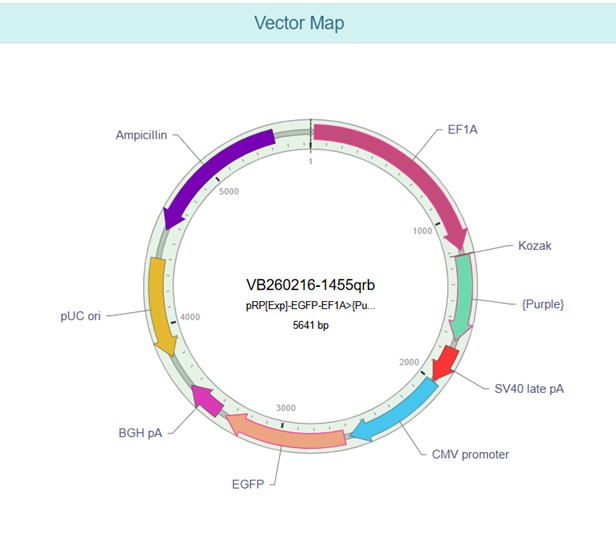
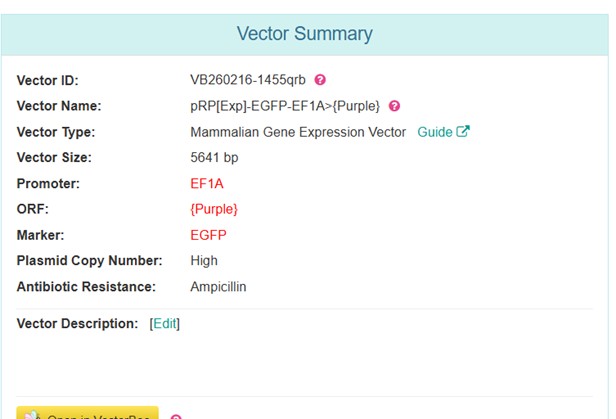
Vector Sequence:
Part 5: DNA Read/Write/Edit
- DNA read:
What DNA would you want to sequence and why? I would focus on sequencing DNA from microbial communities capable of degrading crustacean shell waste, with the goal of identifying novel chitinase genes. Chitinases are enzymes that break down chitin, a major structural component of shrimp and crab shells. Large amounts of crustacean waste are generated annually, creating environmental burdens, yet this material can be converted into value-added products such as chitin oligosaccharides with therapeutic potential (antimicrobial, anti-inflammatory, wound-healing properties). By sequencing environmental DNA from marine sediments, compost systems, or shell-processing waste streams, I aim to discover efficient chitinase genes that could be used for sustainable bioconversion and circular bioeconomy applications.
My input would be total environmental DNA (eDNA) extracted from samples rich in chitin-degrading microorganisms, such as shrimp shell waste piles or marine sediments. Preparation steps:
- DNA extraction from environmental samples.
- DNA fragmentation (if needed) to generate short fragments (~300–500 bp).
- End repair and A-tailing to prepare fragments for ligation.
- Adapter ligation, attaching sequencing adapters required for Illumina platforms.
- PCR amplification to enrich properly ligated fragments.
- Library quality control (quantification and fragment size verification).
This produces a sequencing-ready DNA library:
Essential steps of the sequencing technology & base calling (Illumina example) Illumina sequencing works through sequencing-by-synthesis:
- DNA fragments bind to a flow cell coated with complementary adapter sequences.
- Bridge amplification creates clusters of identical DNA fragments.
- Fluorescently labeled nucleotides are added one cycle at a time.
- Each nucleotide (A, T, C, G) emits a specific fluorescent signal.
- A camera captures the signal after each incorporation cycle.
- The system converts fluorescence signals into base calls (A, T, C, or G). Base calling is performed computationally by interpreting the fluorescent intensity at each cycle, reconstructing the nucleotide sequence.
The output consists of: • Millions of short DNA reads (FASTQ files) • Each read contains: o The nucleotide sequence o A quality score (Phred score) for each base These reads can then be: • Assembled into longer contigs • Compared against databases to identify chitinase genes • Analyzed using bioinformatics pipelines for gene annotation
Through metagenomic sequencing, I could identify novel chitinase genes without needing to culture the microorganisms. This expands access to hidden microbial diversity and enables the engineering of optimized enzymes for waste valorization and therapeutic oligosaccharide production. This approach transforms crustacean waste from an environmental liability into a biotechnological resource.
- DNA write What DNA would you want to synthesize and why? If I could write DNA, I would design a chromoprotein-based genetic circuit capable of transforming invisible molecular signals into visible color. My goal would be to engineer a responsive DNA construct that can function inside a microfluidic diagnostic device, producing a clear color output in the presence of disease-associated biomarkers. Conceptually, I am interested in encoding perception into biology — designing DNA that allows cells or cell-free systems to “sense” inflammation, metabolic imbalance, or pathogen-derived molecules and translate that information into pigment production. Instead of relying on fluorescence or complex instrumentation, the output would be visible to the naked eye. Color becomes data. The DNA I would synthesize would not be just a gene, but a functional module: • A disease-responsive promoter • A regulatory sequence tuned for sensitivity • A chromoprotein coding sequence • A terminator This would allow biological information to flow through a designed pathway: detection → transcription → translation → color. In this way, synthetic DNA becomes a communication interface between biology and human interpretation.
Sequencing as a design-feedback tool
To validate and refine this system, I would use next-generation sequencing (NGS), particularly Illumina sequencing, to verify construct integrity and evaluate mutations or circuit stability.
Essential steps of the sequencing method:
- DNA extraction or plasmid purification
- Fragmentation of DNA
- Adapter ligation
- PCR amplification to prepare sequencing libraries
- Cluster generation on a flow cell
- Sequencing-by-synthesis using fluorescent nucleotides
- Base calling via optical detection and computational analysis
Sequencing works by detecting fluorescent signals emitted when nucleotides are incorporated during DNA synthesis. Each cycle adds one base, and the emitted fluorescence is translated into a digital readout of A, T, C, or G. Biology becomes light; light becomes code. The output is millions of short sequence reads, which can be assembled and compared to the intended design. This creates a feedback loop between writing and reading DNA.
Limitations of the sequencing method While powerful, this technology has limitations: • Speed: Library preparation and sequencing runs require hours to days. Rapid iteration in design-build-test cycles can be slowed by this step. • Read length limitations: Illumina produces short reads (typically 150–300 bp), which can complicate assembly of longer constructs or repetitive regions. • Accuracy trade-offs: Although highly accurate (~99.9%), errors can still occur, especially in homopolymer regions. • Scalability constraints: While massively parallel, sequencing still requires centralized infrastructure, expensive instruments, and computational resources. These limitations shape how we design biological systems. Writing DNA is increasingly accessible. Reading it, interpreting it, and integrating it meaningfully remains a technical and infrastructural challenge.
What interests me most is the dialogue between synthesis and sequencing. We write DNA to create function. We sequence DNA to verify and understand it. The future of biofabrication lies in tightening this loop — accelerating the translation between digital design and biological behavior. In designing a chromoprotein-based diagnostic system, I am not only engineering color production. I am exploring how biology can become a programmable material — responsive, interpretable, and embedded into devices that expand access to healthcare.
- DNA edit
What DNA would you want to edit and why? I would like to edit the DNA encoding chitinases in environmentally relevant microorganisms to enhance their efficiency in degrading crustacean shell waste. Specifically, I would aim to improve catalytic activity, substrate specificity, or stability under industrial conditions such as variable pH, salinity, or temperature. The broader goal is to optimize biological systems for circular bioeconomy applications — transforming shell waste into high-value chitin oligosaccharides with therapeutic potential. Editing DNA in this context is not about creating something artificial, but about refining natural enzymatic pathways to increase sustainability and scalability.
How does your technology of choice edit DNA? What are the essential steps? My technology of choice would be CRISPR-based genome editing, particularly CRISPR-Cas systems. Conceptually, CRISPR editing works by:
- Designing a guide RNA (gRNA) that matches a specific DNA sequence in the target gene.
- The Cas enzyme (such as Cas9) binds to the guide RNA.
- The complex scans the genome and binds to the complementary DNA sequence.
- The Cas enzyme introduces a cut (double-strand break) at that location.
- The cell repairs the break through natural repair mechanisms:
- Non-homologous end joining (NHEJ), which may introduce small insertions or deletions.
- Homology-directed repair (HDR), if a repair template is provided, allowing precise sequence changes.
Through this process, specific mutations, insertions, or replacements can be introduced into the chitinase gene.
What preparation is needed and what is the input? Before editing, several design and preparation steps are required: Design steps: • Identify the target gene sequence. • Analyze protein structure and catalytic residues to determine desired mutations. • Design guide RNAs specific to the target region. • Design a repair template (if precise edits are desired). • Evaluate potential off-target sites computationally. Input materials: • DNA sequence of the target organism. • CRISPR-associated enzyme (e.g., Cas9). • Guide RNA(s). • Optional repair template DNA. • Delivery system (e.g., plasmid, ribonucleoprotein complex, or viral vector). • Host cells for transformation or transfection. Editing is essentially a programmed intervention into the genome, guided by sequence complementarity.
What are the limitations of your editing method? Despite its precision, CRISPR-based editing has important limitations:
- Efficiency Editing efficiency can vary widely depending on: • The organism • The delivery method • The repair pathway preference (NHEJ vs HDR) Some cells repair DNA in ways that reduce precise editing success.
- Off-target effects Although guide RNAs are designed to be specific, unintended cuts can occur at similar sequences elsewhere in the genome.
- Repair unpredictability When relying on NHEJ, insertions or deletions can be variable and difficult to control.
- Scalability Editing at industrial scale (e.g., engineering production strains) requires extensive screening and validation to ensure stability across generations.
Editing DNA is a powerful intervention. It allows us not just to read or write genetic information, but to reshape biological function. However, the process is probabilistic rather than perfectly deterministic. Even precise tools operate within the complexity of cellular repair systems. For me, editing chitinase genes is about aligning biological potential with environmental need — improving enzyme performance to reduce waste and generate therapeutic molecules. The challenge lies not only in making edits, but in ensuring they are precise, stable, and ethically deployed.