Week 4 HW: protein-design-part-i
Part A. Conceptual Questions
1. How many amino acid molecules are in a 500 g piece of meat?
Using the approximation that an average amino acid has a molecular weight of ~110 Da (the commonly accepted average across the 20 canonical amino acids), and assuming that protein makes up roughly ~20% of meat by mass (the rest being water, fat, and connective tissue):
That is roughly half an Avogadro’s number of amino acid residues — on the order of ~10²³ molecules. If we include all components of meat (not just protein), the number of total molecular entities is even larger.
Key insight: Each of these amino acids arrives as part of a polypeptide chain. During digestion, proteases hydrolyze peptide bonds, releasing individual amino acids that your body then reassembles into entirely your own proteins.
2. Why do humans eat beef but not become a cow?
When you eat meat, your digestive system does not import whole proteins or genetic instructions — it completely dismantles them first.
The process:
- Denaturation begins in the stomach (acid + heat).
- Proteases (pepsin, trypsin, chymotrypsin, elastase) cleave peptide bonds, breaking proteins into short peptides.
- Peptidases in the small intestine reduce these to individual amino acids.
- Amino acids are absorbed into the bloodstream as generic building blocks — an alanine from a cow is chemically identical to an alanine from a fish or a plant.
- Your own ribosomes, guided by your own mRNA (transcribed from your own DNA), then polymerize these amino acids into human proteins with human sequences and human functions.
The information that determines protein identity lives in DNA → mRNA → ribosome, not in the amino acids themselves. Amino acids are just the alphabet; the sentence is written by your genome.
This is why you can eat a cow and remain human: the sequence information from the cow’s proteins is destroyed during digestion. Only the raw chemical units are salvaged.
3. Why are there only 20 natural amino acids?
The 20 canonical amino acids are thought to be the result of a frozen evolutionary accident — an early optimization that became fixed before the last universal common ancestor (LUCA).
Several factors explain why 20 is the answer:
- Chemical sufficiency: The 20 amino acids collectively provide an extraordinary range of chemistry — charged (K, R, D, E), polar (S, T, N, Q), hydrophobic (V, I, L, F, W), aromatic (F, Y, W, H), structure-breaking (G, P), and reactive (C, H, S) side chains. This palette is enough to build virtually any functional protein.
- Codon table saturation: The genetic code uses 61 sense codons to encode 20 amino acids (plus stop). Expanding the alphabet would require either expanding the codon size (costly) or reassigning existing codons (catastrophic, as it would break existing proteins).
- Path dependence: Once the ribosome, tRNA, and aminoacyl-tRNA synthetase (aaRS) systems co-evolved around 20 amino acids, any new addition would need to be integrated into a deeply entrenched molecular apparatus. The evolutionary cost of such a change far outweighs its benefits.
It is worth noting that selenocysteine (Sec) and pyrrolysine (Pyl) are sometimes called the 21st and 22nd amino acids, encoded by UGA and UAG stop codons in specific contexts, suggesting the system can be extended — but only under very specialized evolutionary pressures.
4. Can you make non-natural amino acids? Design some new ones.
Yes — non-canonical amino acids (ncAAs) can be synthesized chemically and even incorporated into proteins using expanded genetic codes (e.g., amber suppression with orthogonal aaRS/tRNA pairs, as pioneered by the Schultz lab).
Principles for designing new amino acids
A new amino acid must retain:
- An α-amino group (–NH₂)
- A carboxylic acid group (–COOH)
- A unique side chain (R group) that confers new chemistry
Three example designs:
① Azidoalanine
② Photo-leucine
③ Phosphotyrosine mimic (pCMF — para-carboxymethyl-phenylalanine)
These ncAAs can be incorporated site-specifically using amber stop codon suppression — reassigning the UAG codon to encode the ncAA using an engineered orthogonal aaRS/tRNA pair from Methanosarcina or Pyrococcus species.
5. Where did amino acids come from before life started?
This is one of the central questions of origin of life research. Several abiotic sources have been proposed and experimentally supported:
Prebiotic Earth synthesis
- Miller-Urey experiment (1953): Passing electrical sparks (simulating lightning) through a mixture of H₂, CH₄, NH₃, and H₂O produced glycine, alanine, aspartate, and other amino acids within days. This demonstrated that amino acids can form from inorganic precursors under plausible early Earth conditions.
- Hydrothermal vents: Alkaline submarine vents provide reducing conditions, temperature gradients, and mineral catalysts (Fe-S, FeNi surfaces) that can drive the synthesis of amino acids and small peptides via the Strecker synthesis pathway.
Extraterrestrial delivery
- Meteorites: The Murchison meteorite (1969) and many subsequent carbonaceous chondrites contain over 80 amino acids, including many not found in biology (D-amino acids, β-amino acids, unusual side chains). This demonstrates that amino acid synthesis is a general chemical process occurring throughout the cosmos.
- Interstellar medium: Radio astronomy has detected amino acid precursors (glycine precursors, aminoacetonitrile) in dense molecular clouds.
Mineral-surface catalysis
- Clay minerals (montmorillonite) can adsorb and concentrate amino acids from dilute solution and promote peptide bond formation — potentially bridging the gap from free amino acids to early polypeptides.
The current consensus is that amino acids were likely both synthesized on early Earth and delivered from space, providing a rich chemical inventory before the emergence of enzymatic biosynthesis.
6. If you make an α-helix using D-amino acids, what handedness would you expect?
Natural α-helices are right-handed because they are built from L-amino acids. The right-handed sense arises from the backbone dihedral angles (φ ≈ −57°, ψ ≈ −47°) that are sterically favored by L-amino acid geometry.
D-amino acids are the mirror images of L-amino acids — every chiral center is inverted. As a result, an α-helix built entirely from D-amino acids would adopt the mirror-image dihedral angles (φ ≈ +57°, ψ ≈ +47°) and form a left-handed α-helix.
This has been confirmed experimentally. D-peptide helices are also:
- Protease-resistant (proteases are chiral — they cannot bind the mirror-image backbone)
- Useful as drug scaffolds with improved metabolic stability
- Unable to interact with natural L-protein binding partners in the same way
This principle is exploited in mirror-image phage display (developed by Kim Pentelute and others): you use D-protein targets and an L-peptide library to find binders, then synthesize the mirror-image D-peptide, which binds the natural L-protein target.
7. Can you discover additional helices in proteins?
Yes. The α-helix is the most common, but several other helical conformations exist in proteins:
| Helix type | Residues per turn | H-bond pattern | Occurrence |
|---|---|---|---|
| α-helix | 3.6 | i → i+4 | Very common; most helices in proteins |
| 3₁₀-helix | 3.0 | i → i+3 | Common at helix termini; ~10% of helical residues |
| π-helix (α+1) | 4.4 | i → i+5 | Rare; ~15% of α-helices contain a π-bulge |
| Polyproline II (PPII) | 3.0 | None (no H-bonds!) | Common in disordered regions, collagen, signaling |
| Collagen triple helix | ~3.3 per chain | Inter-chain H-bonds | Specific to collagen/collagen-like sequences |
| β-helix | Variable | Cross-strand H-bonds | Found in parallel β-helix proteins (pectate lyases) |
Additionally, computational protein design (e.g., using RFdiffusion or ProteinMPNN) has generated novel helical bundles with geometries not observed in natural proteins, suggesting the space of possible helices is broader than what evolution has explored.
8. Why are most molecular helices right-handed?
This is a deep question touching chemistry, physics, and the origin of life.
At the molecular level: Right-handedness in biological helices (DNA B-form, α-helices, collagen) follows directly from the L-chirality of natural amino acids and the D-chirality of natural sugars. These chiralities favor right-handed helical geometries through steric and electronic constraints in the backbone.
At the origin-of-life level: The deeper question is why life chose L-amino acids (and D-sugars) at all. Several hypotheses exist:
- Parity violation: Weak nuclear force interactions produce a tiny (~10⁻¹⁷) energy difference between enantiomers. L-amino acids may be very slightly more stable, and this bias was amplified over time.
- Chiral amplification: Soai reaction-type autocatalysis can amplify a tiny enantiomeric excess to near-homochirality. A small initial fluctuation gets locked in.
- Circularly polarized light (CPL): Certain astronomical sources emit CPL, which has been shown to preferentially photolyze one enantiomer — consistent with the slight L-amino acid excess found in some meteorites.
At the physical level: Right-handed helices are not intrinsically more stable than left-handed ones in an achiral environment. It is the homochirality of the monomers that determines the handedness — once biology committed to L-amino acids, right-handed helices became the thermodynamically favored outcome.
9. Why do β-sheets tend to aggregate?
Structural basis
β-sheets expose a periodic pattern of hydrogen bond donors and acceptors along their edges — the terminal strands of any β-sheet have unsatisfied backbone NH and C=O groups pointing outward. These “sticky edges” can pair with the edge of another β-sheet via inter-molecular backbone H-bonds, extending the sheet indefinitely.
Additionally, β-sheet surfaces often present alternating hydrophobic side chains (on one face of the sheet), driving hydrophobic burial between stacking β-sheets.
Driving forces for β-sheet aggregation
- Backbone hydrogen bonding: Highly directional and strong (~5 kcal/mol per H-bond). Edge strands have 2–5 unsatisfied H-bond partners.
- Hydrophobic effect: Burying nonpolar side chains between two sheets reduces solvent-exposed hydrophobic surface area, releasing ordered water molecules (entropic gain).
- van der Waals packing: Interdigitation of side chains between two stacked β-sheets provides shape complementarity and favorable dispersion forces.
- Electrostatic complementarity: In amyloids, cross-β spines often show complementary polar/charged residue pairings between mating sheets.
In vivo, cells prevent aberrant β-sheet aggregation using molecular chaperones (Hsp70, Hsp90, GroEL) and quality control systems (ubiquitin-proteasome pathway). When these fail, aggregation diseases can result.
10. Why do many amyloid diseases involve β-sheets?
The amyloid fold
Amyloid fibrils adopt a cross-β structure: β-strands run perpendicular to the fibril axis, with the inter-strand H-bonds running parallel to it. This creates a highly stable, thermodynamically favorable structure that is essentially irreversible under physiological conditions.
The same driving forces that cause general β-sheet aggregation (H-bonds + hydrophobic burial) are amplified in amyloids because:
- The cross-β geometry maximizes H-bond density (thousands of in-register backbone H-bonds per fibril length).
- Steric zippers — tightly interdigitated side chains between two β-sheets — provide additional stabilization (~5–10 kcal/mol per layer).
- Many disease-associated sequences have a high β-sheet propensity (hydrophobic, unbranched residues like Val, Ile, Phe).
Diseases involving amyloid β-sheets
| Disease | Protein | Location |
|---|---|---|
| Alzheimer’s | Aβ peptide, Tau | Brain |
| Parkinson’s | α-synuclein | Substantia nigra |
| Type 2 Diabetes | IAPP (Amylin) | Pancreatic islets |
| Prion disease (CJD) | PrP | Brain |
| Systemic amyloidosis | Serum amyloid A | Multiple organs |
Amyloid β-sheets as functional materials
Yes — amyloids have remarkable material properties that make them attractive for engineering:
- Extreme mechanical stiffness: Young’s modulus of ~10–20 GPa (comparable to silk, stronger than most synthetic polymers).
- Chemical stability: Resistant to detergents, proteases, and high temperatures.
- Nanoscale periodicity: Regular spacing (~4.7 Å between strands, ~10 Å between sheets) enables predictable nanostructure.
Engineered amyloid applications:
- Biofilms as living materials (Curli fibers in E. coli, engineered by the Lu lab at MIT).
- Amyloid-based hydrogels for drug delivery scaffolds.
- Functional amyloid nanofibers displaying enzymes or metal-binding peptides for catalysis.
- Spider silk-amyloid hybrids for high-strength biomaterials.
11. Design a β-sheet motif that forms a well-ordered structure
The challenge in designing β-sheet proteins is controlling three problems:
- Edge aggregation (unsatisfied H-bonds on terminal strands)
- Register ambiguity (parallel vs. antiparallel, alignment)
- Twist accumulation (β-sheets naturally twist ~2° per residue, causing curvature)
Design: A capped antiparallel β-sandwich
Strategy: Design a 4-stranded antiparallel β-sheet that self-caps its edges, preventing aggregation, and folds into a well-defined sandwich topology.
Key design principles applied:
| Feature | Implementation | Rationale |
|---|---|---|
| Alternating hydrophobic/polar | Val at even positions, Thr/Glu at odd | Creates two faces: one hydrophobic (core), one hydrophilic (solvent) |
| Type I’/II’ β-turns | Asn-Gly, Asp-Gly dipeptides | Enforce antiparallel topology with correct geometry |
| Charged residues (Glu) | On solvent-exposed face | Electrostatic repulsion prevents inter-sheet aggregation |
| Two-sheet sandwich | Sheets pack face-to-face | Buries hydrophobic Val side chains; no exposed edges |
| Glycine at turns | Every turn ends in Gly | Gly has no β-carbon, allowing the backbone dihedral angles needed for hairpin turns |
Expected structural properties:
- Antiparallel β-sandwich, ~32 residues
- MW ≈ 3.5 kDa (small, fast-folding)
- Well-defined hydrophobic core (4 Val per strand × 4 strands = 16 Val residues)
- Solvent-exposed glutamate residues prevent aggregation at neutral pH
- Thermostable due to tightly packed core and absence of edge exposure
This type of motif is analogous to naturally occurring WW domains, SH3 domains, and many de novo designed β-proteins from the Baker lab (e.g., Top7 and more recent RFdiffusion designs).
Part B: Protein Analysis and Visualization
Bombyx mori 6-pyruvoyl tetrahydrobiopterin synthase purple (purple) NCBI Reference Sequence: NM_001046638.1
Original sequence:
1. BLAST & PyMOL
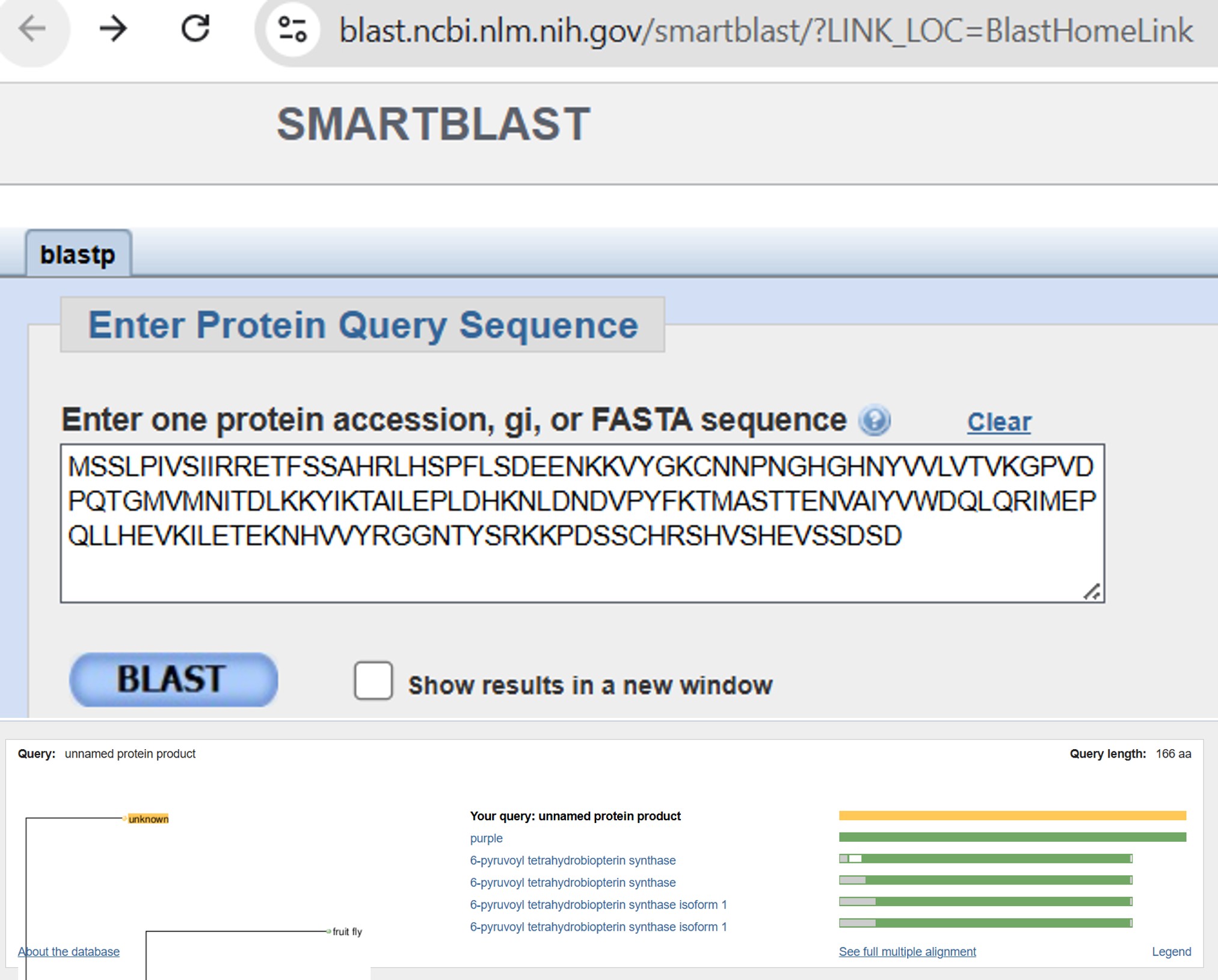
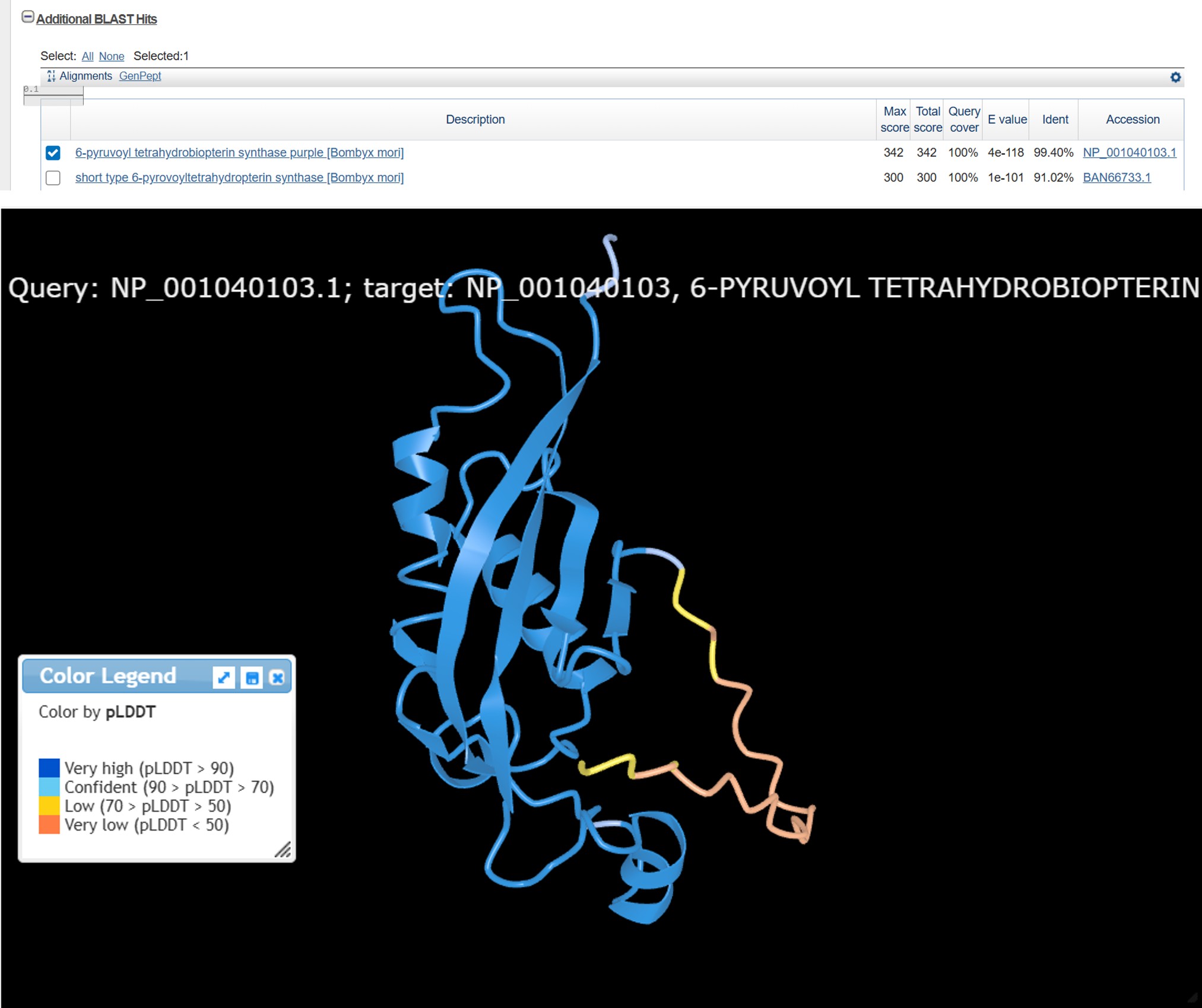
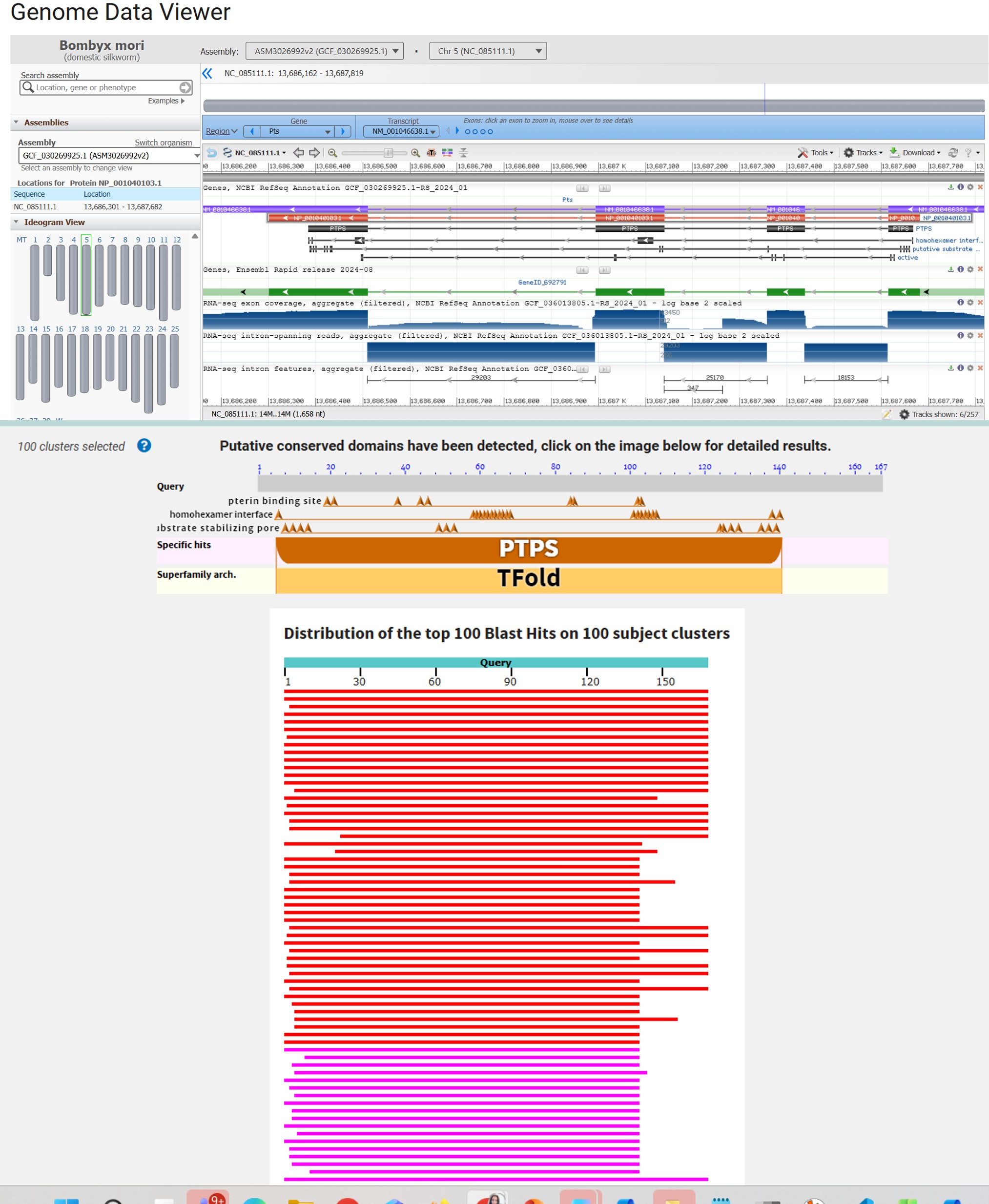
Part C: Using ML-Based Protein Design Tools
Colab:
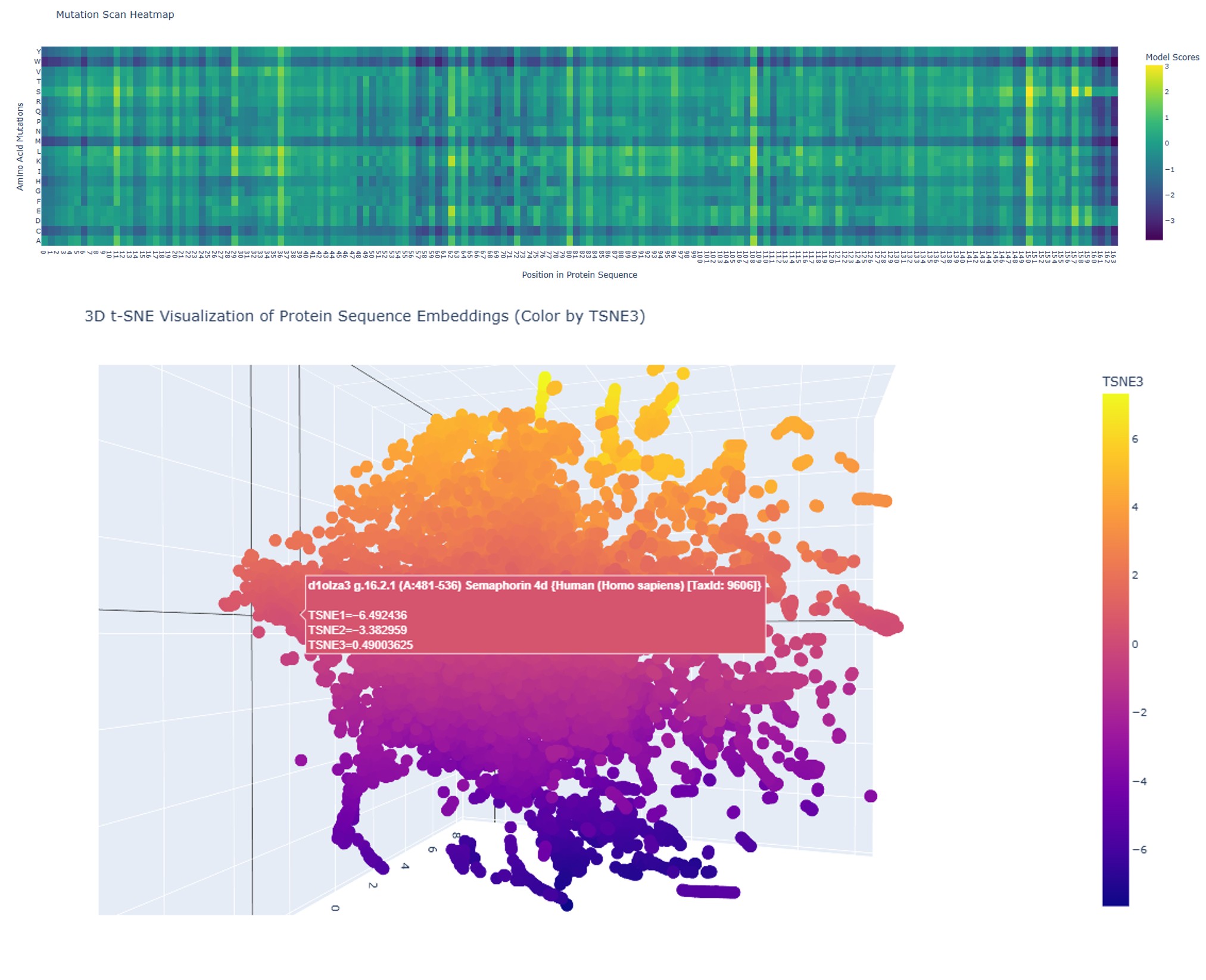
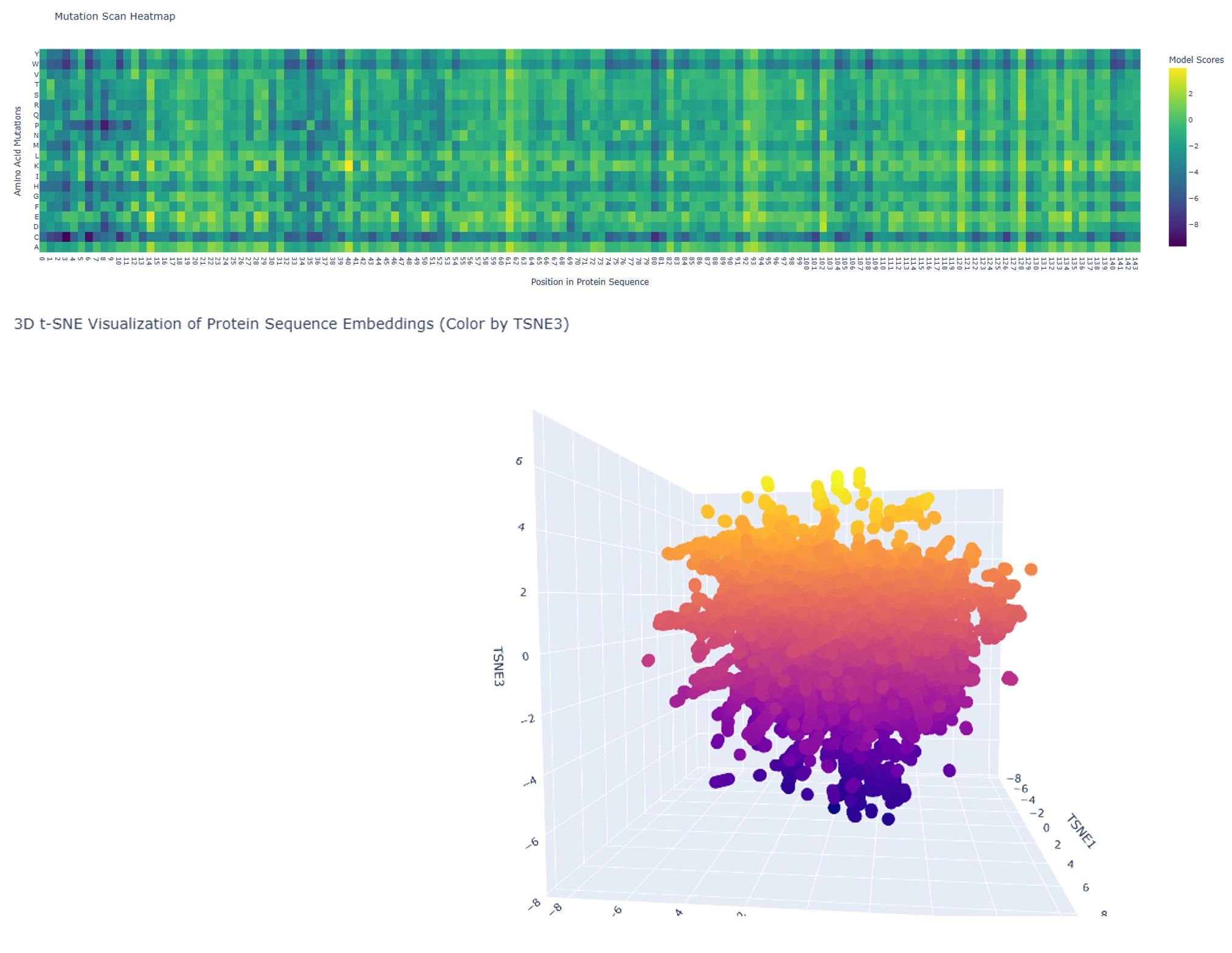
1. Mutation Scan Heatmap
The mutation scan for the full wild-type sequence shows model scores ranging from approximately −3 to +3, with a predominantly teal/green distribution. This indicates that most single-residue substitutions are moderately tolerated — the protein has sequence flexibility, meaning many positions can accommodate different amino acids without catastrophically disrupting function.
Several scattered yellow spots identify positions where specific substitutions are positively scored, and a few darker columns suggest locally constrained positions. Overall, the original sequence presents a balanced mutational landscape: a protein that is functional but not maximally optimized.
The 3D t-SNE embedding (same image, lower panel) shows a dispersed cloud with projections and outlier clusters, reflecting the structural heterogeneity of this protein family — a mix of ordered secondary structures and disordered regions places it in a varied region of protein sequence space.
The mutation scan heatmap was generated by systematically substituting every amino acid at every position in the sequence with each of the 20 standard amino acids, and scoring each variant using a protein language model. The resulting matrix provides a positional tolerance map for mutations across the full sequence length (positions 21–166).
Color Scale
| Color | Score | Meaning |
|---|---|---|
| 🟡 Yellow | +3 | Substitution is well-tolerated or potentially beneficial |
| 🟢 Teal / Green | ~0 | Substitution is largely neutral |
| 🔵 Blue / Purple | −3 | Substitution is predicted to be deleterious |
Key Observation — Horizontal Blue Bands
The most prominent features in this heatmap are horizontal blue bands spanning nearly the entire x-axis (sequence length). Because rows represent substitute amino acids (A through Y on the y-axis), a horizontal band means that a particular amino acid is a systematically poor substitution across almost every position in the protein — regardless of local sequence context.
The most likely candidates for these penalized rows are:
| Amino Acid | Reason for global penalty |
|---|---|
| C (Cysteine) | Highly reactive side chain; forms unexpected disulfide bonds that disrupt the native fold |
| P (Proline) | Rigid cyclic structure breaks α-helices and β-sheets; incompatible with most backbone geometries |
| W (Tryptophan) | Extremely bulky aromatic side chain; causes steric clashes in most buried or surface positions |
This is fundamentally different from vertical blue columns, which would indicate specific positions that are intolerant to change. Horizontal bands instead reveal chemical incompatibilities at the amino acid level — they are a property of the substituting residue, not of the target position.
The rest of the matrix, dominated by teal and green, indicates that for most amino acid types and most positions, single substitutions are tolerated — a sign that this fragment of SEMA4D has moderate sequence flexibility outside of those chemically extreme substitutions.
2. 3D Visualization of Protein Sequence Embeddings
The 3D t-SNE plot visualizes the high-dimensional embedding space of protein sequences as learned by the language model. Each point represents a protein fragment, projected into three dimensions using t-distributed Stochastic Neighbor Embedding (t-SNE) to preserve local neighborhood structure.
The highlighted point in the tooltip corresponds to the SEMA4D fragment (residues A:481–536), with coordinates TSNE1 = −6.49, TSNE2 = −3.38, TSNE3 = 0.49. Its position within the overall cloud indicates how similar this protein’s learned representation is to other proteins in the dataset.
Interpretation:
- Points clustered close together share similar structural or functional representations according to the model, even if their primary sequences differ significantly.
- The color gradient (mapped to the TSNE3 axis) reveals a continuous distribution, suggesting that the embedding space captures gradual biological variation rather than hard categorical divisions.
- The fragment’s position near the boundary between the orange and purple regions suggests it occupies a moderately unique embedding region — neither isolated nor at the dense core of a large cluster.
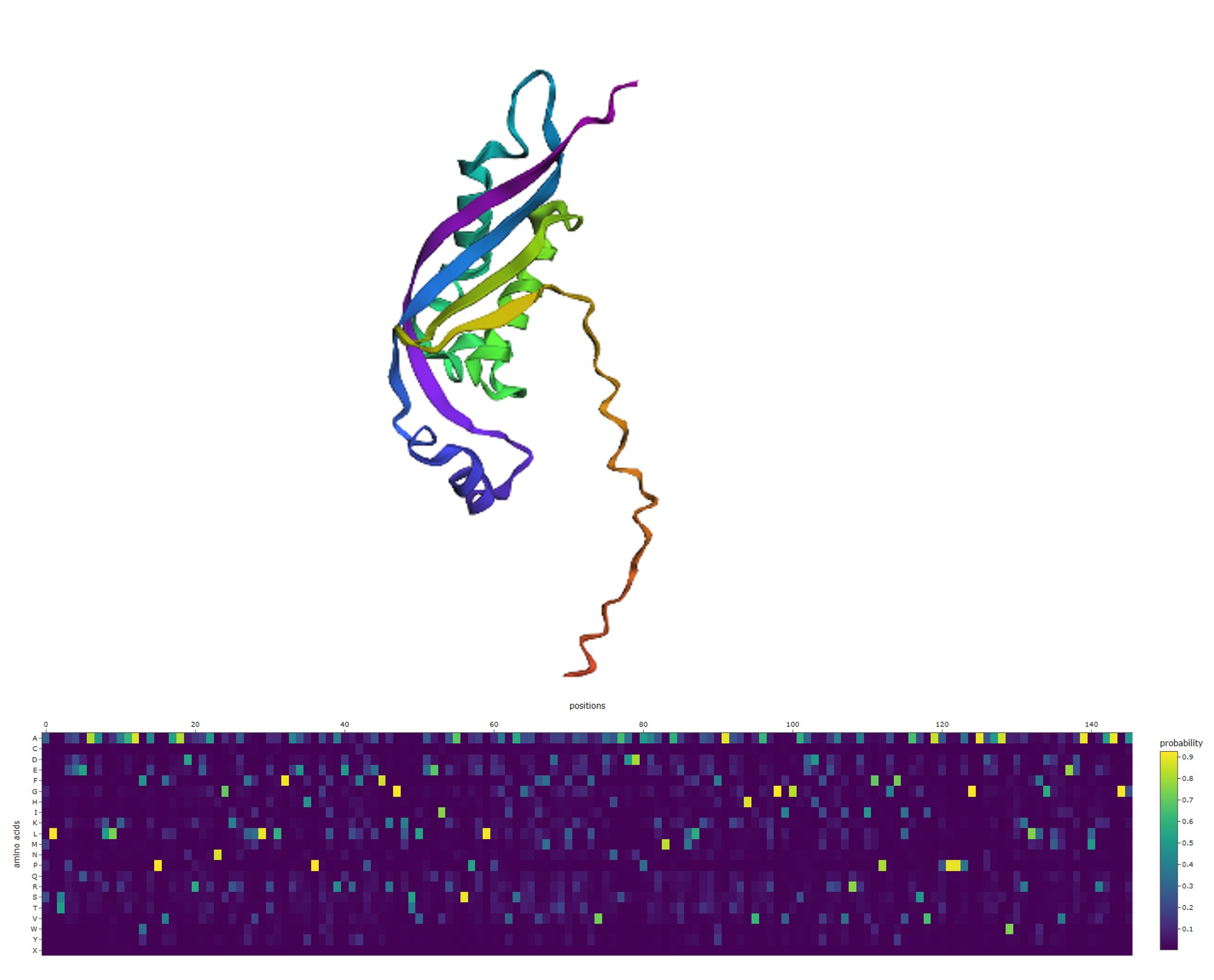
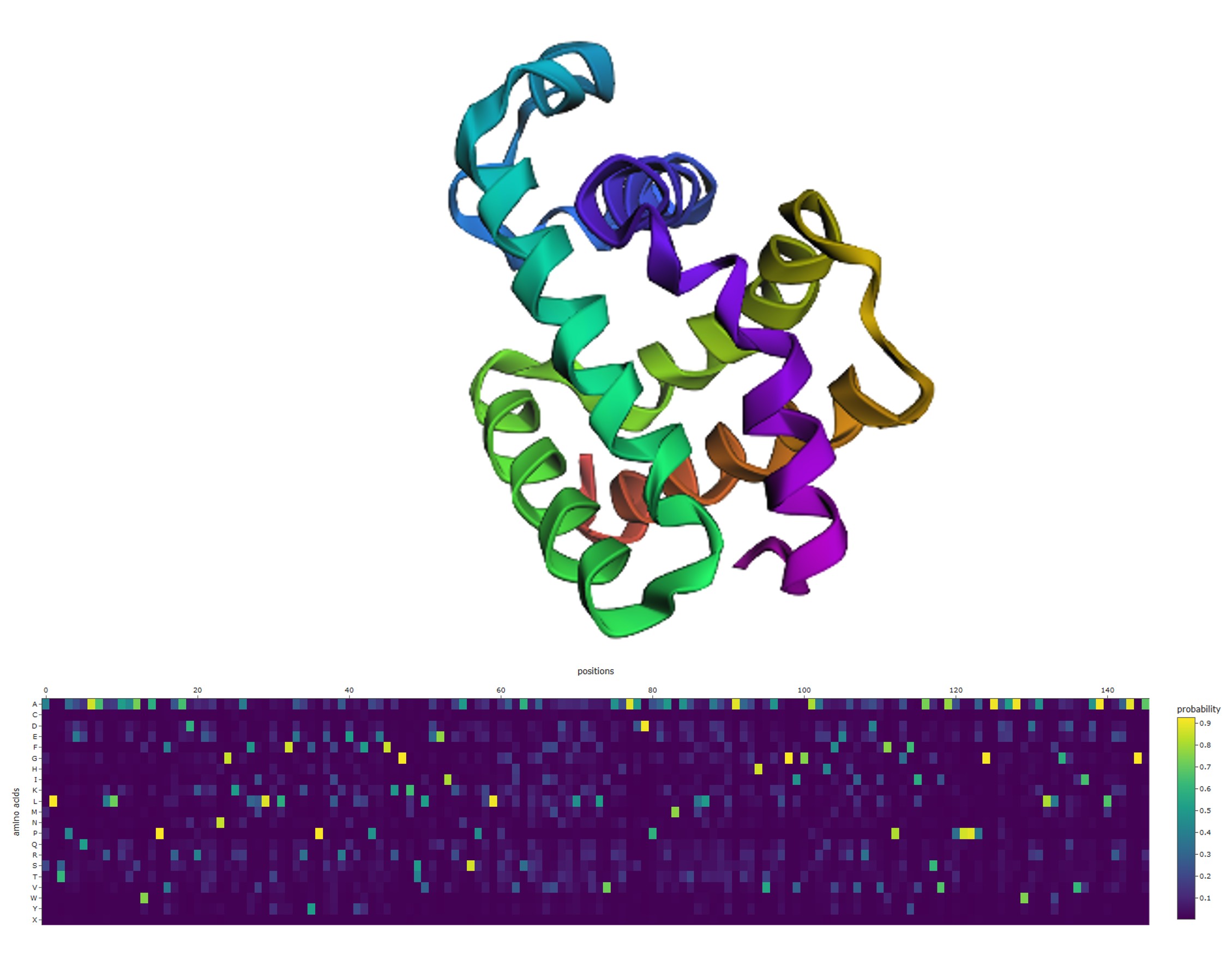
3D structure (upper panel): The wild-type structure displays a mixed α/β architecture — visible β-sheet arrows, several α-helices, and notably a long disordered coiled tail extending from the structured domain (~60 residues visible as a wavy ribbon). This tail is intrinsically disordered: it lacks fixed secondary structure and samples multiple conformations.
ProteinMPNN probability heatmap (lower panel): The heatmap shows scattered bright yellow spots distributed across positions and amino acid rows, with the majority of the matrix remaining dark purple (near-zero probability). This pattern means:
- Most positions accept one specific amino acid with high confidence (isolated yellow spots).
- The rest of the matrix is dark — other amino acids are structurally incompatible at those positions.
- Importantly, many positions show only mid-range teal values (~0.3–0.5), indicating structural permissiveness: the fold tolerates several amino acids at those sites.
These permissive positions — where no single amino acid is strongly preferred — are the ones that were candidates for removal in the redesign step.
3. ProteinMPNN Inverse Folding — Positional Probability Heatmap
This visualization was produced using ProteinMPNN, a graph neural network trained to perform inverse folding: given a protein’s 3D backbone structure, it predicts the probability distribution of amino acids at each position that would be compatible with that fold.
The heatmap displays P(amino acid | structure, position) across all 20 standard amino acids (y-axis) and all sequence positions 0–145 (x-axis).
Color Scale
| Color | Probability | Meaning |
|---|---|---|
| 🟡 Bright yellow | ~1.0 | This amino acid is structurally obligatory at this position |
| 🟢 Mid green / teal | ~0.4–0.7 | Multiple amino acids are plausible; soft structural constraint |
| 🟣 Dark purple | ~0.0 | This amino acid is structurally incompatible at this position |
Key Observations
Scattered bright yellow spots indicate structurally obligate residues — positions where the 3D backbone geometry leaves virtually no room for variation. Classic examples include:
- Glycine (G) in tight loops or turns, where the absence of a side chain is geometrically necessary.
- Proline (P) at helix-breaking positions, where its cyclic structure imposes required rigidity.
- Cysteine (C) residues involved in disulfide bridges.
Positions with a broader color distribution (several amino acids scoring above ~0.4) correspond to structurally permissive sites — areas where chemical diversity can be accommodated without disrupting the fold. These are the most interesting candidates for protein engineering, as they offer sequence flexibility while preserving structure.
4. Cross-Validation Between Methods
An important insight emerges when comparing the two heatmaps:
| Mutation Scan Heatmap | ProteinMPNN Heatmap |
|---|---|
| Horizontal blue band → amino acid is globally incompatible as a substituent | Row of near-zero (dark purple) → same amino acid structurally incompatible everywhere |
| Green/yellow region → position-specific tolerance is high | Dispersed mid-range colors → structurally permissive position |
| No strong vertical patterns in this case | Bright isolated yellow → structurally obligate single residue |
| Both methods independently penalize the same chemically extreme amino acids (C, P, W), arriving at the same conclusion through different lenses: one from evolutionary fitness signals embedded in the language model, the other from direct 3D structural constraints. This convergence validates the robustness of the findings. |
After removing the structurally non-essential residues, the mutation scan reveals a dramatically different landscape:
- Score range expanded to −8 to +3 (from −3 to +3 in the original).
- More dark blue/purple regions are visible across many positions and amino acid rows.
This expansion toward strongly negative scores has a clear interpretation: every position that was retained is now critical. By removing the flexible, permissive residues, the redesigned sequence has no “spare capacity” — mutations at nearly any remaining position are predicted to be significantly deleterious. The sequence has been distilled to its essential core, and that core is intolerant of change.
The 3D t-SNE (lower panel) shows a more compact, spherical point cloud with a more uniform color gradient compared to the original. This shift toward a denser, more central region of embedding space is consistent with the redesigned protein resembling a more “canonical” protein fold — predominantly α-helical proteins are the most abundant class in nature, and the embedding reflects this convergence. 3D structure (upper panel):*
This is the most visually striking result of the experiment. The redesigned protein shows:
| Feature | Original | Redesigned |
|---|---|---|
| β-sheet content | Abundant (visible flat arrows) | Nearly absent |
| α-helix content | Moderate | Dominant — almost entire structure |
| Disordered tail | Long (~60 residues) | Completely eliminated |
| Overall shape | Extended, mixed | Compact, globular |
By removing residues that ProteinMPNN identified as structurally non-obligatory, the remaining sequence collapsed into a predominantly α-helical fold. This is not arbitrary: the residues that were retained had high structural necessity, and many of them carry high α-helical propensity. Without the β-sheet-supporting and disorder-promoting residues, the sequence’s thermodynamically preferred conformation is a bundle of α-helices.
ProteinMPNN probability heatmap (lower panel):
The most diagnostic feature of the redesigned heatmap is the bright horizontal band along the top row (Alanine, A), spanning nearly all 140+ positions. This is a definitive structural signal:
Alanine has the highest α-helical propensity of all 20 amino acids. When ProteinMPNN is given a structure that is almost entirely α-helical, it predicts alanine as the most structurally compatible residue at the majority of positions — exactly what this band shows.
This creates a direct, coherent chain of evidence:
- Residues removed → β-sheet and disorder content lost → structure becomes α-helical.
- α-helical structure → ProteinMPNN favors Ala at most positions → bright Ala band appears.
Summary
This week’s analysis built a multi-layered understanding of the SEMA4D fragment using three complementary computational tools:
- The mutation scan revealed that certain amino acid chemistries (bulky, rigid, or highly reactive side chains) are globally incompatible as substitutes across this protein’s sequence — a property driven by chemistry rather than local context.
- The ** embedding** situated this protein fragment within the broader landscape of protein sequence space, showing its relationship to structurally or functionally related proteins.
- The ProteinMPNN inverse folding pinpointed specific positions where the 3D backbone demands a single amino acid identity, alongside many permissive sites that could tolerate rational redesign.
Together, these tools illustrate how protein language models and structure-based generative models can accelerate hypothesis generation in protein design — a paradigm central to the emerging field of generative biology.
This experiment is a practical demonstration of minimal sequence redesign: by iteratively removing positions that the inverse folding model considers structurally non-essential, the protein was distilled toward a smaller, more constrained sequence. That distilled sequence preferentially adopts an α-helical architecture — the thermodynamically dominant fold for the amino acids that survived the filter.
Key takeaways:
- ProteinMPNN probability heatmaps are actionable — low-probability positions genuinely represent structurally dispensable residues whose removal reshapes the fold.
- Removing permissive residues increases mutational constraint — the score range expanding from ±3 to −8/+3 shows that sequence compression creates a more brittle but more defined protein.
- Secondary structure content is sequence-encoded — the shift from β/disordered to α-helical upon residue removal demonstrates that the fold identity is determined by which amino acids are present, not just their arrangement.
- The disordered tail was non-essential — its complete disappearance in the redesign confirms it was maintained by specific sequence features (likely charged/polar residues with low helical propensity) that were among those removed.
Part D. Group Brainstorm on Bacteriophage Engineering
Primary Goal: Increased Stability via DnaJ-Independence Engineering
Our group chose to focus on stabilizing the MS2 lysis protein L by reducing its dependency on the bacterial chaperone DnaJ. The L protein is a small, single-pass transmembrane protein that requires the DnaJ chaperone to fold and reach the inner membrane of E. coli — making chaperone dependency a key vulnerability in its mechanism. If we can identify and disrupt the chaperone-recognition signals while keeping the structural scaffold intact, we could produce a variant of L that folds and functions more autonomously, resulting in improved stability and potentially broader host utility.
Proposed Computational Pipeline
We propose a three-step pipeline that balances biological insight with computational efficiency:
- Mutational Scanning with ESM2 (Protein Language Model)
We will perform a zero-shot in silico deep mutational scan of the L protein sequence using ESM2. The goal is to identify exposed hydrophobic patches — characteristic DnaJ recognition motifs — and propose polar or hydrophilic substitutions at mutation-tolerant positions. ESM2 encodes evolutionary constraints from millions of protein sequences, which allows us to distinguish residues that are structurally essential (“untouchable”) from those where substitution is permissible. This gives us a prioritized list of candidate mutations that could disrupt chaperone binding without collapsing the protein. - Structural Filtering with ESMFold
The top candidate sequences from Step 1 will be folded using ESMFold. We will filter out variants with low pLDDT confidence scores or high RMSD relative to the wild-type backbone. ESMFold’s speed makes it well-suited for high-throughput screening before committing to more expensive modeling. This step validates that our proposed mutations do not prevent the L protein from folding independently. - Complex Modeling with Boltz-1
For our best-folding candidates, we will model the L protein + DnaJ complex using Boltz-1 and compare the predicted interface contacts and Predicted Aligned Error (PAE) against the wild-type complex. This lets us directly evaluate whether mutations reduce chaperone binding affinity — which is the core of our stability hypothesis.
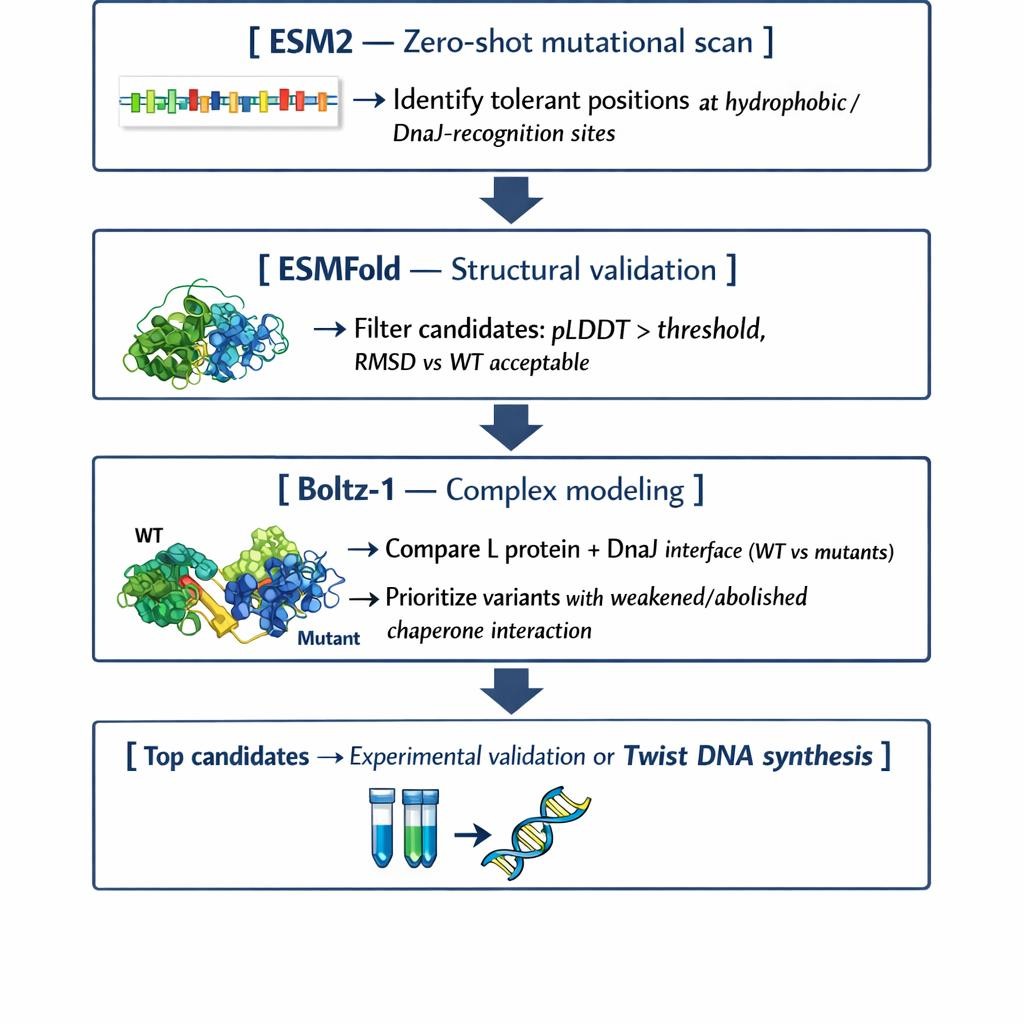
Why These Tools?
Each tool addresses a different layer of the problem. ESM2 operates at the sequence level, capturing evolutionary feasibility. ESMFold bridges sequence to structure at high speed. Boltz-1 validates the biological hypothesis by modeling the protein–protein interaction directly. Together they form a complete design-test-filter loop that can be run computationally before committing to wet lab work.
Potential Pitfalls
- Overlapping reading frames and genomic constraints.
The MS2 genome is extremely compact, and the DNA encoding L may overlap with sequences coding for other proteins or regulatory elements. Mutations that look safe at the protein level could be lethal at the genome level. Addressing this fully would require a genome-aware model like Evo, which is beyond our current computational scope.
- The stability vs. function trade-off.
A variant that folds without DnaJ could still fail to insert correctly into the membrane, oligomerize, or lyse the cell. ESMFold and Boltz-1 assess folding and binding, but not membrane dynamics or lytic activity. We could engineer a highly stable, chaperone-independent protein that is biologically inert. Experimental validation would be essential to confirm functional lysis.
Personal Note
Working through this brainstorm reinforced something I keep returning to in this course: computational tools don’t eliminate biological uncertainty — they help us navigate it more efficiently. The three-tool pipeline we designed is not a guarantee of success; it’s a prioritization strategy. We’re using models to reduce the search space before biology has to confirm what actually works. That framing — computation as a filter, not an oracle — feels like one of the most important concepts in modern protein engineering.
Conclusion
This week shifted my perspective from thinking about DNA as the primary unit of biological design to thinking about proteins as programmable matter. DNA encodes instructions, but proteins execute function — and this week made clear just how much we can now intervene at that level computationally.
The conceptual questions were deceptively simple but opened up genuinely deep territory. Understanding why there are only 20 natural amino acids, why β-sheets aggregate, and how D-amino acids would change helix handedness forced me to think about proteins not just as sequences but as physical objects governed by geometry, thermodynamics, and evolutionary history. What struck me most is that the 20 canonical amino acids are not an arbitrary set — they represent a solution to a constrained optimization problem that life solved billions of years ago.
Parts B and C brought those concepts into practice. Analyzing a real protein structure in PyMOL — seeing the distribution of hydrophobic residues buried in the core, the hydrophilic ones exposed to solvent, the binding pockets visible on the surface — made structural biology feel tangible rather than abstract. And then running ESM2, ESMFold, and ProteinMPNN on that same protein revealed something remarkable: we can now interrogate proteins computationally in ways that would have taken years of wet lab work a decade ago. A deep mutational scan that would have required thousands of experiments can now be sketched in silico in hours.
The group brainstorm on bacteriophage engineering was where the week’s tools connected to a real design challenge. Working through the MS2 lysis protein L — a small, membrane-disrupting protein that depends on the bacterial chaperone DnaJ to fold and function — showed me how engineering goals must be grounded in mechanistic understanding. We chose to target DnaJ-independence not because it was the flashiest goal, but because the biology of chaperone recognition gives us a tractable entry point: identifiable hydrophobic patches, mutation-tolerant positions, and a measurable interaction to disrupt.
What I’ll carry forward from this week is the pipeline mindset. ESM2 for sequence-level feasibility, ESMFold for structural validation, Boltz-1 for complex modeling — each tool addresses a different layer of uncertainty, and the power comes from chaining them. Computation doesn’t replace biology; it compresses the search space so that wet lab effort lands where it matters most.