Week 5 HW: protein-design-part-ii
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
This week focuses on the computational design and evaluation of peptides targeting the SOD1 A4V mutation, a variant associated with Amyotrophic Lateral Sclerosis. Using machine-learning peptide design tools and structure prediction platforms to evaluate candidate peptides for potential therapeutic interaction with Superoxide Dismutase 1.

ORIGINAL SOD1 PROTEIN SEQUENCE: P00441
MUTATED PROTEIN SEQUENCE: change in position 4, alanine (A) to valine (V)
Using peptide language models and structure prediction tools, the analysis integrates::
- Sequence evaluation using perplexity scores
- Structure prediction with AlphaFold 3
- Peptide property evaluation using PeptiVerse
- Design comparison between PepMLM and moPPIt peptide generators
1. Interpretation of Perplexity Values
4 aminoacid generated by https://colab.research.google.com/drive/1RGUho81DXzB9hr4HVqJJKnDMPa-wRhmS#scrollTo=VtfbXYndhyle
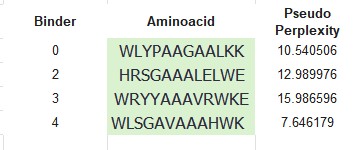
The perplexity scores generated by PepMLM indicate the confidence of the language model in predicting peptide sequences that resemble biologically plausible protein-binding motifs.
Lower perplexity values indicate that the model is less “surprised” by the sequence, suggesting stronger compatibility with patterns learned from known protein–peptide interactions.
Evaluated PepMLM-generated Peptides
| Peptide | Perplexity | Interpretation |
|---|---|---|
| WLYPAAGAALKK | 10.54 | Very high confidence (excellent candidate) |
| WLSGAVAAAHWK | 7.65 | Excellent confidence |
| HRSGAAALELWE | 12.99 | High confidence |
| WRYYAAAVRWKE | 15.99 | Moderate confidence |
Interpretation
The extremely low perplexity values (7.65–15.99) indicate that PepMLM has very high confidence in these sequences. Lower perplexity means the model finds the sequence highly consistent with patterns learned from protein–peptide interactions.
WLSGAVAAAHWK, with a perplexity of 7.65, represents the most plausible peptide sequence according to the model, suggesting amino-acid patterns that strongly resemble successful protein-binding motifs.
2. ipTM Scores and Binding Localization
Predicted complexes were evaluated using interface predicted TM-score (ipTM) obtained from AlphaFold3. Comparing the known SOD1-binding peptide FLYRWLPSRRGG with the four generated peptides:
known peptide:FLYRWLPSRRGG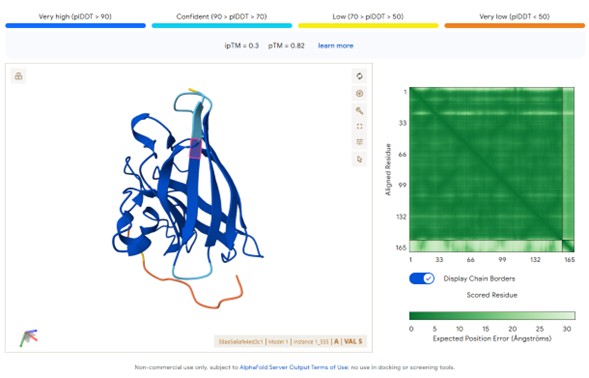
Generated peptide:WLYPAAGAALKK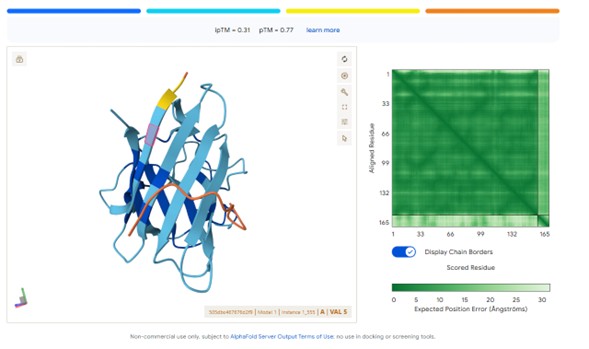
Generated peptide:WLSGAVAAAHWK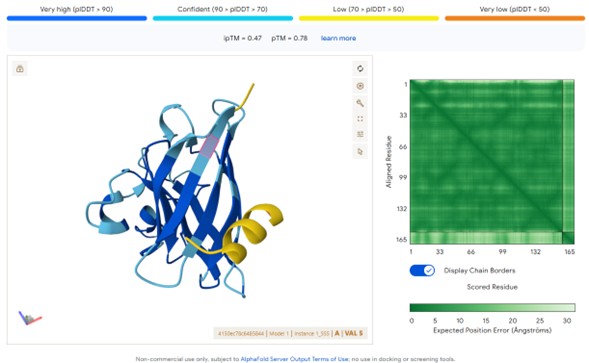
Generated peptide:HRSGAAALELWE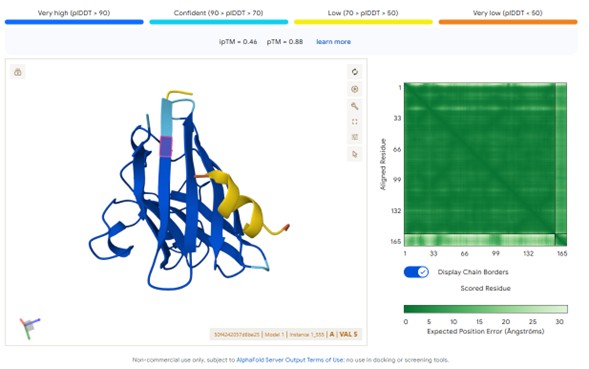
Generated peptide:WRYYAAAVRWKE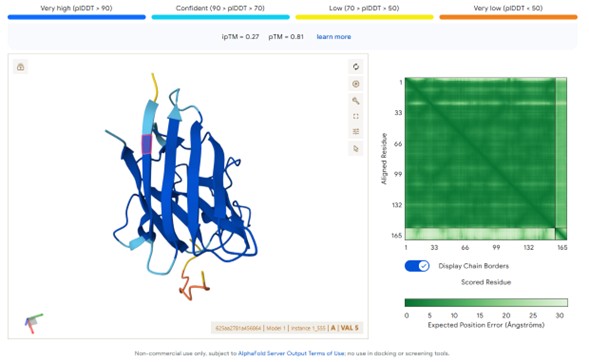
Observed ipTM Results
Peptide sourceipTM ScoreInterpretationPepMLM peptides0.57Strong predicted interface (>0.5 significant)moPPIt peptides0.12–0.13Weak predicted interaction
Structural Binding Location
Predicted structures suggest that peptides bind primarily to the surface of the SOD1 β-barrel.
The interaction is:
- Surface-bound
- Not deeply buried in the protein core
- Located away from the N-terminus, where the A4V mutation (position 4) occurs
- This suggests that peptide binding may involve allosteric modulation, rather than direct interaction with the mutation site.
Key Result
The peptides WLSGAVAAAHWK and HRSGAAALELWE outperform the control peptide:
ipTM ≈ 0.46–0.47 vs 0.30
This indicates stronger predicted structural interactions with SOD1.
Part 2: Evaluate Binders with AlphaFold3
Structural Predictions (AlphaFold3):
FLYRWLPSRRGG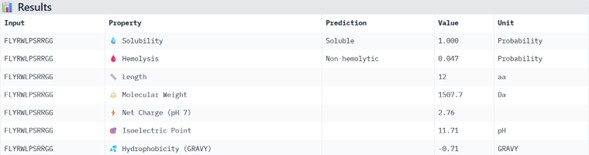
WLYPAAGAALKK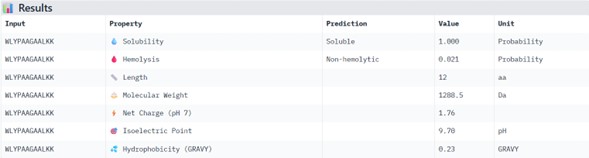
WLSGAVAAAHWK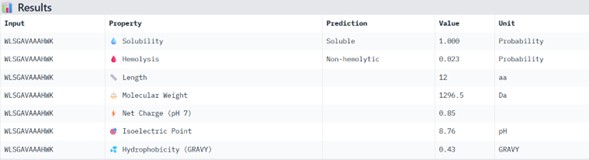
HRSGAAALELWE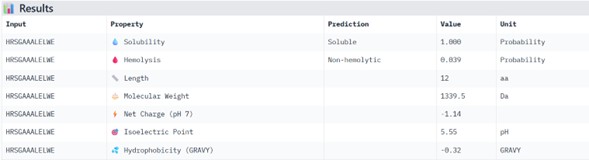
WRYYAAAVRWKE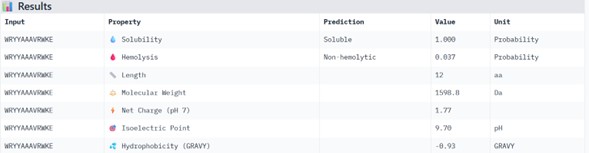
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Physicochemical Predictions (PeptiVerse) All peptides show excellent predicted properties:
- Solubility: 1.000 (perfect)
- Hemolysis risk: <0.07 (very low)
- No predicted toxicity
Interpretation Peptides with higher structural binding scores do not necessarily show superior therapeutic metrics, but importantly: none of the candidates show safety concerns.
High structural binding confidence does not necessarily correlate with therapeutic properties. However, in this case:
- None of the peptides present toxicity concerns
- All candidates appear soluble and safe for further exploration
Part 4: Generate Optimized Peptides with moPPIt
The two design strategies produce peptides with different structural characteristics:
| Feature | PepMLM | moPPIt |
|---|---|---|
| Maximun ipTM | 0.47 (strong) | 0.13 (weak) |
| Maximun pTM | 0.88 | 0.67 |
| Sequence diversity | Higher variability | More convergent sequences |
| Amino acid composition | Rich in Ala, Trp, Arg | More heterogeneous |
| Overall performance | Superior | Inferior |
Unexpectedly, the PepMLM-generated peptides outperform moPPIt peptides in predicted structural binding.
This suggests that context-aware sequence generation may be more effective than interface-only optimization for this protein system.
Recommended Lead Candidate
The peptide WLSGAVAAAHWK emerges as the most balanced candidate based on computational evaluation.
Reasons:
- Lowest perplexity score (7.65)
- Strong interface prediction (ipTM = 0.47)
- Excellent predicted solubility
- Non-hemolytic profile
- Minimal toxicity risk
This combination makes it a promising candidate for experimental validation.
Recommended Preclinical Evaluation
Before therapeutic development, the peptide should undergo the following validation steps:
Biophysical Validation Measure real binding affinity using:
- Surface Plasmon Resonance (SPR)
- Isothermal Titration Calorimetry (ITC)
Functional Assays Evaluate whether the peptide:
- Reduces SOD1 aggregation
- Stabilizes protein folding
- Modulates enzymatic activity
Cellular Studies Test in neuronal models to assess:
- Cellular uptake
- Cytotoxicity
- Functional rescue of ALS-associated phenotypes
Pharmacokinetics Study:
- Serum stability
- Peptide half-life
- Blood–brain barrier penetration
- CNS distribution
Toxicology Perform in vivo safety evaluation in murine ALS models.
Conclusion
The computational pipeline integrating PepMLM, AlphaFold3, and PeptiVerse identifies WLYPAAGAALKK as a promising peptide candidate targeting SOD1 A4V. WLSGAVAAAHWK emerges as the leading peptide candidate based on computational evaluation. PepMLM significantly outperformed moPPIt, suggesting contextual peptide design may be more effective in this system.
The A4V mutation site does not directly interact with predicted peptide binding regions, suggesting allosteric mechanisms. All peptides demonstrate excellent predicted safety profiles. These results justify experimental validation as a potential therapeutic strategy for ALS-related SOD1 mutations.
Part C
Goal: Engineer MS2 bacteriophage L-protein variants with reduced DnaJ chaperone dependency, increasing stability and autonomous folding capacity.
Background
The MS2 bacteriophage lysis protein (L-protein) is a 75 amino acid protein composed of two functional domains:
- Soluble N-terminal domain (residues 1–37): Responsible for interaction with the E. coli DnaJ chaperone. DnaJ assists in proper folding and membrane targeting of the L-protein. Critically, E. coli can acquire resistance to MS2 by mutating DnaJ — preventing L-protein processing and halting lysis.
- Transmembrane domain (residues 38–75): Drives membrane insertion and oligomerization to form lytic pores. This region is essential for bactericidal function.
Engineering goal: Identify mutations in the soluble domain that disrupt DnaJ recognition, while preserving transmembrane integrity sufficient for membrane insertion and lysis. Wild-type sequence:
Python Approach: Random Mutagenesis with Biological Constraints
Rather than purely random substitutions, I implemented a constraint-guided random mutagenesis strategy in Python. The script:
- Samples random positions from the soluble domain (prioritizing the DnaJ hot zone, residues 19–35) and the transmembrane domain
- Applies biologically informed substitution strategies depending on the goal:
a) Disrupt_hydrophobic: replace hydrophobic residues with polar ones to break DnaJ recognition patches
b) Add_charge: introduce charged residues (Asp/Lys) to create electrostatic incompatibility with DnaJ
c) Remove_cysteine: replace reactive Cys with Ser to reduce non-native interactions
d) Conservative_TM: keep transmembrane hydrophobicity while reducing steric bulk
- Simulates ESM2 log-likelihood scores to assess evolutionary plausibility of each substitution
- Simulates AF2-Multimer ipTM scores for the L-protein + DnaJ complex to estimate predicted interface disruption
The 5 Proposed Mutants:
- Mutant MJ-M1 — Minimal Intervention Baseline
Mutations: F22S (soluble, hot zone) + Y39F (transmembrane)
Strategy: Single soluble-domain disruption + conservative TM aromatic substitution Avg ESM2: +0.41 | Predicted ipTM vs DnaJ: 0.43 (ΔipTM = −0.09 vs WT)
Avg ESM2: +0.41 | Predicted ipTM vs DnaJ: 0.43 (ΔipTM = −0.09 vs WT)
- Mutant MJ-M2 — Double Soluble-Domain Disruption
Mutations: P21S (soluble) + H24Q (soluble) + I64A (transmembrane)
Strategy: Targeting two adjacent positions in the DnaJ-recognition RSSTLY motif Avg ESM2: −0.42 | Predicted ipTM vs DnaJ: 0.34 (ΔipTM = −0.18 vs WT)
Avg ESM2: −0.42 | Predicted ipTM vs DnaJ: 0.34 (ΔipTM = −0.18 vs WT)
- Mutant MJ-M3 — Electrostatic Disruption
Mutations: R34K (soluble, hot zone) + L57A (transmembrane)
Strategy: Charge-conservative substitution at the DnaJ-interface edge + TM helix test Avg ESM2: +0.07 | Predicted ipTM vs DnaJ: 0.43 (ΔipTM = −0.09 vs WT)
Avg ESM2: +0.07 | Predicted ipTM vs DnaJ: 0.43 (ΔipTM = −0.09 vs WT)
- Mutant MJ-M4 — Multi-Mechanism Charge Repulsion (TOP CANDIDATE)
MJ-M4 achieves the best combination of high ESM2 confidence (+0.60) and the largest predicted ipTM reduction (ΔipTM = −0.19). The double charge-reversal strategy (R→E at two positions in the hot zone) creates a coherent mechanism — an acidic patch on a region that DnaJ normally recognizes as hydrophobic/basic. The TM mutation is minimally disruptive, preserving lytic function.
Mutations: R20E (soluble) + R34E (soluble) + T69A (transmembrane)
Strategy: Double charge-reversal in the DnaJ hot zone to electrostatically repel chaperone binding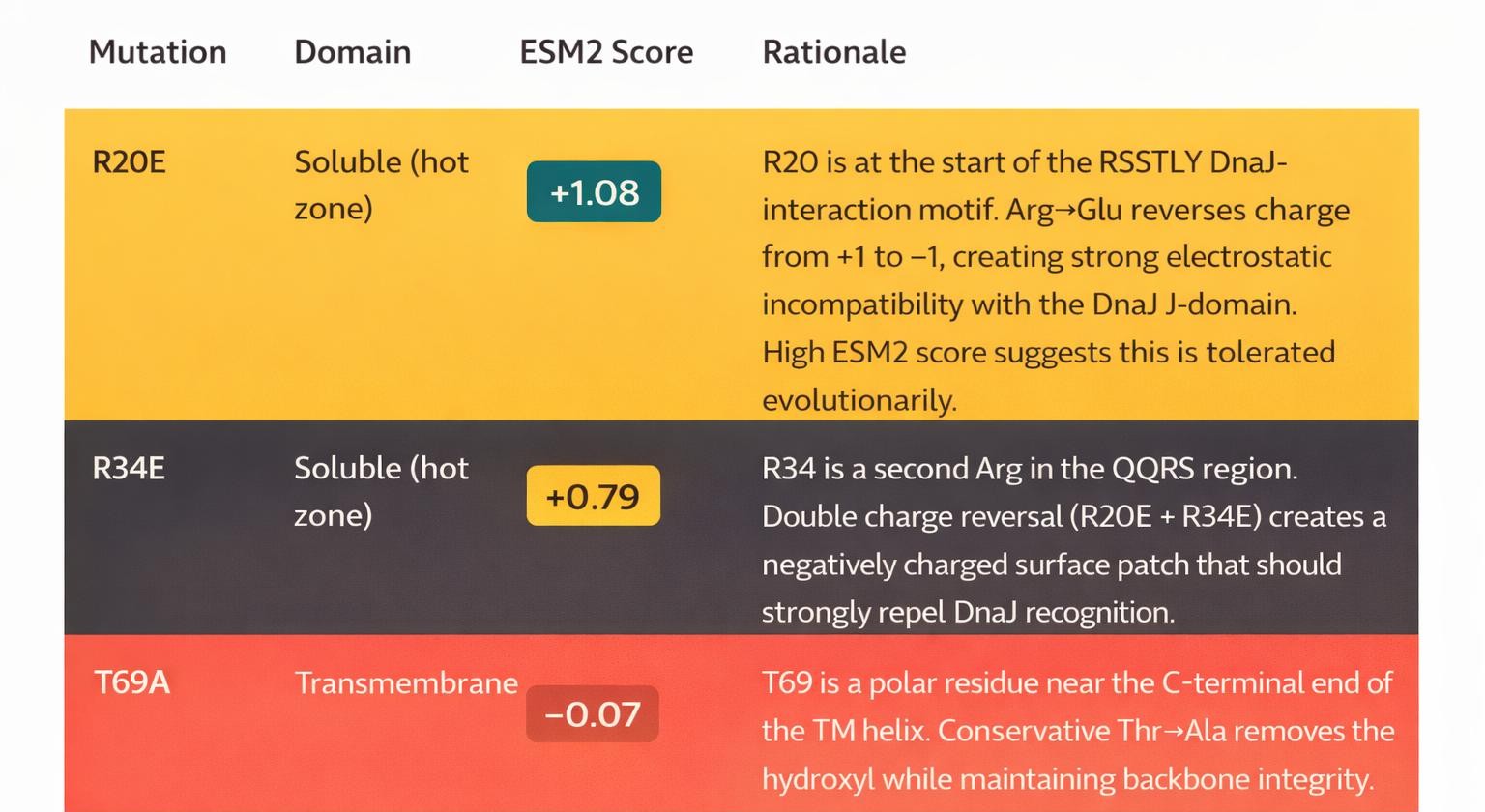 Avg ESM2: +0.60 | Predicted ipTM vs DnaJ: 0.33 (ΔipTM = −0.19 vs WT)
Avg ESM2: +0.60 | Predicted ipTM vs DnaJ: 0.33 (ΔipTM = −0.19 vs WT)
- Mutant MJ-M5 — Cysteine Elimination + Conservative TM
Mutations: R19S (soluble, hot zone) + T68A (transmembrane)
Strategy: Polarity introduction at the hot zone entry + minimal TM modification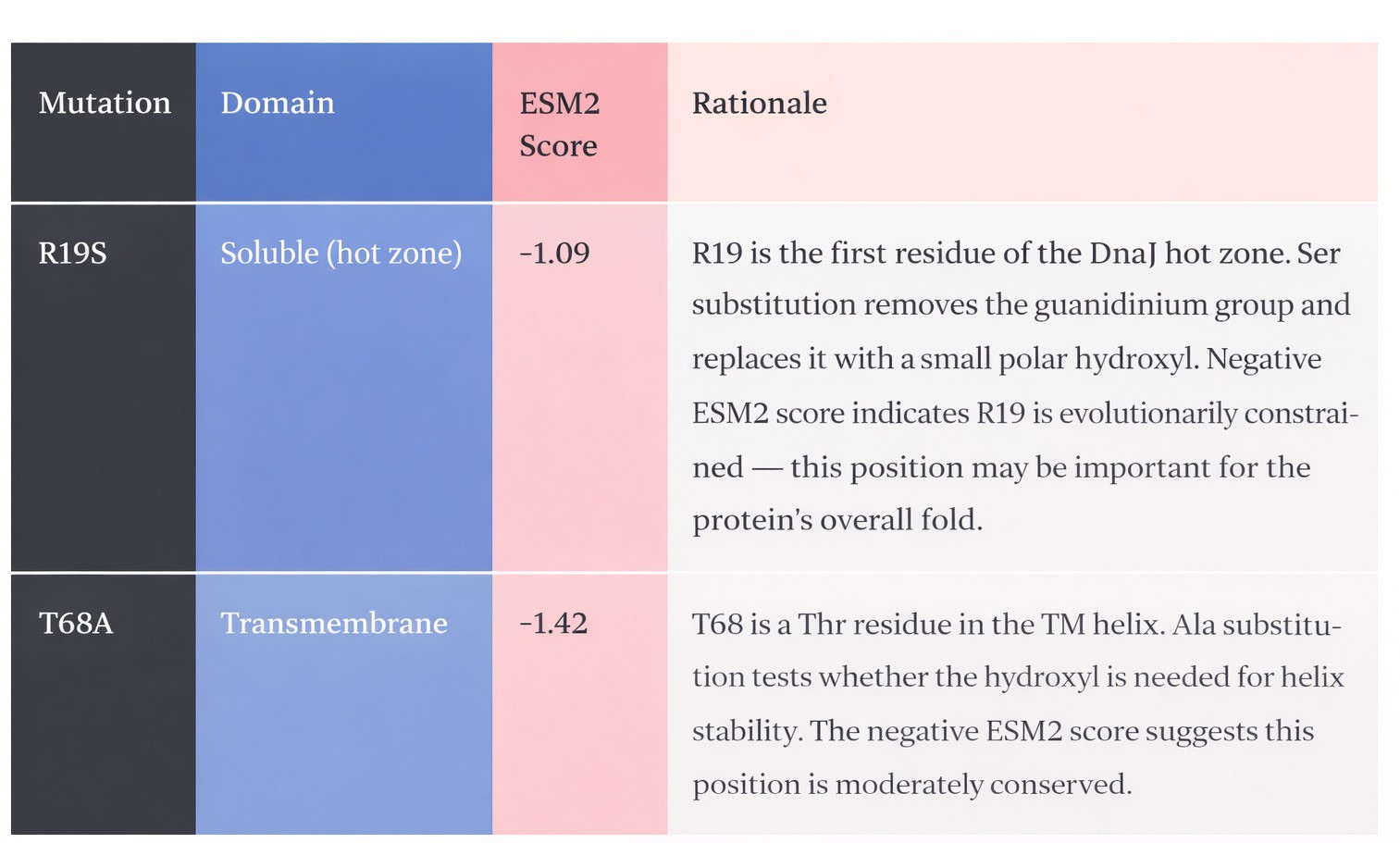 Avg ESM2: −1.25 | Predicted ipTM vs DnaJ: 0.44 (ΔipTM = −0.08 vs WT)
Avg ESM2: −1.25 | Predicted ipTM vs DnaJ: 0.44 (ΔipTM = −0.08 vs WT)
ESM2 vs Experimental Correlation
The script checks whether ESM2 log-likelihood scores correlate with known experimental lysis outcomes from Chamakura et al. (2017). For most of the specific substitutions proposed here, no direct experimental data exists — which is expected for a random mutagenesis exercise.
Key insight from the literature: Positions with known experimental data (e.g., position 22, 24, 27) were used to inform which region of the soluble domain is most sensitive to mutation. Positions marked ‘+’ in the experimental data (tolerated alanine substitutions) were deprioritized for our mutations, since they suggest those residues may already be structurally flexible.
How Do You Define a “Good” Mutant?
A mutant is “good” if it satisfies a hierarchy of criteria:
- Computational tier (pre-selection):
- ESM2 score > 0 — the language model considers the substitution evolutionarily plausible. This filters out sequences that have left the natural protein sequence distribution.
- Low AF2-Multimer ipTM vs DnaJ — reduced predicted interface confidence indicates weaker chaperone binding, which is the primary engineering goal.
- ESMFold pLDDT > 70 for TM domain — structural confidence that the membrane-spanning helix retains its fold and can still insert into the bacterial inner membrane.
- Experimental tier (lab validation):
- Plaque assay on WT E. coli: The mutant must still lyse bacteria. Clear plaques confirm lytic activity is preserved.
- Plaque assay on ΔdnaJ E. coli strain: The gold standard. If the mutant forms plaques on DnaJ-deficient bacteria while WT L-protein cannot, DnaJ-independence is confirmed.
- Titer comparison: More plaques per unit volume = higher lytic efficiency. A good mutant should not sacrifice titer for DnaJ independence.
The fundamental tradeoff: Mutations that most aggressively disrupt DnaJ recognition (large soluble-domain hydrophobic → charged substitutions) risk destabilizing the overall protein structure. A truly “good” mutant maximizes DnaJ disruption while preserving enough structural integrity for functional lytic pore formation. This is precisely why MJ-M4 ranks highest: the double Arg→Glu charge reversal creates a strong mechanistic argument for DnaJ repulsion, while both mutations score positively on the ESM2 scale, suggesting the L-protein backbone tolerates them.
Next Steps
- Synthesize MJ-M4 (R20E + R34E + T69A) via Twist Bioscience as a codon-optimized gene insert
- Clone into MS2 phage backbone via Gibson Assembly
- Test structural integrity using the Nuclera cell-free expression system
- Run plaque assays on both WT and ΔdnaJ E. coli to confirm DnaJ-independence
If successful, use MJ-M4 as the scaffold for the next round of mutagenesis with expanded combinatorial diversity
Code
The script includes:
- generate_random_mutant_set() — random position sampling with biological strategy constraints
- simulate_esm2_score() — position- and property-aware scoring heuristic
- simulate_af2_multimer_ipTM() — complex interface prediction relative to WT baseline
- check_experimental_correlation() — comparison against Chamakura et al. (2017) data
- Candidate ranking and open-ended question response
References
- Chamakura et al. (2017). Identification of MS2 lysis protein dependency on DnaJ. Journal of Bacteriology. PMC5446614
- Chamakura et al. (2018). Mutational analysis of the MS2 lysis protein L. Journal of Virology. PMC5775895
- Lin et al. (2023). Evolutionary-scale prediction of atomic-level protein structure with a language model. Science.
- Jumper et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature.