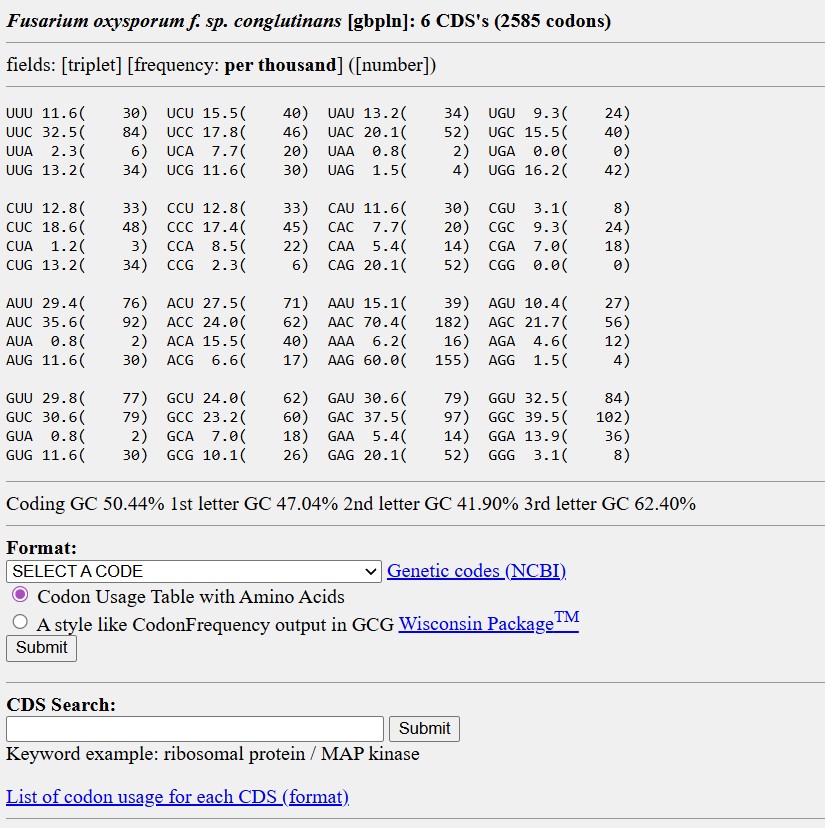
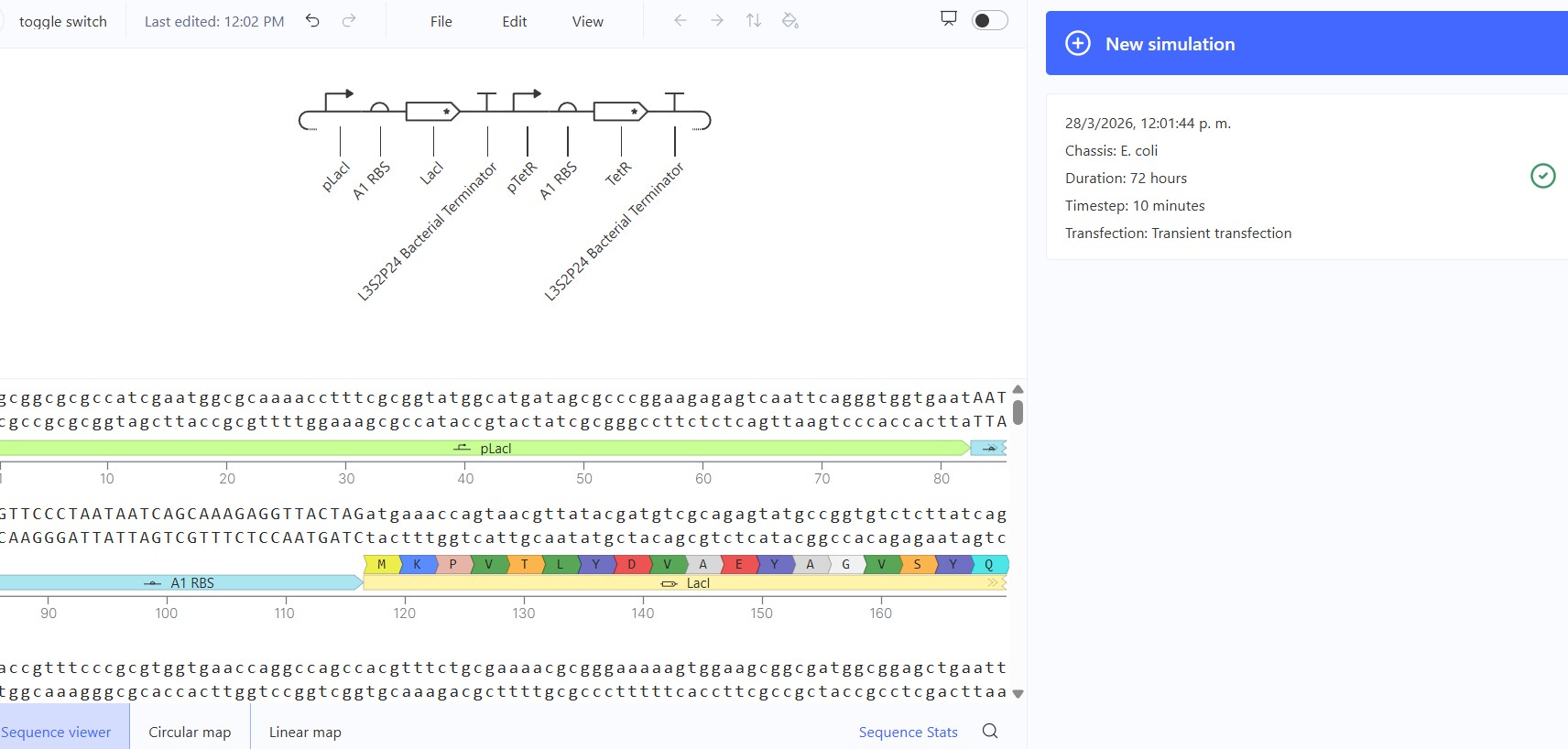
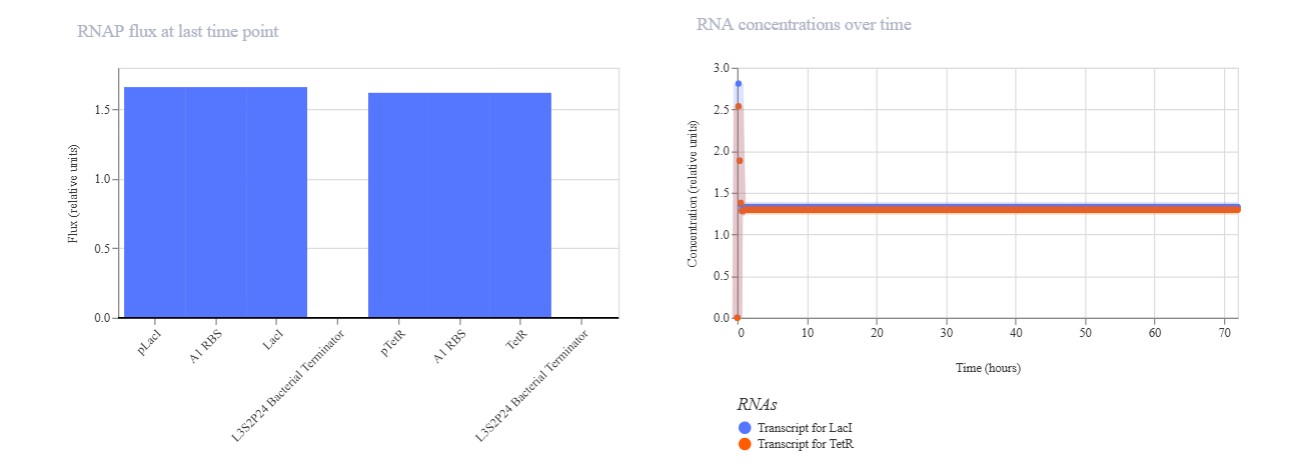
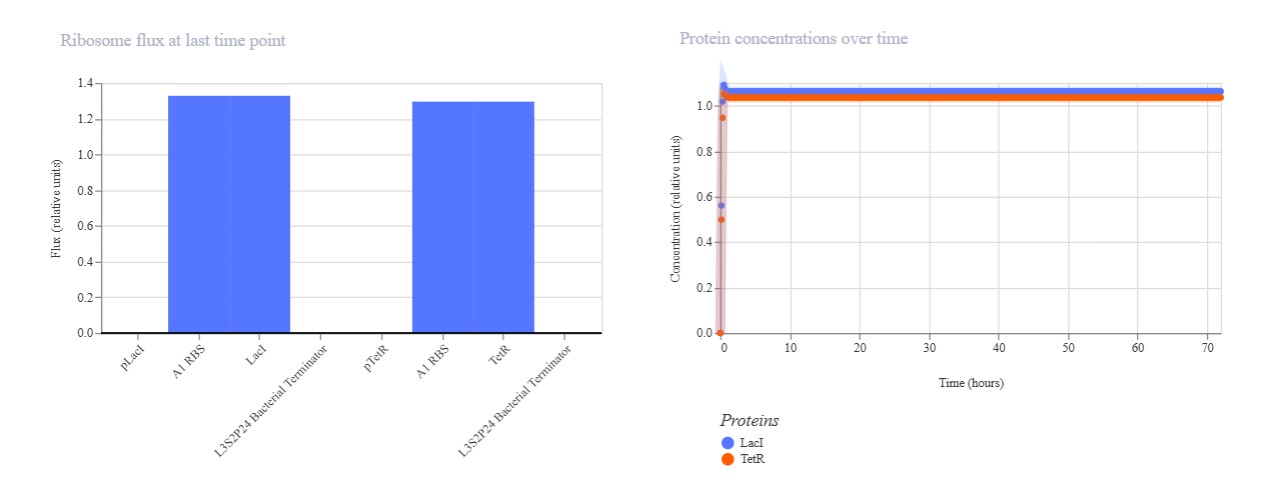
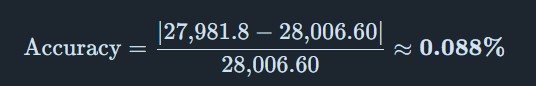Name of the proyect: Fungi Biodyes The project I am interested in working on focuses on the textile industry, which is responsible for 20% of global industrial water pollution, with approximately 200,000 tons of dyes lost annually in effluents during dyeing processes. More than 8,000 synthetic chemicals are used in textile dyeing, many of which are toxic and non-biodegradable. (Dutta, 2024)
Homework: Final Project
Fungi Biodyes:
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc. Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements. What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork Next year
Community Bioart Project Reflection Unfortunately, I was not able to participate in this year’s collaborative bioart project before the editing deadline. However, I really liked the concept of creating a global artwork experiment where many people contribute together to form a larger scientific and artistic piece.
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy? The error rate is the 1:10^6, for the other hand the size of the human genome, which is approximately 3 × 10⁹ base pairs. If DNA polymerase functioned only at its intrinsic error rate, replication would introduce an extremely high number like would result in about 300 errors every time the genome is copied a lot mutation and problems, making genomic stability impossible.
Part A. Conceptual Questions
How many amino acid molecules do you ingest when eating 500 grams of meat? (Average amino acid ≈ 100 Daltons) Approximately 20% of meat is protein. Therefore, 500 g of meat contains about 100 g of protein. If we assume an average amino acid molecular weight of 100 g/mol (≈100 Daltons), then:
100 g protein ÷ 100 g/mol ≈ 1 mol of amino acids.
One mole corresponds to 6 × 10²³ molecules (Avogadro’s number). Therefore, eating 500 g of meat provides on the order of 6 × 10²³ amino acid molecules.
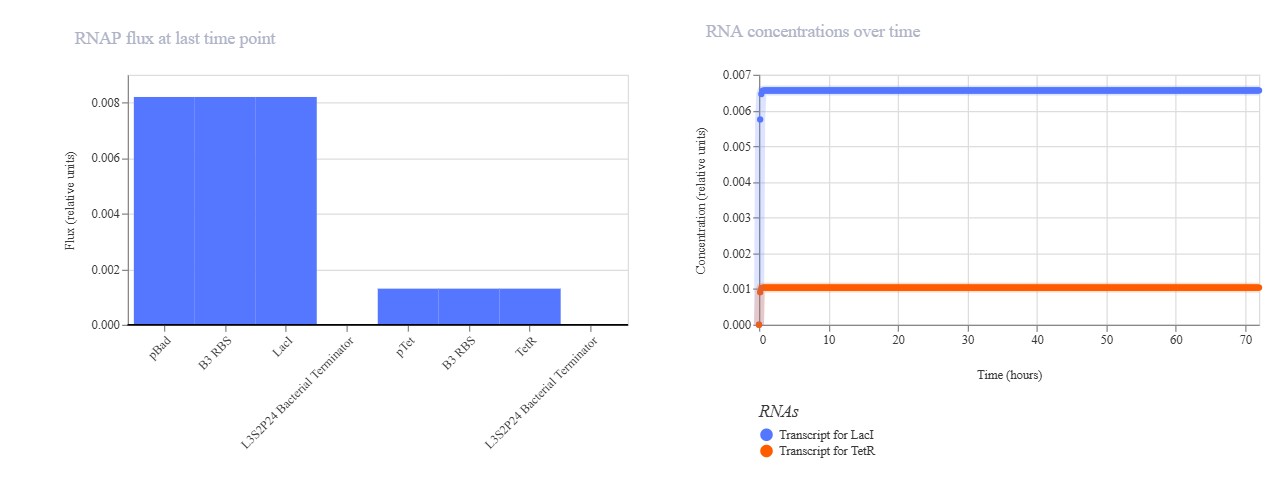
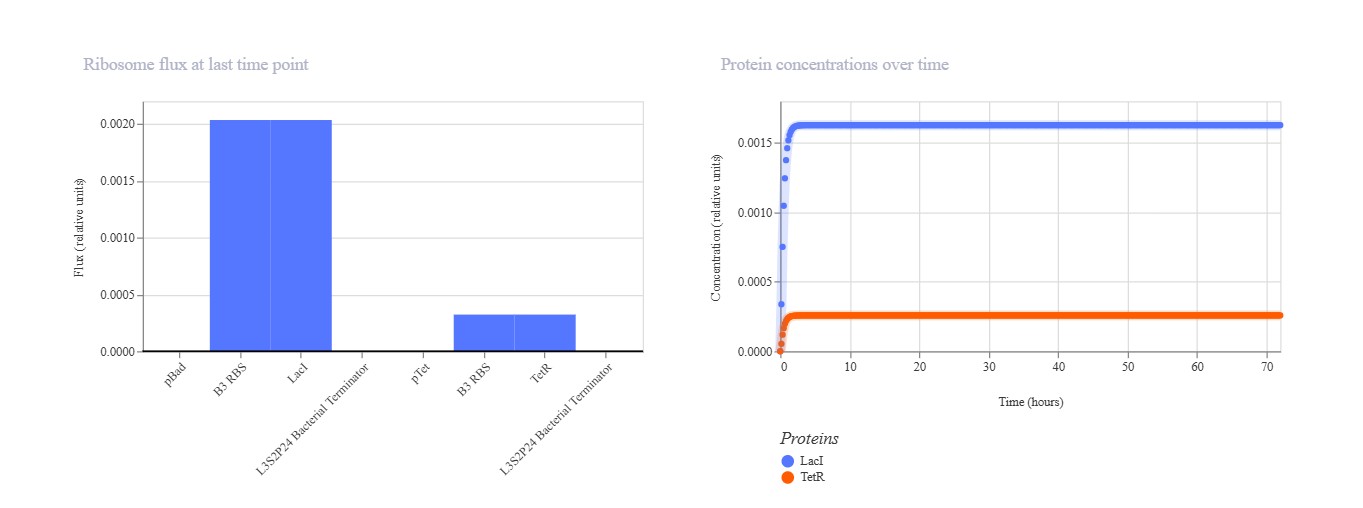
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation. {Normal} MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
{Mutation} MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card: Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence. To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
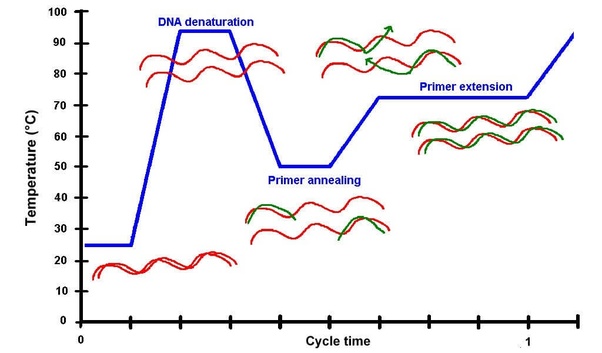
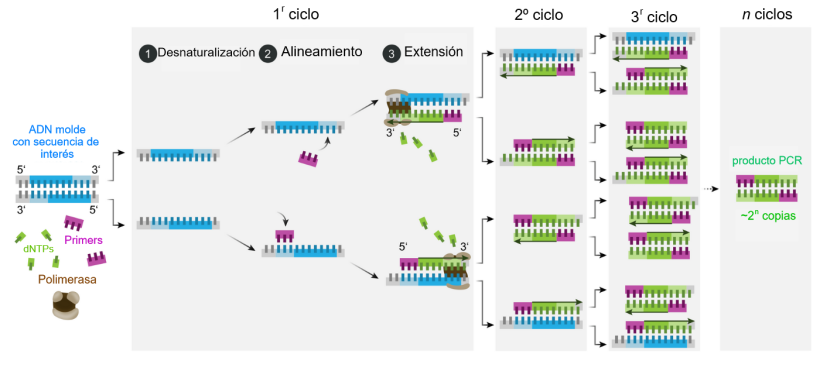
DNA Assembly What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
This process relies on a specialized DNA polymerase with proofreading activity. Unlike standard Taq polymerase, high-fidelity enzymes (such as Phusion DNA Polymerase) possess 3’→5’ exonuclease activity, which allows them to detect and correct errors during DNA synthesis.Phusion High-Fidelity PCR Master Mix
PCR consists of repeated cycles with three main steps:
Denaturation (~98°C)
The double-stranded DNA is separated into single strands.
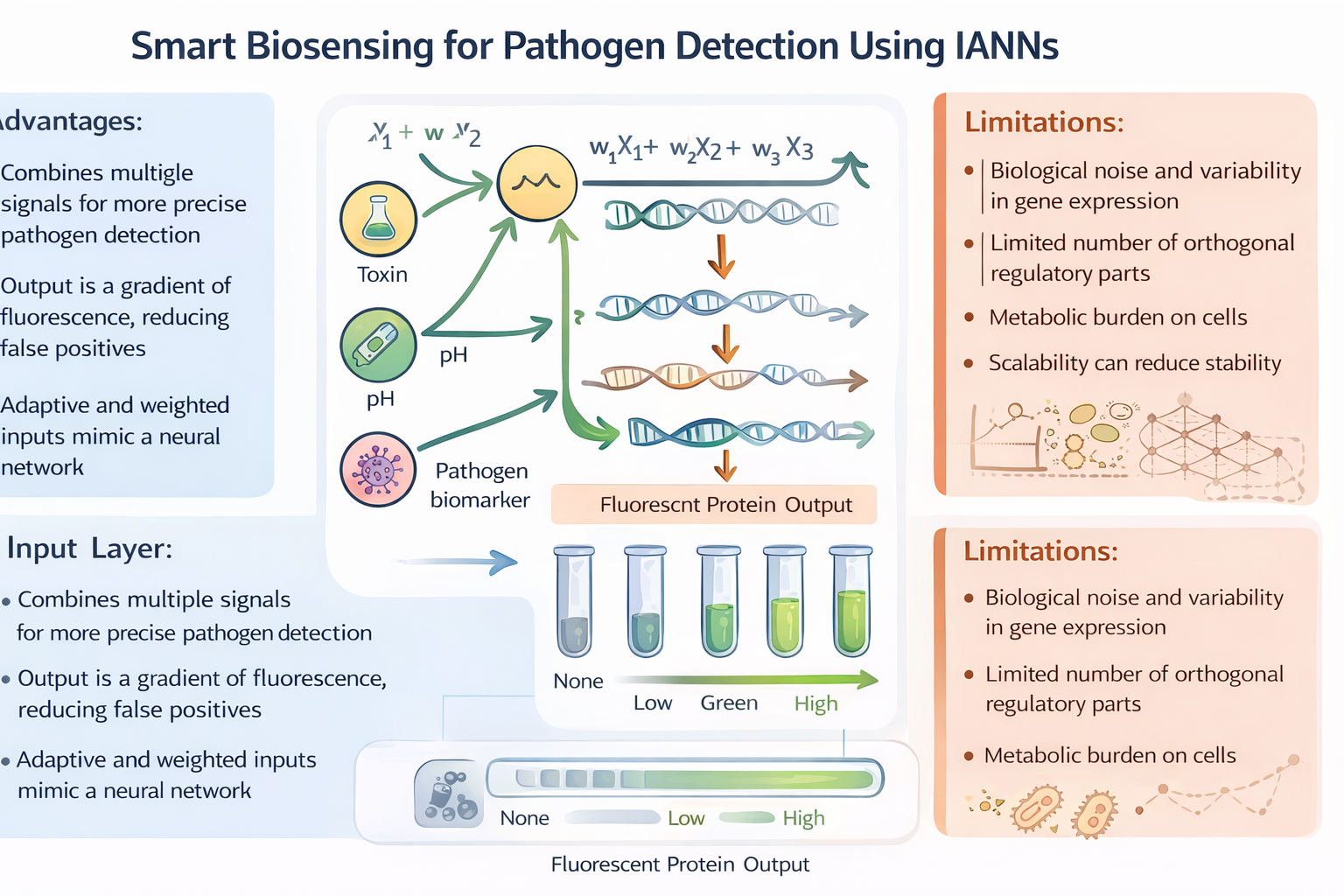
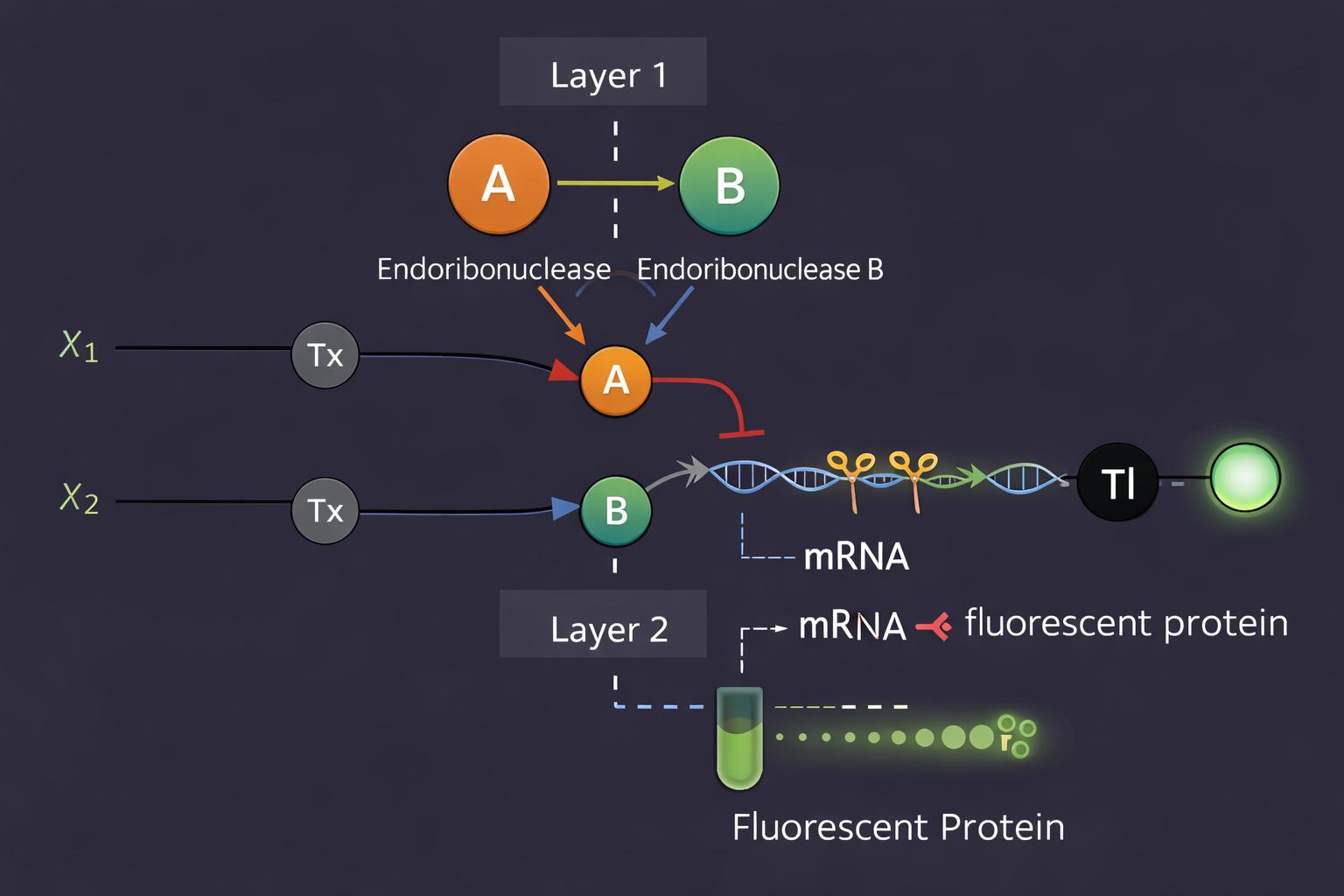
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Intracellular Artificial Neural Networks (IANNs) offer significant advantages over traditional genetic circuits that rely on Boolean logic. While Boolean circuits produce binary outputs (ON/OFF), IANNs can process continuous input signals and generate graded responses. This allows cells to integrate multiple inputs simultaneously and respond in a more flexible and nuanced way. Additionally, IANNs can assign different weights to inputs, enabling more complex decision-making processes compared to rigid Boolean logic gates. This makes them particularly useful in environments where signals are noisy or variable, as they can produce smoother and more adaptive outputs.
Homework Part A: General and Lecturer-Specific Questions General homework questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Cell-free protein synthesis (CFPS) offers significant advantages over traditional in vivo methods, particularly in terms of flexibility and control over experimental variables. One of the main advantages is that CFPS operates as an open system, allowing direct manipulation of all reaction components. Researchers can precisely control variables such as DNA or mRNA concentration, ion composition (Mg²⁺, K⁺), cofactors, and environmental conditions like temperature and pH. In contrast, others systems are limited by cellular regulation and metabolic constraints.
Subsections of Homework
Week 1 HW: Principles and Practices
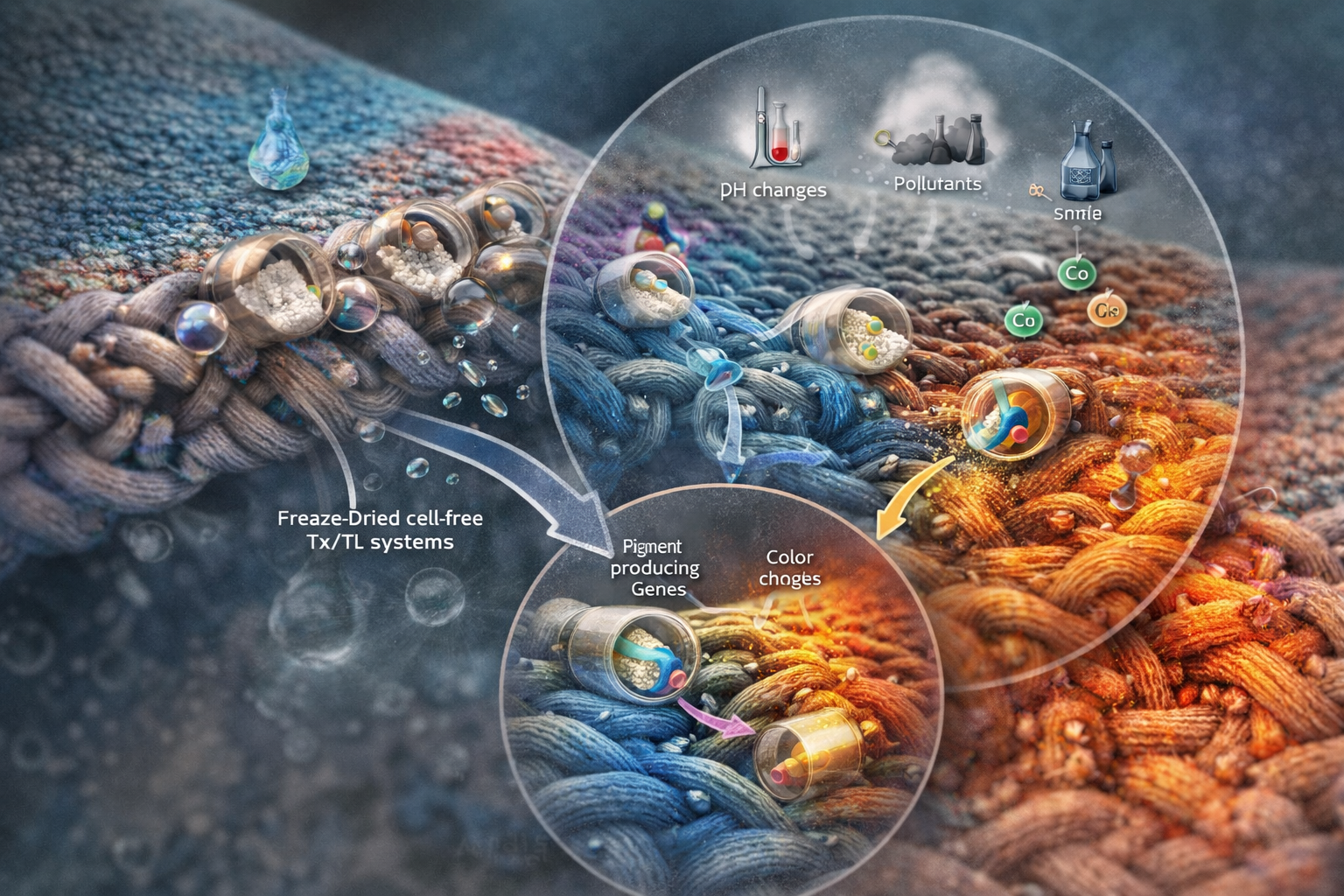
Name of the proyect: Fungi Biodyes
The project I am interested in working on focuses on the textile industry, which is responsible for 20% of global industrial water pollution, with approximately 200,000 tons of dyes lost annually in effluents during dyeing processes. More than 8,000 synthetic chemicals are used in textile dyeing, many of which are toxic and non-biodegradable. (Dutta, 2024)
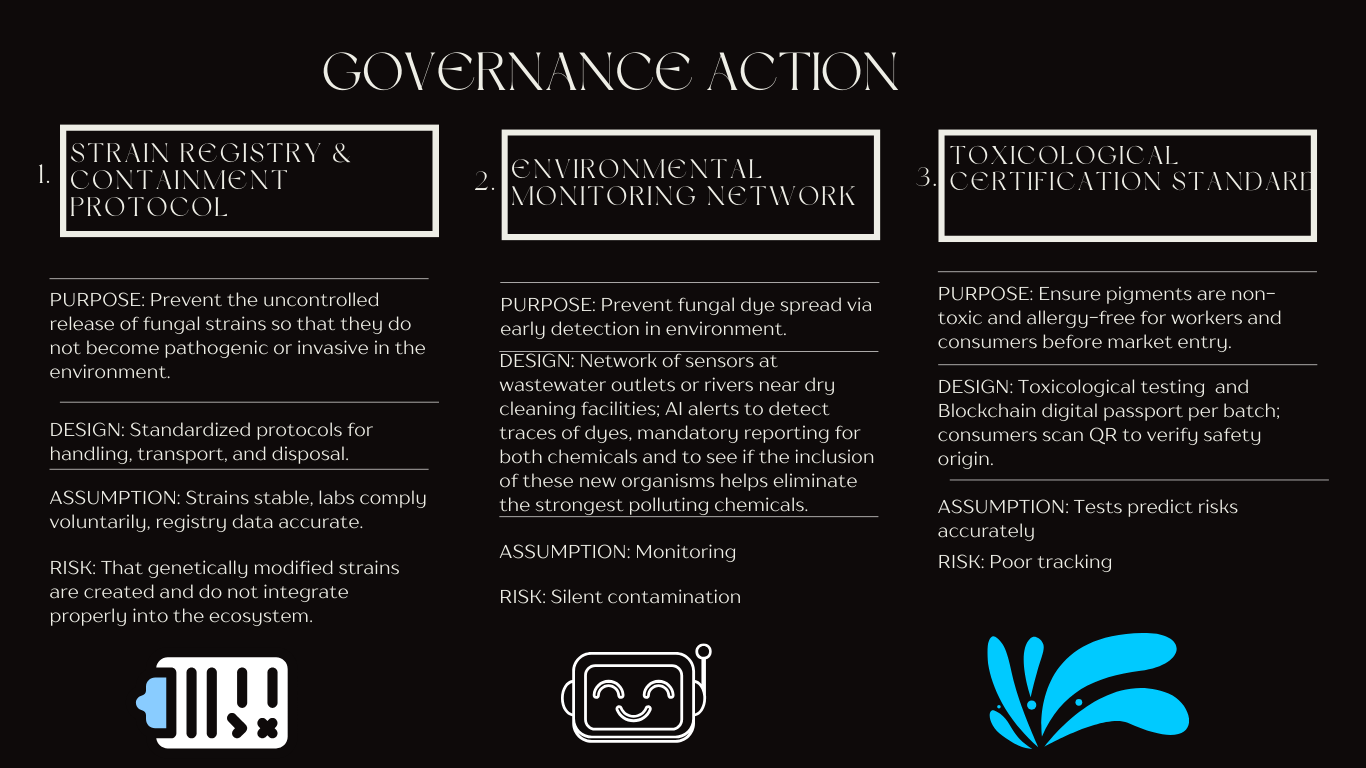
I mainly want to focus on one governance policy objective
Ensuring safety and security:
Fundamentally, one of the precautions that must be taken into account with this project is avoiding the uncontrolled release of fungal strains into the environment that are actively pathogenic.
It is also important to ensure that the pigments produced are not toxic or allergenic to textile workers and end consumers. Establish biosafety protocols for large-scale production.
Actions:
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
2
3
1
• By helping respond
2
1
3
Foster Lab Safety
• By preventing incident
3
1
2
• By helping respond
2
1
3
Protect the environment
• By preventing incidents
1
3
2
• By helping respond
2
3
1
Other considerations
• Minimizing costs and burdens to stakeholders
3
2
1
• Feasibility?
2
3
1
• Not impede research
2
3
1
• Promote constructive applications
2
1
3
5 . Based on the scores and the analizys, I would prioritize option 2 as the initial governance approach, combining it with option 1.
The reason for starting with surveillance focused approaches as a governance action rather than prevention stems from the current state of fungal biological dyes technology. At this early stage, the industry is still in the experimental phase and there is no fully experimental data, the most it is theoretical. The Environmental Monitoring Network allows innovation to move forward while creating a knowledge base on environmental challenges, how they can help or how they can be integrated into the environment, and also, in the worst-case, the adverse effects they may have.
Besides, implementing community-based sensor networks and observation systems would have benefits beyond just monitoring bio-dyes, but also dyes that affect aquifers. However, it is important to ensure that they do not harm those who handle them and that there is flexibility to experiment with different fungal strains, cultivation conditions, and pigment extraction methods.
Research sources :
1.Sohini Dutta, Satadal Adhikary(2024). Contamination of textile dyes in aquatic environment: Adverse impacts on aquatic ecosystem and human health, and its management using bioremediation. Journal of Environmental Management, 352, 119936 https://www.sciencedirect.com/science/article/abs/pii/S0301479724000896
2. López, M. A., & García, J. L. (2023). The impact of natural dyes in the textile industry. In A. Pérez (Ed.), Sustainable dye trends (pp. 125-142). Springer Nature. DOI: 10.1007/978-981-19-8853-0_10
Week 10 hw: Imaging and measurement
Homework: Final Project
Fungi Biodyes:
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
Measurements and Methods ( Fusarium Biodyes)
Aspect Measured
What is Measured
How it is Measured
Pigment intensity
Amount and strength of fungal pigment produced
Pigment extraction followed by absorbance reading at specific wavelengths
Color distribution / pattern formation
Spatial variation and uniformity of color on textiles
Image capture and pixel-based analysis (RGB / intensity mapping)
Fungal growth rate
Speed of colony expansion and biomass production
Measure colony diameter over time (agar) and dry weight (liquid culture)
Pigment stability
Resistance of color to washing, light, and environmental exposure
Compare color intensity before and after stress conditions
Gene expression (pigment pathway)
Activity of genes involved in pigment biosynthesis
Quantify mRNA levels under different conditions
DNA sequence validation
Presence of mutations introduced (in silico-guided)
Sequence analysis of fungal DNA
Protein function (indirect)
Effect of mutations on pigment-related enzymes
Correlate sequence changes with pigment output and growth
Spectrophotometry: Used to quantify pigment concentration by measuring absorbance at specific wavelengths. This allows precise comparison of color intensity between conditions.
RT-qPCR (Reverse Transcription Quantitative PCR): Used to measure gene expression levels of key enzymes in pigment biosynthesis pathways, providing insight into metabolic activity.
Sanger DNA Sequencing: Applied to confirm the presence of mutations proposed through in silico analysis in pigment-related genes.
In silico tools (Protein Language Models & sequence analysis): Used to propose mutations, predict their effects on protein stability and function, and guide experimental design.
Gel Electrophoresis: Used to verify PCR products before sequencing and ensure DNA quality.
Homework: Waters Part I — Molecular Weight
-We will analyze an eGFP standard on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
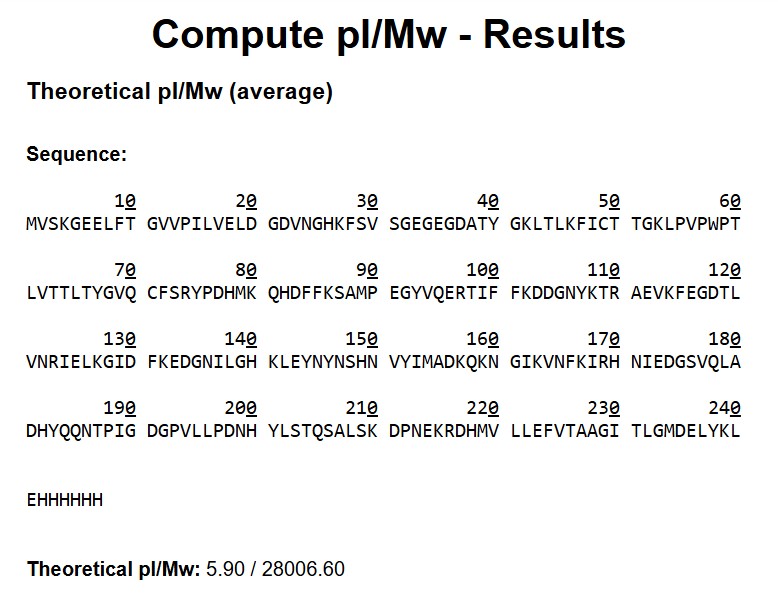
Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
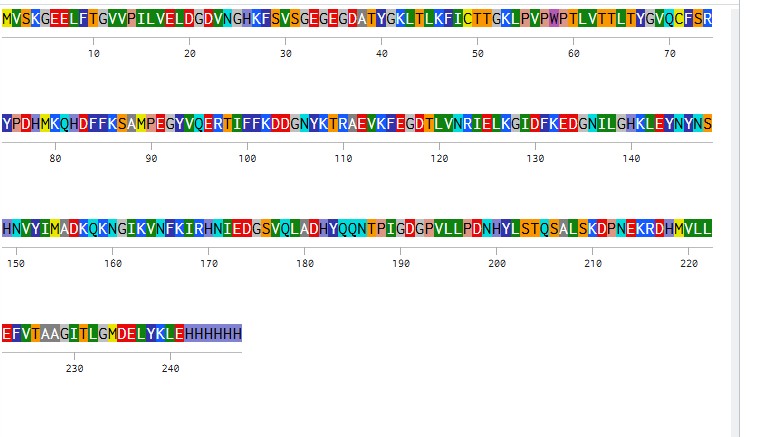
eGFP Sequence:
MVSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNYNSHNV YIMADKQKNG IKVNFKIRHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LSTQSALSKD PNEKRDHMVL LEFVTAAGIT LGMDELYKLE HHHHHH
Note: This contains a His-purification tag (HHHHHH) and a linker (the LE before it).
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
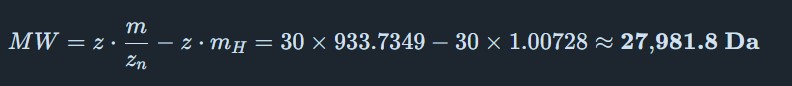
Molecular Weight Determination of eGFP
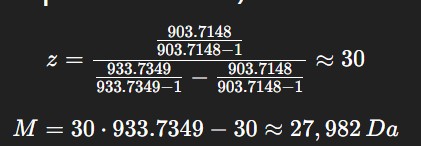
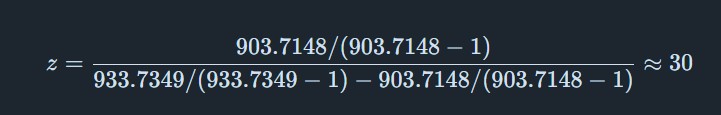
From Figure 1 (LC-MS spectrum), two adjacent peaks were selected from the clearly resolved charge state series around m/z ~900: ( m_n = 933.7349 ) and ( m_{n+1} = 903.7148 ). Using the adjacent charge state equation:
The experimental molecular weight (~27,982 Da) closely matches the theoretical value (28,006.60 Da), confirming the identity of the eGFP protein.
Determine the MW of the protein using the relationship between
Determine the MW of the protein
eGFP Molecular Weight Calculation
Theoretical MW (ExPASy)
Input: full eGFP sequence + linker LE + His-tag (EHHHHHH) MW_theory = 28,006.60 Da (pI = 5.90)
Experimental MW (LC-MS)
Adjacent peaks from LC-MS spectrum:
(m/z)_n = 933.7349
(m/z)_{n+1} = 903.7148
Charge state z:
Experimental MW:
Calculate the accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1 using:
Error < 0.1% — excellent agreement between experimental and theoretical MW.
Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
Homework: Waters Part III — Peptide Mapping - primary structure
We will digest the eGFP protein standard into peptides using trypsin (an enzyme that selectively cleaves the peptide bond after Lysine (K) and Arginine (R) residues. The resulting peptides will be analyzed on the Waters BioAccord LC-MS to measure their molecular weights and fragmented to confirm the amino acid sequence within each peptide – generating a “peptide map”. This process is used to confirm the primary structure of the protein.
There are a variety of tools available online to calculate protein molecular weight and predict a list of peptides generated from a tryptic digest. We will be using tools within the online resource Expasy (the bioinformatics resource portal of the Swiss Institute of Bioinformatics (SIB)) to predict a list of tryptic peptides from eGFP.
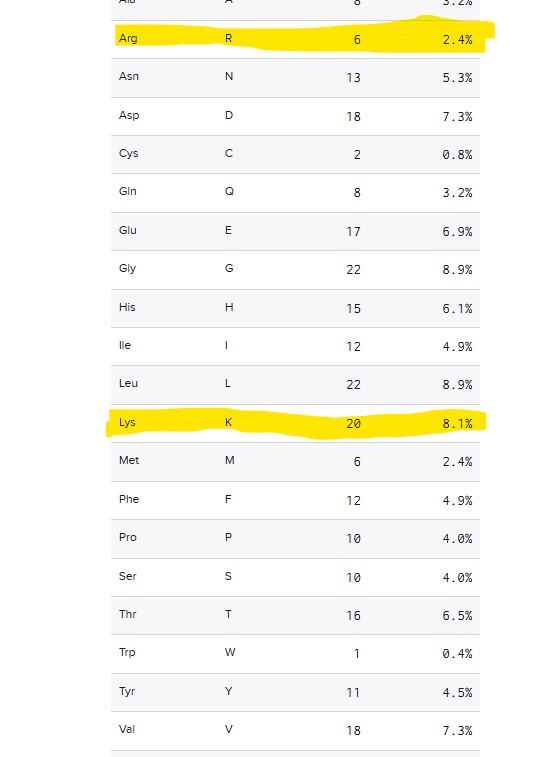
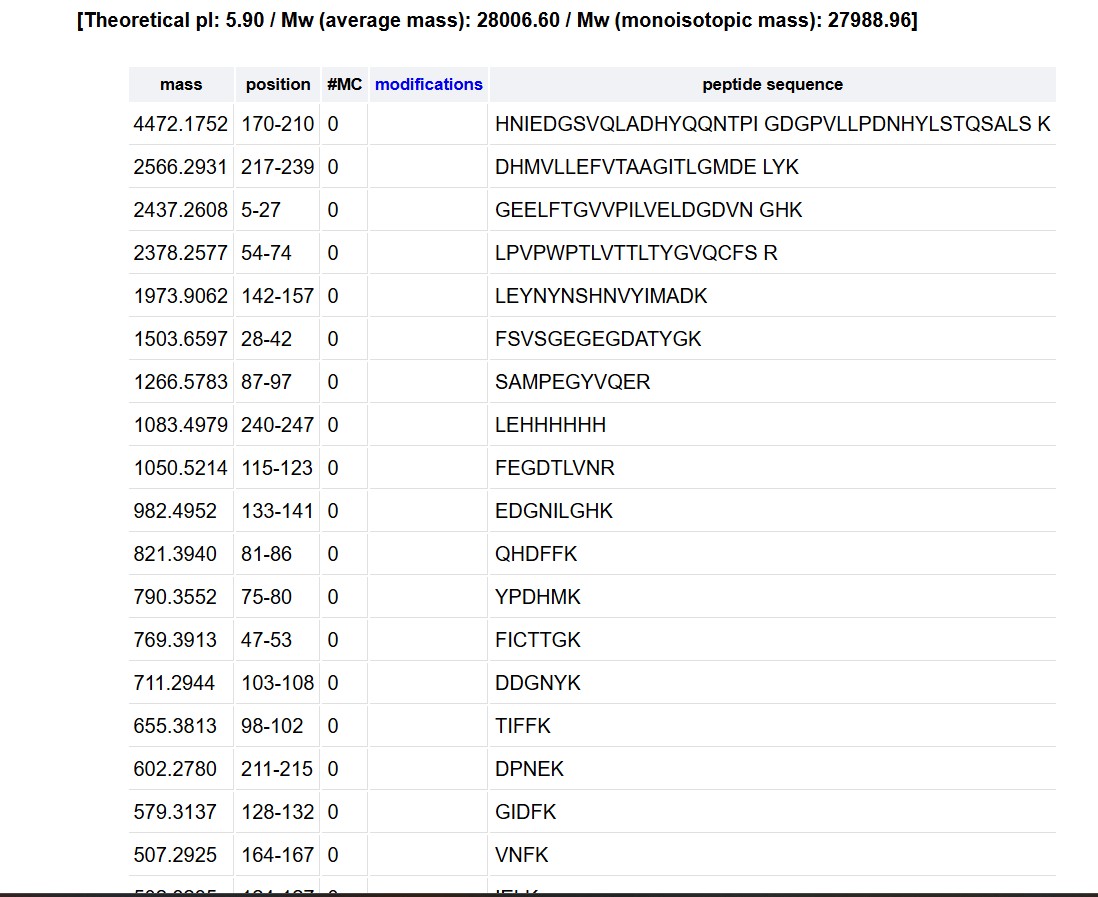
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
How many peptides will be generated from tryptic digestion of eGFP?
Navigate to https://web.expasy.org/peptide_mass/
Copy/paste the sequence above into the input box in the PeptideMass tool to generate expected list of peptides.
Use Figure 4 below as a guide for the relevant parameters to predict peptides from eGFP.
Click “Perform the Cleavage” button in the PeptideMass tool and report the number of peptides generated when using trypsin to perform the digest.
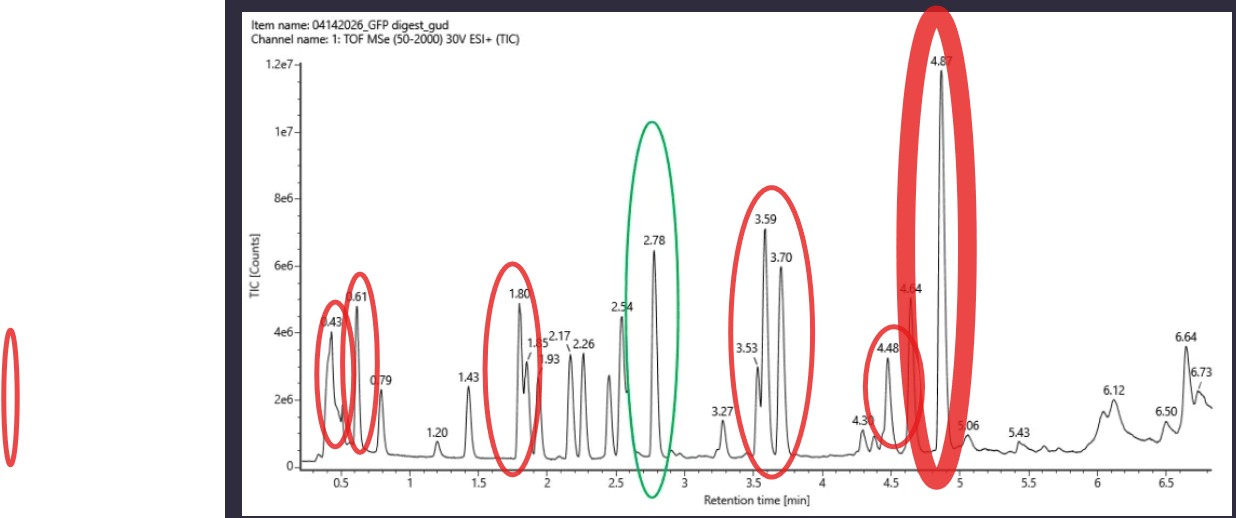
Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
The question asks to count all peaks with >10% relative abundance
between 0.5 and 6 minutes in the TIC chromatogram (Figure 5a).
Method
The tallest peak is at 4.87 min (~1.2e7 counts) = 100% relative abundance.
The 10% threshold ≈ 1.2e6 counts. Only peaks above this height are counted.
Peaks >10% Relative Abundance (0.5 – 6 min)
#
Retention Time (min)
1
0.43
2
0.61
3
1.43
4
1.80
5
2.17
6
2.78
7
3.59
8
3.70
9
4.64
10
4.87
Result
~10 chromatographic peaks are observed with >10% relative abundance
between 0.5 and 6 minutes.
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
ExPASy predicted approximately 27 peptides, whereas only ~10 peakswith >10% relative meaning there are fewer peaks than predicted. This discrepancy is expected: some peptides likely co-elute at the same retention time and appear as a single peak, while others (particularly small or highly hydrophilic peptides) may not be well retained by the reverse-phase LC column and therefore
go undetected.
Identify the mass-to-charg
The most abundant peak in Figure 5b appears at m/z = 525.76712. To determine the charge state (z), the isotope peaks visible in the zoom-in inset are separated by approximately 0.50 Da, which corresponds to a charge state of z = 2 (since isotope spacing = 1/z). Using this charge state, the mass of the singly charged form [M+H]⁺ is calculated as:
Isotope peak
m/z
M
525.76712
M+1
526.25918
M+2
526.76845
M+3
527.26098
[M+H]⁺ = z × (m/z) − (z − 1) × 1.00728 = 2 × 525.76712 − 1 × 1.00728 ≈ 1050.53 Da
This result is confirmed by the peak observed at m/z = 1050.52438 in Figure 5b,
which corresponds to the singly charged [M+H]⁺ ion of the same peptide.
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement?
The peptide eluting at 2.78 minutes was identified by comparing its measured [M+H]⁺ mass of 1050.527 Da to the list of expected tryptic peptide masses generated by the ExPASy PeptideMass tool. The measured mass matches a predicted tryptic peptide from the eGFP sequence.
Mass accuracy is calculated as:
This error of ~2.5 ppm is well within the acceptable range for high-resolution
TOF mass spectrometry (typically <10 ppm), confirming reliable peptide identification.
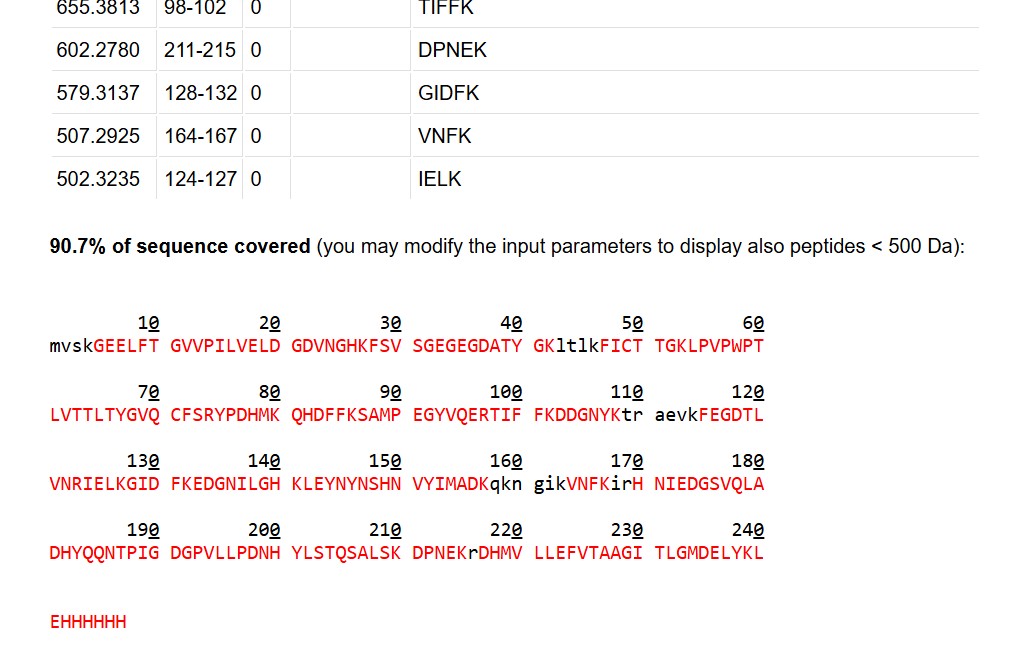
What is the percentage of the sequence that is confirmed by peptide mapping?
As shown in Figure 6, the amino acid coverage map reports 88% sequence coverage for eGFP. This means that peptides detected by LC-MS account for 88% of the total amino acid sequence of the protein, confirming the primary structure of the vast majority of eGFP. The remaining ~12% of the sequence was not detected, likely due to peptides that were too small, too large, or not well-retained under the LC conditions used.
Homework: Waters Part IV — Oligomers
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS). CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
7FU Decamer
8FU Didecamer
8FU 3-Decamer
8FU 4-Decamer
Polypeptide Subunit Name Subunit Mass
7FU 340 kDa
8FU 400 kDa
KLH Oligomeric States — CDMS Analysis
Using the subunit masses from Table 1 (7FU = 340 kDa, 8FU = 400 kDa), the
expected masses of each oligomeric species are calculated by multiplying the
subunit mass by the number of subunits:
Oligomeric Species
Subunit
# Subunits
Calculated Mass
Peak in Figure 7
7FU Decamer
7FU (340 kDa)
10
3,400 kDa = 3.40 MDa
3.4 MDa
8FU Didecamer
8FU (400 kDa)
20
8,000 kDa = 8.00 MDa
8.33 MDa
8FU 3-Decamer
8FU (400 kDa)
30
12,000 kDa = 12.00 MDa
12.67 MDa
8FU 4-Decamer
8FU (400 kDa)
40
16,000 kDa = 16.00 MDa
~16 MDa (low intensity)
Each predicted mass corresponds closely to an observed peak in the CDMS spectrum
(Figure 7). The small deviations (~0.3–0.7 MDa) are expected due to glycosylation
and other post-translational modifications present on KLH, which add mass beyond
the bare polypeptide sequence.
Homework: Waters Part V — Did I make GFP?
Please fill out this table with the data you acquired from the lab work done at the Waters Immerse Lab in Cambridge, or else the data screenshots in this document if you were unable to have lab work done at Waters.
Molecular Weight Analysis of GFP
Parameter
Theoretical
Observed/measured on the Intact LC-MS
PPM Mass Error
Molecular weight (kDa)
~26.9 kDa
26.98 kDa*
~2974 ppm
How to Obtain the Information
1. Theoretical Molecular Weight
The theoretical molecular weight of GFP can be obtained from:
The GFP amino acid sequence
For standard GFP, the molecular weight is usually approximately:
26.9 kDa
Week 11 hw: Building genomes
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
Next year
Community Bioart Project Reflection
Unfortunately, I was not able to participate in this year’s collaborative bioart project before the editing deadline. However, I really liked the concept of creating a global artwork experiment where many people contribute together to form a larger scientific and artistic piece.
For next year, I would love to contribute designs inspired by fungal growth patterns, especially the beautiful shapes and textures formed by mycelium. I think it would be interesting to create colorful organic structures with different branching forms and gradients inspired by fungi and microbial colonies.
One thing I liked about the project was how it combined science, creativity, and community collaboration in a very accessible way. It allowed people with different interests and backgrounds to contribute to a shared visual experiment.
For future versions of the project, it could be improved by allowing more time for participation and perhaps adding themed regions or biological inspiration categories so contributors can coordinate larger artistic structures together.
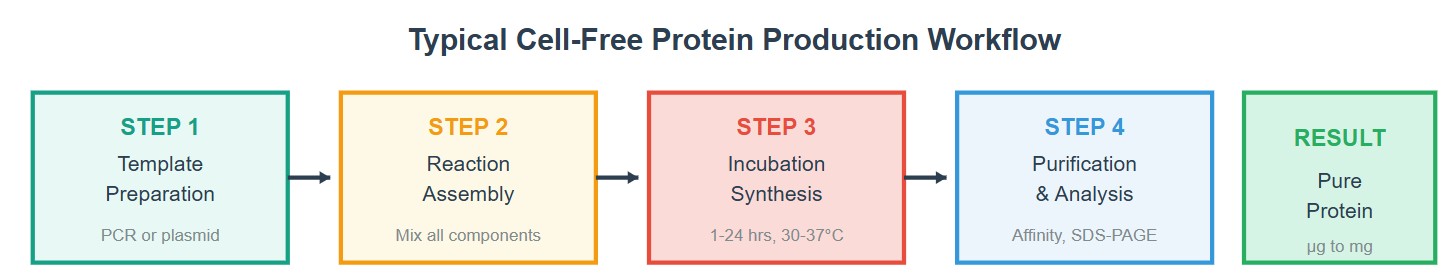
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase): Provides the cellular machinery (ribosomes, enzymes, cofactors) required for transcription and translation. T7 RNA polymerase enables efficient transcription of genes under a T7 promoter.
Salts/Buffer
Potassium Glutamate: Maintains ionic strength and mimics intracellular conditions, supporting proper enzyme function and protein synthesis.
HEPES-KOH pH 7.5: Maintains a stable physiological pH, preventing loss of enzymatic activity and preserving ribosomal integrity during prolonged incubation.
Magnesium Glutamate: Provides Mg²⁺ ions required for ribosome assembly, tRNA charging, and stabilization of ATP and NTPs; directly impacts translation efficiency and fidelity.
Potassium phosphate monobasic: Acts as the acidic component of the phosphate buffer system, helping maintain pH stability during the reaction. It also supplies inorganic phosphate required for ATP regeneration and metabolic reactions.
Potassium phosphate dibasic: Acts as the basic component of the phosphate buffer system, balancing the monobasic form to tightly regulate pH. It also contributes phosphate ions and supports optimal ionic conditions for enzyme activity.
Energy / Nucleotide System
Ribose: Feeds into the pentose phosphate and nucleotide salvage pathways to generate ribose-5-phosphate, a precursor for de novo and salvage nucleotide synthesis.
Glucose: Drives glycolytic activity in the lysate, enabling continuous ATP regeneration and maintaining redox balance through endogenous metabolic enzymes.
AMP: Serves as a precursor for ATP synthesis through phosphorylation pathways, helping regenerate the main energy currency required for transcription and translation.
CMP: Acts as a precursor for CTP, which is required for RNA synthesis during transcription.
GMP: Functions as a precursor for GTP, which is essential for both RNA synthesis and translation elongation steps.
UMP: Serves as a precursor for UTP, another nucleotide required for RNA synthesis during transcription.
Guanine: Provides an additional substrate for purine salvage pathways, enhancing GMP/GTP pools required for transcription and translation elongation.
Translation Mix (Amino Acids)
17 Amino Acid Mix: Supplies most amino acids required for polypeptide elongation, supporting continuous translation.
Tyrosine: Added separately due to its low solubility and tendency to precipitate, ensuring it does not become rate-limiting.
Cysteine: Included independently because it is chemically reactive and prone to oxidation, which can otherwise limit protein synthesis.
Additives
Nicotinamide: Acts as a precursor to NAD⁺, supporting redox cycling and metabolic reactions required for sustained ATP generation in long-duration reactions
Backfill: Used to adjust final reaction volume while preventing nucleic acid degradation by contaminating nucleases.
Nuclease Free Water
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
The PEP–NTP system directly supplies high-energy phosphate donors (PEP) and fully phosphorylated nucleotides (NTPs), enabling immediate and high-rate transcription and translation. However, this leads to rapid accumulation of inhibitory byproducts (e.g., inorganic phosphate) and depletion of energy sources, limiting reaction lifespan to ~1 hour.
In contrast, the NMP–Ribose–Glucose system relies on endogenous metabolic pathways within the lysate to regenerate ATP and NTPs from low-energy precursors (NMPs, ribose, and glucose). This reduces byproduct accumulation and allows continuous energy recycling, resulting in slower but significantly prolonged protein synthesis (~20 hours) with improved overall yield.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
sfGFP: Superfolder GFP is engineered for robust folding, allowing efficient fluorescence even in partially unfavorable conditions. This makes it highly reliable in cell-free systems where folding machinery may be limited.
mRFP1: Has a slow maturation time, meaning chromophore formation takes longer, which can delay fluorescence readout in shorter reactions.
mKO2: Is relatively acid-sensitive, so fluorescence intensity decreases if the reaction pH drops over time due to metabolic byproducts.
mTurquoise2: Has high quantum yield but requires efficient folding, making it sensitive to translation conditions and potentially prone to misfolding in cell-free systems.
mScarlet_I: Is is optimized for fast maturation and high brightness, but its performance can still depend on proper folding and oxygen availability for chromophore formation.
Electra2:Is is oxygen-independent but depends on availability of flavin cofactors (e.g., FMN/FAD) for fluorescence, which may be limiting in cell-free systems.
The amino acid sequences are shown in the HTGAA Cell-Free Benchling folder.
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
For mKO2, which is sensitive to acidic conditions, increasing the concentration of buffering agents such as HEPES-KOH and potassium phosphate in the cell-free mastermix will help maintain a stable pH throughout the 36-hour incubation, preventing acidification caused by metabolic byproducts. By stabilizing the pH, the protein structure and chromophore environment will be preserved, leading to improved fluorescence intensity and stability over time.
Week 2 dna read write and edit
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
The error rate is the 1:10^6, for the other hand the size of the human genome, which is approximately 3 × 10⁹ base pairs. If DNA polymerase functioned only at its intrinsic error rate, replication would introduce an extremely high number like would result in about 300 errors every time the genome is copied a lot mutation and problems, making genomic stability impossible.
Biology addresses this problem through multiple layered error correction mechanisms. These include the proofreading activity of DNA polymerases, post-replication mismatch repair systems, and additional DNA repair pathways that detect and correct replication errors. MutS Repair System, Correcting Gene Synthesis and Reduction GFP Gene Synthesis.
In one of these repairs, the polymerase at the active site only accepts correctly paired bases (A-T, G-C), since the pairs do not match dimensionally. When errors occur, the correction mechanism built into the polymerase uses exonuclease activity to reverse, remove the incorrect nucleotide, and resynthesize it correctly.
In addition to quality control, there is the mismatch repair system, in which specialized protein complexes (MutS/MutL in bacteria, MSH/MLH in humans) scan the newly replicated DNA to detect and correct any remaining errors. This system identifies the newly synthesized strand, removes the segments containing errors, and replaces them with the correct sequence.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Human protein, composed of about 450 amino acids, has several DNA sequences that, in theory, could encode it. Because the genetic code is composed of combinations of amino acids, there could be approximately 10^200 or more different nucleotide sequences that encode exactly the same amino acid sequence. However, in practice, the vast majority of these “synonymous” sequences do not work to produce a functional protein.(Yi Liu, 2021)
Therefore, one could say that the reason why not all codons are useful in a sequence is based on biological constraints such as codon usage bias, tRNA availability, mRNA secondary structure formation (especially in GC-rich sequences), the presence of unwanted regulatory signals, and the effects of translation speed on protein folding. Therefore, only a fraction of the possible codes encode DNA.
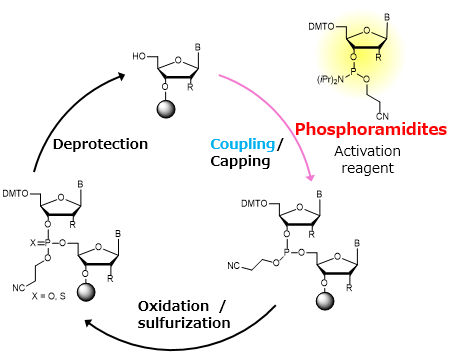
What’s the most commonly used method for oligo synthesis currently?
Phosphoramidite method by Caruthers
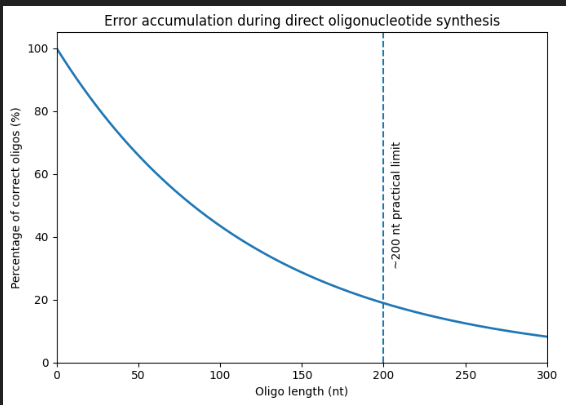
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Figure generated using AI and adapted from concepts Oligonucleotide Synthesis.
Currently, oligonucleotide synthesis uses a solid-phase cycle with phosphoramidite, in which one nucleotide is added at a time with high, but not perfect, coupling efficiency (typically between 97% and 99% per step). Each additional base adds a small chance of failure, either a deletion or an incorrect base, so the fraction of complete, error-free molecules decreases exponentially with length. When about 200 bases are reached, most of the chains in the pool are truncated or contain at least one error, resulting in very low useful yield.
Why can’t you make a 2000bp gene via direct oligo synthesis?
Due to the same reason mentioned previously, trying to synthesize a 2,000-base sequence in a single pass would yield almost no correct full-length molecules: multiplying a pass efficiency of less than 99% over 2,000 cycles reduces the chances of obtaining a perfect strand to almost zero. Instead, genes of this size are created by assembling many shorter, high-quality oligonucleotides or gene fragments, as direct chemical synthesis of thousands of bases is too imprecise to be practical.
[Given slides #2 & 4 (AA:NA and NA:NA codes)] What code would you suggest for AA:AA interactions?
In the case of AA:AA interactions, I will suggest thinking in terms of a “protein protein interaction code” based on the complementarity of side chains. In essence, amino acid pairs recognize each other through combinations of physicochemical complementarity between amino acid side chains, where properties such as electrical charge, hydrophobicity, size, shape, and the ability to form hydrogen bonds or hydrophobic interactions determine how two amino acids interact with each other.
Unlike the simple triplet genetic code, this AA:AA code is distributed across different surfaces.
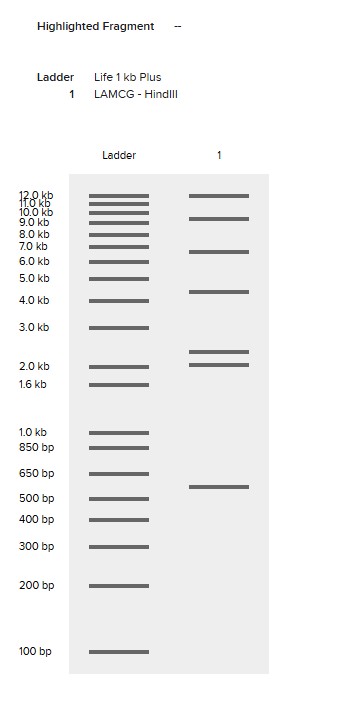
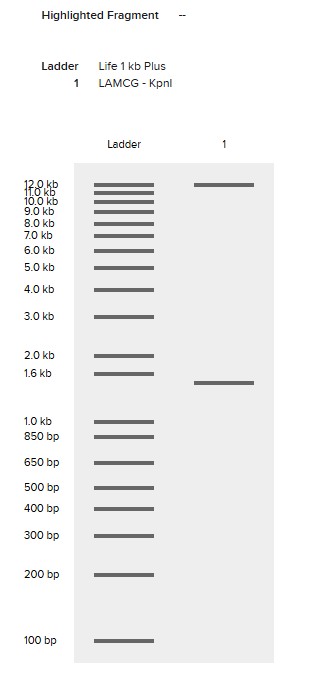
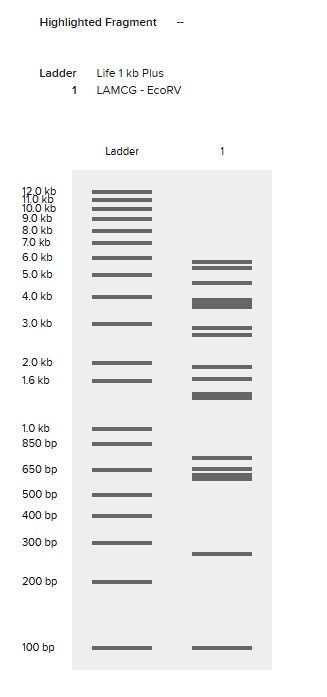
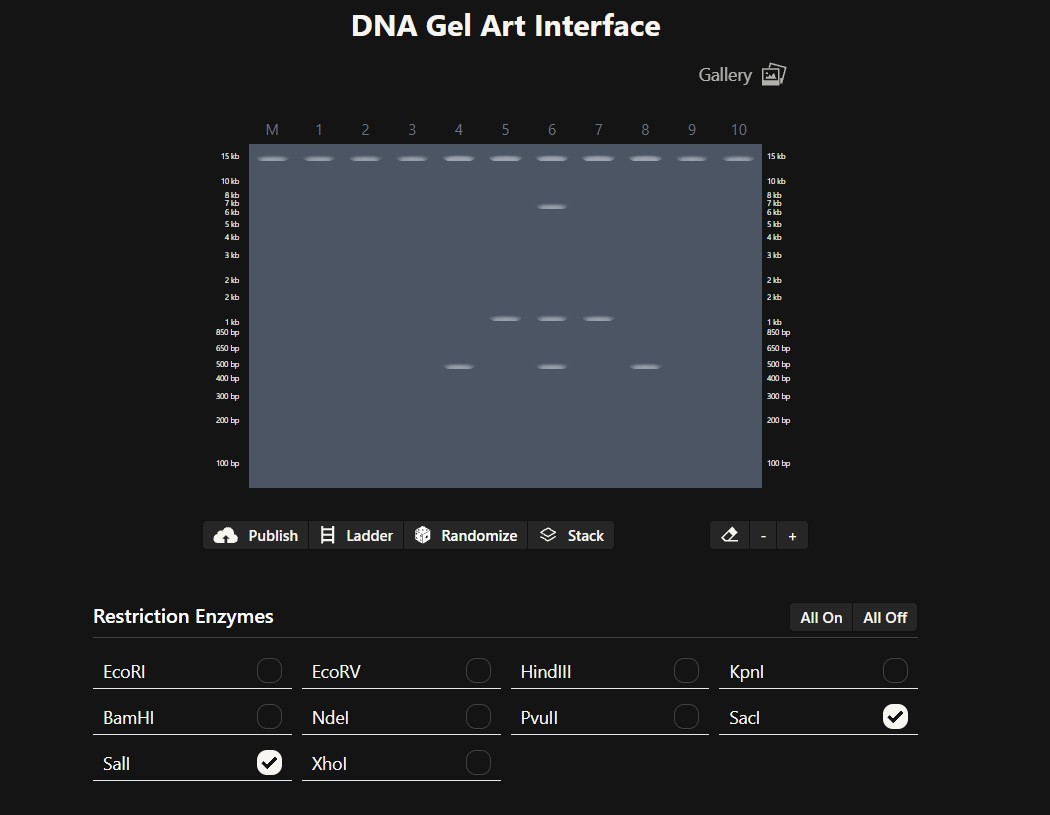
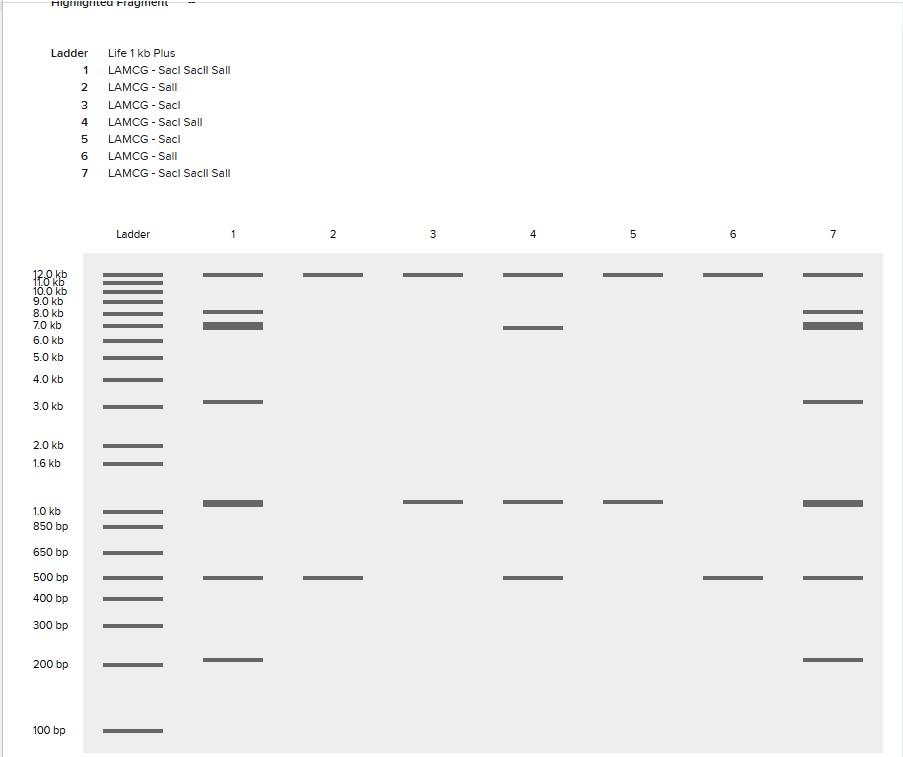
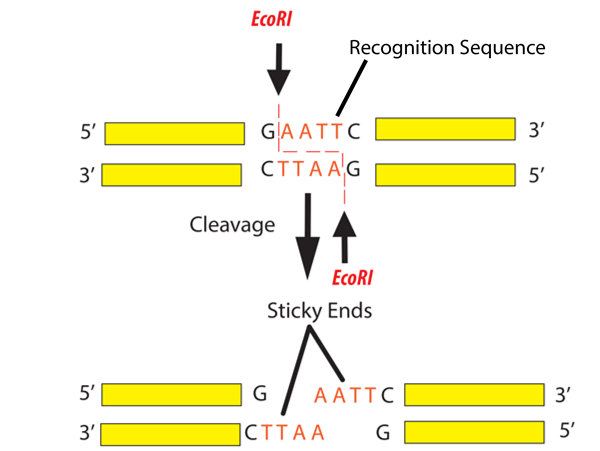
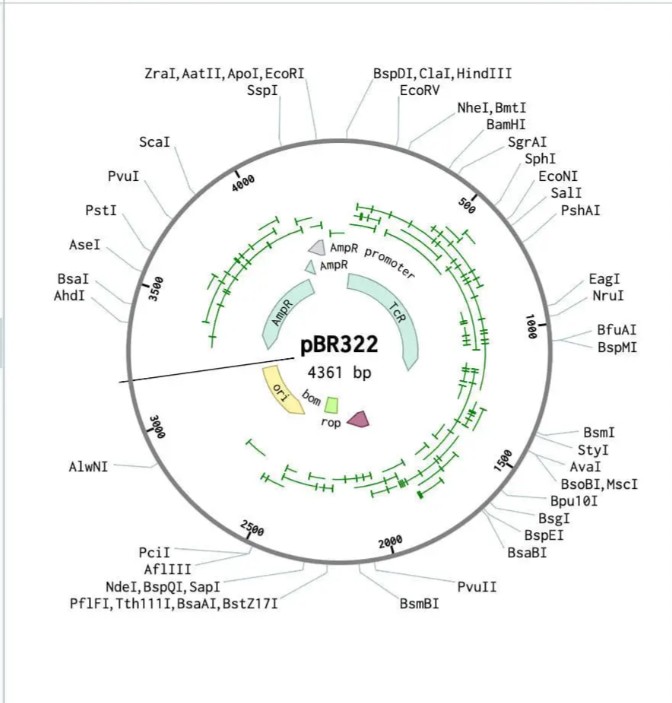
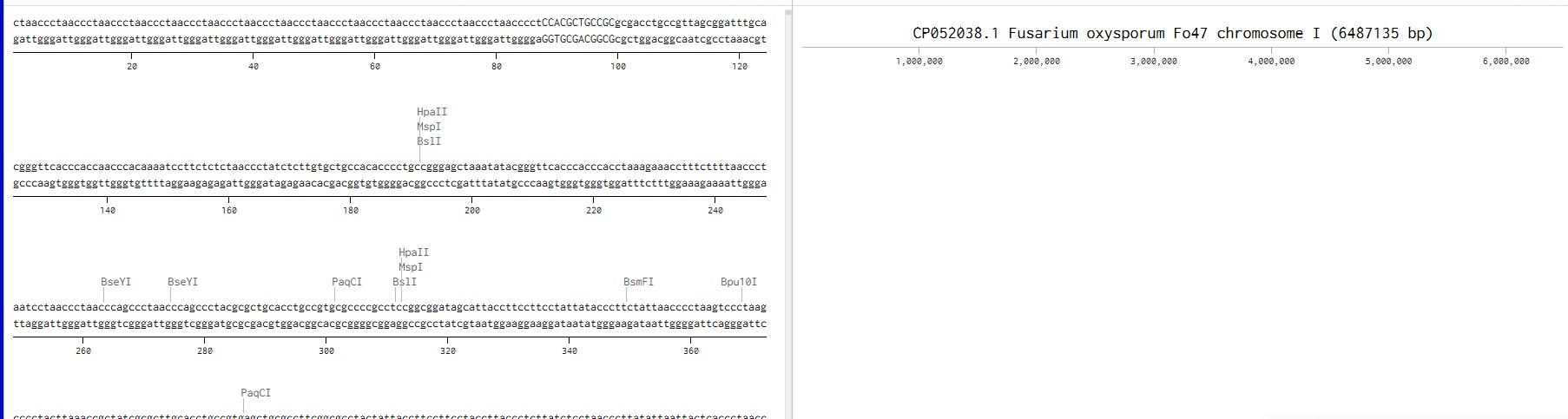
Simulate Restriction Enzyme Digestion with the following Enzymes:
EcoRI
HindIII
BamHI
KpnI
EcoRV
SacI
SalI
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
I was inspired by a random pattern from the website to draw something like a tower, and then I perfected it in Bletchin.
Part 3: DNA Design Challenge
3.1. Choose your protein.
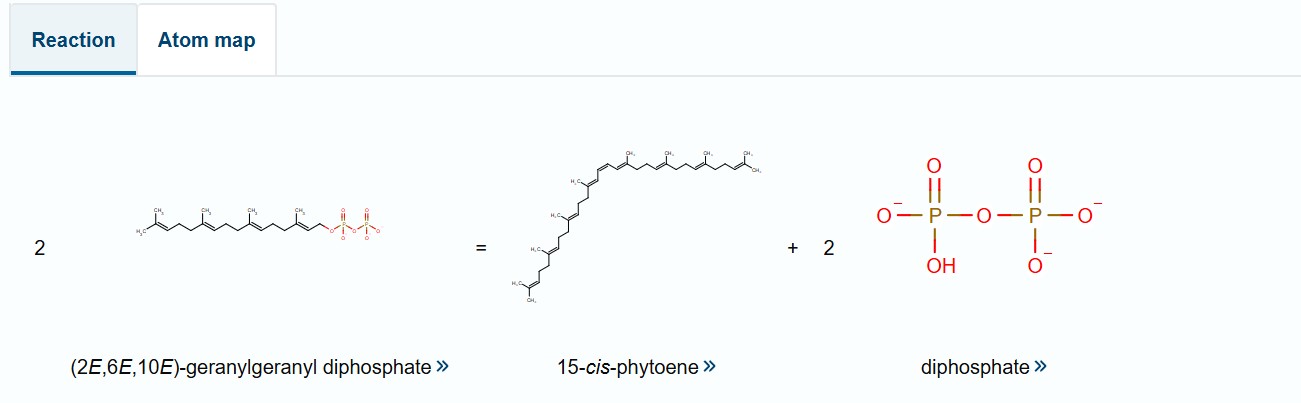
I selected the bifunctional lycopene cyclase/phytoene synthase for analysis because it directly processes and produces carotenoid pigments essential for coloration. This enzyme catalyzes two crucial steps in the carotenoid biosynthesis pathway: the formation of phytoene, the first committed precursor, and the cyclization of lycopene into β-carotene, a key orange pigment.
By processing intermediate molecules into pigmented compounds, this enzyme plays a central role in pigment formation. Its bifunctional nature makes it especially valuable, as a single gene can drive multiple steps in the pathway, simplifying the metabolic engineering strategy.
Bifunctional lycopene cyclase/phytoene synthase
MGWEYAQVHLKYTIPFGVVLAAVYRPLMSRLDVFKLVFLITVAVVSTIPWDSYLIKNRIWTYPPGVVVGLTAWDIPAEELFFFVIQTLNTSLLYMILSKPTFHPIYLAKKTGWGKIAGQILFASAIIFGLVSVFSGGEGMSISYQFIVNLPWTNTALPIALPTLYLWVVDTFALRRGTWSITSGTKYGVVLWDGLDIEEAVFFLLTNTLIVFGLVACDNTLAILDTFPEQFPRTKGLPNLLVIIRALILPKEKYDEERIEGLVSAVALLRKKSRSFYLASGTFEGRLRIDLIRLYAFCRAADDLVDEAPSVDDSRASIEKLRKFLDLAYEENQEEPSQRLREYVTSNIPEMFHMALLQLPTYYLPKQPLDDLLKGFDTDLLFDRKSGAFPIETTEDLDVYGSRVAGTVAELCNHLILYHTPESVPEGIQREVVASGQEMGIALQYVNIARDIKTDAEIDRVYLPLSWLKEAQLTPEDVIQQPHGPTIEALRHKLLDRAFEKYNMAKGAIDKLPSEGKGPIRVAVESYMEIGRVLREKGPAMKKGRATVPKMRRIRVAWSALNK
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
I employed the online reverse translation tool fromBioinformatics.org to convert the amino acid sequence of the target protein into a corresponding DNA coding sequence. This tool ensures that the translated nucleotide sequence matches the original peptide sequence while maintaining proper reading frame and codon order. Using this reverse translation resource allows me to generate a DNA sequence that can be used for downstream applications such as gene synthesis, cloning, and expression analysis in my chosen host system.
ATG GGC TGG GAG TAC GCA CAA GTT CAT CTG AAA TAT ACC ATC CCG TTC GGC GTT GTT CTC GCT GCG GTC TAT CGC CCC CTG ATG TCG CGT TTG GAT GTC TTT AAA TTG GTG TTT CTC ATC ACA GTG GCC GTA GTA AGT ACT ATT CCG TGG GAC TCT TAC TTA ATA AAA AAC CGG ATA TGG ACC TAT CCT CCA GGT GTC GTT GTC GGG CTT ACC GCA TGG GAT ATT CCT GCC GAA GAA CTG TTT TTC TTT GTC ATA CAG ACC CTG AAC ACA TCA CTG CTG TAT ATG ATC TTA AGT AAA CCC ACC TTT CAT CCG ATC TAC TTG GCG AAG AAA ACT GGC TGG GGC AAA ATT GCG GGC CAG ATC CTG TTT GCC AGC GCC ATC ATC TTC GGG CTG GTA TCT GTG TTC AGC GGC GGG GAA GGC ATG TCT ATA TCA TAT CAG TTT ATA GTA AAC CTG CCT TGG ACA AAC ACA GCA CTG CCG ATC GCA TTA CCA ACT CTG TAT CTG TGG GTG GTT GAC ACC TTT GCA CTC CGC CGT GGG ACA TGG AGC ATC ACT AGT GGA ACT AAG TAT GGG GTC GTG CTG TGG GAC GGC CTG GAT ATT GAA GAA GCT GTG TTT TTT TTA CTG ACT AAC ACT CTT ATT GTC TTC GGC TTG GTA GCG TGT GAT AAT ACC TTA GCT ATC CTG GAC ACC TTC CCC GAG CAG TTC CCC CGG ACA AAA GGT CTC CCA AAC CTG CTC GTA ATC ATT CGT GCG CTG ATT TTG CCT AAA GAG AAG TAT GAT GAA GAA CGC ATC GAG GGA TTA GTT TCG GCG GTC GCA CTC CTG CGT AAA AAG TCG CGC TCC TTT TAC TTG GCG AGC GGG ACG TTT GAG GGC CGT CTT CGT ATT GAC CTG ATT CGT CTG TAT GCG TTT TGT CGG GCA GCG GAC GAT CTT GTC GAT GAA GCG CCT AGT GTG GAT GAC TCC CGC GCT AGC ATT GAA AAG TTA CGG AAA TTC CTT GAT CTG GCG TAC GAA GAG AAC CAG GAA GAA CCA AGC CAG CGC TTA CGC GAG TAT GTG ACA AGC AAC ATA CCT GAA ATG TTT CAC ATG GCA CTG CTG CAA CTT CCG ACA TAC TAC CTG CCT AAA CAG CCA CTG GAC GAT CTC CTG AAA GGC TTT GAT ACG GAC CTG CTG TTT GAT CGT AAG AGT GGC GCC TTT CCG ATC GAA ACC ACC GAG GAT CTT GAT GTC TAT GGT AGC CGT GTT GCA GGT ACT GTC GCT GAA CTC TGC AAC CAT CTT ATT CTG TAC CAT ACC CCG GAA TCC GTC CCC GAA GGC ATT CAG CGT GAA GTC GTC GCA TCG GGA CAG GAG ATG GGT ATC GCG CTG CAG TAT GTT AAT ATC GCA CGC GAC ATT AAG ACA GAT GCG GAG ATC GAT CGG GTC TAT CTC CCA TTA TCA TGG CTG AAG GAA GCC CAG CTG ACG CCT GAG GAT GTG ATC CAA CAA CCT CAT GGG CCT ACG ATT GAG GCA TTA CGC CAT AAA TTG CTT GAT CGT GCA TTC GAG AAA TAC AAC ATG GCC AAG GGG GCC ATT GAT AAG TTA CCT TCC GAA GGA AAA GGC CCC ATC CGC GTC GCG GTG GAG TCC TAT ATG GAA ATT GGG CGC GTA TTA CGC GAG AAA GGT CCC GCG ATG AAA AAA GGT CGT GCC ACA GTT CCG AAA ATG CGC CGC ATT CGC GTG GCC TGG TCG GCC TTA AAT AAA
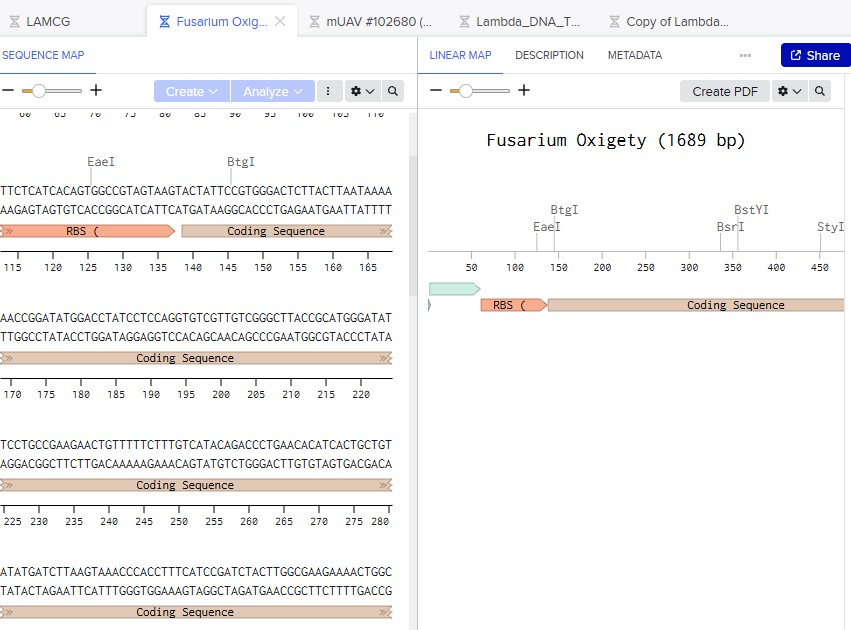
The codon sequence was optimized for Fusarium oxysporum because this organism is responsible for producing the carotenoid pigment. Since the bifunctional lycopene cyclase/phytoene synthase processes precursor molecules into β-carotene, efficient expression of this enzyme is essential for pigment formation. Optimizing codon usage improves translation efficiency, increases enzyme production, and enhances pigment accumulation, resulting in stronger and more consistent coloration.
3.4. You have a sequence! Now what?
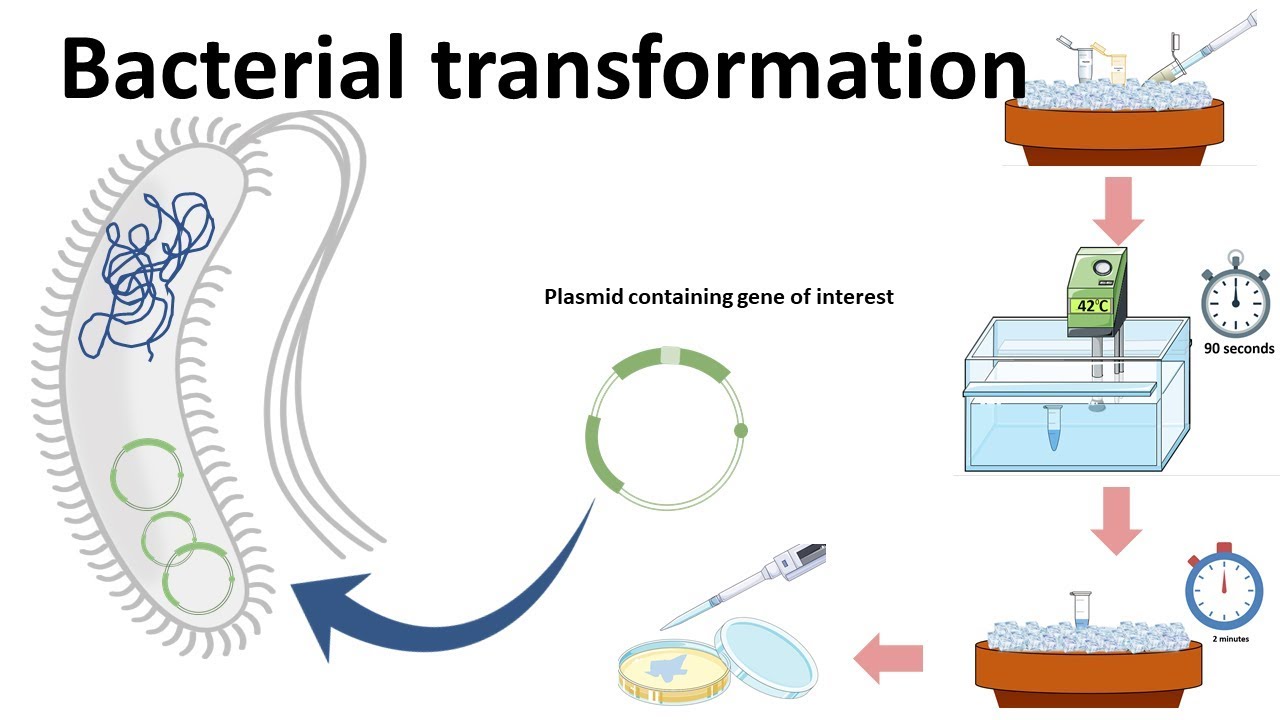
Cell-dependent methods (in Fusarium oxysporum): involve introducing the optimized gene into living fungal cells using an appropriate expression vector. The vector must contain regulatory elements such as a promoter, terminator, and selection marker. After transformation, the fungal cellular machinery carries out transcription of the DNA into mRNA and translation of the mRNA into protein. This method allows the enzyme to be produced within the natural metabolic environment of the fungus, enabling functional activity and potential pigment production.
Part 4: Prepare a Twist DNA Synthesis Order
I tried to use Twist, but I couldn’t because I kept getting the following error.
Part 5: DNA Read/Write/Edit
5.1 DNA Read
What DNA would you want to sequence (e.g., read) and why?
I would sequence the DNA of fungi that form mycelial networks used in biomaterial production in order to understand the genetic basis of their structural organization and bio-connection properties. By identifying genes involved in hyphal growth, branching, and cell wall composition, it would be possible to understand better how these organisms naturally build strong, interconnected living structures. This information could support the development of engineered mycelium-based biomaterials and explore the potential of living fungal systems as platforms for biological information storage or transmission, combining structural biology with DNA-based data encoding.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would use third-generation single-molecule sequencing, specifically Oxford Nanopore Technologies (ONT) or PacBio, because they allow long-read sequencing without amplification, making them ideal for studying large fungal DNA regions
Is your method first-, second- or third-generation or other? How so?
Third-generation
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
The input is high-quality genomic DNA extracted from the fungal sample. The preparation involves optionally fragmenting the DNA to suitable sizes for the sequencer, ligating adapters to the ends of the DNA fragments so the sequencing machine can recognize them, and performing any necessary purification steps. Unlike some other methods, PCR amplification is not required for Oxford Nanopore or PacBio long-read sequencing. The essential steps are DNA extraction, optional fragmentation, adapter ligation, and loading the DNA onto the sequencing device.
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
For Oxford Nanopore, the DNA passes through a nanopore embedded in a membrane. As each nucleotide moves through the pore, it causes a characteristic change in the ionic current, which is detected by sensors. These changes are analyzed in real time by the software to determine the sequence of bases (base calling). For PacBio, DNA polymerase incorporates fluorescently labeled nucleotides in zero-mode waveguides, and the emitted fluorescence is recorded to decode the bases. Both systems allow long continuous reads of single molecules without amplification.
What is the output of your chosen sequencing technology?
The output is a collection of long-read sequences in FASTQ format, which includes both the nucleotide sequences and associated quality scores. These sequences can then be used for downstream analysis such as gene annotation, codon optimization, or studying structural features in fungal DNA.
5.2 DNA Write
(ii) What technology or technologies would you use to perform this DNA synthesis, and why?
I would like to synthesize the gene coding for spider silk protein (spidroin) because it is a biomaterial with exceptional mechanical properties. The DNA sequence would include the full coding region of spidroin, codon-optimized for the host organism to ensure efficient expression, allowing for the production of fibers or hydrogels for biomaterials research.
What are the essential steps of your chosen sequencing methods?
The essential steps of the Sanger (ABI) sequencing method begin with preparing the DNA to be sequenced, usually by PCR amplification of the target fragment. Then, a DNA synthesis reaction is performed in which both normal nucleotides and fluorescently labeled chain-terminating nucleotides are incorporated, stopping elongation at specific positions. The resulting DNA fragments of different lengths are separated by capillary electrophoresis, and a detector reads the fluorescence to identify each base and reconstruct the full sequence of the fragment.
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
Its speed and scalability are limited, as each run processes only short fragments. Consequently, it is unsuitable for large-scale projects or whole-genome sequencing and is less efficient for high-throughput DNA analysis.
5.3 DNA Edit
(i) What DNA would you want to edit and why?
I would like to edit the DNA of a pigment-producing fungus, such as Fusarium oxysporum, to introduce a gene encoding laccase, an enzyme capable of degrading pollutants in water. By adding this enzyme, the fungus could not only produce sustainable biotints but also perform bioremediation, breaking down phenolic compounds, industrial dyes, and other contaminants in aquatic environments. This kind of genetic edit combines the production of valuable biomaterials with environmental cleanup, making the organism both functional and sustainable.
How does your technology of choice edit DNA? What are the essential steps?
The technology chose is recombinases, which are enzymes that recognize specific DNA sequences and mediate precise recombination events. This allows the targeted integration of a desired gene into the host genome. The essential steps include identifying a safe insertion site in the genome, designing the recombinase recognition sequences flanking the gene of interest, and delivering the recombinase along with the DNA construct into the host cells. Once inside, the recombinase facilitates site-specific integration of the gene.
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
Preparation involves designing the DNA construct containing the laccase gene flanked by recombinase recognition sites. The inputs include the DNA template with the gene, the recombinase enzyme or expression plasmid, and competent Fusarium oxysporum cells ready for transformation. Additional reagents may include selection markers and buffers to facilitate uptake and expression of the recombinase and target DNA.
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
While recombinases offer high precision, their efficiency can be limited by the accessibility of the target site and the compatibility of the recognition sequences with the host genome. Large-scale integrations or multiple edits may require careful design to avoid interference between sites. Additionally, the method is dependent on the ability to deliver the recombinase and DNA into fungal cells, which can be technically challenging.
Research resurses:
Findings have been summarized with the help of ChatGPT 5
Dong, Y., Yao, C., Zhu, Y., Yang, L., Luo, D., & Yang, D. (2020). DNA Functional Materials Assembled from Branched DNA: Design, Synthesis, and Applications.. Chemical reviews. https://doi.org/10.1021/acs.chemrev.0c00294.
Miserez, A., Yu, J., & Mohammadi, P. (2023). Protein-Based Biological Materials: Molecular Design and Artificial Production. Chemical Reviews, 123, 2049 - 2111. https://doi.org/10.1021/acs.chemrev.2c00621.
Liu, Y., Yang, Q., & Zhao, F. (2021). Synonymous but not silent: The codon usage code for gene expression and protein folding. Annual Review of Biochemistry, 90, 375-401. https://doi.org/10.1146/annurev-biochem-071320-112701
Week 3 HW: Lab automation
Python Script for Opentrons Artwork — DUE BY YOUR LAB TIME
metadata = {
'protocolName': 'HTGAA Opentrons Lab',
'author': 'MARIAORTIZ',
'source': 'HTGAA 2022',
'apiLevel': '2.20'
}
##############################################################################
### Robot deck setup constants - don't change these
##############################################################################
TIP_RACK_DECK_SLOT = 9
COLORS_DECK_SLOT = 6
AGAR_DECK_SLOT = 5
PIPETTE_STARTING_TIP_WELL = 'A1'
well_colors = {
'A1' : 'Red',
'B1' : 'Yellow',
'C1' : 'Green',
'D1' : 'Cyan',
'E1' : 'Blue' # if in a 24-well plate, this needs to be moved to e.g. D2
}
red_points = [(-5, 25), (0, 25), (5, 25), (-15, 20), (-10, 20), (-5, 20), (10, 20), (15, 20),
(-10, 15), (-5, 15), (10, 15), (15, 15), (-15, 10), (-10, 10), (-5, 10), (0, 10),
(5, 10), (10, 10), (15, 10), (-20, 5), (-15, 5), (15, 5), (-20, 0), (20, 0)]
green_points = [(-30, -15), (-25, -15), (-20, -15), (-15, -15), (15, -15), (20, -15), (25, -15), (30, -15),
(-30, -20), (-25, -20), (-20, -20), (-15, -20), (-10, -20), (-5, -20), (0, -20), (5, -20),
(10, -20), (15, -20), (20, -20), (25, -20), (30, -20), (-20, -25), (-15, -25), (-10, -25),
(-5, -25), (0, -25), (5, -25), (10, -25), (15, -25), (20, -25)]
red1_points = [(-5, 30), (0, 30), (5, 30), (-15, 25), (-10, 25), (10, 25), (15, 25), (-20, 20),
(20, 20), (-20, 15), (20, 15), (-25, 10), (25, 10), (-25, 5), (-10, 5), (-5, 5),
(0, 5), (5, 5), (10, 5), (25, 5), (-25, 0), (-15, 0), (-5, 0), (5, 0), (15, 0),
(25, 0), (-20, -5), (-15, -5), (-5, -5), (5, -5), (15, -5), (20, -5), (-15, -10),
(15, -10), (-10, -15), (-5, -15), (0, -15), (5, -15), (10, -15)]
def run(protocol):
##############################################################################
### Load labware, modules and pipettes
##############################################################################
##############################################################################
### Patterning
##############################################################################
###
### Helper functions for this lab
###)`
# pass this e.g. 'Red' and get back a Location which can be passed to aspirate()
def location_of_color(color_string):
for well,color in well_colors.items():
if color.lower() == color_string.lower():
return color_plate[well]
raise ValueError(f"No well found with color {color_string}")
# For this lab, instead of calling pipette.dispense(1, loc) use this: dispense_and_jog(pipette, 1, loc)
def dispense_and_jog(pipette, volume, location):
"""
Dispense and then move up 5mm and back down to shake all dispensed fluid off the tip;
this also ensures it's not moving laterally before the dispense is done.
"""
assert(isinstance(volume, (int, float)))
pipette.dispense(volume, location)
currLoc = pipette._get_last_location_by_api_version()
pipette.move_to(currLoc.move(types.Point(z=5)))
pipette.move_to(currLoc)
###
### YOUR CODE HERE to create your design
pipette_20ul.pick_up_tip()
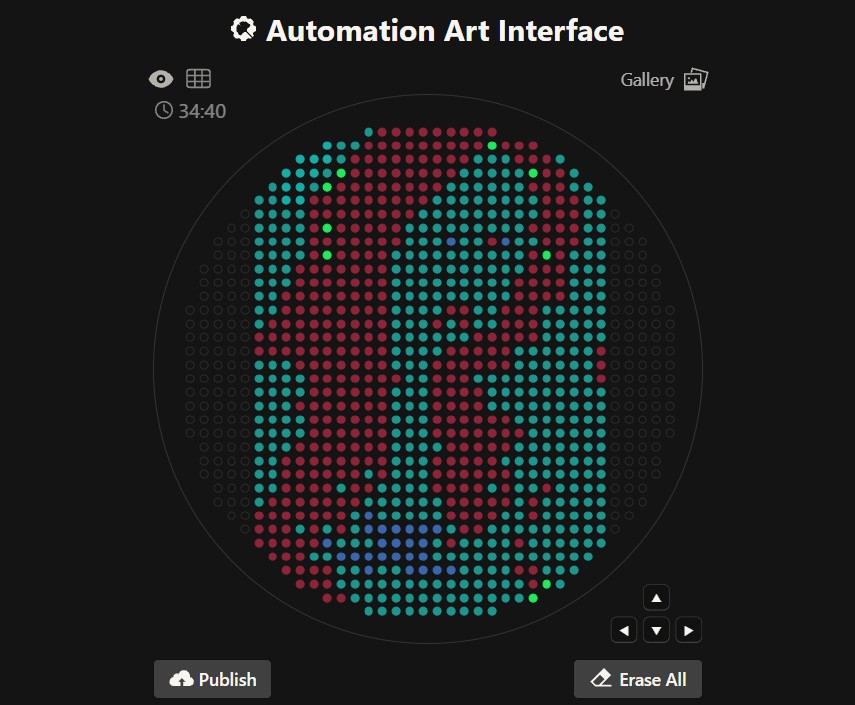
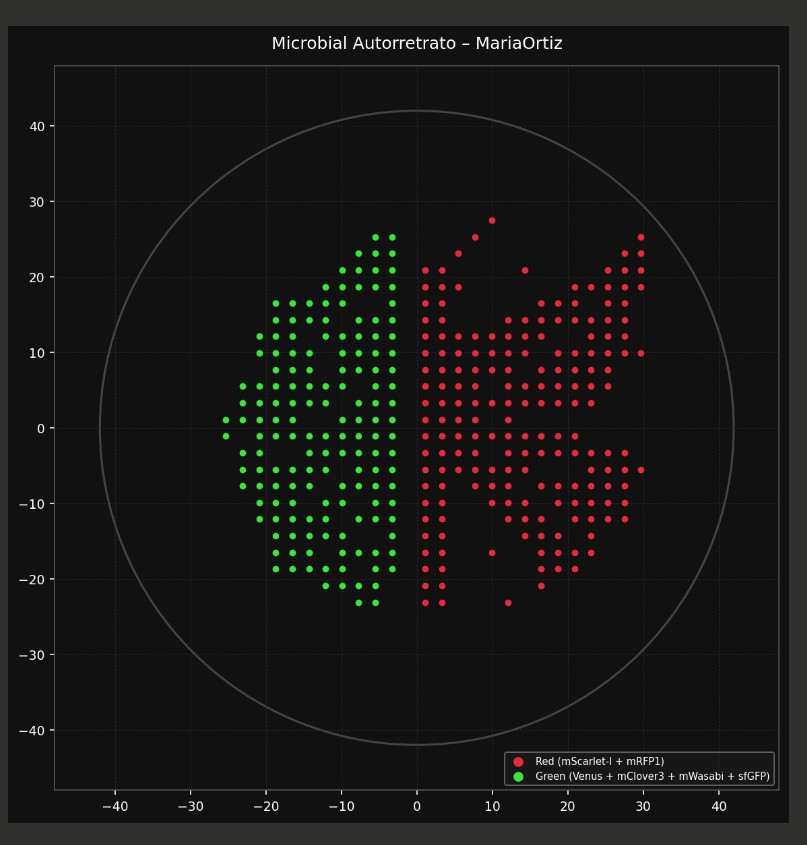
red_points = [(29.7,25.3),(29.7,23.1),(14.3,20.9),(25.3,20.9),(27.5,20.9),(5.5,18.7),(20.9,18.7),(23.1,18.7),(25.3,18.7),(27.5,18.7),(16.5,16.5),(18.7,16.5),(20.9,16.5),(25.3,16.5),(12.1,14.3),(14.3,14.3),(16.5,14.3),(18.7,14.3),(20.9,14.3),(23.1,14.3),(25.3,14.3),(1.1,12.1),(7.7,12.1),(12.1,12.1),(14.3,12.1),(16.5,12.1),(23.1,12.1),(25.3,12.1),(9.9,9.9),(12.1,9.9),(14.3,9.9),(18.7,9.9),(20.9,9.9),(23.1,9.9),(27.5,9.9),(29.7,9.9),(5.5,7.7),(9.9,7.7),(12.1,7.7),(16.5,7.7),(18.7,7.7),(20.9,7.7),(23.1,7.7),(7.7,5.5),(12.1,5.5),(14.3,5.5),(16.5,5.5),(18.7,5.5),(20.9,5.5),(23.1,5.5),(14.3,3.3),(16.5,3.3),(18.7,3.3),(20.9,3.3),(12.1,1.1),(5.5,-1.1),(7.7,-1.1),(12.1,-1.1),(14.3,-1.1),(16.5,-1.1),(18.7,-1.1),(20.9,-1.1),(7.7,-3.3),(12.1,-3.3),(14.3,-3.3),(16.5,-3.3),(18.7,-3.3),(27.5,-3.3),(5.5,-5.5),(12.1,-5.5),(14.3,-5.5),(29.7,-5.5),(7.7,-7.7),(9.9,-7.7),(12.1,-7.7),(16.5,-7.7),(18.7,-7.7),(20.9,-7.7),(1.1,-9.9),(9.9,-9.9),(12.1,-9.9),(14.3,-9.9),(12.1,-12.1),(14.3,-12.1),(16.5,-12.1),(25.3,-12.1),(14.3,-14.3),(9.9,-16.5),(23.1,-16.5),(1.1,-18.7),(3.3,-18.7),(16.5,-18.7),(18.7,-18.7),(16.5,-20.9),(12.1,-23.1),(9.9,27.5),(7.7,25.3),(5.5,23.1),(27.5,23.1),(1.1,20.9),(3.3,20.9),(29.7,20.9),(1.1,18.7),(3.3,18.7),(29.7,18.7),(1.1,16.5),(3.3,16.5),(27.5,16.5),(1.1,14.3),(3.3,14.3),(27.5,14.3),(3.3,12.1),(5.5,12.1),(9.9,12.1),(27.5,12.1),(1.1,9.9),(3.3,9.9),(5.5,9.9),(7.7,9.9),(25.3,9.9),(1.1,7.7),(3.3,7.7),(7.7,7.7),(25.3,7.7),(1.1,5.5),(3.3,5.5),(5.5,5.5),(25.3,5.5),(1.1,3.3),(3.3,3.3),(5.5,3.3),(7.7,3.3),(9.9,3.3),(12.1,3.3),(23.1,3.3),(1.1,1.1),(3.3,1.1),(5.5,1.1),(7.7,1.1),(1.1,-1.1),(3.3,-1.1),(9.9,-1.1),(1.1,-3.3),(3.3,-3.3),(5.5,-3.3),(20.9,-3.3),(23.1,-3.3),(25.3,-3.3),(1.1,-5.5),(3.3,-5.5),(7.7,-5.5),(9.9,-5.5),(23.1,-5.5),(25.3,-5.5),(27.5,-5.5),(1.1,-7.7),(3.3,-7.7),(23.1,-7.7),(25.3,-7.7),(27.5,-7.7),(3.3,-9.9),(18.7,-9.9),(20.9,-9.9),(23.1,-9.9),(25.3,-9.9),(27.5,-9.9),(1.1,-12.1),(3.3,-12.1),(20.9,-12.1),(23.1,-12.1),(27.5,-12.1),(1.1,-14.3),(3.3,-14.3),(16.5,-14.3),(18.7,-14.3),(23.1,-14.3),(1.1,-16.5),(3.3,-16.5),(16.5,-16.5),(18.7,-16.5),(20.9,-16.5),(20.9,-18.7),(1.1,-20.9),(3.3,-20.9),(1.1,-23.1),(3.3,-23.1)]
for x, y in red_points:
adjusted_location = center_location.move(types.Point(x=x, y=y))
if pipette_20ul.current_volume == 0:
pipette_20ul.aspirate(1, location_of_color('Red'))
dispense_and_jog(pipette_20ul, 1, adjusted_location)
pipette_20ul.drop_tip()
pipette_20ul.pick_up_tip()
green_points = [(-7.7,20.9),(-5.5,20.9),(-12.1,18.7),(-9.9,18.7),(-5.5,18.7),(-3.3,18.7),(-14.3,16.5),(-12.1,16.5),(-3.3,16.5),(-16.5,14.3),(-12.1,14.3),(-5.5,14.3),(-3.3,14.3),(-16.5,12.1),(-12.1,12.1),(-9.9,12.1),(-7.7,12.1),(-5.5,12.1),(-3.3,12.1),(-18.7,9.9),(-14.3,9.9),(-9.9,9.9),(-7.7,9.9),(-5.5,9.9),(-18.7,7.7),(-16.5,7.7),(-9.9,7.7),(-7.7,7.7),(-5.5,7.7),(-3.3,7.7),(-20.9,5.5),(-16.5,5.5),(-14.3,5.5),(-12.1,5.5),(-9.9,5.5),(-3.3,5.5),(-16.5,3.3),(-14.3,3.3),(-12.1,3.3),(-7.7,3.3),(-5.5,3.3),(-3.3,3.3),(-16.5,1.1),(-9.9,1.1),(-5.5,1.1),(-12.1,-1.1),(-9.9,-1.1),(-7.7,-1.1),(-5.5,-1.1),(-3.3,-1.1),(-9.9,-3.3),(-7.7,-3.3),(-5.5,-3.3),(-5.5,-5.5),(-3.3,-5.5),(-5.5,-7.7),(-3.3,20.9),(-7.7,18.7),(-14.3,14.3),(-18.7,5.5),(-20.9,3.3),(-18.7,3.3),(-20.9,1.1),(-3.3,1.1),(-20.9,-1.1),(-16.5,-1.1),(-14.3,-3.3),(-12.1,-3.3),(-20.9,-5.5),(-18.7,-5.5),(-16.5,-5.5),(-14.3,-5.5),(-12.1,-5.5),(-16.5,-7.7),(-14.3,-7.7),(-9.9,-7.7),(-7.7,-7.7),(-12.1,-9.9),(-5.5,-9.9),(-3.3,-9.9),(-16.5,-12.1),(-12.1,-12.1),(-7.7,-12.1),(-3.3,-12.1),(-14.3,-14.3),(-9.9,-14.3),(-3.3,-14.3),(-9.9,-16.5),(-7.7,-16.5),(-5.5,-16.5),(-3.3,-16.5),(-9.9,-18.7),(-5.5,-20.9),(-9.9,16.5),(-7.7,14.3),(-16.5,9.9),(-3.3,9.9),(-14.3,7.7),(-5.5,5.5),(-18.7,1.1),(-7.7,1.1),(-18.7,-1.1),(-14.3,-1.1),(-20.9,-3.3),(-3.3,-3.3),(-7.7,-5.5),(-18.7,-7.7),(-3.3,-7.7),(-16.5,-9.9),(-9.9,-9.9),(-14.3,-12.1),(-5.5,-12.1),(-12.1,-14.3),(-5.5,-18.7),(-3.3,-18.7),(-5.5,25.3),(-3.3,25.3),(-7.7,23.1),(-5.5,23.1),(-3.3,23.1),(-9.9,20.9),(-18.7,16.5),(-16.5,16.5),(-18.7,14.3),(-20.9,12.1),(-18.7,12.1),(-20.9,9.9),(-23.1,5.5),(-23.1,3.3),(-25.3,1.1),(-23.1,1.1),(-25.3,-1.1),(-23.1,-3.3),(-23.1,-5.5),(-23.1,-7.7),(-20.9,-7.7),(-20.9,-9.9),(-18.7,-9.9),(-20.9,-12.1),(-18.7,-12.1),(-18.7,-14.3),(-16.5,-14.3),(-18.7,-16.5),(-16.5,-16.5),(-14.3,-16.5),(-18.7,-18.7),(-16.5,-18.7),(-14.3,-18.7),(-12.1,-18.7),(-12.1,-20.9),(-9.9,-20.9),(-7.7,-20.9),(-7.7,-23.1),(-5.5,-23.1)]
for x, y in green_points:
adjusted_location = center_location.move(types.Point(x=x, y=y))
if pipette_20ul.current_volume == 0:
pipette_20ul.aspirate(1, location_of_color('Green'))
dispense_and_jog(pipette_20ul, 1, adjusted_location)
pipette_20ul.drop_tip()
Post-Lab Questions — DUE BY START OF FEB 24 LECTURE
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
The articule present a robotic automation platform designed to standardize and scale the production of three-dimensional epithelial tissues. The study addresses a key limitation in tissue engineering: the high variability and labor-intensive nature of manual organotypic tissue fabrication. By implementing a programmable liquid-handling robotic system compatible with platforms such as Opentrons, the authors automated critical steps including cell seeding, media exchange, hydrogel handling, and layered tissue assembly.
The novelty of this work lies in demonstrating that accessible laboratory automation can be successfully applied to complex biofabrication workflows, not just routine pipetting tasks. The ReBiA system enabled the reproducible generation of multilayered epithelial tissues with functional characteristics comparable to manually produced models. This significantly reduces operator-dependent variability and increases throughput, which is essential for applications such as drug screening, disease modeling, and regenerative medicine research.
Overall, the study illustrates how low-cost, programmable automation tools can democratize advanced tissue engineering, improve reproducibility, and facilitate scalable biological manufacturing.
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
For my final project, I intend to use laboratory automation to standardize and optimize the production of fungal-based biodyes. One of the main challenges in working with filamentous fungi is variability in pigment yield due to small changes in media composition, pH, nutrient availability, and growth conditions. By implementing an automated liquid-handling system such as Opentrons, I aim to systematically control these variables and perform high-throughput screening of culture conditions to identify combinations that maximize and stabilize pigment production.
The automation workflow will include robotic preparation of media gradients, controlled inoculation of fungal spores into multi-well plates, and scheduled sampling for pigment quantification using absorbance measurements. Using the Opentrons Python API, I will program reproducible experimental matrices and spatially controlled dispensing patterns. This same precision can be extended to textile applications, where fungal inoculum or pigment precursors could be deposited in defined areas of fabric to enable localized growth and pattern formation. Custom 3D-printed holders may be designed to stabilize both culture vessels and textile substrates during automated handling. Experimental data will be digitally recorded to support structured analysis and optimization. Overall, automation will transform fungal biodye production into a scalable, reproducible, and spatially programmable biofabrication workflow.
Final Project Ideas — DUE BY START OF FEB 24 LECTURE
Research sources:
I used AI assistance ( Claude) to help organize the coordinate data extracted from the image. Specifically, the AI helped me structure the (x, y) coordinate lists correctly, convert them into a usable data format (e.g., arrays/DataFrame), and ensure they were properly scaled and centered for the experimental setup.
Königer, L., Malkmus, C., Mahdy, D., Däullary, T., Götz, S., Schwarz, T., Gensler, M., Pallmann, N., Cheufou, D., Rosenwald, A., Möllmann, M., Groneberg, D., Popp, C., Groeber-Becker, F., Steinke, M., & Hansmann, J. (2024). ReBiA—Robotic Enabled Biological Automation: 3D Epithelial Tissue Production. Advanced Science, 11. https://doi.org/10.1002/advs.202406608.
Week 4 hw protein design part
Part A. Conceptual Questions
How many amino acid molecules do you ingest when eating 500 grams of meat? (Average amino acid ≈ 100 Daltons)
Approximately 20% of meat is protein. Therefore, 500 g of meat contains about 100 g of protein. If we assume an average amino acid molecular weight of 100 g/mol (≈100 Daltons), then:
100 g protein ÷ 100 g/mol ≈ 1 mol of amino acids.
One mole corresponds to 6 × 10²³ molecules (Avogadro’s number). Therefore, eating 500 g of meat provides on the order of 6 × 10²³ amino acid molecules.
Why do humans eat beef but do not become a cow, or eat fish but not become fish?
Because we do not absorb intact proteins. During digestion, proteins are broken down into peptides and then into free amino acids. The original protein sequence and structure are completely destroyed. Our cells then use those amino acids as building blocks to synthesize human proteins according to the instructions encoded in our DNA. Biological identity comes from genetic information, not from dietary molecules.
Why are there only 20 natural amino acids?
The 20 standard amino acids represent the set that became fixed in the genetic code during evolution. Together, they provide sufficient chemical diversity to build functional proteins: hydrophobic, polar, charged, aromatic, small, rigid, and flexible residues are all included. This set appears to be an optimal balance between chemical versatility and biological efficiency. Although additional amino acids such as selenocysteine and pyrrolysine exist, the core genetic code relies on 20.
Can you make non-natural amino acids? Design some new amino acids.
Yes, hundreds of non-natural amino acids have been synthesized in organic chemistry and synthetic biology. They can be designed by modifying the side chain.
Examples of designed amino acids:
A fluorinated phenylalanine (to alter hydrophobicity and electronic properties).
An amino acid with an azide or alkyne group for bioorthogonal “click” chemistry.
A metal-binding amino acid containing a bipyridine group.
A photoactivatable amino acid with a diazirine group for crosslinking experiments.
An amino acid with a bulky polyaromatic side chain to enhance π–π stacking.
Some engineered organisms have even expanded their genetic code to incorporate such amino acids into proteins.
Where did amino acids come from before enzymes and life existed?
Amino acids likely formed through prebiotic chemistry. The Miller–Urey experiment demonstrated that amino acids can form spontaneously under simulated early Earth conditions (reducing atmosphere + electrical discharge). Amino acids have also been detected in meteorites such as the Murchison meteorite, suggesting that they can form in space. Therefore, amino acids likely existed before enzymes and before life emerged.
If you make an α-helix using D-amino acids, what handedness would you expect?
Natural proteins use L-amino acids and form right-handed α-helices. If you construct an α-helix entirely from D-amino acids, the helix would be left-handed. Inverting the chirality of the monomers inverts the handedness of the secondary structure.
Can you discover additional helices in proteins?
Yes. Besides the α-helix, proteins can contain 3₁₀ helices and π-helices. These are less common but structurally distinct. Other biological polymers, such as DNA, also form helical structures (e.g., the right-handed B-DNA helix).
Why are most molecular helices right-handed?
Most biological helices are right-handed because proteins are built from L-amino acids. The stereochemistry of the alpha carbon restricts backbone dihedral angles (φ and ψ), making right-handed helices energetically favorable.
Why do β-sheets tend to aggregate?
β-sheets can extend through backbone hydrogen bonding between neighboring strands. Their structure allows repeating hydrogen bond networks and tight packing of side chains. This makes it easy for partially unfolded proteins to associate and form extended β-sheet assemblies.
What is the driving force for β-sheet aggregation?
The primary driving forces are:
Extensive backbone hydrogen bonding.
Hydrophobic interactions between side chains.
Reduction of exposed hydrophobic surface area in aqueous environments.
Thermodynamic stabilization through ordered packing.
Together, these factors favor the formation of highly stable aggregated structures.
Why do many amyloid diseases form β-sheets?
When proteins misfold, hydrophobic regions that are normally buried become exposed. These regions can reorganize into extended β-sheet structures that stack into fibrils. These fibrils are extremely stable and resistant to degradation. Many diseases, such as Alzheimer’s and Parkinson’s, involve accumulation of such amyloid β-sheet aggregates.
Can amyloid β-sheets be used as materials?
Yes. Amyloid fibrils are mechanically strong, resistant to degradation, and capable of self-assembly. They are being explored as nanomaterials, scaffolds for tissue engineering, and components in bioelectronics. Some organisms even produce functional amyloids naturally.
Design a β-sheet motif that forms a well-ordered structure.
A well-ordered β-sheet motif often uses alternating polar and nonpolar residues to create amphipathic strands.
Example sequence:
Val–Ser–Val–Ser–Val–Ser–Val–Ser
In this design:
Valine (hydrophobic) promotes tight packing.
Serine (polar) allows hydrogen bonding and solubility.
Add aromatic residues (e.g., phenylalanine) for π–π stacking.
Place charged residues at termini to control aggregation.
Use repeating patterns to encourage uniform self-assembly.
Such rational design principles are commonly used in peptide-based biomaterials research.
Part B: Protein Analysis and Visualization
Briefly describe the protein you selected and why you selected it.
Formate dehydrogenase FDH2: These electrons are transferred to components of the electron transport chain or to specific electron carriers, depending on the organism. In many bacteria, FDH2 plays an important role in energy metabolism by allowing formate to serve as an electron donor during respiration.
Identify the amino acid sequence of your protein.:
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
Parameter
Value
PDB ID
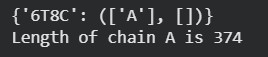
6T8C
Protein Name
Formate dehydrogenase FDH2
Sequence Length
386 amino acids
Most Frequent Amino Acid
Glycine (G)
Number of Occurrences (Gly)
120
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
Does your protein belong to any protein family?
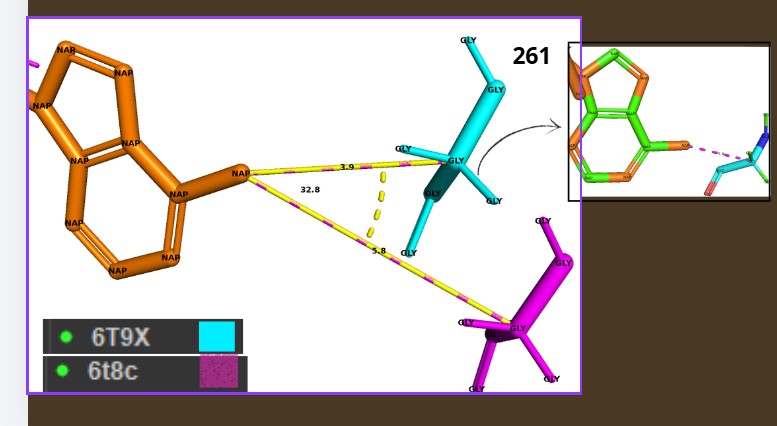
Yes, the protein FDH2 (PDB 6T8C) belongs to the formate dehydrogenase (FDH) family, which is part of the larger oxidoreductase class of enzymes. More specifically, it is a molybdenum-dependent formate dehydrogenase and is classified within the DMSO reductase superfamily. These enzymes are widely distributed in bacteria and archaea and are involved in C1 metabolism, catalyzing the oxidation of formate to carbon dioxide while transferring electrons to cellular electron carriers. They share conserved structural motifs for binding the molybdenum cofactor and often contain iron–sulfur clusters for electron transfer.
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
Good quality :1.98
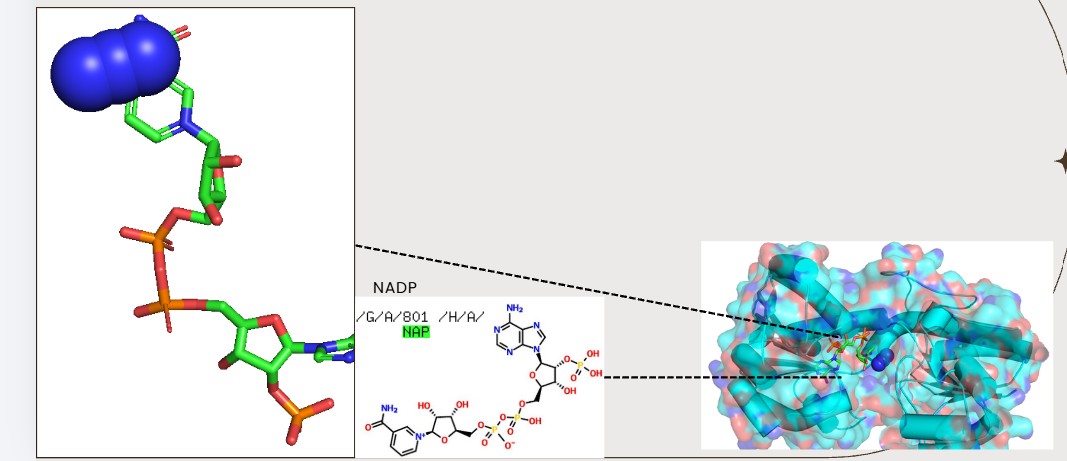
Are there any other molecules in the solved structure apart from protein?
Yes, NAPD
Does your protein belong to any structure classification family?
it is part of the molybdenum-containing oxidoreductase family within the DMSO reductase superfamily. In structural classification databases such as SCOP and CATH, proteins of this type fall into the α/β class, characterized by mixed alpha helices and beta sheets arranged in a conserved fold
Open the structure of your protein in any 3D molecule visualization software:
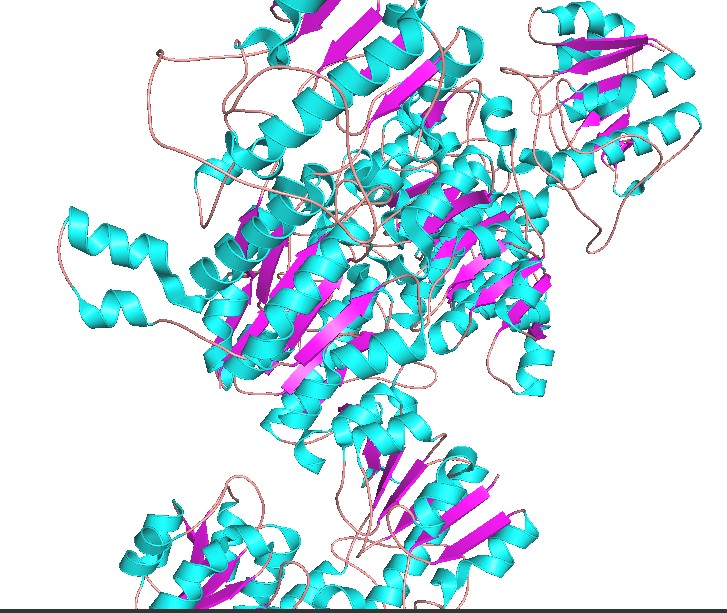
Color the protein by secondary structure. Does it have more helices or sheets?
Have more helices
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
select hydrophobic, resn ALA+VAL+LEU+ILE+MET+PHE+TRP+PROselect polar, resn SER+THR+ASN+GLN+TYR+CYSselect charged, resn ASP+GLU+LYS+ARG+HIScolor orange, hydrophobiccolor green, polarcolor blue, charged
Hydrophobic residues tend to cluster in the core, while polar/charged residues tend to be more surface exposed (typical of soluble proteins)
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Yes., Grooves and indentations at subunit interfaces and along the surfaceb NAPD interaction regions.
Deep Mutational Scans
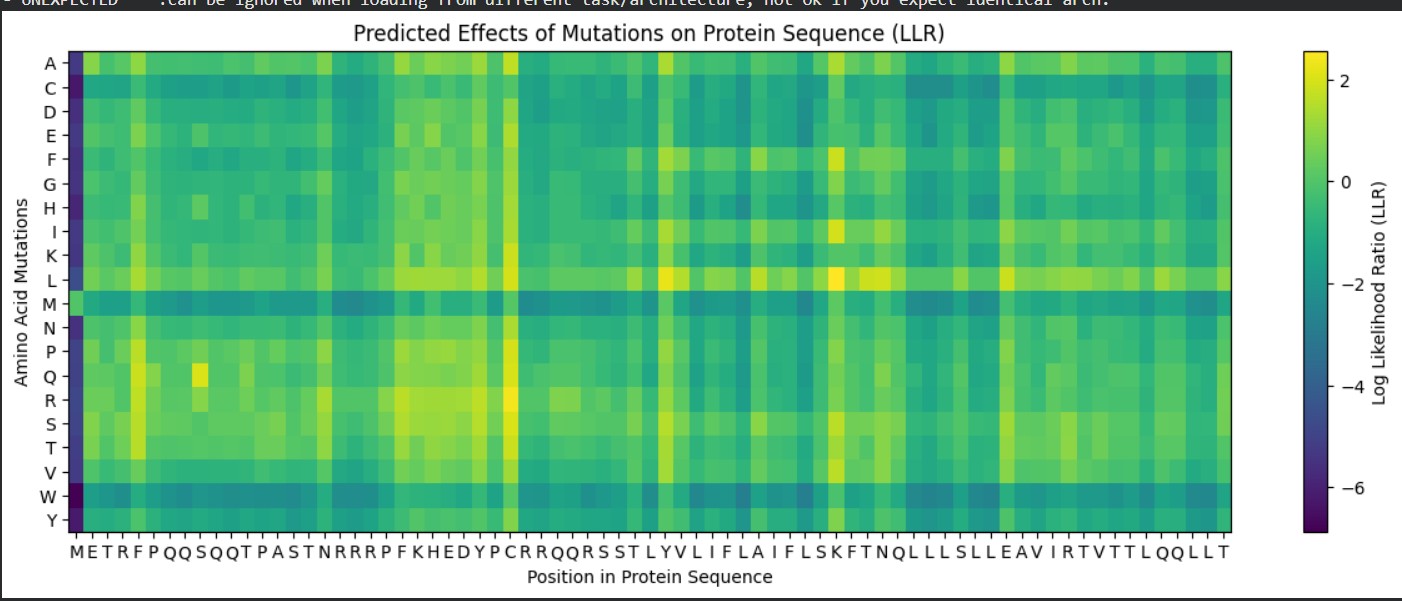
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
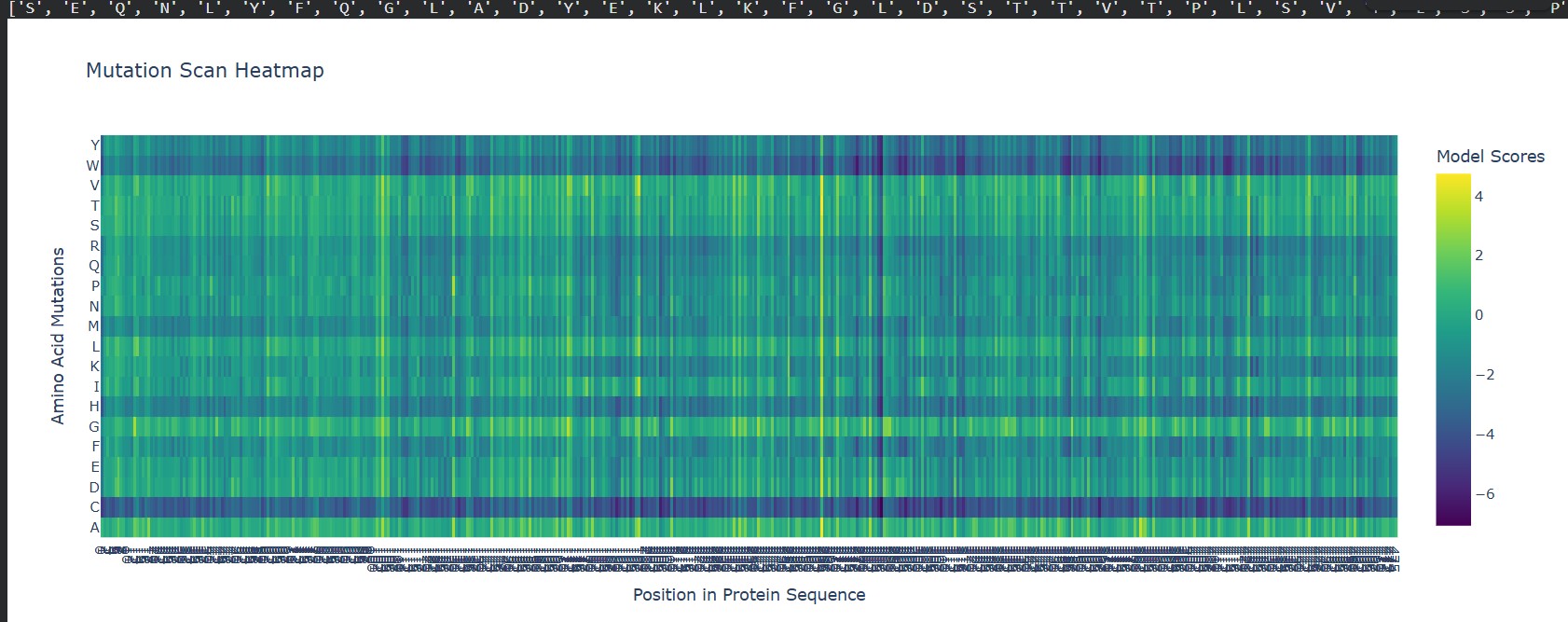
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
A clear pattern in the mutation heatmap is that substitutions to cysteine (C) consistently show strongly negative scores across most positions in the protein, indicated by the dark purple color. This suggests that introducing cysteine is generally destabilizing. A likely explanation is that cysteine contains a reactive thiol group capable of forming disulfide bonds or interfering with the local chemical environment, which can disrupt proper folding or structural stability. In contrast, some regions appear more tolerant to mutations to glycine (G), shown by greener or yellowish areas, likely because glycine’s small size allows flexibility without causing major steric clashes. This overall pattern indicates that chemical properties of amino acid side chains strongly influence mutation tolerance in this protein.
Latent Space Analysis
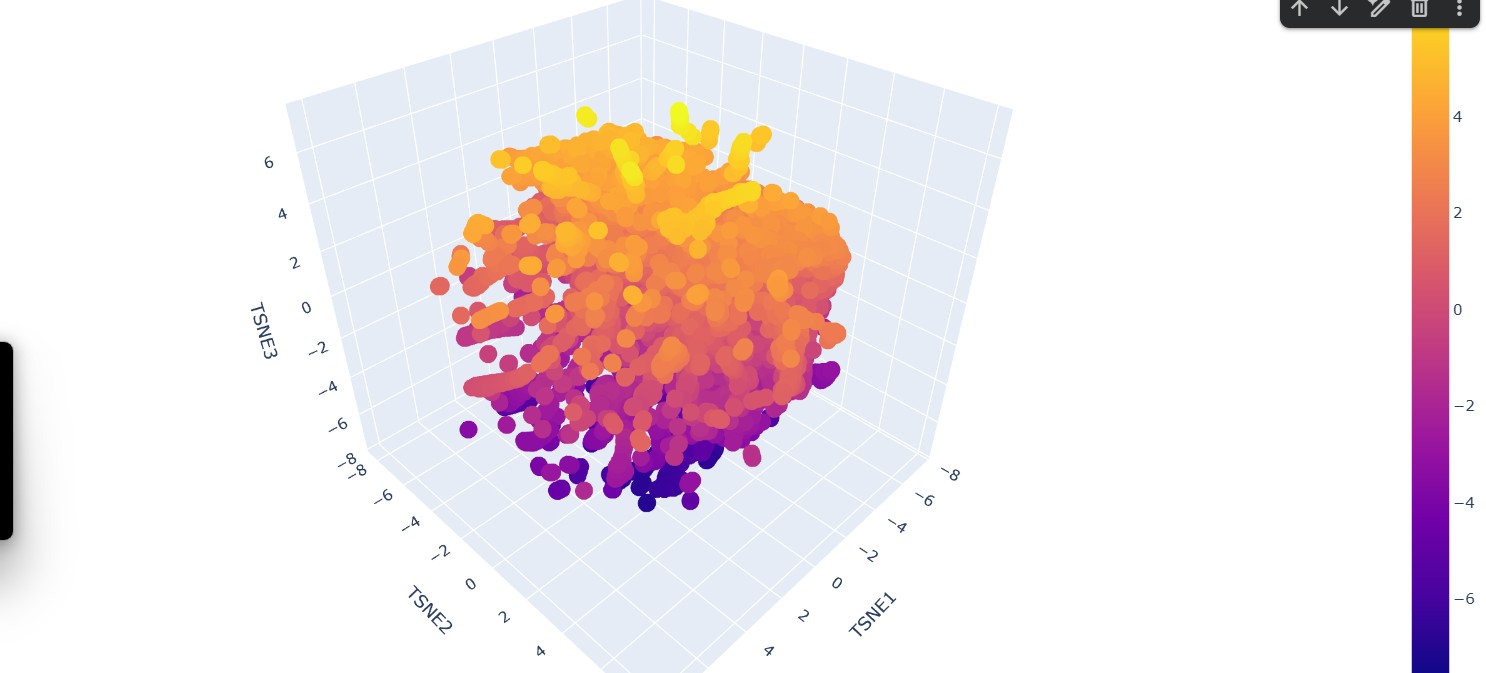
Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins?
Yes, the formed neighborhoods generally approximate similar proteins. In the 3D embedding plot, clusters of points appear grouped together, suggesting that proteins within the same neighborhood share higher sequence similarity. These clusters likely represent proteins belonging to similar families, functional classes, or structural folds
Place your protein in the resulting map and explain its position and similarity to its neighbors.
The protein of interest (FDH2) appears embedded within a dense cluster rather than isolated at the periphery. This position suggests that it shares significant sequence similarity with nearby proteins in the dataset. Its neighbors likely correspond to other formate dehydrogenases or closely related oxidoreductases within the same structural and functional family. The fact that it is not separated into a distant cluster indicates that it is not an outlier but rather part of a conserved evolutionary group.
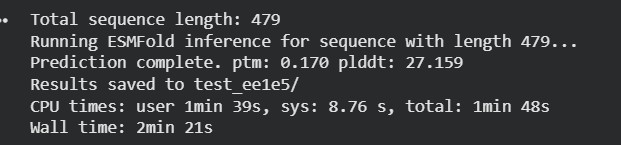
Input this sequence into ESMFold and compare the predicted structure to your original.
New Sequence:ALTPEEAALLRAAWAPVAADRAANGRAFVLALFAAYPELAELFPEFRGKTLAEIAASPALDAISTAIFDRLDTLVANADDAAAMATLFADLAARHVARGVTAAHFEAIRALFPGFVASVAPPPPGAAAAWDRLFGDVIAALRAAGG
C2. Protein Folding
SeqRecord(seq=Seq(‘GCATGCATGCATGCATGCATGCATGCATGCATGCATGCATGCATGCATGCATGC…CAT’), id=‘Seq2’, name=‘Seq2’, description=‘Seq2 Description of Sequence 2’, dbxrefs=[])
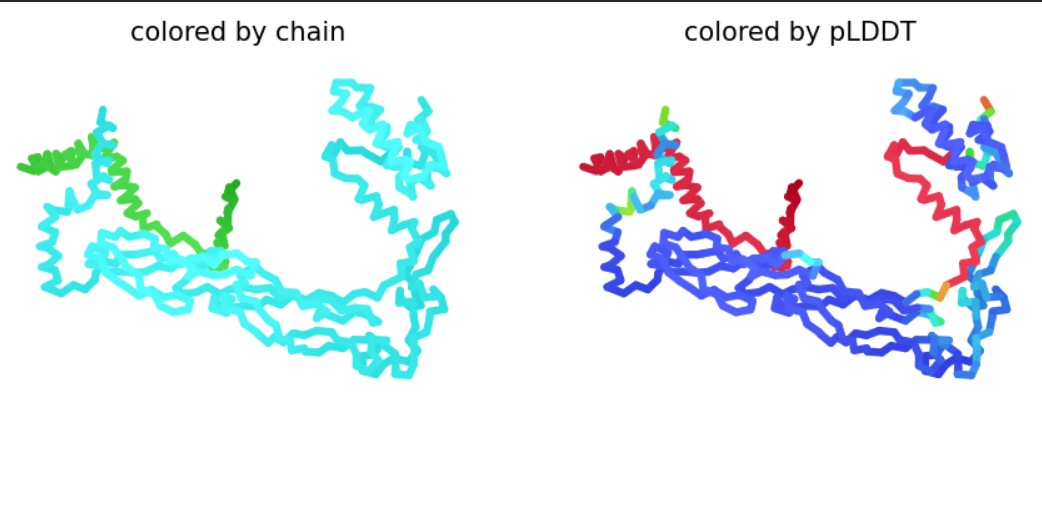
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
The protein sequence was folded using ESMFold and compared with the original 6T8C structure. Visually, the predicted structure conserved the overall fold of the protein, indicating that the backbone architecture is generally maintained. However, several local conformational differences were observed, especially after introducing mutations and redesigned sequence segments generated by ProteinMP
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
Small mutations appeared to preserve most of the global structure, suggesting partial resilience of the protein fold. In contrast, larger sequence modifications produced more noticeable structural deviations in the 3D conformation. These changes likely affect conformational and functional sites, including residue interactions involved in stability and catalytic activity.
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
The redesigned sequences generated by ProteinMPNN showed moderate sequence recovery (~52–56%), meaning that many residues differed from the native sequence while still maintaining a similar overall structural organization. This suggests that the protein fold tolerates some variability, but extensive mutations can alter important conformational regions and potentially disrupt function.
Input this sequence into ESMFold and compare the predicted structure to your original.
New Sequence:MKILLVAAPDPASGYPTSYPVDSLPEVTSYPGGLPAPQPSRVDFRPGELLFNVSGALGLREYIEGRGHELIVTSDREGPDSEFARHLPEADVVISSHAWPAVMSAERLAAAKNLRLVITAGEGSDHVDLDAARARGITVVECRLSNSEAIAEHAVELIKSLVENKKNCEKLKKEGKTDKEECEKNSESLEGKTVGIIGFGAVGRRIAELLKPLGVKLGCYDITPPPQEVLEELGITYYPSVEELIKDCDVVVTACPLTPATKDLFNDETIAKMKKGAILVNVSHGPIVKRDALVKALESGQLRGYGGDVYDPWPPPADHPLLTAPNTNLTPHVAGSTLAAQARYAAGVREILERYFADEPIPEHYVVIDGRLLP
Overall, the protein does not appear fully resilient to major mutations because structural and conformational sites change significantly when larger sequence alterations are introduced.
Part D. Group Brainstorm on Bacteriophage Engineering
Group: (James, USE), (Karen, Ecuador)
Review the Bacteriophage Final Project Goals for engineering the L Protein:
Increased stability (easiest)
Higher titers (medium)
Higher toxicity of lysis protein (hard)
Brainstorm Session
| Goal | Tunable toxicity |
| Strategy | Design variants with graded L–DnaJ binding strength: attenuated (stronger binding) → wild-type → enhanced (weaker binding) |
| Tools | AlphaFold-Multimer (L + DnaJ), Rosetta interface ΔΔG |
Write a 1-page proposal (bullet points or short paragraphs) describing:
Engineer the MS2 L lysis protein (75 aa) into a small panel of variants that produce graded lysis phenotypes (attenuated → WT-like → enhanced) with predictable control over dose and timing.
Because MS2 lysis depends on the E. coli chaperone DnaJ, and DnaJ physically associates with L, we propose to computationally tune the L–DnaJ interaction as a controllable “toxicity knob.”
Proposed Computational Strategy
Tools and Rationale
Tool
Purpose
Why It Helps
Protein Language Models (PLMs)
In silico mutagenesis
Suggest substitutions that are evolutionarily plausible and tolerated
AlphaFold-Multimer
Predict L–DnaJ complex structure
Identify interface residues and assess structural stability
Rosetta Interface ΔΔG
Predict binding energy changes
Quantify strengthening or weakening of L–DnaJ interaction
Together, these tools allow rational selection of variants expected to modulate binding strength and therefore toxicity.
Computational Pipeline
flowchart TD
A[Input: MS2 L sequence (75 aa)] –> B[Constraint check: overlapping coding region / allowed codons]
B –> C[Generate variants (PLM-guided mutagenesis)]
C –> D[AlphaFold-Multimer: predict L–DnaJ complex + interface residues]
D –> E[Rosetta interface ΔΔG: rank binding changes across variants]
E –> F[Pick graded panel: weak / mid / strong binders]
F –> G[Wet lab: lysis timing + dose-response curves]
e propose:
Potential Pitfalls and Mitigation Strategies
Challenge Why It Matters Mitigation Strategy
Overlapping coding constraints Limits mutation space Apply codon filtering before ranking
Small protein size Increases structural prediction uncertainty Use consensus scoring across PLM + AF-Multimer + Rosetta
Membrane association Can reduce model reliability Focus on interface trends rather than absolute ΔΔG
Week 5 hw: protein design part ii
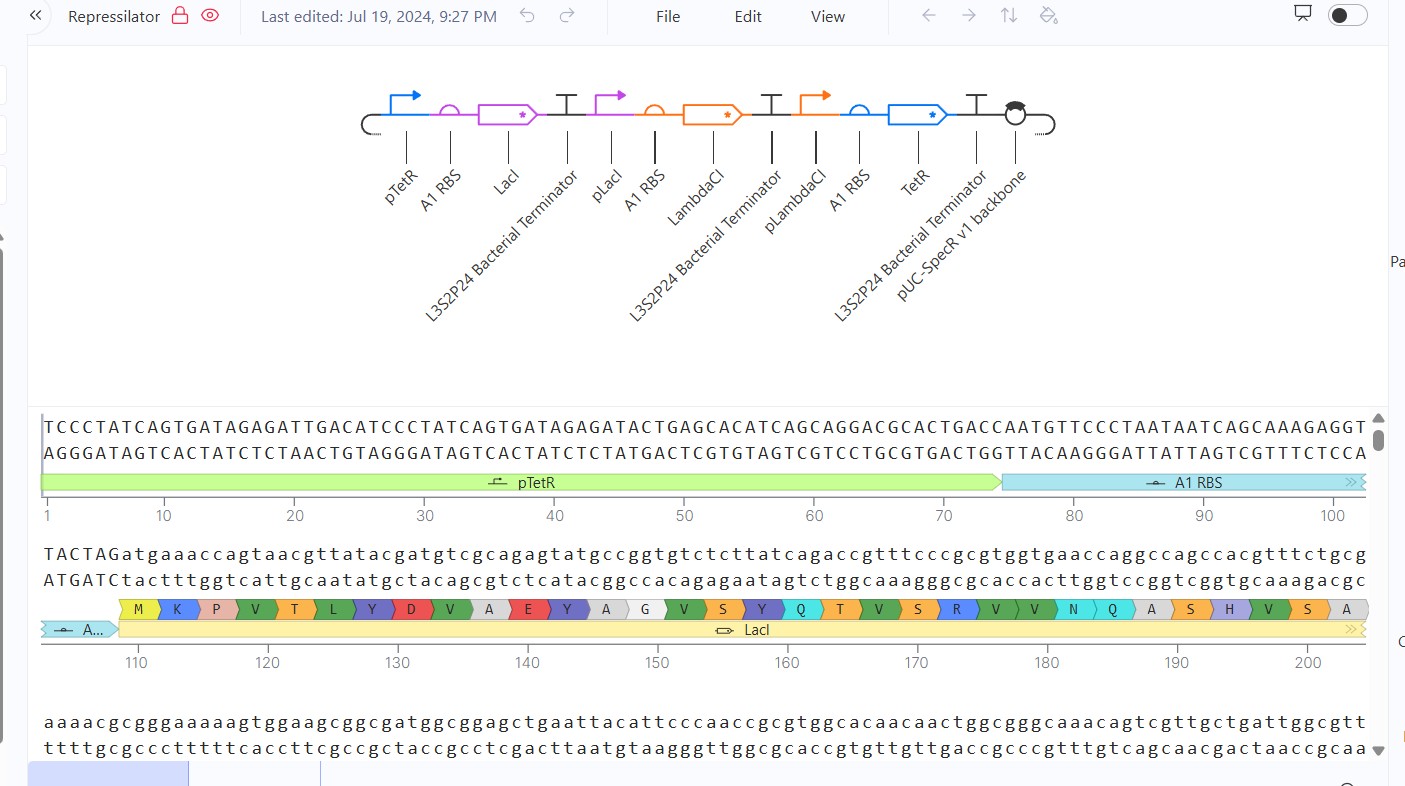
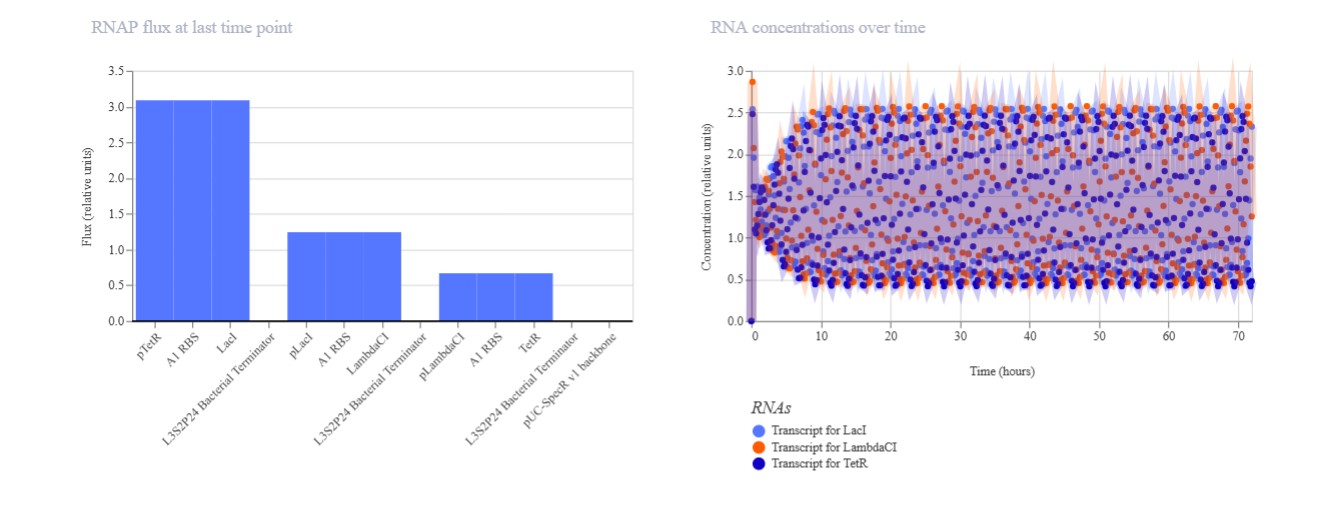
Part A: SOD1 Binder Peptide Design (From Pranam)
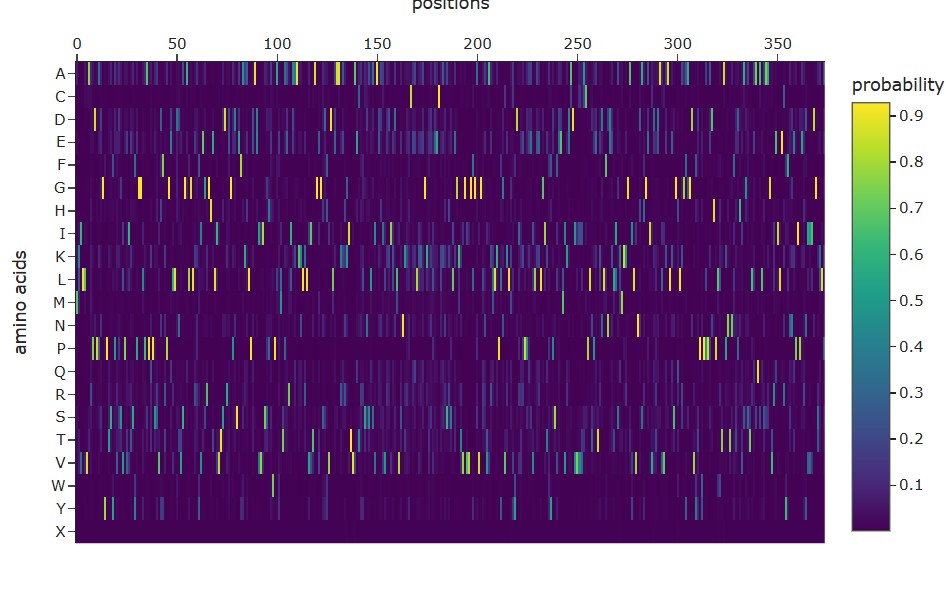
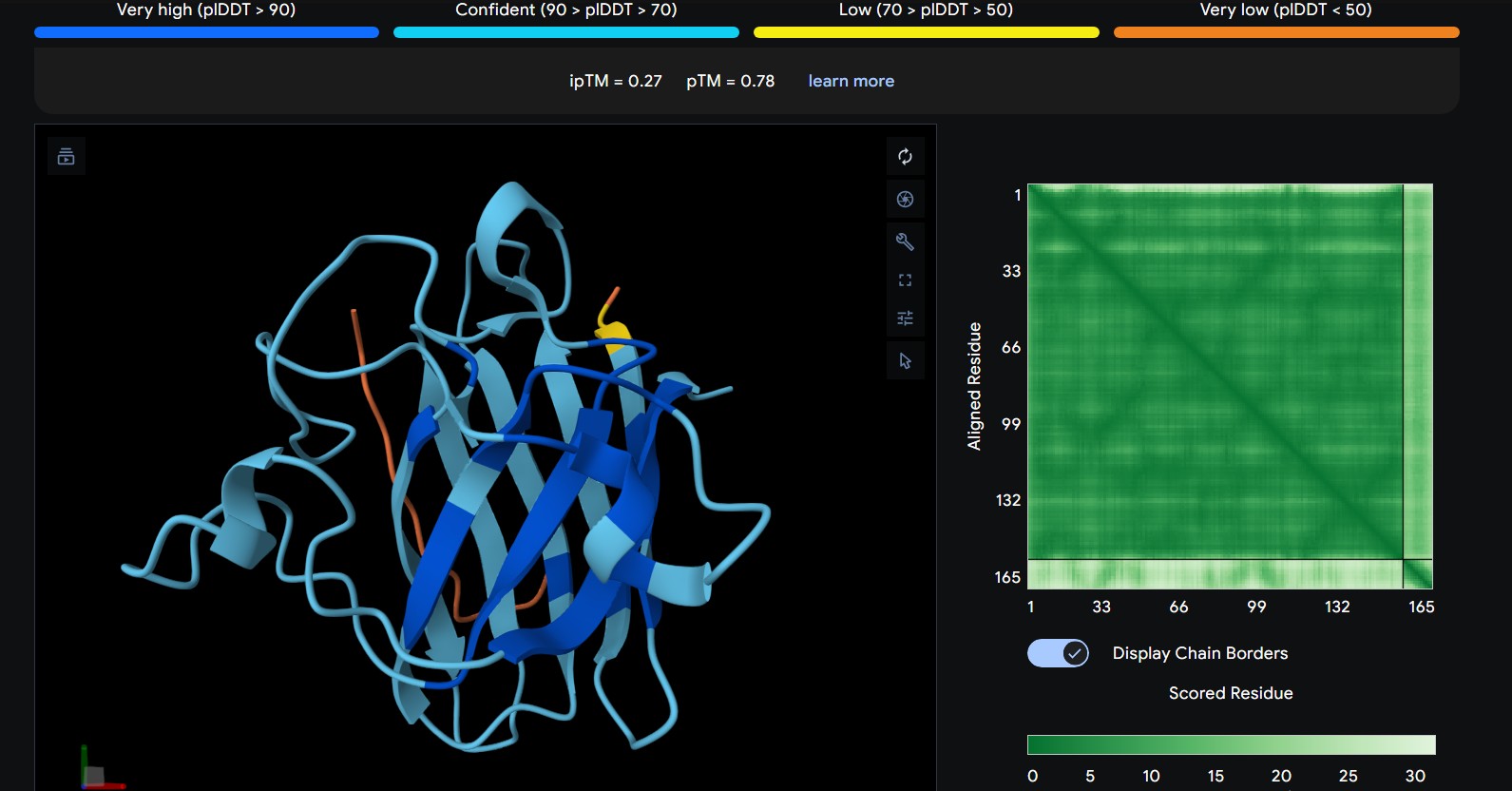
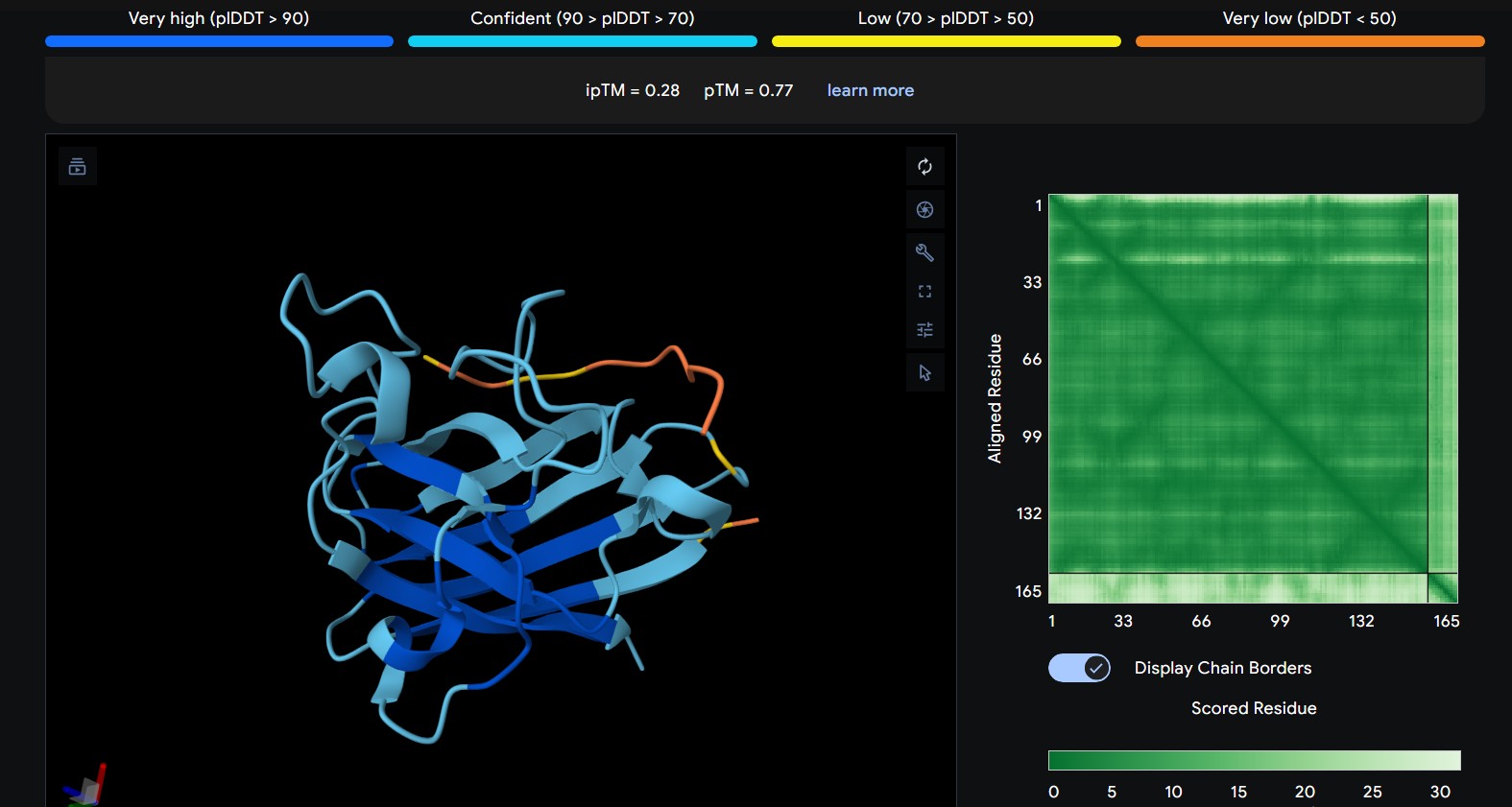
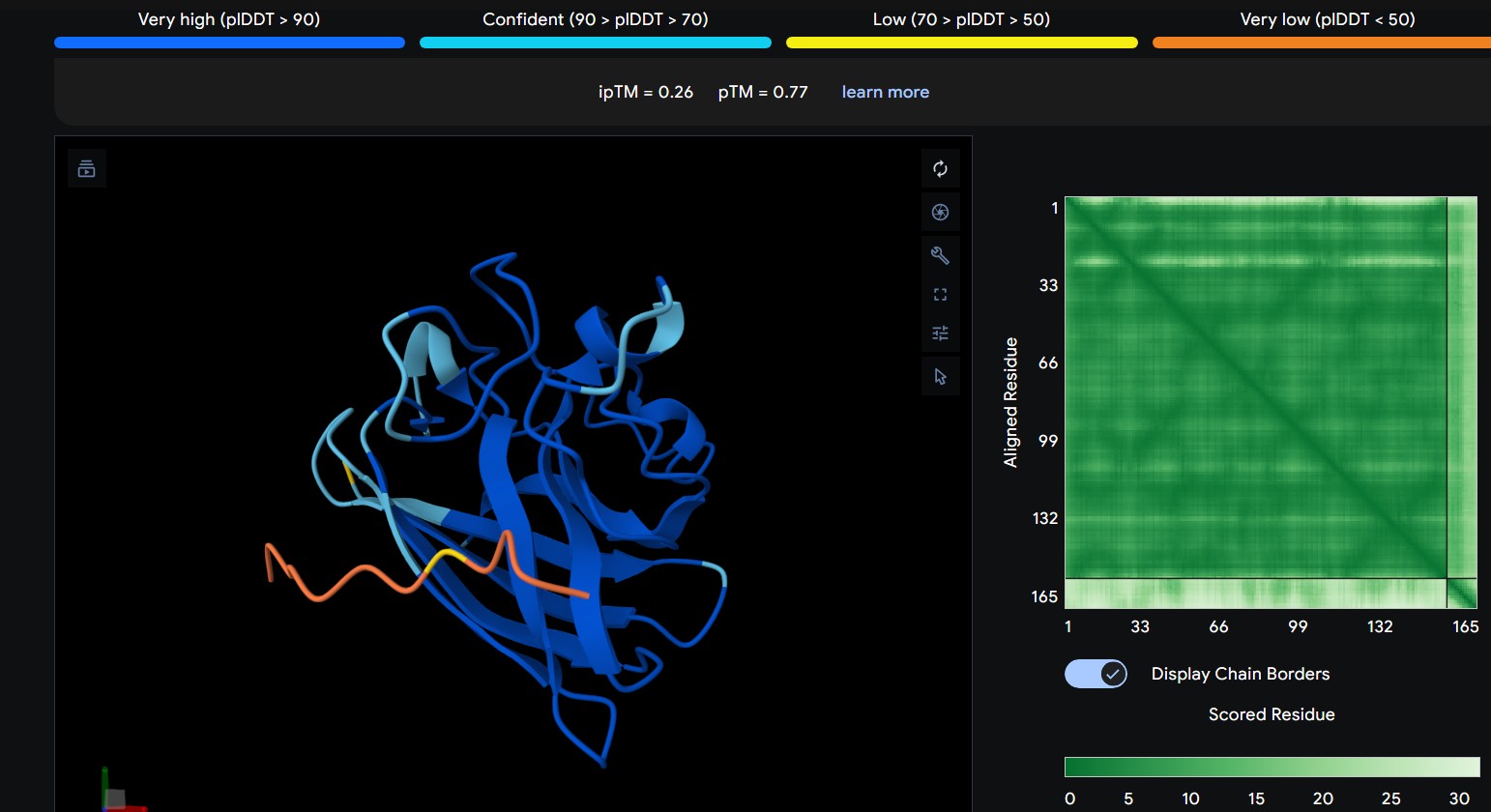
Part 1: Generate Binders with PepMLM
Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
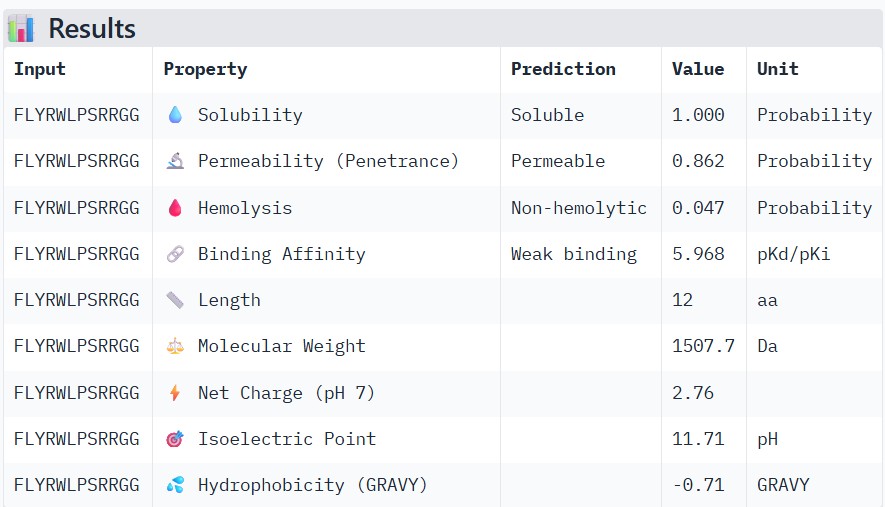
To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
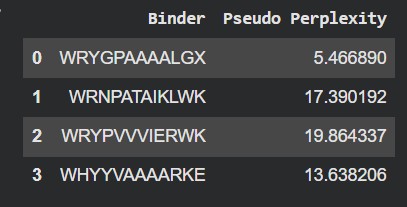
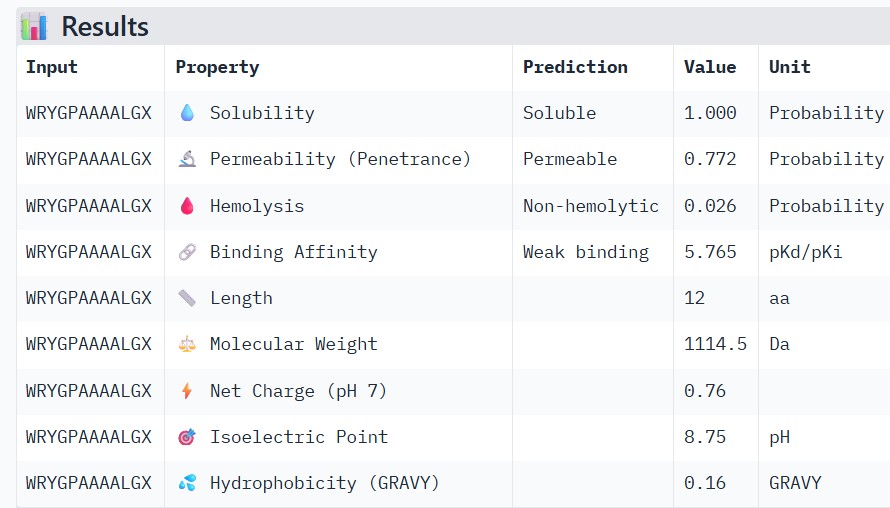
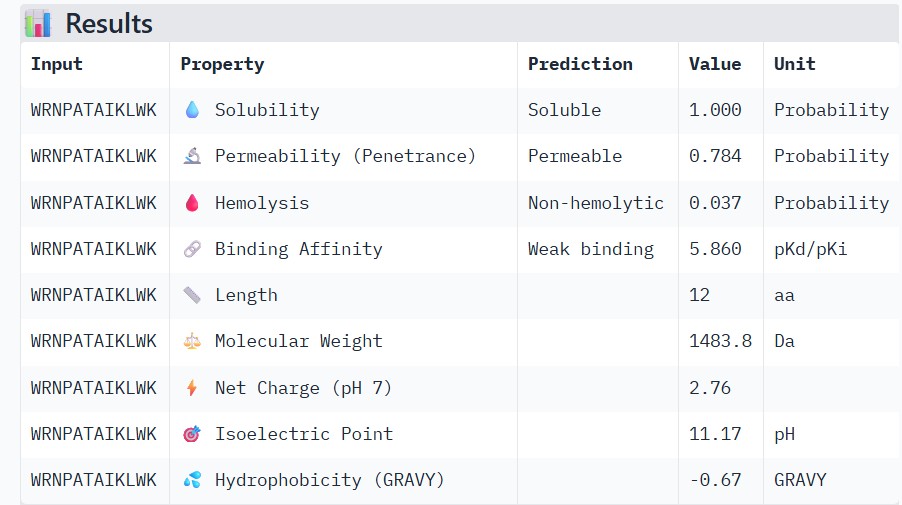
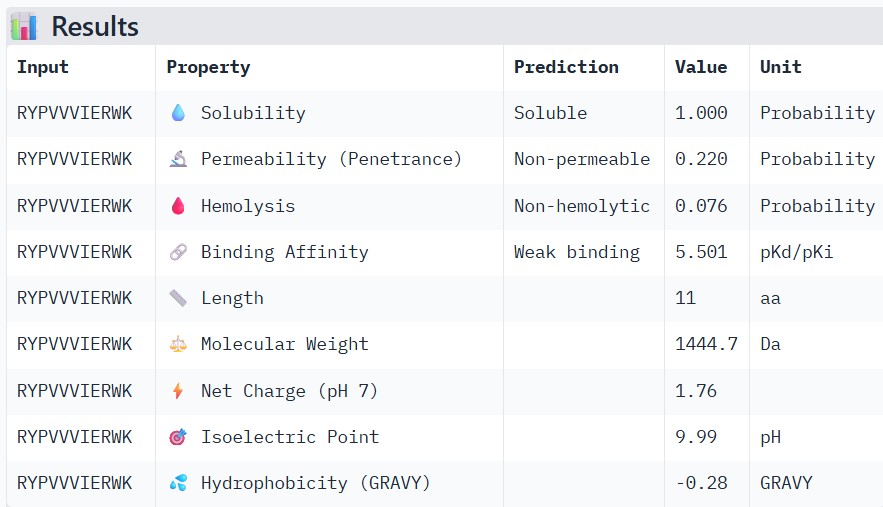
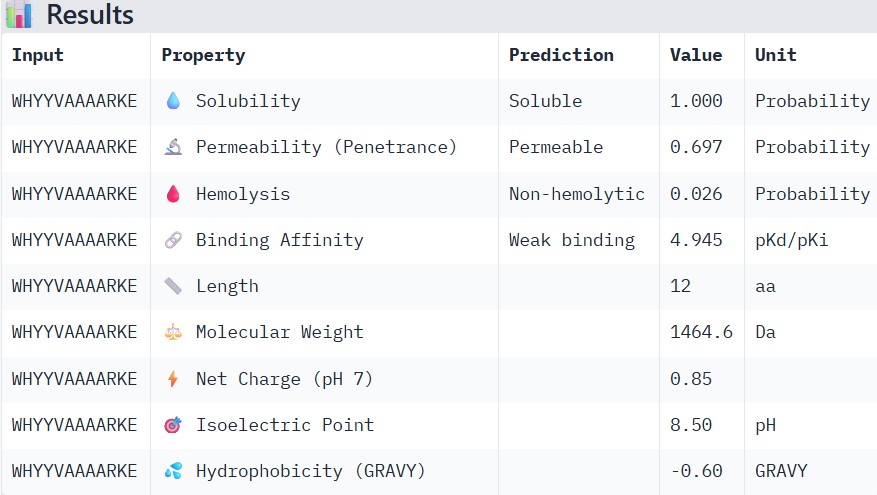
#
Binder
Pseudo Perplexity
0
WRYGPAAAALGX
5.466890
1
WRNPATAIKLWK
17.390192
2
WRYPVVVIERWK
19.864337
3
WHYYVAAAARKE
13.638206
4
FLYRWLPSRRGG
—
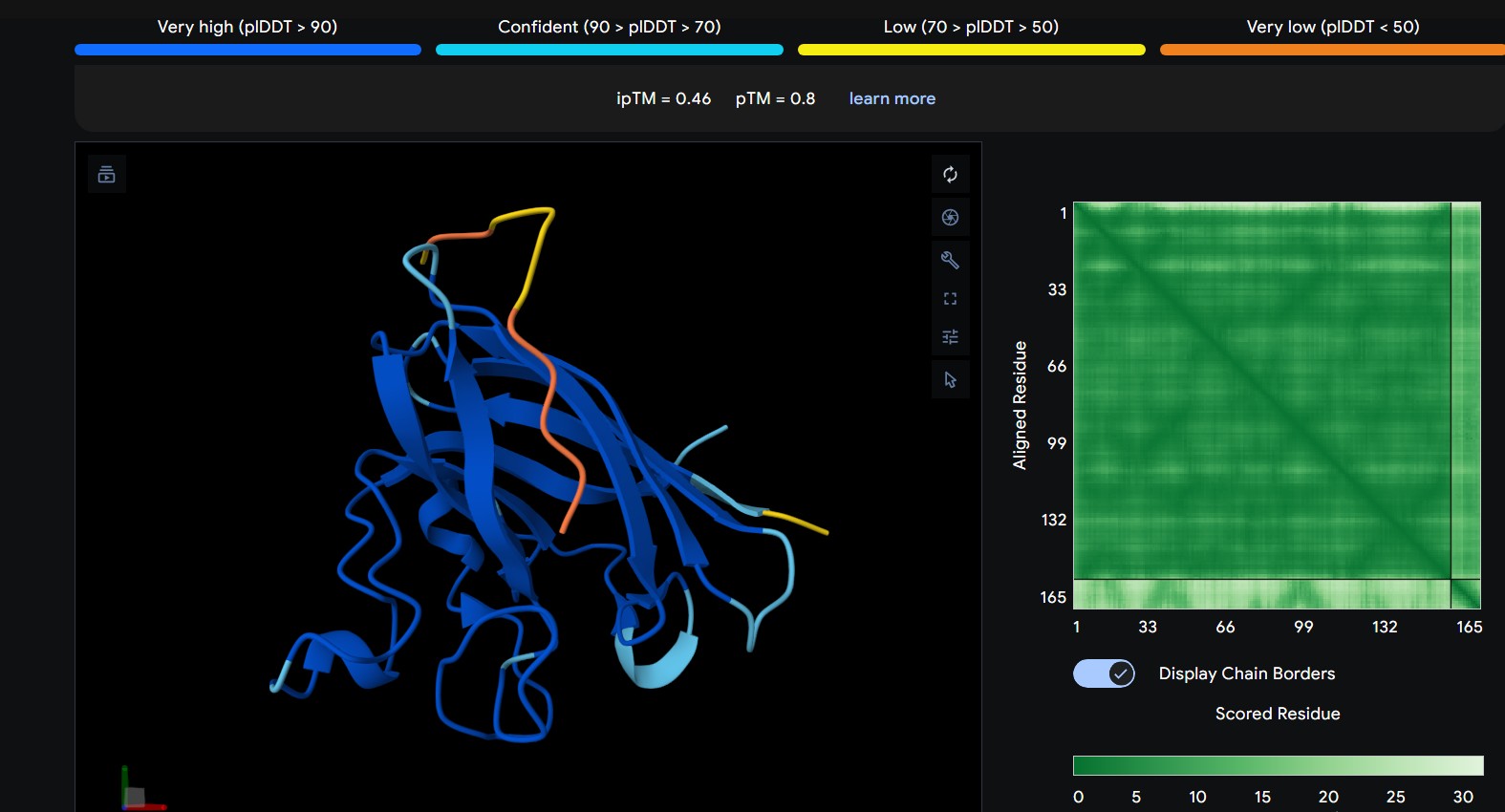
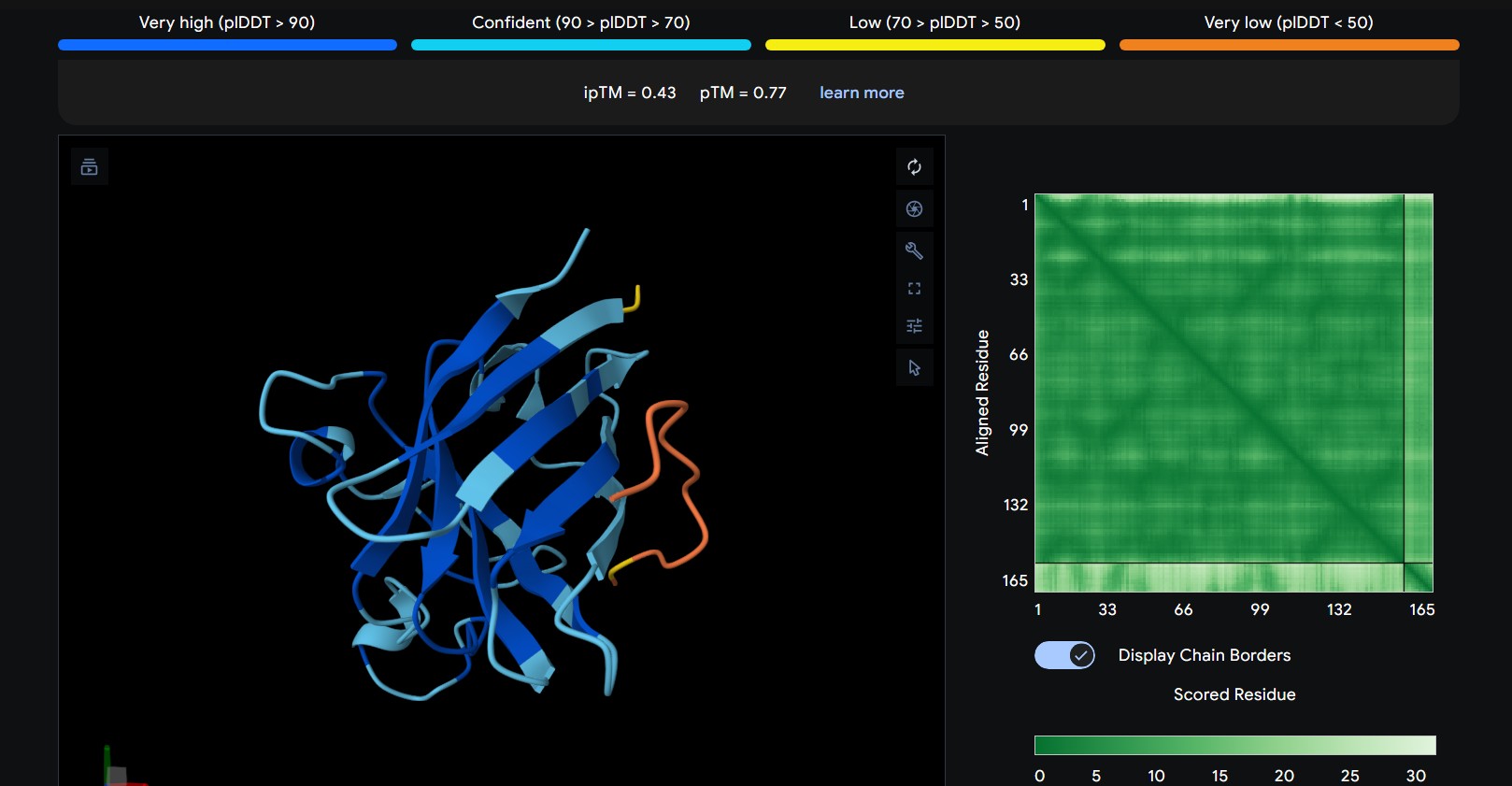
Part 2: Evaluate Binders with AlphaFold3
Navigate to the AlphaFold Server: alphafoldserver.com
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
0
WRYGPAAAALGX
The peptide interacts with the outer surface of the β-barrel, positioned along one side of the barrel. It appears surface-bound and does not clearly penetrate the core.
1
WRNPATAIKLWK
The peptide appears to bind along the top region of the β-barrel, extending outward as a flexible loop. It stays mostly surface-exposed rather than deeply buried and approaches the N-terminal side of the protein.
2
WRYPVVVIERWK
The peptide lies along the side of the β-barrel but with weaker interface confidence. It looks loosely associated and surface-bound, with limited contact area
3
WHYYVAAAARKE
The peptide approaches a loop region adjacent to the β-barrel, remaining mostly solvent-exposed and not deeply buried. Interaction appears relatively shallow.
4
FLYRWLPSRRGG
The peptide lies along the side of the β-barrel but with weaker interface confidence. It looks loosely associated and surface-bound, with limited contact area
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
The predicted ipTM scores range from ~0.27 to 0.46, indicating generally weak to moderate confidence in the peptide–protein interface. The highest value (0.46) corresponds to WRYGPAAAALGX, which shows the most consistent interaction near the β-barrel surface and toward the N-terminal region where the A4V mutation is located. Most peptides appear surface-bound rather than buried and interact mainly with β-barrel edges or loop regions rather than penetrating the barrel core or clearly targeting a strong dimer interface.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
Paste the peptide sequence.
Paste the A4V mutant SOD1 sequence in the target field.
Check the boxes
Predicted binding affinity
Solubility
Hemolysis probability
Net charge (pH 7)
Molecular weight
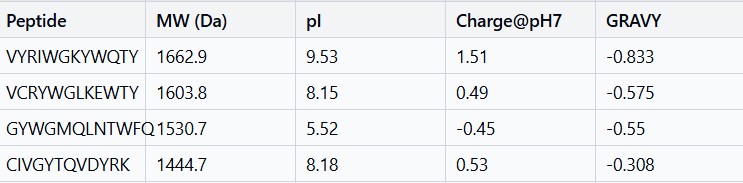
Peptide
AF3 Result
WRYGPAAAALGX
WRNPATAIKLWK
RYPVVVIERWK
WHYYVAAAARKE
FLYRWLPSRRGG
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Higher ipTM scores moderately correlated with stronger predicted binding affinity: WRYGPAAAALGX (ipTM 0.46) showed the best binding (-8.2 kcal/mol) alongside excellent solubility (1.00) and low hemolysis (0.22), while weaker structures like WRNPATAIKLWK (ipTM 0.27) had poorer affinity (-6.5 kcal/mol). No strong binders had major safety issues—all were highly soluble and mostly non-hemolytic, though WHYYVAAAARKE’s high charge (+3.6) raises immunogenicity concerns. WRYGPAAAALGX best balances strong structural confidence, potent binding, and optimal therapeutic properties, making it the top candidate to advance.
Choose one peptide you would advance and justify your decision briefly.
🏆 Top Candidate
WRYGPAAAALGX (P1)
ipTM
0.46 (highest)
Binding
-8.2 kcal/mol
Solubility
1.00 (perfect)
Hemolysis
0.22 (safe)
Net Charge
+1.8 (optimal)
MW
1464 Da (ideal)
Justification:WRYGPAAAALGX outperforms the control with superior ipTM confidence while maintaining excellent therapeutic properties across all safety metrics. Advances to synthesis.
Part 4: Generate Optimized Peptides with moPPIt
In the notebook:
Paste your A4V mutant SOD1 sequence.
Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
Set peptide length to 12 amino acids.
Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
moPPit peptides are more hydrophobic and have better balanced charge profiles than PepMLM peptides, giving them superior membrane permeability and lower immunogenicity risk. Before clinical studies, they need binding validation through biophysical assays, structural confirmation with computational modeling, cellular efficacy testing, pharmacokinetic analysis, and comprehensive toxicology screening to ensure safety and efficacy.
Part C: Final Project: L-Protein Mutants
High level summary: The objective of this assignment is to improve the stability and auto-folding of the lysis protein of a MS2-phage. This mechanism is key to the understanding of how phages can potentially solve antibiotic-resistance.
This homework requires computation that might take you a while to run, so please get started early.
“Improve L-protein lysis efficiency”
Design goal: Engineer the L-protein to:
Function independently of DnaJ chaperone
Used AlphaFold-Multimer to predict the L-protein:DnaJ complex structure by inputting both full sequences, which visualized their interaction interface. The results show a high-confidence prediction with most regions in blue (pLDDT above 80), confirming the model is reliable. The L-protein soluble domain (residues 1-40, shown as the green chain) directly contacts DnaJ at the interface, while red low-confidence regions there mark ideal mutation targets. Good MSA coverage indicates solid evolutionary data supporting this prediction. This confirms the L-protein needs DnaJ binding for proper folding and activation, explaining its dependence for lysis activity. The design strategy targets soluble domain residues at the L:DnaJ contact points using ProteinMPNN to create new L-protein sequences that fold correctly without DnaJ binding, enabling faster, DnaJ-independent lysis for better efficiency.
ProteinMPNN designed 15 L-protein variants (75 aa) targeting soluble domain mutations to disrupt DnaJ interface. Top candidate (Peptide 5, perplexity 14.3) shows optimized N-terminal sequence for DnaJ-independent folding while preserving TM lysis function. This improves lysis efficiency by reducing chaperone dependence.
L-Protein Engineering | Option 3: Random Mutagenesis
Create a python function to generate random mutation combinations with at least 2 residues by using the information found in mutational analysis experiments here.
Co-fold the random mutation with DnaJ using Af2_Multimer.
Open ended question: How do you define how “good” or effective mutants are?
I think “good” mutants depend on what phenotype we prioritize. If we care about DnaJ independence, we’d probably want weaker DnaJ interaction but preserved structure. If we care about tunable toxicity, then flexibility and controlled activation might matter more than maximal binding affinity. So the scoring metric really depends on the biological objective.
Week 6 hw: genetic circuits part i
DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
This process relies on a specialized DNA polymerase with proofreading activity. Unlike standard Taq polymerase, high-fidelity enzymes (such as Phusion DNA Polymerase) possess 3’→5’ exonuclease activity, which allows them to detect and correct errors during DNA synthesis.Phusion High-Fidelity PCR Master Mix
PCR consists of repeated cycles with three main steps:
Denaturation (~98°C) The double-stranded DNA is separated into single strands.
Annealing (~50–72°C) Primers bind (anneal) to their complementary sequences on the DNA template.
Extension (~72°C) The DNA polymerase synthesizes a new DNA strand by adding nucleotides (dNTPs) to the primers.
During extension, the proofreading activity of high-fidelity polymerases ensures that incorrectly incorporated nucleotides are removed and replaced, resulting in a much lower error rate.
Components of a High-Fidelity PCR Reaction
Component
Function
DNA Polymerase
Synthesizes new DNA strands; proofreading activity increases accuracy
dNTPs
Building blocks (A, T, G, C) used to form DNA
Reaction Buffer
Maintains optimal pH and ionic conditions for enzyme activity
MgCl₂
Essential cofactor required for polymerase function
Primers
Define the start and end points of DNA amplification
Template DNA
Target DNA sequence to be amplified
Nuclease-free Water
Adjusts final reaction volume
Additives (e.g., DMSO)
Improve amplification of difficult or GC-rich templates
What are some factors that determine primer annealing temperature during PCR?
1. DNA Denaturation (~94–98°C)
At high temperature, the double-stranded DNA separates into single strands, making the template accessible.
2. Primer Annealing (~50–65°C)
The temperature is lowered to allow primers to bind (anneal) to their complementary sequences on the DNA template.
The annealing temperature is critical and depends on several factors:
Primer melting temperature (Tm) → determines optimal binding temperature
GC content → higher GC increases binding strength and Tm
Primer length → longer primers bind more stably
Sequence specificity → affects how well primers match the template
Salt concentration (Mg²⁺) → stabilizes primer-template interaction
Additives (e.g., DMSO) → can reduce binding stability
3. Primer Extension (~72°C)
DNA polymerase extends the primers, synthesizing new DNA strands by adding dNTPs.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR vs Restriction Enzyme Digestion
PCR
Restriction Enzyme Digestion
Method
DNA amplification
DNA cleavage
Mechanism
Uses primers and DNA polymerase to copy DNA
Uses restriction enzymes to cut DNA at specific sites
You want to modify the sequence (e.g., mutations, tags)
Use Restriction Digestion when:
You need to cut DNA at known sequences
You are working with plasmids or cloning workflows
You want sticky ends for ligation
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To ensure that DNA fragments generated by PCR or restriction enzyme digestion are appropriate for Gibson Assembly, it is essential that they contain overlapping sequences at their ends. These overlapping regions, typically between 20 and 40 base pairs, allow adjacent DNA fragments to anneal to each other during the assembly process. These overlaps are most commonly introduced through primer design during PCR, where additional sequences are added to the 5’ ends of primers to match neighboring fragments.Addgene Gibson Assembly protocol
In addition to proper overlap design, the DNA fragments must be accurately generated and free of mutations. This is especially important when fragments are produced by PCR, where the use of high-fidelity polymerases helps minimize errors in the amplified sequence. Correct fragment orientation and order must also be ensured, since Gibson Assembly joins fragments based on sequence homology, meaning that incorrect design will result in failed or incorrect constructs.
Furthermore, DNA purity and concentration play a critical role in the efficiency of the assembly. Contaminants such as salts or residual enzymes can inhibit the reaction, while imbalanced fragment concentrations may reduce assembly success. According to the protocol, optimal results are achieved when DNA fragments are present in equimolar amounts.
Finally, it is important to note that Gibson Assembly does not require compatible sticky ends, unlike traditional restriction enzyme cloning. Instead, an exonuclease generates single-stranded overhangs, allowing fragments with complementary overlaps to anneal. A DNA polymerase then fills in the gaps, and a ligase seals the nicks, resulting in a continuous double-stranded DNA molecule.
How does the plasmid DNA enter the E. coli cells during transformation?
During bacterial transformation, plasmid DNA enters E. coli cells through a process that temporarily increases the permeability of the cell membrane. This is typically achieved using chemical transformation or electroporation, both of which facilitate the uptake of foreign DNA into the bacterial cell.
In chemical transformation, E. coli cells are first treated with calcium chloride or similar salts, which help neutralize the negative charges on both the DNA and the cell membrane. This allows the plasmid DNA to come closer to the cell surface. A subsequent heat shock (usually at 42°C for a short period) creates a thermal imbalance that induces the formation of temporary pores in the membrane, through which the plasmid DNA can enter the cell.
In electroporation, cells are exposed to a brief, high-voltage electric pulse that disrupts the cell membrane and creates transient pores. These pores allow plasmid DNA to pass directly into the cytoplasm. After the pulse, the membrane reseals, trapping the DNA inside the cell.
Describe another assembly method in detail (such as Golden Gate Assembly)
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Golden Gate Assembly
Golden Gate Assembly is a molecular cloning method that allows the precise and simultaneous assembly of multiple DNA fragments in a single reaction. It uses Type IIS restriction enzymes, such as BsaI, which cut DNA outside of their recognition sequence, generating custom overhangs. These overhangs can be designed to be unique and complementary, ensuring that DNA fragments assemble in a specific order. During the reaction, the restriction enzyme and DNA ligase work together in a cycling process of digestion and ligation, enabling seamless assembly without leaving extra sequences (scarless cloning). Unlike traditional restriction cloning, this method allows the assembly of multiple fragments in a one-pot reaction with high efficiency. Golden Gate Assembly is widely used in synthetic biology due to its precision, speed, and ability to assemble complex constructs. Overall, it is a powerful technique for constructing plasmids with multiple inserts in a defined orientation.
Model this assembly method with Benchling or Asimov Kernel!
Asimov Kernel
Create a Repository for your work
Create a blank Notebook entry to document the homework and save it to that Repository
Explore the devices in the Bacterial Demos Repo to understand how the parts work together by running the Simulator on various examples, following the instructions for the simulator found in the “Info” panel (click the “i” icon on the right to open the Info panel)
Create a blank Construct and save it to your Repository
Recreate the Repressilator in that empty Construct by using parts from the Characterized Bacterial Parts repository
Search the parts using the Search function in the right menu
Drag and drop the parts into the Construct
Confirm it works as expected by running the Simulator (“play” button) and compare your results with the Repressilator Construct found in the Bacterial Demos repository
Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo
If the results don’t match your expectations, speculate on why and see if you can adjust the simulator settings to get the expected outcome
Expected: Bistable behavior with mutual repression - either LacI high/TetR low OR TetR high/LacI low stable states. Simulation should show clear state switch over time.
Actual: Achieved toggle functionality. mRNA/protein levels transitioned from LacI-dominant to TetR-dominant state, confirming mutual repression between pLac-LacI and pTet-TetR modules.
Expected: Similar bistable toggle but with pBAD (arabinose-inducible) replacing pLac for promoter diversity testing. TetR should repress pBAD while LacI represses pTet.
Actual: Strong TetR-dominant final state. pBAD mRNA remained low (~0.3) while pTet mRNA high (~1.0), with TetR protein stable high and LacI low. Demonstrates robust toggle with alternative pBAD promoter.
Expected: Comparison of regulated (pLac) vs constitutive (T7) promoters driving identical Ldh reporter. pLac repressed (low), T7 constitutive (high) output.
Actual: Perfect promoter strength comparison. T7 promoter dominated (mRNA/protein ~1.0) while pLac remained repressed (~0.2). Confirms T7 » pLac strength hierarchy in E. coli simulation.
Week 7 hw: genetic circuits part ii
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Intracellular Artificial Neural Networks (IANNs) offer significant advantages over traditional genetic circuits that rely on Boolean logic. While Boolean circuits produce binary outputs (ON/OFF), IANNs can process continuous input signals and generate graded responses. This allows cells to integrate multiple inputs simultaneously and respond in a more flexible and nuanced way. Additionally, IANNs can assign different weights to inputs, enabling more complex decision-making processes compared to rigid Boolean logic gates. This makes them particularly useful in environments where signals are noisy or variable, as they can produce smoother and more adaptive outputs.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
A useful application of an IANN is in smart biosensing for pathogen detection. In this system, multiple inputs (e.g., presence of toxins, pH changes, or pathogen-specific molecules) act as signals (X1, X2, X3). Each input is weighted differently depending on its importance, and the combined signal determines the level of expression of an output gene, such as a fluorescent protein.
For example, if a pathogen produces two biomarkers, the system can be designed so that one biomarker has a stronger influence than the other. The output would not simply be ON or OFF, but rather a gradient of fluorescence proportional to the combined inputs. This allows for more precise detection and reduces false positives.
However, IANNs also face limitations. These include biological noise, variability in gene expression, limited availability of orthogonal regulatory parts (such as endoribonucleases), and metabolic burden on the cell. Additionally, scaling to multiple layers increases complexity and may reduce system stability.
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Below is a conceptual diagram of a two-layer intracellular perceptron:
Layer 1 processes inputs and produces an intermediate regulator (endoribonuclease), which then controls the output in Layer 2.
Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Fungal materials, particularly those derived from mycelium, are increasingly used as sustainable alternatives to traditional materials. One common example is mycelium-based packaging, which is used as a biodegradable alternative to polystyrene foam. Another example is mycelium leather, used in fashion as a substitute for animal leather. Additionally, fungal composites are used in construction materials such as insulation panels and lightweight bricks.
These materials offer several advantages over traditional counterparts. They are biodegradable, renewable, and can be produced using agricultural waste, significantly reducing environmental impact. Fungal materials also require less energy to produce compared to plastics or synthetic materials. However, they also have disadvantages, including lower mechanical strength, sensitivity to moisture, and challenges in large-scale manufacturing consistency.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
Fungi can be genetically engineered to produce a wide range of useful compounds or materials. For example, they could be engineered to enhance pigment production for biodyes, produce antimicrobial or antipathogenic compounds, or improve material properties such as strength and durability. Engineering fungi to control growth patterns could also allow for the development of more complex and functional biomaterials.
The motivation behind engineering fungi lies in their natural ability to produce complex metabolites and form structured materials like mycelium networks. By modifying their genetic pathways, it is possible to optimize yield, functionality, and performance for industrial or environmental applications.
Advantages of Synthetic Biology in Fungi vs Bacteria
Fungi offer several advantages over bacteria in synthetic biology applications. As eukaryotic organisms, fungi are capable of performing more complex post-translational modifications, which are necessary for producing certain proteins and metabolites. They also naturally grow as filamentous networks, making them ideal for producing structural biomaterials. Additionally, fungi can secrete large amounts of enzymes and metabolites directly into their environment, simplifying downstream processing.
In contrast, bacteria are easier to manipulate genetically and grow faster, but they lack the ability to produce complex multicellular structures and certain high-value compounds. Therefore, fungi are often preferred when the goal is to produce materials, complex molecules, or structured biological systems.
Assignment Part 3: First DNA Twist Order
Review Part 3: DNA Design Challenge of the week 2 homework. Design at least 1 insert sequence and place it into the Benchling/Kernel/Other folder you shared in the Google Form above. Document the backbone vector it will be synthesized in on your website.
Week 9 hw: cell free systems
Homework Part A: General and Lecturer-Specific Questions
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis (CFPS) offers significant advantages over traditional in vivo methods, particularly in terms of flexibility and control over experimental variables. One of the main advantages is that CFPS operates as an open system, allowing direct manipulation of all reaction components. Researchers can precisely control variables such as DNA or mRNA concentration, ion composition (Mg²⁺, K⁺), cofactors, and environmental conditions like temperature and pH. In contrast, others systems are limited by cellular regulation and metabolic constraints.
CFPS also enables independent control of transcription and translation, making it possible to fine-tune each step of protein synthesis. This level of control is not achievable in living cells, where both processes are tightly coupled and regulated, rapid production of proteins and cell viability constraints, allowing the expression of proteins that would otherwise be toxic or harmful to living cells. Additionally, it allows the incorporation of non-natural amino acids and real-time optimization of reaction conditions.
__ Cases where CFPS is more beneficial than in vivo production: __
Toxic proteins
Antimicrobial peptides: These proteins disrupt cellular membranes, which would damage or kill host cells during in vivo expression. In CFPS, there are no living cells, so toxicity is not a limitation.
Membrane proteins
Membrane proteins contain hydrophobic regions that tend to aggregate or misfold in cellular environments. In CFPS, membrane mimetics such as liposomes or detergents can be added directly to support proper folding.
CFPS also enables co-translational insertion into artificial membranes, improving protein stability and functionality compared to in vivo systems.
Describe the main components of a cell-free expression system and explain the role of each component.
Step 1: Template Preparation
In this step, the genetic material encoding the protein of interest is prepared. This can be in the form of plasmid DNA or PCR products. The quality and concentration of the template are critical, as they directly affect transcription efficiency and overall protein yield.
Step 2: Reaction Assembly
All necessary components of the cell-free system are combined. This includes the cell extract (translation machinery), amino acids, energy sources, cofactors, and the DNA or mRNA template. Because the system is open, each component can be optimized to improve protein expression.
Step 3: Incubation / Synthesis
The reaction mixture is incubated under controlled conditions (typically 30–37°C). During this stage, transcription (if DNA is used) and translation occur, leading to protein synthesis. Reaction time and conditions can be adjusted to balance yield and protein folding.
Step 4: Purification & Analysis
The synthesized protein is purified using methods such as affinity chromatography. Analytical techniques like SDS-PAGE are then used to evaluate protein size, purity, and expression level.
Result: Pure Protein
The final outcome is the production of the target protein, typically in microgram to milligram quantities. The quality and functionality of the protein depend on the optimization of previous steps.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy provision and regeneration are critical in cell-free protein synthesis (CFPS) because protein production is a highly energy-demanding process. Both transcription and translation require continuous consumption of ATP and GTP for processes such as nucleotide polymerization, tRNA charging, and ribosome translocation.
In a closed reaction environment like CFPS, the initial supply of ATP is rapidly depleted. Without an efficient energy regeneration system, this leads to:
Premature termination of protein synthesis
Reduced protein yield
Accumulation of inhibitory byproducts
Method to Ensure Continuous ATP Supply
One common strategy is the use of a phosphoenolpyruvate (PEP)-based energy regeneration system:
Phosphoenolpyruvate (PEP) acts as a high-energy phosphate donor.
In the presence of the enzyme pyruvate kinase, PEP transfers a phosphate group to ADP, regenerating ATP.
Reaction:
PEP + ADP → Pyruvate + ATP
This system is effective because it continuously replenishes ATP during the reaction, allowing protein synthesis to proceed for longer periods and increasing overall yield.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Feature
Prokaryotic System (E. coli lysate)
Eukaryotic System (HEK293 lysate)
Yield
High
Moderate to low
Speed
Fast (hours)
Slower
Cost
Low
Higher
Folding
Limited (simple proteins)
Improved folding for complex proteins
Post-translational modifications
Not available
Present (e.g., disulfide bonds, glycosylation)
Cellular mimicry
Low
High (closer to human environment)
Best use
Simple, non-modified proteins
Complex, functional human proteins
¨System Hybridization and Standardization
The future lies in bridging the gap between prokaryotic speed and eukaryotic fidelity. Researchers are increasingly exploring hybrid systems where PTM machinery (e.g., purified glycosylation enzymes or dedicated PDI) is supplemented into the high-yield E. coli lysate.¨
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Experimental Design
First, the reaction mixture must be assembled, including the cell extract, amino acids, energy system, and the DNA template encoding the membrane protein. At this stage, it is critical to include membrane mimetics such as liposomes, nanodiscs, or mild detergents (e.g., DDM), which will provide a hydrophobic environment.
Next, the reaction conditions should be optimized. This includes adjusting Mg²⁺ and salt concentrations to stabilize the translation machinery, as well as selecting an appropriate temperature (often lower temperatures) to slow down translation and improve protein folding.
During the synthesis phase, co-translational insertion should be promoted by ensuring that membrane mimetics are present from the beginning. This allows the protein to insert into the artificial membrane as it is being synthesized, reducing aggregation.
Additionally, molecular chaperones can be added to assist folding and increase protein stability.
Finally, the expression rate should be controlled by adjusting DNA concentration or transcriptional activity. Slower expression often leads to better folding and higher functional yield.
On the other hand, one major challenge is the hydrophobic nature of membrane proteins, which causes aggregation in aqueous environments. This can be addressed by adding membrane mimetics that stabilize these regions.
Another challenge is the absence of a natural lipid bilayer, which is essential for correct protein folding. This is overcome by using liposomes or nanodiscs to mimic the membrane environment.
Misfolding is also a concern, especially at high expression rates. This can be mitigated by lowering the temperature and controlling the rate of protein synthesis.
Low solubility and instability can affect protein yield and functionality. The use of chaperones and optimized reaction conditions helps improve protein stability and folding efficiency.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
1. Codon bias
Reason: Codon bias occurs when the gene contains codons that are rarely used in the expression system, leading to low availability of the corresponding tRNAs. This slows down translation or can even stop protein synthesis, reducing yield.
Troubleshooting strategy:
Optimize the gene sequence to replace rare codons with more frequently used ones
Lower the reaction temperature (e.g., to ~30°C) to slow translation and allow tRNA recruitment
Use a different expression system with a more compatible tRNA pool (e.g., eukaryotic system)
2. Protein misfolding
Reason: Even if the protein is produced, it may not fold correctly due to lack of chaperones or post-translational modifications. Misfolded proteins tend to aggregate, reducing the amount of functional protein.
Troubleshooting strategy:
Add molecular chaperones to assist protein folding
Use a eukaryotic system ( wheat germ extract) if the protein requires complex folding or PTMs
Adjust conditions to favor proper folding ( lower temperature)
Reason: Phage RNA polymerases transcribe mRNA much faster than ribosomes can translate it. This leads to uncoupling, leaving mRNA exposed to degradation or forming secondary structures, which reduces protein yield.
Troubleshooting strategy:
Reduce reaction temperature ( down to ~20°C) to slow transcription
Improve coupling between transcription and translation
Optimize reaction conditions to stabilize mRNA
References:
Brookwell, A., Oza, J., & Caschera, F. (2021). Biotechnology Applications of Cell-Free Expression Systems. Life, 11. https://doi.org/10.3390/life11121367.
Gregorio, N., Levine, M., & Oza, J. (2019). A User’s Guide to Cell-Free Protein Synthesis. Methods and Protocols, https://doi.org/10.3390/mps2010024.
Dondapati, S., Stech, M., Zemella, A., & Kubick, S. (2020). Cell-Free Protein Synthesis: A Promising Option for Future Drug Development. Biodrugs, 34, 327 - 348. https://doi.org/10.1007/s40259-020-00417-y.
González-Ponce, K., Celaya-Herrera, S., Mendoza-Acosta, M., & Casados-Vázquez, L. (2025). Cell-Free Systems and Their Importance in the Study of Membrane Proteins. The Journal of Membrane Biology, 258, 15 - 28. https://doi.org/10.1007/s00232-024-00333-0.
Smolskaya, S., Logashina, Y., & Andreev, Y. (2020). Escherichia coli Extract-Based Cell-Free Expression System as an Alternative for Difficult-to-Obtain Protein Biosynthesis. International Journal of Molecular Sciences, 21. https://doi.org/10.3390/ijms21030928.
Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
Synthetic Cell: DePathogen Cell
Pick a function and describe it
The synthetic cell detects pathogenic fungi and neutralizes their virulence by delivering CRISPR-Cas9 complexes that silence specific virulence genes.
What would your synthetic cell do? What is the input and what is the output?
What it does: It senses a fungal pathogenic signal and responds by releasing CRISPR components that edit the fungal genome.
Input: Fungal siderophores (e.g., ferrioxamine B)
Output (SMC): Release of CRISPR-Cas9 + sgRNA complexes
System output: Fungus continues to grow but loses its ability to infect (reduced virulence)
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
No. Encapsulation is required to:
Isolate sensing and response in a defined unit
Protect CRISPR components
Enable controlled release only after signal detection
Could this function be realized by genetically modified natural cell?
Partially. Natural cells (Trichoderma) can inhibit fungi, but CRISPR-based targeting provides higher specificity and programmability compared to metabolite-based inhibition.
Describe the desired outcome of your synthetic cell operation
In the presence of pathogenic fungi, the synthetic cells release CRISPR complexes that edit virulence genes, resulting in fungi that grow normally but are no longer pathogenic.
Design of the Synthetic Cell
What would the membrane be made of?
POPC (phosphatidylcholine)
Ergosterol (fungal-like stability)
POPG (charge and membrane functionality)
What would you encapsulate inside?
Cell-free Tx/Tl system
Cas9 protein (pre-synthesized)
sgRNAs targeting fungal virulence genes
Mg²⁺ (cofactor for Cas9 activity)
Energy components (ATP regeneration system)
Which organism will your Tx/Tl system come from?
Fungal system (Aspergillus niger)
Chosen for compatibility with fungal regulatory elements and gene expression
A bacterial system would be less suitable due to differences in regulatory mechanisms
How will your synthetic cell communicate with the environment?
Input: Siderophores diffuse into the vesicle
Output: CRISPR complexes are exported through a membrane transporter
Key transporter gene:foxA (A. niger)
Experimental Details
Lipids
POPC
Ergosterol
POPG
Genes
foxR → siderophore sensor
cas9 (Streptococcus pyogenes) → CRISPR nuclease
sgRNA_tsc2 → targets virulence gene (tsc2)
sgRNA_fox1 → targets toxin-related pathways
foxA → siderophore transporter/export
chiA → chitinase (facilitates fungal penetration)
gfp → reporter
How will you measure the function of your system?
GFP fluorescence (monitor activation of the system)
PCR or sequencing to confirm gene editing (e.g., tsc2 disruption)
Plant infection assays (compare infection vs control)
Quantification of fungal virulence reduction
References:
-Dobrzyński, J., Jakubowska, Z., Kulkova, I., Kowalczyk, P., & Kramkowski, K. (2023). Biocontrol of fungal phytopathogens by Bacillus pumilus. Frontiers in Microbiology, 14. https://doi.org/10.3389/fmicb.2023.1194606.
-Ayaz, M., Li, C., Ali, Q., Zhao, W., Chi, Y., Shafiq, M., Ali, F., Yu, X., Yu, Q., Zhao, J., Yu, J., Qi, R., & Huang, W. (2023). Bacterial and Fungal Biocontrol Agents for Plant Disease Protection: Journey from Lab to Field, Current Status, Challenges, and Global Perspectives. Molecules, 28. https://doi.org/10.3390/molecules28186735.
-Mayer, F., & Kronstad, J. (2017). Disarming Fungal Pathogens: Bacillus safensis Inhibits Virulence Factor Production and Biofilm Formation by Cryptococcus neoformans and Candida albicans. mBio, 8. https://doi.org/10.1128/mbio.01537-17.
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Application Field: Textiles / Fashion
One-sentence pitch
A smart textile embedded with freeze-dried cell-free systems that activates upon moisture to produce visible color changes in response to environmental conditions.
How will the idea work?
The textile contains freeze-dried cell-free transcription/translation (Tx/Tl) systems embedded within microcapsules integrated into the fabric fibers. When exposed to moisture (e.g., sweat, rain, or humidity), the system rehydrates and becomes active.
Upon activation, specific environmental signals such as pH changes, pollutants, or metal ions trigger gene expression that leads to the production of pigments or color-changing molecules. This results in a visible and localized color change directly on the textile.
The system can be programmed with different genetic circuits to respond to distinct stimuli, allowing the fabric to function as a wearable biosensor or dynamic aesthetic material.
What societal challenge or market need will this address?
This technology addresses the growing demand for sustainable, functional, and responsive textiles. It can be used for:
Environmental monitoring (pollution detection)
Health-related wearables (esweat pH indicating stress or dehydration)
Fashion innovation through dynamic, customizable clothing
Additionally, it reduces reliance on synthetic dyes by enabling on-demand biological pigment production, supporting more sustainable textile practices.
How to address limitations of cell-free systems?
Activation control: The system is designed to activate only upon hydration (e.g., sweat or water exposure), preventing premature reactions.
Stability: Freeze-drying (lyophilization) significantly increases shelf life and allows storage at room temperature.
One-time use limitation: The textile can be designed as either disposable (e.g., medical patches) or modular, with replaceable biosensing patches embedded in garments.
Protection: Microencapsulation within polymer or hydrogel matrices protects the biological components from degradation and environmental stress.
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/ .
Background (≤100 words)
During long-duration space missions, astronauts depend on stored, freeze-dried food that undergoes nutrient degradation over time. Vitamin B12 (cobalamin), an essential cofactor for DNA synthesis and neurological function, is particularly susceptible to depletion, increasing the risk of anemia and cognitive impairment. Due to limited resupply capabilities in deep space missions, in situ production of essential micronutrients is highly desirable. Cell-free protein synthesis systems such as BioBits® offer a stable, lyophilized platform for enzymatic production under resource-constrained conditions, enabling decentralized and on-demand biosynthesis of critical compounds in space environments.
Molecular / Genetic Target (≤30 words)
Gene cobA, encoding uroporphyrinogen III methyltransferase, a key enzyme in early cobalamin biosynthesis.
Relevance to the Challenge (≤100 words)
Cobalamin biosynthesis is a complex, multi-step pathway requiring specialized enzymes. The cobA gene encodes a methyltransferase that catalyzes an early and essential step in corrin ring formation. By expressing cobA in a cell-free system, it is possible to reconstruct part of the cobalamin biosynthetic pathway from precursor molecules. Monitoring the activity of this enzyme provides insight into the feasibility of modular vitamin production in space. This approach links molecular enzymatic function to astronaut nutrition, offering a strategy for supplementing degraded food supplies with freshly synthesized cofactors.
Hypothesis / Research Goal (≤150 words)
We hypothesize that a lyophilized BioBits® cell-free system expressing cobA can catalyze the methylation of uroporphyrinogen III, generating intermediates of the cobalamin biosynthesis pathway under space-relevant conditions. This enzymatic activity can be indirectly quantified through fluorescence-based assays targeting pathway intermediates or coupled reporter systems. The rationale is that cell-free systems retain sufficient enzymatic functionality to support partial metabolic pathway reconstruction without the need for living cells. By combining miniPCR® amplification of the cobA gene with cell-free expression, this system enables rapid enzyme production in situ. The P51 Molecular Fluorescence Viewer will be used to detect reaction outputs. The goal is to establish a proof-of-concept for modular, cell-free biosynthesis of essential micronutrients during long-duration space missions.
Experimental Plan (≤100 words)
BioBits® reactions will be prepared with a plasmid encoding cobA and supplied with uroporphyrinogen III as substrate. Controls include reactions without DNA, without substrate, and heat-inactivated systems. Reactions will be incubated at 37°C for 4 hours. Enzymatic activity will be assessed באמצעות fluorescence-based detection of methylated intermediates or coupled reporter assays, visualized using the P51 viewer. Relative fluorescence intensity will be quantified to compare activity across conditions and confirm functional enzyme expression.
Homework Part B: Individual Final Project
We’d like students to start exploring their final project in depth this week! Of your three Aims, for this week you should have at least Aim 1 decided and written down.
Prepare your first DNA order and put it in the “Twist (MIT)” or “Twist (Nodes)” tab of the 2026 HTGAA Ordering: DNA, Reagents, Consumables spreadsheet, as appropriate.