Week 2 dna read write and edit
- Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
The error rate is the 1:10^6, for the other hand the size of the human genome, which is approximately 3 × 10⁹ base pairs. If DNA polymerase functioned only at its intrinsic error rate, replication would introduce an extremely high number like would result in about 300 errors every time the genome is copied a lot mutation and problems, making genomic stability impossible.
Biology addresses this problem through multiple layered error correction mechanisms. These include the proofreading activity of DNA polymerases, post-replication mismatch repair systems, and additional DNA repair pathways that detect and correct replication errors. MutS Repair System, Correcting Gene Synthesis and Reduction GFP Gene Synthesis.
In one of these repairs, the polymerase at the active site only accepts correctly paired bases (A-T, G-C), since the pairs do not match dimensionally. When errors occur, the correction mechanism built into the polymerase uses exonuclease activity to reverse, remove the incorrect nucleotide, and resynthesize it correctly.
In addition to quality control, there is the mismatch repair system, in which specialized protein complexes (MutS/MutL in bacteria, MSH/MLH in humans) scan the newly replicated DNA to detect and correct any remaining errors. This system identifies the newly synthesized strand, removes the segments containing errors, and replaces them with the correct sequence.
- How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Human protein, composed of about 450 amino acids, has several DNA sequences that, in theory, could encode it. Because the genetic code is composed of combinations of amino acids, there could be approximately 10^200 or more different nucleotide sequences that encode exactly the same amino acid sequence. However, in practice, the vast majority of these “synonymous” sequences do not work to produce a functional protein.(Yi Liu, 2021)
Therefore, one could say that the reason why not all codons are useful in a sequence is based on biological constraints such as codon usage bias, tRNA availability, mRNA secondary structure formation (especially in GC-rich sequences), the presence of unwanted regulatory signals, and the effects of translation speed on protein folding. Therefore, only a fraction of the possible codes encode DNA.
- What’s the most commonly used method for oligo synthesis currently?
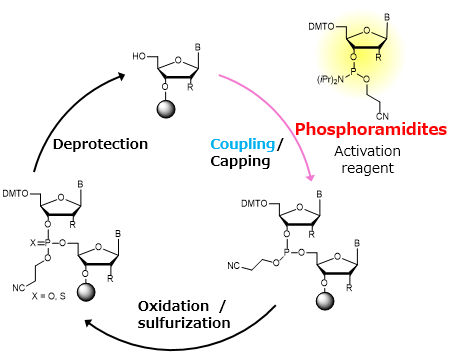
Phosphoramidite method by Caruthers
- Why is it difficult to make oligos longer than 200nt via direct synthesis?
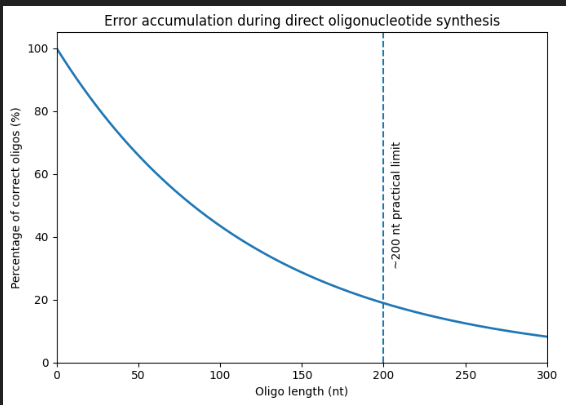
Figure generated using AI and adapted from concepts Oligonucleotide Synthesis.
Currently, oligonucleotide synthesis uses a solid-phase cycle with phosphoramidite, in which one nucleotide is added at a time with high, but not perfect, coupling efficiency (typically between 97% and 99% per step). Each additional base adds a small chance of failure, either a deletion or an incorrect base, so the fraction of complete, error-free molecules decreases exponentially with length. When about 200 bases are reached, most of the chains in the pool are truncated or contain at least one error, resulting in very low useful yield.
- Why can’t you make a 2000bp gene via direct oligo synthesis?
Due to the same reason mentioned previously, trying to synthesize a 2,000-base sequence in a single pass would yield almost no correct full-length molecules: multiplying a pass efficiency of less than 99% over 2,000 cycles reduces the chances of obtaining a perfect strand to almost zero. Instead, genes of this size are created by assembling many shorter, high-quality oligonucleotides or gene fragments, as direct chemical synthesis of thousands of bases is too imprecise to be practical.
- [Given slides #2 & 4 (AA:NA and NA:NA codes)] What code would you suggest for AA:AA interactions? In the case of AA:AA interactions, I will suggest thinking in terms of a “protein protein interaction code” based on the complementarity of side chains. In essence, amino acid pairs recognize each other through combinations of physicochemical complementarity between amino acid side chains, where properties such as electrical charge, hydrophobicity, size, shape, and the ability to form hydrogen bonds or hydrophobic interactions determine how two amino acids interact with each other. Unlike the simple triplet genetic code, this AA:AA code is distributed across different surfaces.
Part 2
Part 1: Benchling & In-silico Gel Art
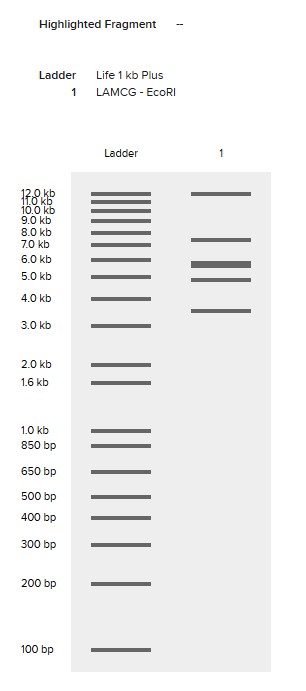
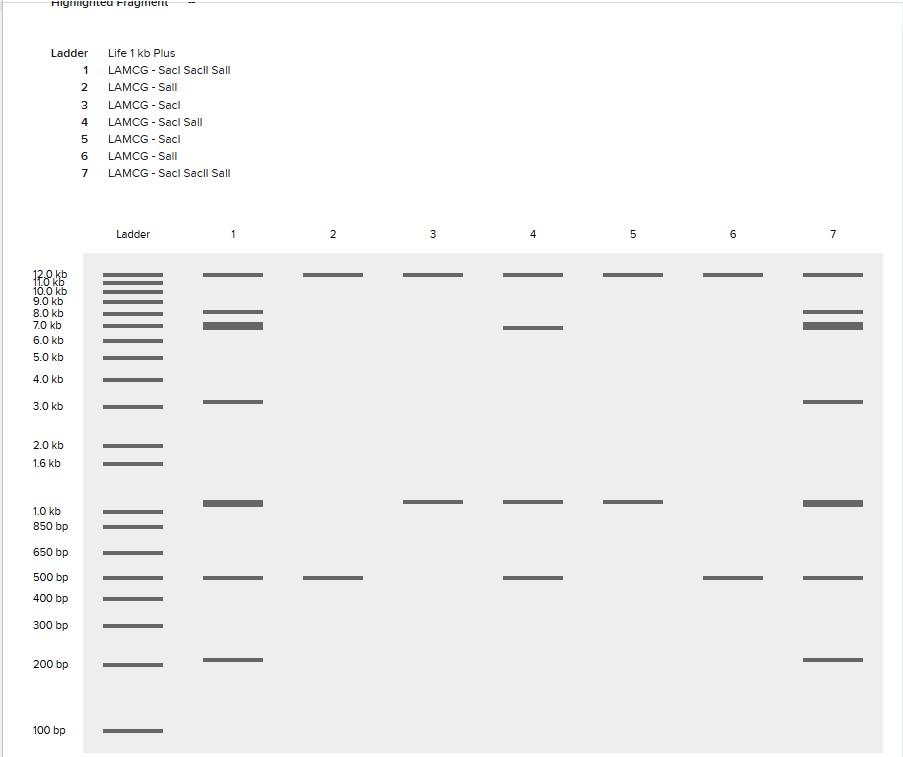
See the Gel Art: Restriction Digests and Gel Electrophoresis protocol for details.
Overview:
Simulate Restriction Enzyme Digestion with the following Enzymes:
EcoRI
HindIII
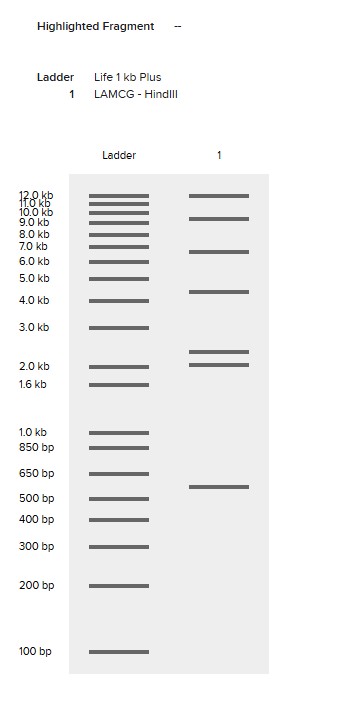
BamHI

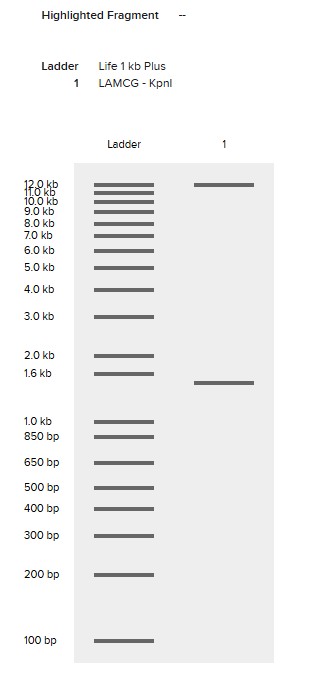
KpnI
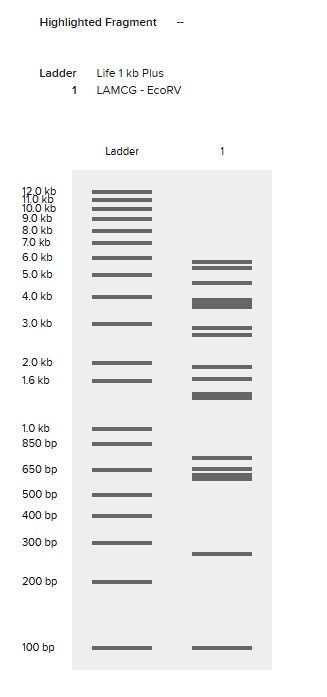
EcoRV
SacI

SalI

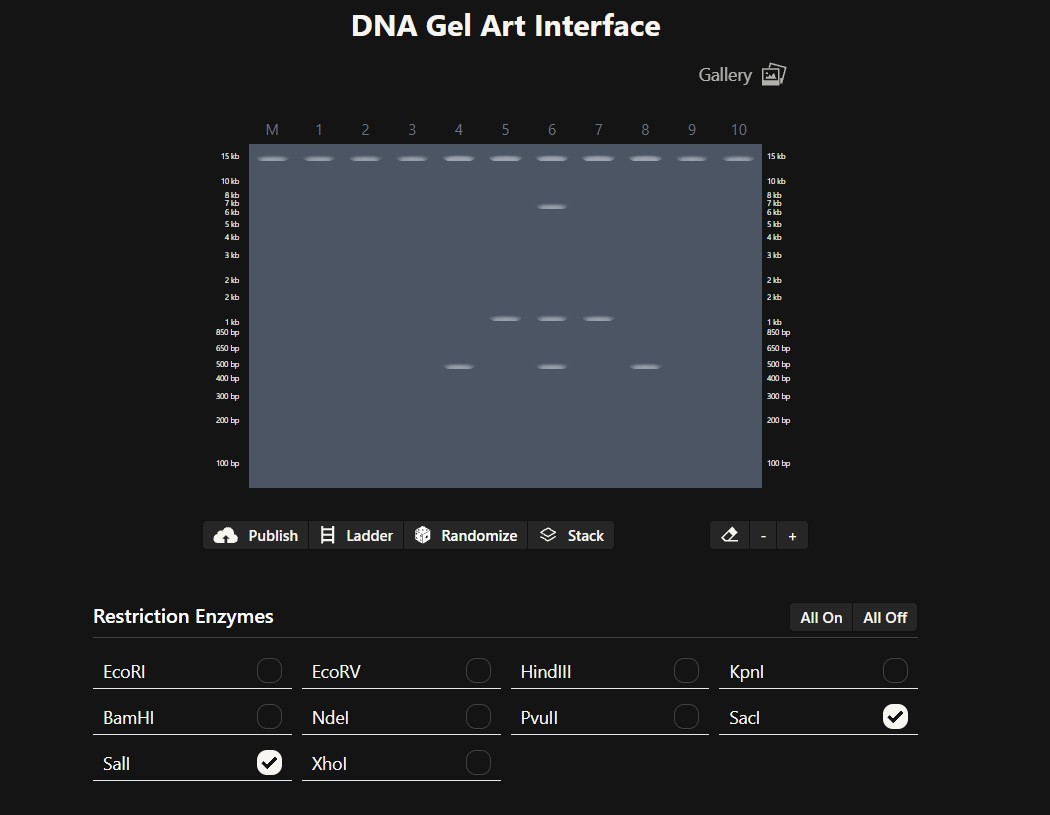
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
I was inspired by a random pattern from the website to draw something like a tower, and then I perfected it in Bletchin.
Part 3: DNA Design Challenge
3.1. Choose your protein.
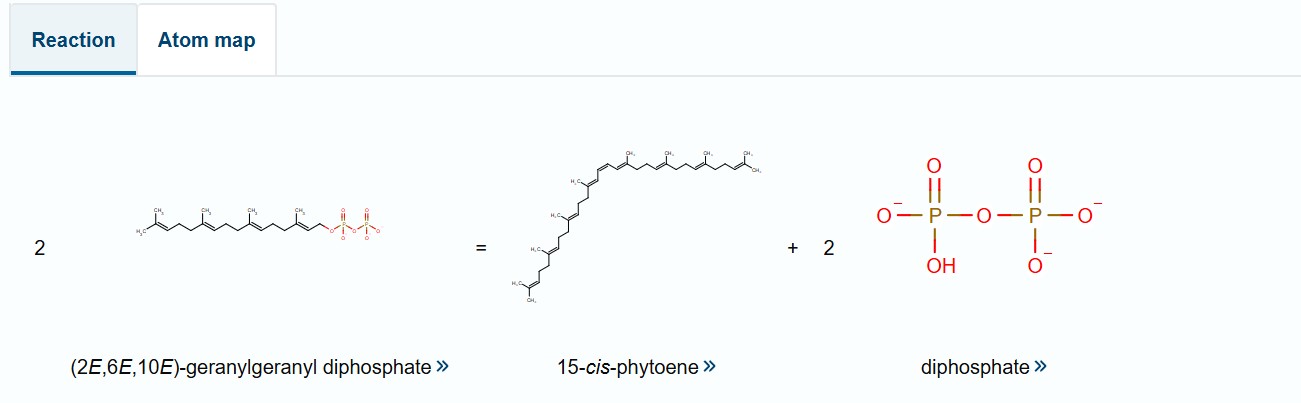
I selected the bifunctional lycopene cyclase/phytoene synthase for analysis because it directly processes and produces carotenoid pigments essential for coloration. This enzyme catalyzes two crucial steps in the carotenoid biosynthesis pathway: the formation of phytoene, the first committed precursor, and the cyclization of lycopene into β-carotene, a key orange pigment.
By processing intermediate molecules into pigmented compounds, this enzyme plays a central role in pigment formation. Its bifunctional nature makes it especially valuable, as a single gene can drive multiple steps in the pathway, simplifying the metabolic engineering strategy.
Bifunctional lycopene cyclase/phytoene synthase

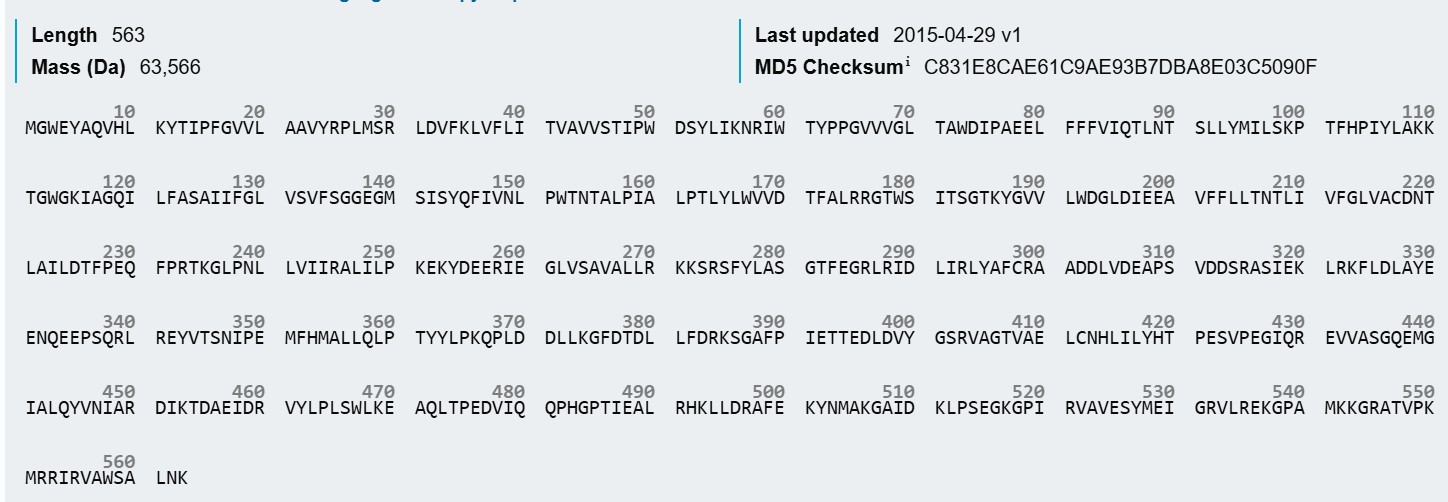 MGWEYAQVHLKYTIPFGVVLAAVYRPLMSRLDVFKLVFLITVAVVSTIPWDSYLIKNRIWTYPPGVVVGLTAWDIPAEELFFFVIQTLNTSLLYMILSKPTFHPIYLAKKTGWGKIAGQILFASAIIFGLVSVFSGGEGMSISYQFIVNLPWTNTALPIALPTLYLWVVDTFALRRGTWSITSGTKYGVVLWDGLDIEEAVFFLLTNTLIVFGLVACDNTLAILDTFPEQFPRTKGLPNLLVIIRALILPKEKYDEERIEGLVSAVALLRKKSRSFYLASGTFEGRLRIDLIRLYAFCRAADDLVDEAPSVDDSRASIEKLRKFLDLAYEENQEEPSQRLREYVTSNIPEMFHMALLQLPTYYLPKQPLDDLLKGFDTDLLFDRKSGAFPIETTEDLDVYGSRVAGTVAELCNHLILYHTPESVPEGIQREVVASGQEMGIALQYVNIARDIKTDAEIDRVYLPLSWLKEAQLTPEDVIQQPHGPTIEALRHKLLDRAFEKYNMAKGAIDKLPSEGKGPIRVAVESYMEIGRVLREKGPAMKKGRATVPKMRRIRVAWSALNK
MGWEYAQVHLKYTIPFGVVLAAVYRPLMSRLDVFKLVFLITVAVVSTIPWDSYLIKNRIWTYPPGVVVGLTAWDIPAEELFFFVIQTLNTSLLYMILSKPTFHPIYLAKKTGWGKIAGQILFASAIIFGLVSVFSGGEGMSISYQFIVNLPWTNTALPIALPTLYLWVVDTFALRRGTWSITSGTKYGVVLWDGLDIEEAVFFLLTNTLIVFGLVACDNTLAILDTFPEQFPRTKGLPNLLVIIRALILPKEKYDEERIEGLVSAVALLRKKSRSFYLASGTFEGRLRIDLIRLYAFCRAADDLVDEAPSVDDSRASIEKLRKFLDLAYEENQEEPSQRLREYVTSNIPEMFHMALLQLPTYYLPKQPLDDLLKGFDTDLLFDRKSGAFPIETTEDLDVYGSRVAGTVAELCNHLILYHTPESVPEGIQREVVASGQEMGIALQYVNIARDIKTDAEIDRVYLPLSWLKEAQLTPEDVIQQPHGPTIEALRHKLLDRAFEKYNMAKGAIDKLPSEGKGPIRVAVESYMEIGRVLREKGPAMKKGRATVPKMRRIRVAWSALNK
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
Bifunctional lycopene
atgggctgggaatatgcgcaggtgcatctgaaatataccattccgtttggcgtggtgctg gcggcggtgtatcgcccgctgatgagccgcctggatgtgtttaaactggtgtttctgatt accgtggcggtggtgagcaccattccgtgggatagctatctgattaaaaaccgcatttgg acctatccgccgggcgtggtggtgggcctgaccgcgtgggatattccggcggaagaactg tttttttttgtgattcagaccctgaacaccagcctgctgtatatgattctgagcaaaccg acctttcatccgatttatctggcgaaaaaaaccggctggggcaaaattgcgggccagatt ctgtttgcgagcgcgattatttttggcctggtgagcgtgtttagcggcggcgaaggcatg agcattagctatcagtttattgtgaacctgccgtggaccaacaccgcgctgccgattgcg ctgccgaccctgtatctgtgggtggtggatacctttgcgctgcgccgcggcacctggagc attaccagcggcaccaaatatggcgtggtgctgtgggatggcctggatattgaagaagcg gtgttttttctgctgaccaacaccctgattgtgtttggcctggtggcgtgcgataacacc ctggcgattctggatacctttccggaacagtttccgcgcaccaaaggcctgccgaacctg ctggtgattattcgcgcgctgattctgccgaaagaaaaatatgatgaagaacgcattgaa ggcctggtgagcgcggtggcgctgctgcgcaaaaaaagccgcagcttttatctggcgagc ggcacctttgaaggccgcctgcgcattgatctgattcgcctgtatgcgttttgccgcgcg gcggatgatctggtggatgaagcgccgagcgtggatgatagccgcgcgagcattgaaaaa ctgcgcaaatttctggatctggcgtatgaagaaaaccaggaagaaccgagccagcgcctg cgcgaatatgtgaccagcaacattccggaaatgtttcatatggcgctgctgcagctgccg acctattatctgccgaaacagccgctggatgatctgctgaaaggctttgataccgatctg ctgtttgatcgcaaaagcggcgcgtttccgattgaaaccaccgaagatctggatgtgtat ggcagccgcgtggcgggcaccgtggcggaactgtgcaaccatctgattctgtatcatacc ccggaaagcgtgccggaaggcattcagcgcgaagtggtggcgagcggccaggaaatgggc attgcgctgcagtatgtgaacattgcgcgcgatattaaaaccgatgcggaaattgatcgc gtgtatctgccgctgagctggctgaaagaagcgcagctgaccccggaagatgtgattcag cagccgcatggcccgaccattgaagcgctgcgccataaactgctggatcgcgcgtttgaa aaatataacatggcgaaaggcgcgattgataaactgccgagcgaaggcaaaggcccgatt cgcgtggcggtggaaagctatatggaaattggccgcgtgctgcgcgaaaaaggcccggcg atgaaaaaaggccgcgcgaccgtgccgaaaatgcgccgcattcgcgtggcgtggagcgcg ctgaacaaa
I employed the online reverse translation tool fromBioinformatics.org to convert the amino acid sequence of the target protein into a corresponding DNA coding sequence. This tool ensures that the translated nucleotide sequence matches the original peptide sequence while maintaining proper reading frame and codon order. Using this reverse translation resource allows me to generate a DNA sequence that can be used for downstream applications such as gene synthesis, cloning, and expression analysis in my chosen host system.
3.3. Codon optimization.
Tool: Codon Optimization IDT https://www.idtdna.com/CodonOpt
ATG GGC TGG GAG TAC GCA CAA GTT CAT CTG AAA TAT ACC ATC CCG TTC GGC GTT GTT CTC GCT GCG GTC TAT CGC CCC CTG ATG TCG CGT TTG GAT GTC TTT AAA TTG GTG TTT CTC ATC ACA GTG GCC GTA GTA AGT ACT ATT CCG TGG GAC TCT TAC TTA ATA AAA AAC CGG ATA TGG ACC TAT CCT CCA GGT GTC GTT GTC GGG CTT ACC GCA TGG GAT ATT CCT GCC GAA GAA CTG TTT TTC TTT GTC ATA CAG ACC CTG AAC ACA TCA CTG CTG TAT ATG ATC TTA AGT AAA CCC ACC TTT CAT CCG ATC TAC TTG GCG AAG AAA ACT GGC TGG GGC AAA ATT GCG GGC CAG ATC CTG TTT GCC AGC GCC ATC ATC TTC GGG CTG GTA TCT GTG TTC AGC GGC GGG GAA GGC ATG TCT ATA TCA TAT CAG TTT ATA GTA AAC CTG CCT TGG ACA AAC ACA GCA CTG CCG ATC GCA TTA CCA ACT CTG TAT CTG TGG GTG GTT GAC ACC TTT GCA CTC CGC CGT GGG ACA TGG AGC ATC ACT AGT GGA ACT AAG TAT GGG GTC GTG CTG TGG GAC GGC CTG GAT ATT GAA GAA GCT GTG TTT TTT TTA CTG ACT AAC ACT CTT ATT GTC TTC GGC TTG GTA GCG TGT GAT AAT ACC TTA GCT ATC CTG GAC ACC TTC CCC GAG CAG TTC CCC CGG ACA AAA GGT CTC CCA AAC CTG CTC GTA ATC ATT CGT GCG CTG ATT TTG CCT AAA GAG AAG TAT GAT GAA GAA CGC ATC GAG GGA TTA GTT TCG GCG GTC GCA CTC CTG CGT AAA AAG TCG CGC TCC TTT TAC TTG GCG AGC GGG ACG TTT GAG GGC CGT CTT CGT ATT GAC CTG ATT CGT CTG TAT GCG TTT TGT CGG GCA GCG GAC GAT CTT GTC GAT GAA GCG CCT AGT GTG GAT GAC TCC CGC GCT AGC ATT GAA AAG TTA CGG AAA TTC CTT GAT CTG GCG TAC GAA GAG AAC CAG GAA GAA CCA AGC CAG CGC TTA CGC GAG TAT GTG ACA AGC AAC ATA CCT GAA ATG TTT CAC ATG GCA CTG CTG CAA CTT CCG ACA TAC TAC CTG CCT AAA CAG CCA CTG GAC GAT CTC CTG AAA GGC TTT GAT ACG GAC CTG CTG TTT GAT CGT AAG AGT GGC GCC TTT CCG ATC GAA ACC ACC GAG GAT CTT GAT GTC TAT GGT AGC CGT GTT GCA GGT ACT GTC GCT GAA CTC TGC AAC CAT CTT ATT CTG TAC CAT ACC CCG GAA TCC GTC CCC GAA GGC ATT CAG CGT GAA GTC GTC GCA TCG GGA CAG GAG ATG GGT ATC GCG CTG CAG TAT GTT AAT ATC GCA CGC GAC ATT AAG ACA GAT GCG GAG ATC GAT CGG GTC TAT CTC CCA TTA TCA TGG CTG AAG GAA GCC CAG CTG ACG CCT GAG GAT GTG ATC CAA CAA CCT CAT GGG CCT ACG ATT GAG GCA TTA CGC CAT AAA TTG CTT GAT CGT GCA TTC GAG AAA TAC AAC ATG GCC AAG GGG GCC ATT GAT AAG TTA CCT TCC GAA GGA AAA GGC CCC ATC CGC GTC GCG GTG GAG TCC TAT ATG GAA ATT GGG CGC GTA TTA CGC GAG AAA GGT CCC GCG ATG AAA AAA GGT CGT GCC ACA GTT CCG AAA ATG CGC CGC ATT CGC GTG GCC TGG TCG GCC TTA AAT AAA
The codon sequence was optimized for Fusarium oxysporum because this organism is responsible for producing the carotenoid pigment. Since the bifunctional lycopene cyclase/phytoene synthase processes precursor molecules into β-carotene, efficient expression of this enzyme is essential for pigment formation. Optimizing codon usage improves translation efficiency, increases enzyme production, and enhances pigment accumulation, resulting in stronger and more consistent coloration.
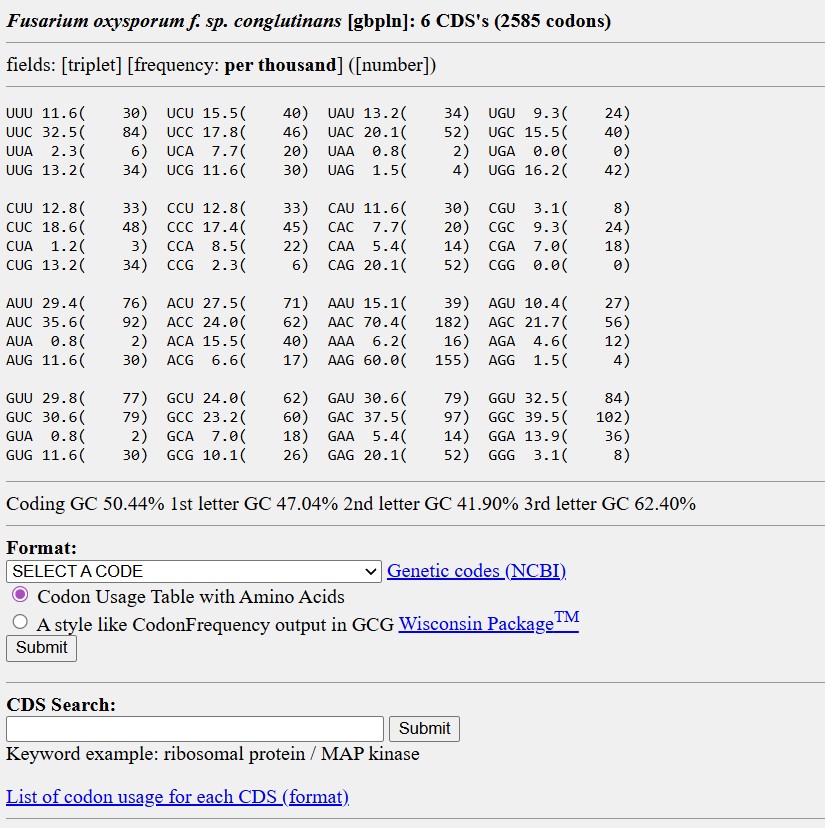
3.4. You have a sequence! Now what?
Cell-dependent methods (in Fusarium oxysporum): involve introducing the optimized gene into living fungal cells using an appropriate expression vector. The vector must contain regulatory elements such as a promoter, terminator, and selection marker. After transformation, the fungal cellular machinery carries out transcription of the DNA into mRNA and translation of the mRNA into protein. This method allows the enzyme to be produced within the natural metabolic environment of the fungus, enabling functional activity and potential pigment production.
Part 4: Prepare a Twist DNA Synthesis Order I tried to use Twist, but I couldn’t because I kept getting the following error.
Part 5: DNA Read/Write/Edit
5.1 DNA Read What DNA would you want to sequence (e.g., read) and why? I would sequence the DNA of fungi that form mycelial networks used in biomaterial production in order to understand the genetic basis of their structural organization and bio-connection properties. By identifying genes involved in hyphal growth, branching, and cell wall composition, it would be possible to understand better how these organisms naturally build strong, interconnected living structures. This information could support the development of engineered mycelium-based biomaterials and explore the potential of living fungal systems as platforms for biological information storage or transmission, combining structural biology with DNA-based data encoding.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why? I would use third-generation single-molecule sequencing, specifically Oxford Nanopore Technologies (ONT) or PacBio, because they allow long-read sequencing without amplification, making them ideal for studying large fungal DNA regions
Is your method first-, second- or third-generation or other? How so? Third-generation What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
The input is high-quality genomic DNA extracted from the fungal sample. The preparation involves optionally fragmenting the DNA to suitable sizes for the sequencer, ligating adapters to the ends of the DNA fragments so the sequencing machine can recognize them, and performing any necessary purification steps. Unlike some other methods, PCR amplification is not required for Oxford Nanopore or PacBio long-read sequencing. The essential steps are DNA extraction, optional fragmentation, adapter ligation, and loading the DNA onto the sequencing device.
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
For Oxford Nanopore, the DNA passes through a nanopore embedded in a membrane. As each nucleotide moves through the pore, it causes a characteristic change in the ionic current, which is detected by sensors. These changes are analyzed in real time by the software to determine the sequence of bases (base calling). For PacBio, DNA polymerase incorporates fluorescently labeled nucleotides in zero-mode waveguides, and the emitted fluorescence is recorded to decode the bases. Both systems allow long continuous reads of single molecules without amplification.
What is the output of your chosen sequencing technology?
The output is a collection of long-read sequences in FASTQ format, which includes both the nucleotide sequences and associated quality scores. These sequences can then be used for downstream analysis such as gene annotation, codon optimization, or studying structural features in fungal DNA.
5.2 DNA Write
(ii) What technology or technologies would you use to perform this DNA synthesis, and why? I would like to synthesize the gene coding for spider silk protein (spidroin) because it is a biomaterial with exceptional mechanical properties. The DNA sequence would include the full coding region of spidroin, codon-optimized for the host organism to ensure efficient expression, allowing for the production of fibers or hydrogels for biomaterials research.
What are the essential steps of your chosen sequencing methods?
The essential steps of the Sanger (ABI) sequencing method begin with preparing the DNA to be sequenced, usually by PCR amplification of the target fragment. Then, a DNA synthesis reaction is performed in which both normal nucleotides and fluorescently labeled chain-terminating nucleotides are incorporated, stopping elongation at specific positions. The resulting DNA fragments of different lengths are separated by capillary electrophoresis, and a detector reads the fluorescence to identify each base and reconstruct the full sequence of the fragment.
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability? Its speed and scalability are limited, as each run processes only short fragments. Consequently, it is unsuitable for large-scale projects or whole-genome sequencing and is less efficient for high-throughput DNA analysis.
5.3 DNA Edit
(i) What DNA would you want to edit and why?
I would like to edit the DNA of a pigment-producing fungus, such as Fusarium oxysporum, to introduce a gene encoding laccase, an enzyme capable of degrading pollutants in water. By adding this enzyme, the fungus could not only produce sustainable biotints but also perform bioremediation, breaking down phenolic compounds, industrial dyes, and other contaminants in aquatic environments. This kind of genetic edit combines the production of valuable biomaterials with environmental cleanup, making the organism both functional and sustainable.
How does your technology of choice edit DNA? What are the essential steps?
The technology chose is recombinases, which are enzymes that recognize specific DNA sequences and mediate precise recombination events. This allows the targeted integration of a desired gene into the host genome. The essential steps include identifying a safe insertion site in the genome, designing the recombinase recognition sequences flanking the gene of interest, and delivering the recombinase along with the DNA construct into the host cells. Once inside, the recombinase facilitates site-specific integration of the gene.
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
Preparation involves designing the DNA construct containing the laccase gene flanked by recombinase recognition sites. The inputs include the DNA template with the gene, the recombinase enzyme or expression plasmid, and competent Fusarium oxysporum cells ready for transformation. Additional reagents may include selection markers and buffers to facilitate uptake and expression of the recombinase and target DNA.
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
While recombinases offer high precision, their efficiency can be limited by the accessibility of the target site and the compatibility of the recognition sequences with the host genome. Large-scale integrations or multiple edits may require careful design to avoid interference between sites. Additionally, the method is dependent on the ability to deliver the recombinase and DNA into fungal cells, which can be technically challenging.
Research resurses: Findings have been summarized with the help of ChatGPT 5
- Dong, Y., Yao, C., Zhu, Y., Yang, L., Luo, D., & Yang, D. (2020). DNA Functional Materials Assembled from Branched DNA: Design, Synthesis, and Applications.. Chemical reviews. https://doi.org/10.1021/acs.chemrev.0c00294.
- Miserez, A., Yu, J., & Mohammadi, P. (2023). Protein-Based Biological Materials: Molecular Design and Artificial Production. Chemical Reviews, 123, 2049 - 2111. https://doi.org/10.1021/acs.chemrev.2c00621.
- Liu, Y., Yang, Q., & Zhao, F. (2021). Synonymous but not silent: The codon usage code for gene expression and protein folding. Annual Review of Biochemistry, 90, 375-401. https://doi.org/10.1146/annurev-biochem-071320-112701