Week 4 hw protein design part
Part A. Conceptual Questions
- How many amino acid molecules do you ingest when eating 500 grams of meat? (Average amino acid ≈ 100 Daltons)
Approximately 20% of meat is protein. Therefore, 500 g of meat contains about 100 g of protein. If we assume an average amino acid molecular weight of 100 g/mol (≈100 Daltons), then:
100 g protein ÷ 100 g/mol ≈ 1 mol of amino acids.
One mole corresponds to 6 × 10²³ molecules (Avogadro’s number). Therefore, eating 500 g of meat provides on the order of 6 × 10²³ amino acid molecules.
- Why do humans eat beef but do not become a cow, or eat fish but not become fish?
Because we do not absorb intact proteins. During digestion, proteins are broken down into peptides and then into free amino acids. The original protein sequence and structure are completely destroyed. Our cells then use those amino acids as building blocks to synthesize human proteins according to the instructions encoded in our DNA. Biological identity comes from genetic information, not from dietary molecules.
- Why are there only 20 natural amino acids?
The 20 standard amino acids represent the set that became fixed in the genetic code during evolution. Together, they provide sufficient chemical diversity to build functional proteins: hydrophobic, polar, charged, aromatic, small, rigid, and flexible residues are all included. This set appears to be an optimal balance between chemical versatility and biological efficiency. Although additional amino acids such as selenocysteine and pyrrolysine exist, the core genetic code relies on 20.
- Can you make non-natural amino acids? Design some new amino acids.
Yes, hundreds of non-natural amino acids have been synthesized in organic chemistry and synthetic biology. They can be designed by modifying the side chain.
Examples of designed amino acids:
A fluorinated phenylalanine (to alter hydrophobicity and electronic properties).
An amino acid with an azide or alkyne group for bioorthogonal “click” chemistry.
A metal-binding amino acid containing a bipyridine group.
A photoactivatable amino acid with a diazirine group for crosslinking experiments.
An amino acid with a bulky polyaromatic side chain to enhance π–π stacking.
Some engineered organisms have even expanded their genetic code to incorporate such amino acids into proteins.
- Where did amino acids come from before enzymes and life existed?
Amino acids likely formed through prebiotic chemistry. The Miller–Urey experiment demonstrated that amino acids can form spontaneously under simulated early Earth conditions (reducing atmosphere + electrical discharge). Amino acids have also been detected in meteorites such as the Murchison meteorite, suggesting that they can form in space. Therefore, amino acids likely existed before enzymes and before life emerged.
- If you make an α-helix using D-amino acids, what handedness would you expect?
Natural proteins use L-amino acids and form right-handed α-helices. If you construct an α-helix entirely from D-amino acids, the helix would be left-handed. Inverting the chirality of the monomers inverts the handedness of the secondary structure.
- Can you discover additional helices in proteins?
Yes. Besides the α-helix, proteins can contain 3₁₀ helices and π-helices. These are less common but structurally distinct. Other biological polymers, such as DNA, also form helical structures (e.g., the right-handed B-DNA helix).
- Why are most molecular helices right-handed?
Most biological helices are right-handed because proteins are built from L-amino acids. The stereochemistry of the alpha carbon restricts backbone dihedral angles (φ and ψ), making right-handed helices energetically favorable.
- Why do β-sheets tend to aggregate?
β-sheets can extend through backbone hydrogen bonding between neighboring strands. Their structure allows repeating hydrogen bond networks and tight packing of side chains. This makes it easy for partially unfolded proteins to associate and form extended β-sheet assemblies.
- What is the driving force for β-sheet aggregation?
The primary driving forces are:
Extensive backbone hydrogen bonding.
Hydrophobic interactions between side chains.
Reduction of exposed hydrophobic surface area in aqueous environments.
Thermodynamic stabilization through ordered packing.
Together, these factors favor the formation of highly stable aggregated structures.
- Why do many amyloid diseases form β-sheets?
When proteins misfold, hydrophobic regions that are normally buried become exposed. These regions can reorganize into extended β-sheet structures that stack into fibrils. These fibrils are extremely stable and resistant to degradation. Many diseases, such as Alzheimer’s and Parkinson’s, involve accumulation of such amyloid β-sheet aggregates.
- Can amyloid β-sheets be used as materials?
Yes. Amyloid fibrils are mechanically strong, resistant to degradation, and capable of self-assembly. They are being explored as nanomaterials, scaffolds for tissue engineering, and components in bioelectronics. Some organisms even produce functional amyloids naturally.
- Design a β-sheet motif that forms a well-ordered structure.
A well-ordered β-sheet motif often uses alternating polar and nonpolar residues to create amphipathic strands.
Example sequence: Val–Ser–Val–Ser–Val–Ser–Val–Ser
In this design:
Valine (hydrophobic) promotes tight packing.
Serine (polar) allows hydrogen bonding and solubility.
Alternating pattern promotes regular β-strand geometry.
To enhance order:
Add aromatic residues (e.g., phenylalanine) for π–π stacking.
Place charged residues at termini to control aggregation.
Use repeating patterns to encourage uniform self-assembly.
Such rational design principles are commonly used in peptide-based biomaterials research.
Part B: Protein Analysis and Visualization
Briefly describe the protein you selected and why you selected it. Formate dehydrogenase FDH2: These electrons are transferred to components of the electron transport chain or to specific electron carriers, depending on the organism. In many bacteria, FDH2 plays an important role in energy metabolism by allowing formate to serve as an electron donor during respiration.
Identify the amino acid sequence of your protein.:

MSEQNLYFQGLADYEKLKFGLDSTTVTPLSVTLSSPSLNDLKANAVVTVPEEGLIIAVADGTNVGKSTTTGRILNAVQQFFETLA ETDGLDAVLRRQLGELAEHPWPMVAVNADGALLPVIDAGGTGTITLHAGATPKFTITDAGYGIPFADKPLEHFLHGEDFVEIIPM GMDHQAVAVTDVKGGTMGGLAVAEANGLNKATGTLAKVIDAQGSLGVSIVGATGAGGIGSAEATYLHDMTVPGHFDLSMAEGLG YVADTGGVNATVWVVPGSRADVAVEGATIVMAXXXXVTLVVATGGNGLANASALDGLGKIIQADGATVLEGASLIGMGEDVMPNV VILDGGEGQSVDALMVDAGFSPALIRAVAGVDGIVATGDSAANLYEKLGKDAVIIAENAGFVATPAGGSMGGLIIADGATNAGT HVTVGGPLGLGGIVAVGTNTGERTLTVADTGPAVIGMSIQAGGEASVIVGTNGTSM
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
| Parameter | Value |
|---|---|
| PDB ID | 6T8C |
| Protein Name | Formate dehydrogenase FDH2 |
| Sequence Length | 386 amino acids |
| Most Frequent Amino Acid | Glycine (G) |
| Number of Occurrences (Gly) | 120 |
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.

Does your protein belong to any protein family?
Yes, the protein FDH2 (PDB 6T8C) belongs to the formate dehydrogenase (FDH) family, which is part of the larger oxidoreductase class of enzymes. More specifically, it is a molybdenum-dependent formate dehydrogenase and is classified within the DMSO reductase superfamily. These enzymes are widely distributed in bacteria and archaea and are involved in C1 metabolism, catalyzing the oxidation of formate to carbon dioxide while transferring electrons to cellular electron carriers. They share conserved structural motifs for binding the molybdenum cofactor and often contain iron–sulfur clusters for electron transfer.
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
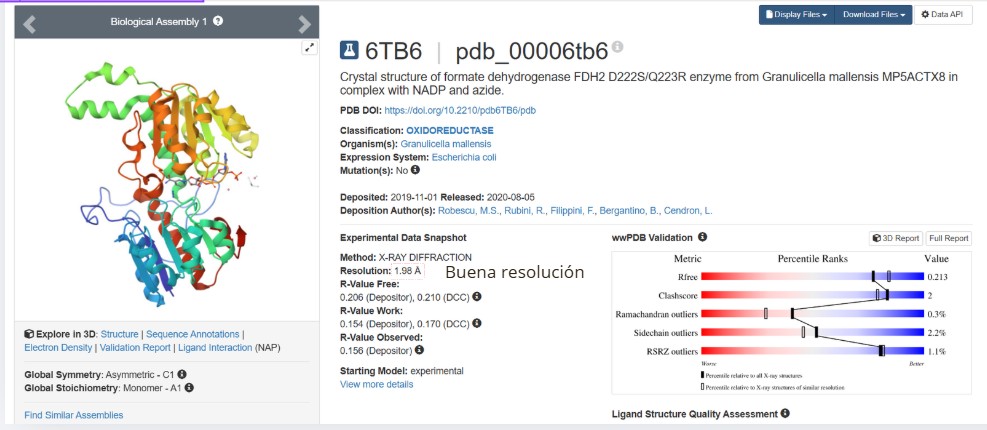
Good quality :1.98
Are there any other molecules in the solved structure apart from protein?
Yes, NAPD
Does your protein belong to any structure classification family?
it is part of the molybdenum-containing oxidoreductase family within the DMSO reductase superfamily. In structural classification databases such as SCOP and CATH, proteins of this type fall into the α/β class, characterized by mixed alpha helices and beta sheets arranged in a conserved fold
Open the structure of your protein in any 3D molecule visualization software:
Color the protein by secondary structure. Does it have more helices or sheets?
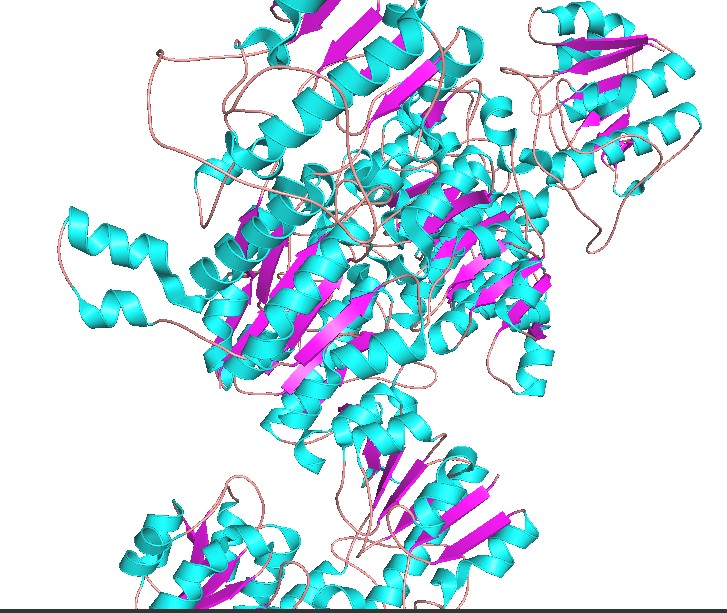
Have more helices
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
select hydrophobic, resn ALA+VAL+LEU+ILE+MET+PHE+TRP+PRO
select polar, resn SER+THR+ASN+GLN+TYR+CYS
select charged, resn ASP+GLU+LYS+ARG+HIS
color orange, hydrophobic
color green, polar
color blue, charged
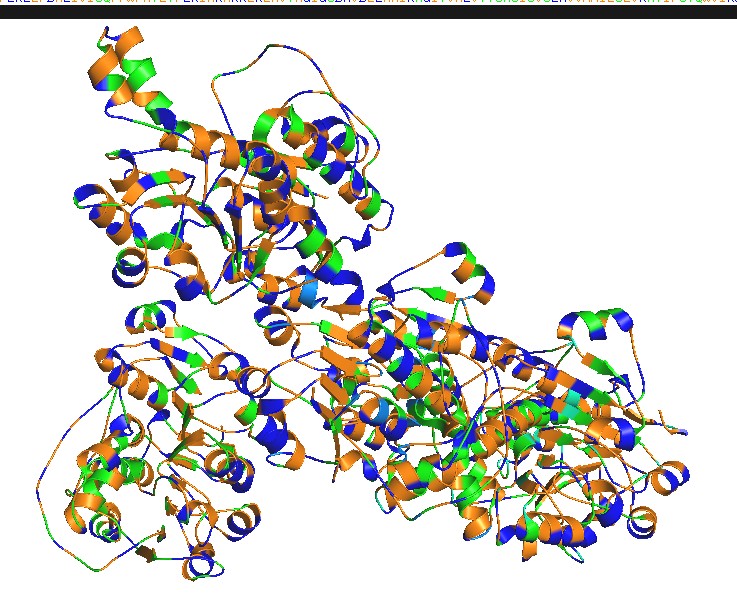
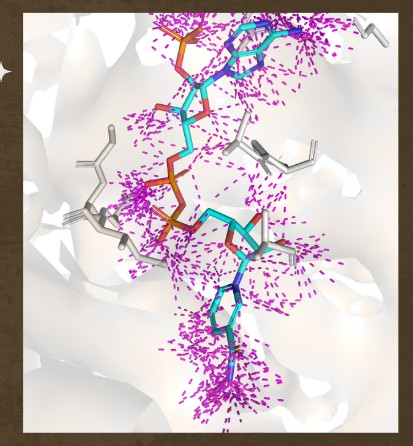
Hydrophobic residues tend to cluster in the core, while polar/charged residues tend to be more surface exposed (typical of soluble proteins) Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
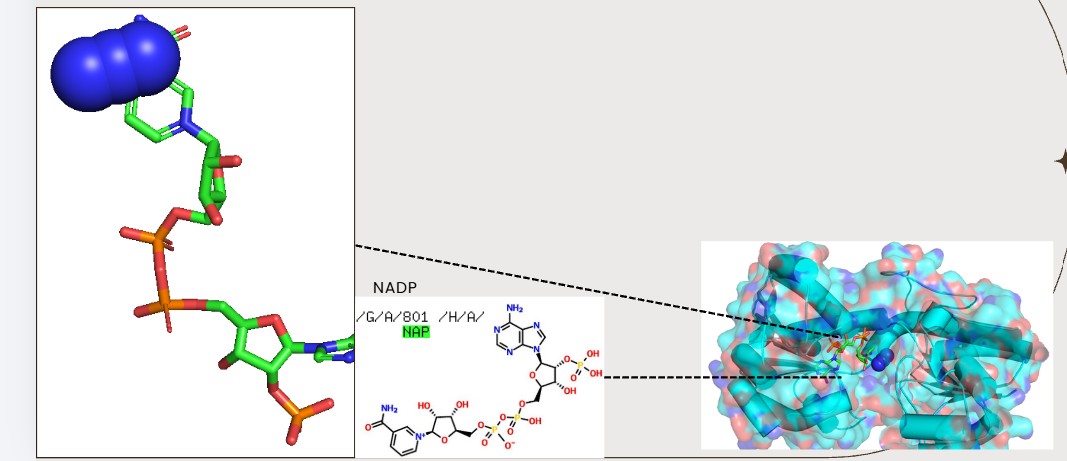
Yes., Grooves and indentations at subunit interfaces and along the surfaceb NAPD interaction regions.
Part C. Using ML-Based Protein Design Tools
Protein Language Modeling
Deep Mutational Scans
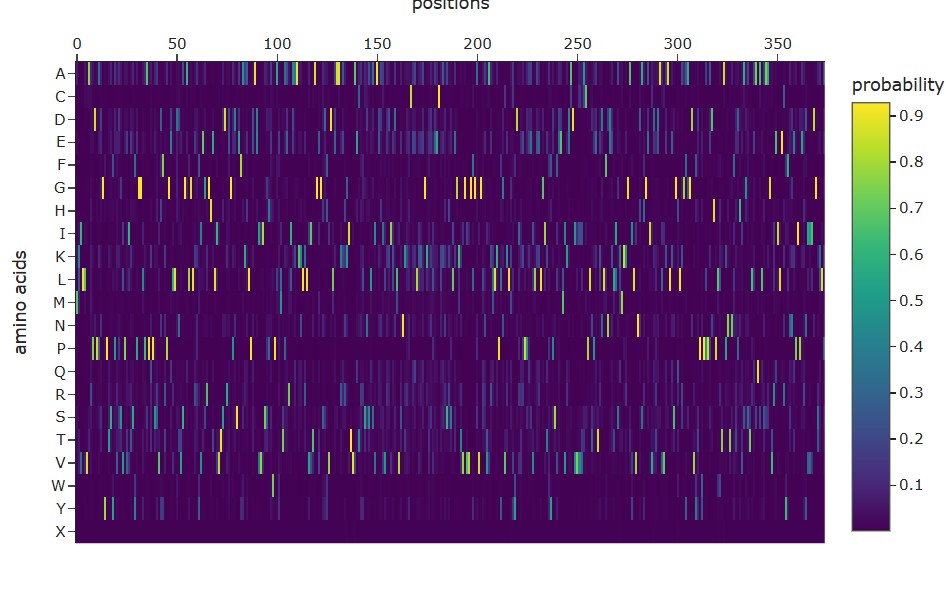
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
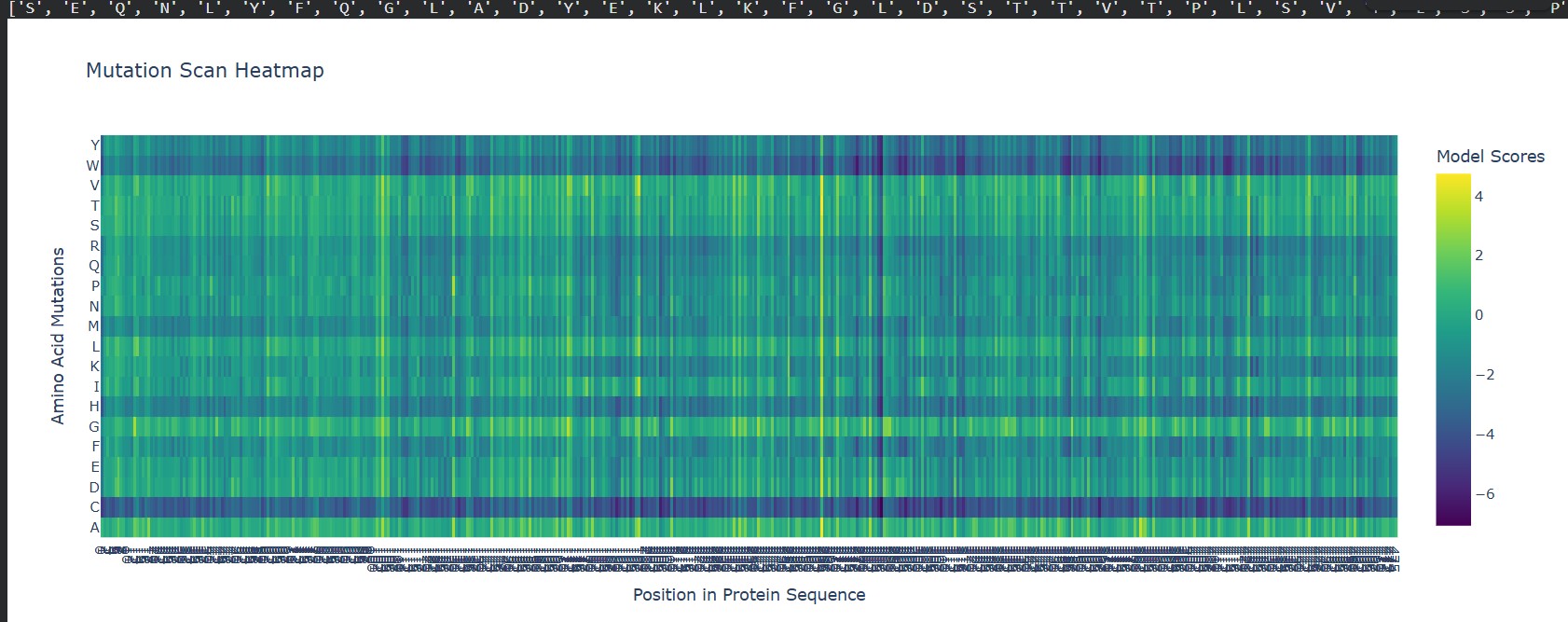
A clear pattern in the mutation heatmap is that substitutions to cysteine (C) consistently show strongly negative scores across most positions in the protein, indicated by the dark purple color. This suggests that introducing cysteine is generally destabilizing. A likely explanation is that cysteine contains a reactive thiol group capable of forming disulfide bonds or interfering with the local chemical environment, which can disrupt proper folding or structural stability. In contrast, some regions appear more tolerant to mutations to glycine (G), shown by greener or yellowish areas, likely because glycine’s small size allows flexibility without causing major steric clashes. This overall pattern indicates that chemical properties of amino acid side chains strongly influence mutation tolerance in this protein.
Latent Space Analysis
Use the provided sequence dataset to embed proteins in reduced dimensionality.
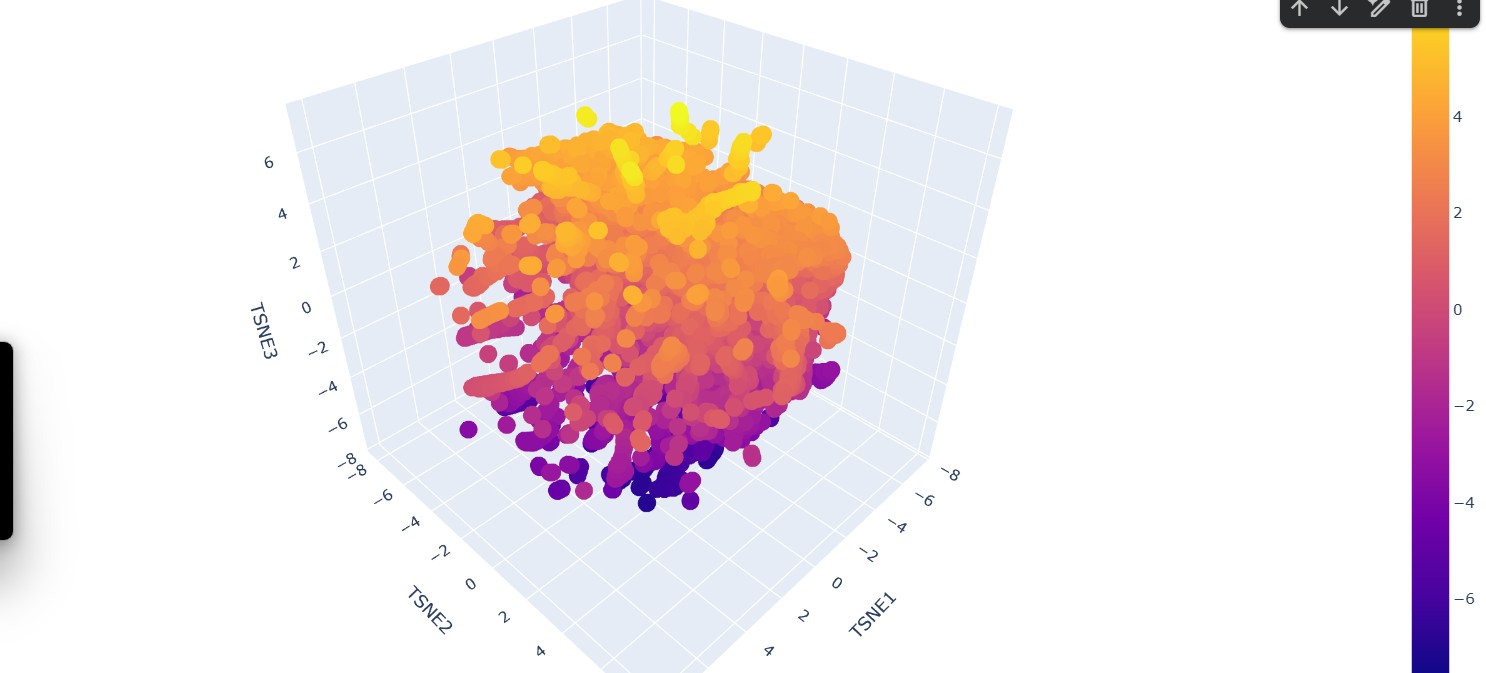
Analyze the different formed neighborhoods: do they approximate similar proteins?
Yes, the formed neighborhoods generally approximate similar proteins. In the 3D embedding plot, clusters of points appear grouped together, suggesting that proteins within the same neighborhood share higher sequence similarity. These clusters likely represent proteins belonging to similar families, functional classes, or structural folds
Place your protein in the resulting map and explain its position and similarity to its neighbors.
The protein of interest (FDH2) appears embedded within a dense cluster rather than isolated at the periphery. This position suggests that it shares significant sequence similarity with nearby proteins in the dataset. Its neighbors likely correspond to other formate dehydrogenases or closely related oxidoreductases within the same structural and functional family. The fact that it is not separated into a distant cluster indicates that it is not an outlier but rather part of a conserved evolutionary group.
Input this sequence into ESMFold and compare the predicted structure to your original. New Sequence:ALTPEEAALLRAAWAPVAADRAANGRAFVLALFAAYPELAELFPEFRGKTLAEIAASPALDAISTAIFDRLDTLVANADDAAAMATLFADLAARHVARGVTAAHFEAIRALFPGFVASVAPPPPGAAAAWDRLFGDVIAALRAAGG
C2. Protein Folding
SeqRecord(seq=Seq(‘GCATGCATGCATGCATGCATGCATGCATGCATGCATGCATGCATGCATGCATGC…CAT’), id=‘Seq2’, name=‘Seq2’, description=‘Seq2 Description of Sequence 2’, dbxrefs=[])
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
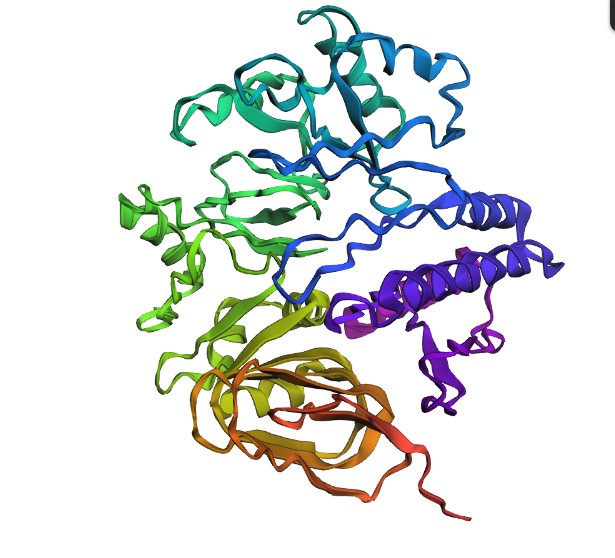
The protein sequence was folded using ESMFold and compared with the original 6T8C structure. Visually, the predicted structure conserved the overall fold of the protein, indicating that the backbone architecture is generally maintained. However, several local conformational differences were observed, especially after introducing mutations and redesigned sequence segments generated by ProteinMP
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
Small mutations appeared to preserve most of the global structure, suggesting partial resilience of the protein fold. In contrast, larger sequence modifications produced more noticeable structural deviations in the 3D conformation. These changes likely affect conformational and functional sites, including residue interactions involved in stability and catalytic activity.
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Generating sequences…
6T8C, score=1.3769, fixed_chains=[], designed_chains=[‘A’], model_name=v_48_020 AKILCVLYDDPITGYPKSYARADVPKIDHYPGGQTAPTPKQIDFTPGELLGSVSGELGLRKYLEGLGHTLVVTSDKEGEDSVFERELPDAEIVISQPFWPAYLTPERIAKAKKLKLAVTAGIGSDHVDLEAAIKNGITVAEVTYSNSISVSEHVVMMILSLVRNYIPSYQWVIKGGWNIADCVERSYDLEAMHVGTVAAGRIGLAVLKRLKPFDVKLHYFDQHRLPESVENELGLTYHPSVEDMVKVCDVVTINAPLHPGTLDLFNDELISKMKRGAYLVNTARGKICNRDAVVRALESGQLAGYAGDVWFPQPAPKDHPWRTMPHHGMTPHISGTSLSAQARYAAGTREILECWFEERPIREEYLIVDGGKLA T=0.1, sample=0, score=0.7038, seq_recovery=0.5561 MKILLVARPDPADGYPTSYPVDSVPVITSYPGGLPAPQPSSVDFRPGELLDNVSGALGLRAYVEGRGHELIVTSDREGPDSEFAKALPEAEIVISSHAWPAEMTAERIAAAKKLKLVITAGEGSDHVDLAAAKAKKITVAETRLSNSLSIAEAIVKDILDLVSNKKKCEALKKEGKTDLEECLANSHSLEGKTVGVVGFGAVGLAVARLLKPLGVKLMCYDIVEPPAEVLEELGITFYPSVEEMVKDADVVVVACPLTPETKDLFNDKLISKMKKGAILVNDAHGEIVDTDAVVKALESGQLKGFAGDVYTPWPPPADHPLLTAPNTNFTPHVAGSTLEAQALSAAGVREILERYFNNEPIPERDIIIDGSKLP
The redesigned sequences generated by ProteinMPNN showed moderate sequence recovery (~52–56%), meaning that many residues differed from the native sequence while still maintaining a similar overall structural organization. This suggests that the protein fold tolerates some variability, but extensive mutations can alter important conformational regions and potentially disrupt function. Input this sequence into ESMFold and compare the predicted structure to your original.
Generating sequences…
6T8C, score=1.3855, fixed_chains=[], designed_chains=[‘A’], model_name=v_48_020 AKILCVLYDDPITGYPKSYARADVPKIDHYPGGQTAPTPKQIDFTPGELLGSVSGELGLRKYLEGLGHTLVVTSDKEGEDSVFERELPDAEIVISQPFWPAYLTPERIAKAKKLKLAVTAGIGSDHVDLEAAIKNGITVAEVTYSNSISVSEHVVMMILSLVRNYIPSYQWVIKGGWNIADCVERSYDLEAMHVGTVAAGRIGLAVLKRLKPFDVKLHYFDQHRLPESVENELGLTYHPSVEDMVKVCDVVTINAPLHPGTLDLFNDELISKMKRGAYLVNTARGKICNRDAVVRALESGQLAGYAGDVWFPQPAPKDHPWRTMPHHGMTPHISGTSLSAQARYAAGTREILECWFEERPIREEYLIVDGGKLA T=0.1, sample=0, score=0.7072, seq_recovery=0.5241 MKILLVAAPDPASGYPTSYPVDSLPEVTSYPGGLPAPQPSRVDFRPGELLFNVSGALGLREYIEGRGHELIVTSDREGPDSEFARHLPEADVVISSHAWPAVMSAERLAAAKNLRLVITAGEGSDHVDLDAARARGITVVECRLSNSEAIAEHAVELIKSLVENKKNCEKLKKEGKTDKEECEKNSESLEGKTVGIIGFGAVGRRIAELLKPLGVKLGCYDITPPPQEVLEELGITYYPSVEELIKDCDVVVTACPLTPATKDLFNDETIAKMKKGAILVNVSHGPIVKRDALVKALESGQLRGYGGDVYDPWPPPADHPLLTAPNTNLTPHVAGSTLAAQARYAAGVREILERYFADEPIPEHYVVIDGRLLP
New Sequence:MKILLVAAPDPASGYPTSYPVDSLPEVTSYPGGLPAPQPSRVDFRPGELLFNVSGALGLREYIEGRGHELIVTSDREGPDSEFARHLPEADVVISSHAWPAVMSAERLAAAKNLRLVITAGEGSDHVDLDAARARGITVVECRLSNSEAIAEHAVELIKSLVENKKNCEKLKKEGKTDKEECEKNSESLEGKTVGIIGFGAVGRRIAELLKPLGVKLGCYDITPPPQEVLEELGITYYPSVEELIKDCDVVVTACPLTPATKDLFNDETIAKMKKGAILVNVSHGPIVKRDALVKALESGQLRGYGGDVYDPWPPPADHPLLTAPNTNLTPHVAGSTLAAQARYAAGVREILERYFADEPIPEHYVVIDGRLLP
Overall, the protein does not appear fully resilient to major mutations because structural and conformational sites change significantly when larger sequence alterations are introduced.
Part D. Group Brainstorm on Bacteriophage Engineering
Group: (James, USE), (Karen, Ecuador)
Review the Bacteriophage Final Project Goals for engineering the L Protein: Increased stability (easiest) Higher titers (medium) Higher toxicity of lysis protein (hard) Brainstorm Session | Goal | Tunable toxicity | | Strategy | Design variants with graded L–DnaJ binding strength: attenuated (stronger binding) → wild-type → enhanced (weaker binding) | | Tools | AlphaFold-Multimer (L + DnaJ), Rosetta interface ΔΔG |
Write a 1-page proposal (bullet points or short paragraphs) describing:
Engineer the MS2 L lysis protein (75 aa) into a small panel of variants that produce graded lysis phenotypes
(attenuated → WT-like → enhanced) with predictable control over dose and timing.
Because MS2 lysis depends on the E. coli chaperone DnaJ, and DnaJ physically associates with L, we propose to computationally tune the L–DnaJ interaction as a controllable “toxicity knob.”
Proposed Computational Strategy
Tools and Rationale
| Tool | Purpose | Why It Helps |
|---|---|---|
| Protein Language Models (PLMs) | In silico mutagenesis | Suggest substitutions that are evolutionarily plausible and tolerated |
| AlphaFold-Multimer | Predict L–DnaJ complex structure | Identify interface residues and assess structural stability |
| Rosetta Interface ΔΔG | Predict binding energy changes | Quantify strengthening or weakening of L–DnaJ interaction |
Together, these tools allow rational selection of variants expected to modulate binding strength and therefore toxicity.
Computational Pipeline
flowchart TD A[Input: MS2 L sequence (75 aa)] –> B[Constraint check: overlapping coding region / allowed codons] B –> C[Generate variants (PLM-guided mutagenesis)] C –> D[AlphaFold-Multimer: predict L–DnaJ complex + interface residues] D –> E[Rosetta interface ΔΔG: rank binding changes across variants] E –> F[Pick graded panel: weak / mid / strong binders] F –> G[Wet lab: lysis timing + dose-response curves] e propose:
Weak binding ───────► Attenuated lysis Medium binding ─────► WT-like phenotype Strong binding ─────► Enhanced / faster lysis
──────────────────────────────── DESIGN STRATEGY ────────────────────────────────
(1) Sequence Level → Protein Language Models • Suggest tolerated mutations • Preserve evolutionary plausibility • Maintain fold stability
│
▼
(2) Structural Level → AlphaFold-Multimer • Model L–DnaJ complex • Identify interface residues • Evaluate model confidence
│
▼
(3) Energetic Level → Rosetta Interface ΔΔG • Quantify binding shifts • Rank weak → strong binders • Select graded panel (~8–15 variants)
Potential Pitfalls and Mitigation Strategies Challenge Why It Matters Mitigation Strategy Overlapping coding constraints Limits mutation space Apply codon filtering before ranking Small protein size Increases structural prediction uncertainty Use consensus scoring across PLM + AF-Multimer + Rosetta Membrane association Can reduce model reliability Focus on interface trends rather than absolute ΔΔG