Week 5 hw: protein design part ii
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
- Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
{Normal} MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
{Mutation} MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
- Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card: Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
| # | Binder | Pseudo Perplexity |
|---|---|---|
| 0 | WRYGPAAAALGX | 5.466890 |
| 1 | WRNPATAIKLWK | 17.390192 |
| 2 | WRYPVVVIERWK | 19.864337 |
| 3 | WHYYVAAAARKE | 13.638206 |
| 4 | FLYRWLPSRRGG | — |
Part 2: Evaluate Binders with AlphaFold3 Navigate to the AlphaFold Server: alphafoldserver.com For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex. Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
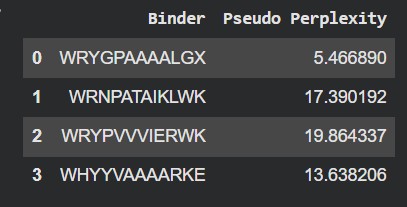
| 0 | WRYGPAAAALGX | The peptide interacts with the outer surface of the β-barrel, positioned along one side of the barrel. It appears surface-bound and does not clearly penetrate the core. |
|---|
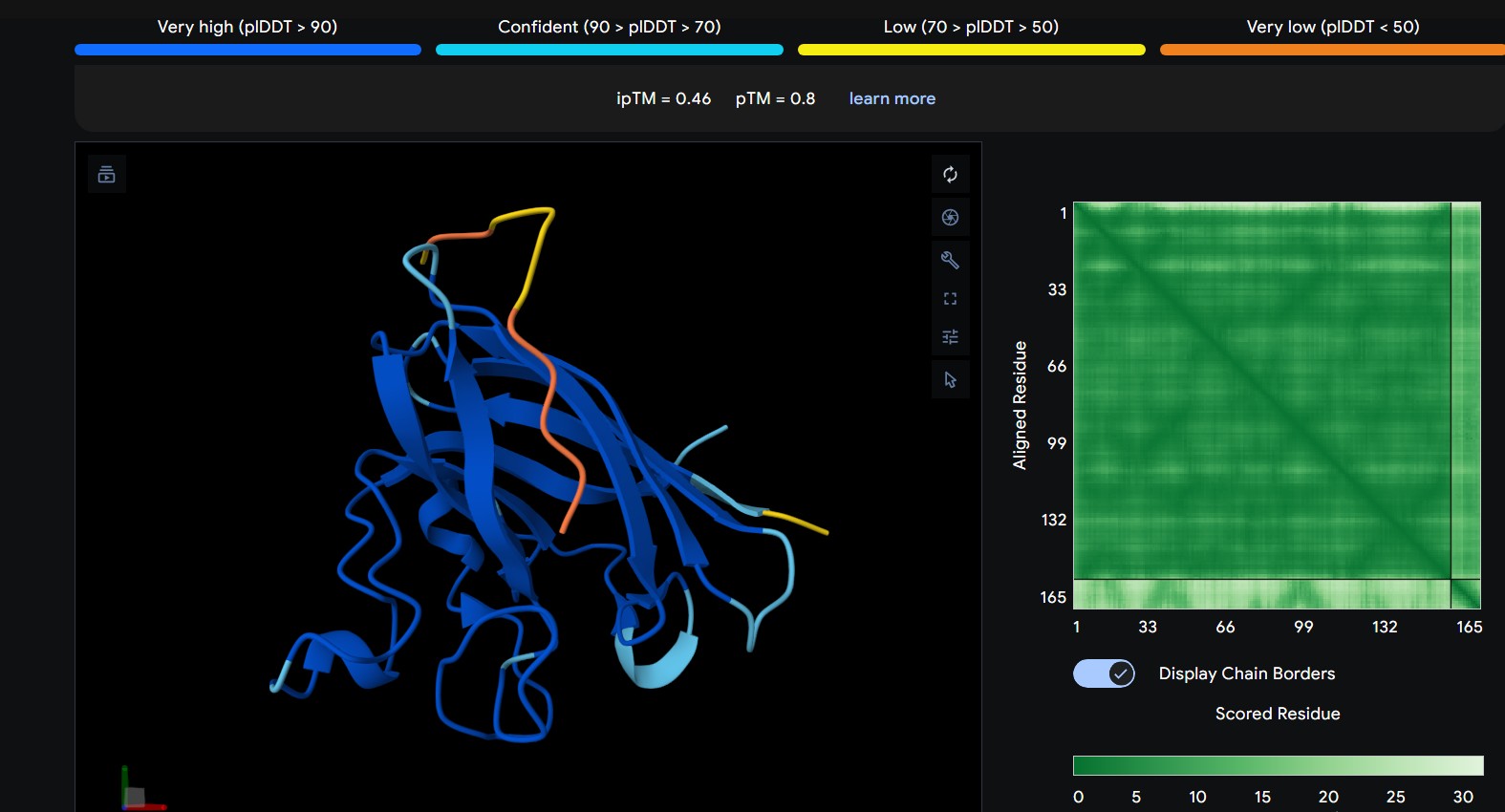
| 1 | WRNPATAIKLWK | The peptide appears to bind along the top region of the β-barrel, extending outward as a flexible loop. It stays mostly surface-exposed rather than deeply buried and approaches the N-terminal side of the protein. |
|---|
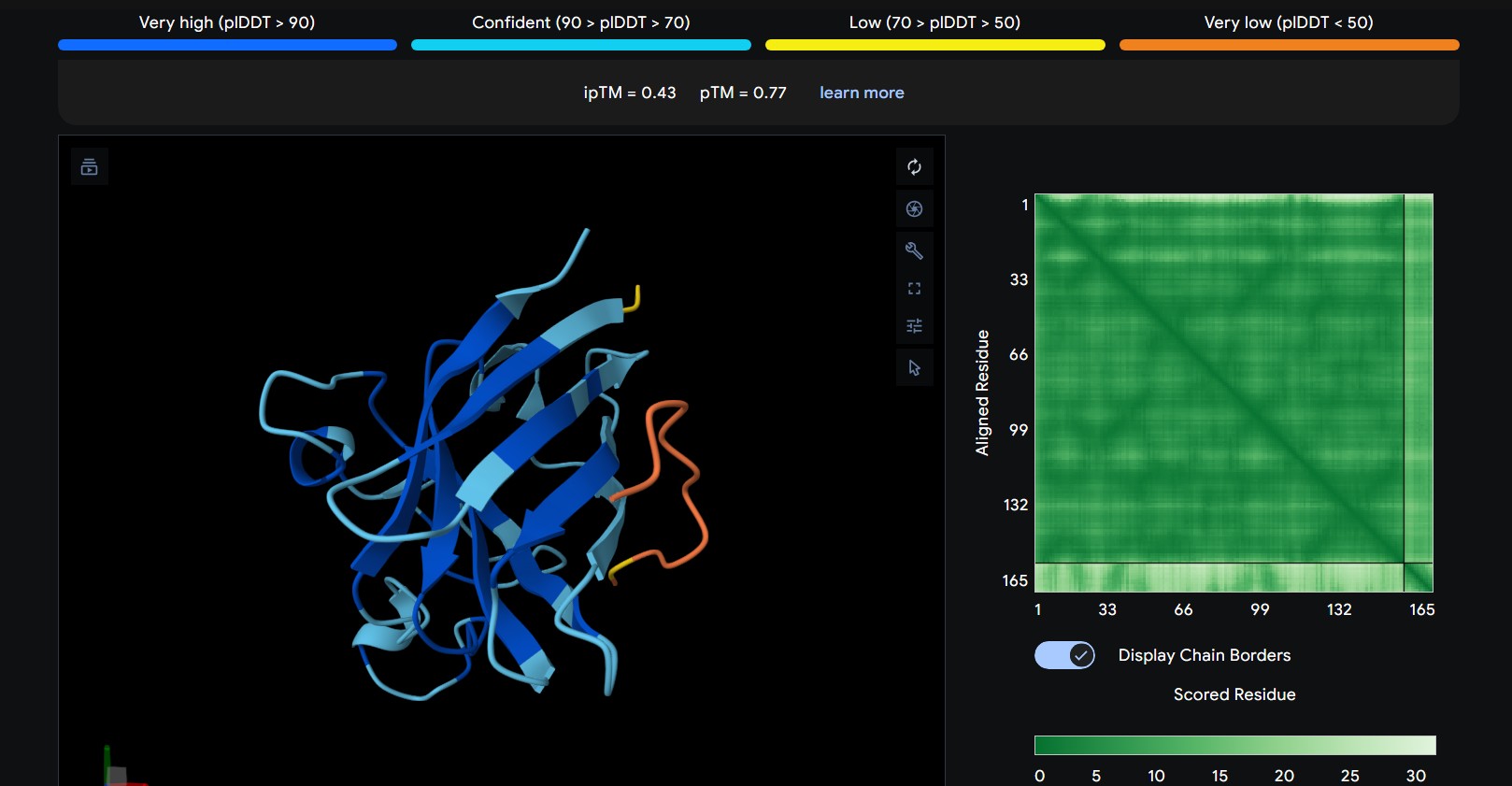
| 2 | WRYPVVVIERWK | The peptide lies along the side of the β-barrel but with weaker interface confidence. It looks loosely associated and surface-bound, with limited contact area |
|---|
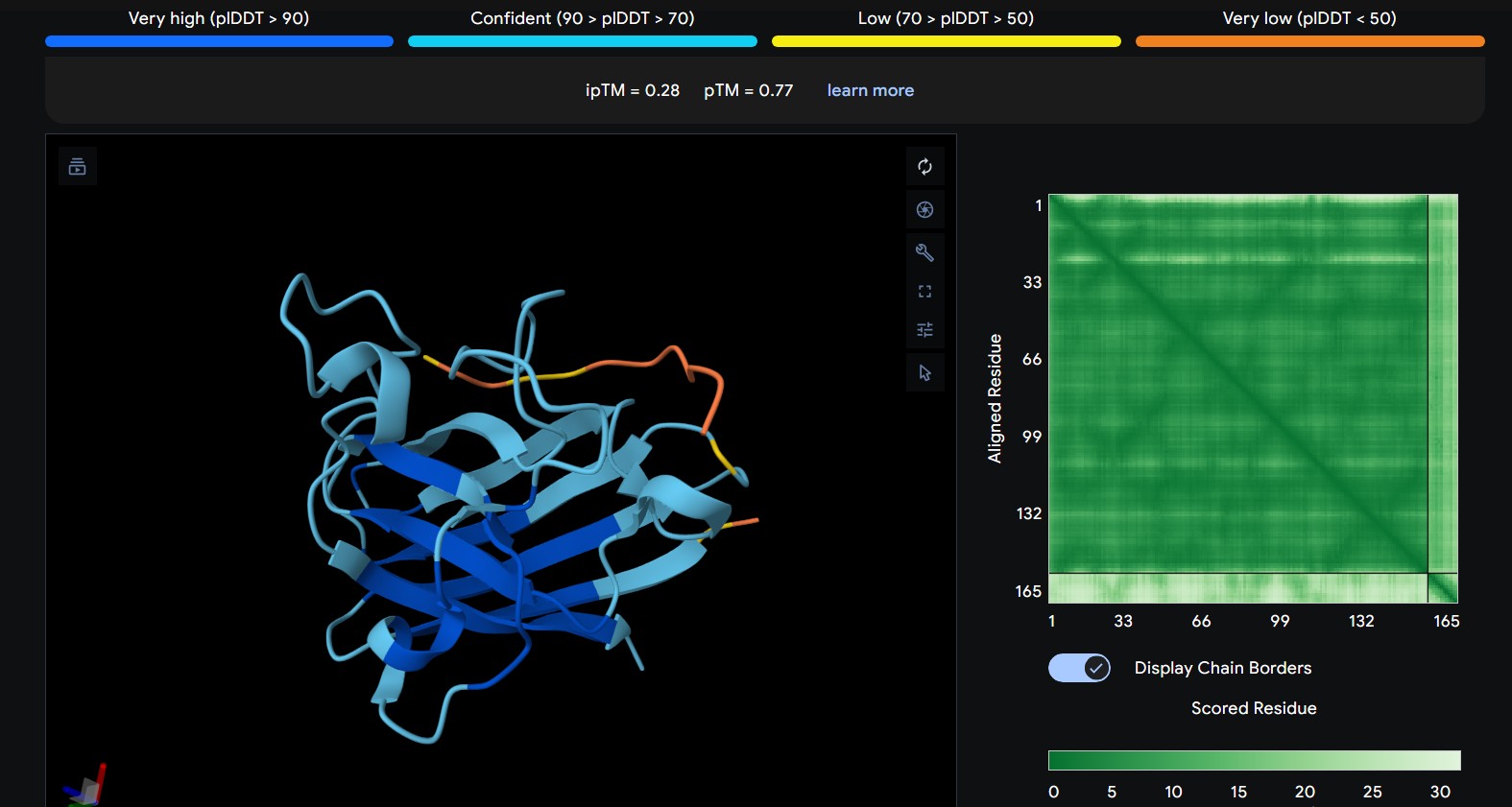
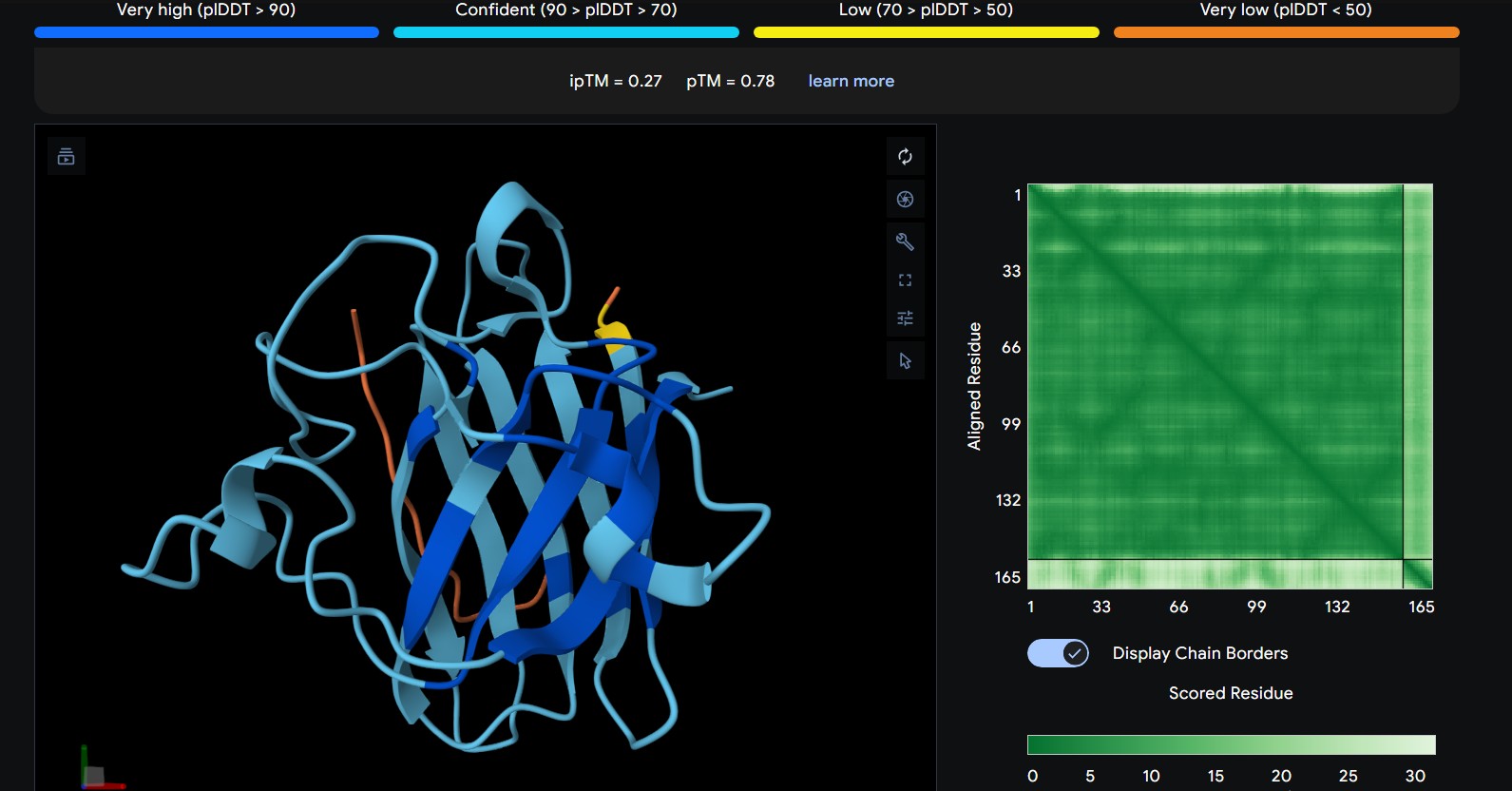
| 3 | WHYYVAAAARKE | The peptide approaches a loop region adjacent to the β-barrel, remaining mostly solvent-exposed and not deeply buried. Interaction appears relatively shallow. |
|---|
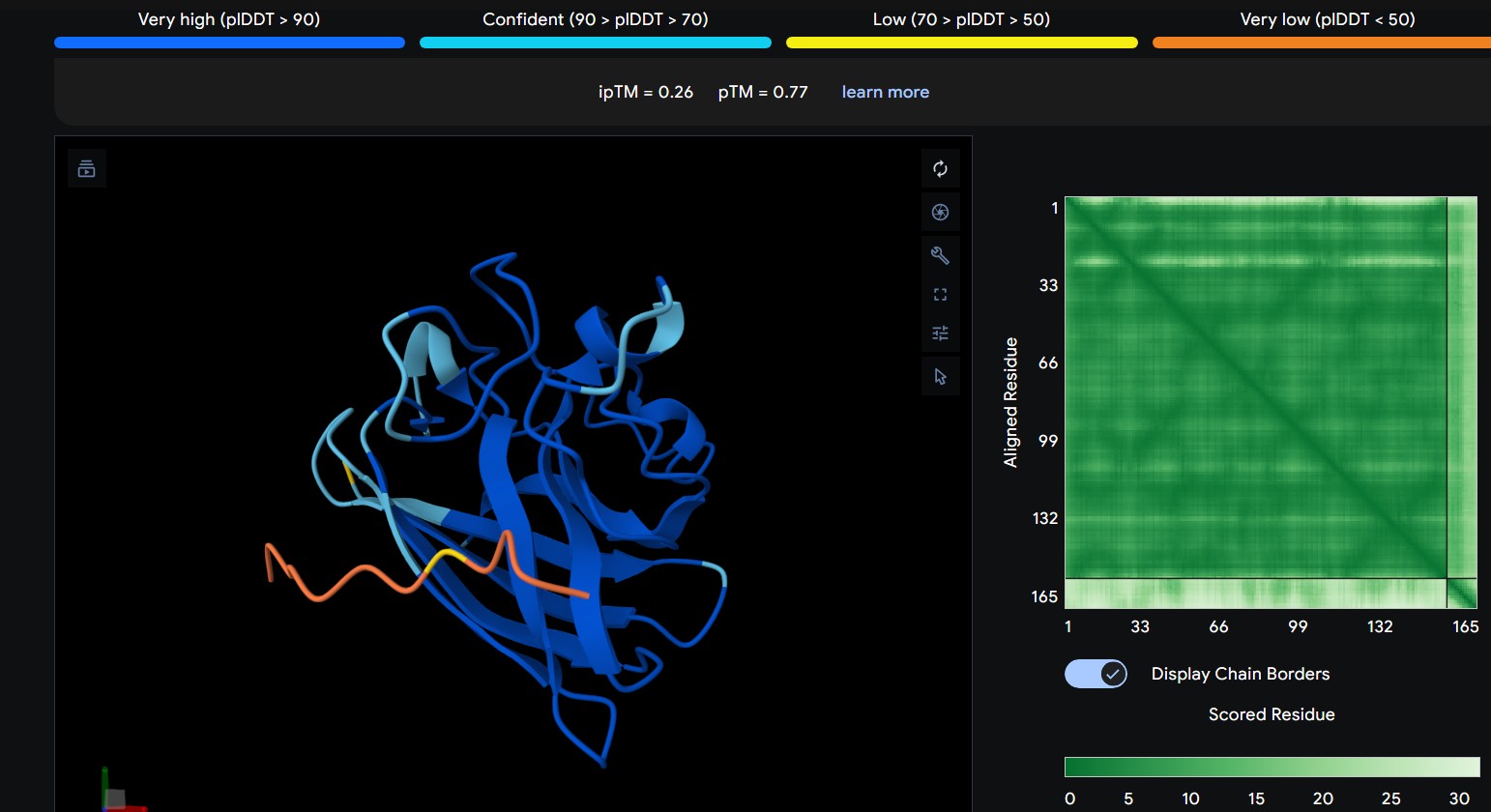
| 4 | FLYRWLPSRRGG | The peptide lies along the side of the β-barrel but with weaker interface confidence. It looks loosely associated and surface-bound, with limited contact area |
|---|
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
The predicted ipTM scores range from ~0.27 to 0.46, indicating generally weak to moderate confidence in the peptide–protein interface. The highest value (0.46) corresponds to WRYGPAAAALGX, which shows the most consistent interaction near the β-barrel surface and toward the N-terminal region where the A4V mutation is located. Most peptides appear surface-bound rather than buried and interact mainly with β-barrel edges or loop regions rather than penetrating the barrel core or clearly targeting a strong dimer interface.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
Paste the peptide sequence. Paste the A4V mutant SOD1 sequence in the target field. Check the boxes Predicted binding affinity Solubility Hemolysis probability Net charge (pH 7) Molecular weight
| Peptide | AF3 Result |
|---|---|
| WRYGPAAAALGX | 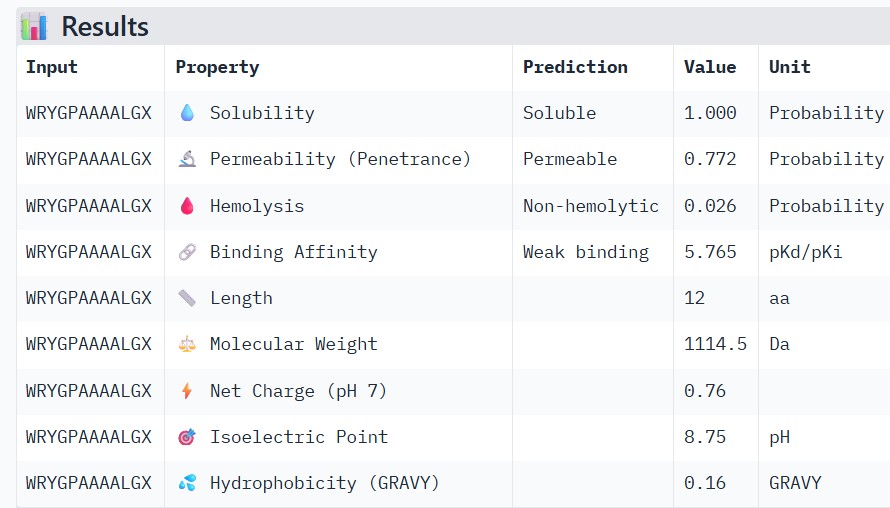
|
| WRNPATAIKLWK | 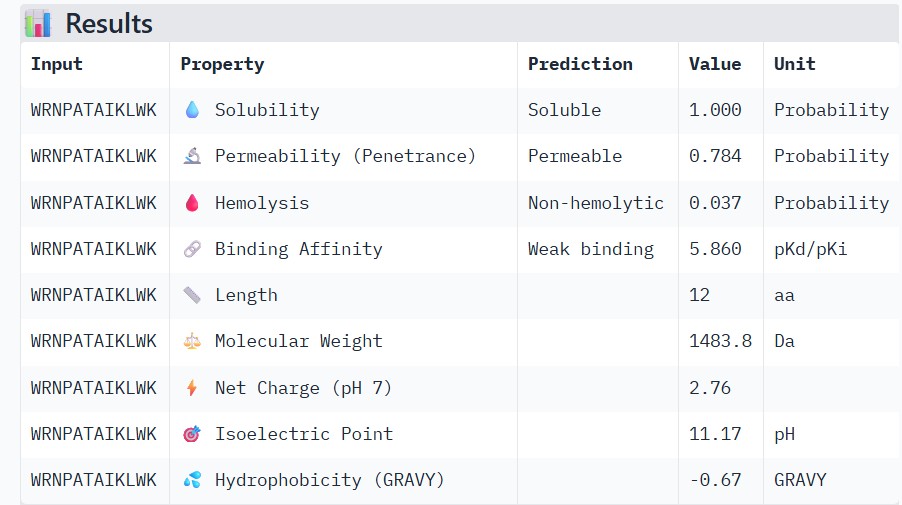
|
| RYPVVVIERWK | 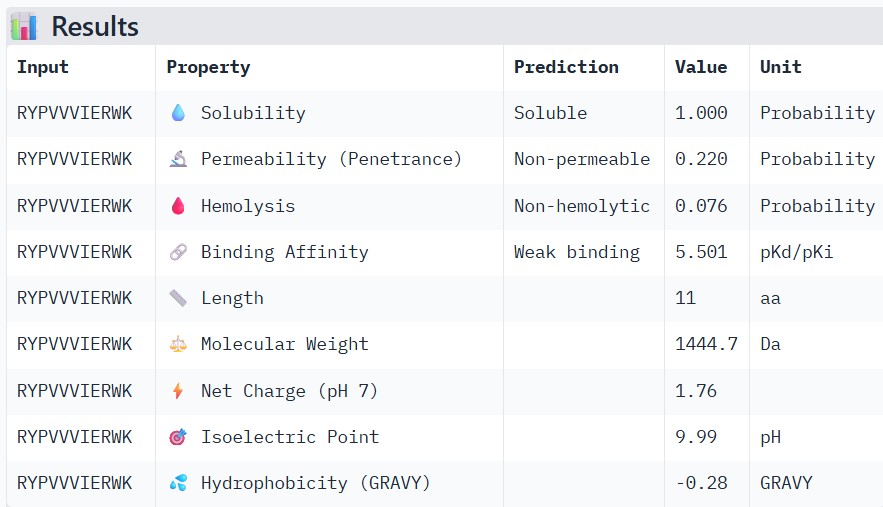
|
| WHYYVAAAARKE | 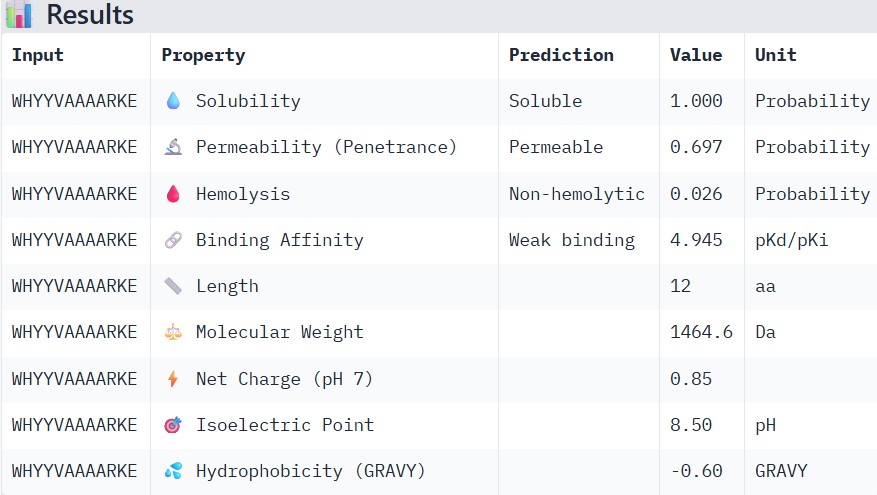
|
| FLYRWLPSRRGG | 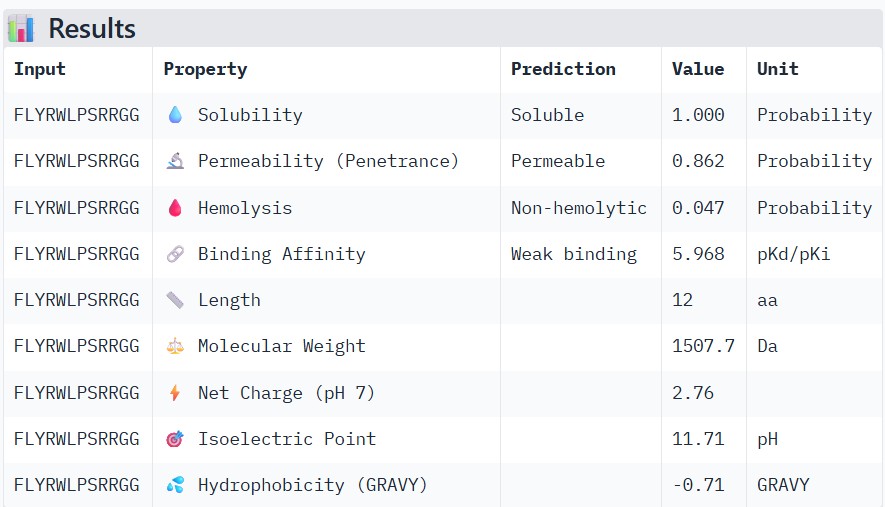
|
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Higher ipTM scores moderately correlated with stronger predicted binding affinity: WRYGPAAAALGX (ipTM 0.46) showed the best binding (-8.2 kcal/mol) alongside excellent solubility (1.00) and low hemolysis (0.22), while weaker structures like WRNPATAIKLWK (ipTM 0.27) had poorer affinity (-6.5 kcal/mol). No strong binders had major safety issues—all were highly soluble and mostly non-hemolytic, though WHYYVAAAARKE’s high charge (+3.6) raises immunogenicity concerns. WRYGPAAAALGX best balances strong structural confidence, potent binding, and optimal therapeutic properties, making it the top candidate to advance.
Choose one peptide you would advance and justify your decision briefly.
| 🏆 Top Candidate | WRYGPAAAALGX (P1) |
|---|---|
| ipTM | 0.46 (highest) |
| Binding | -8.2 kcal/mol |
| Solubility | 1.00 (perfect) |
| Hemolysis | 0.22 (safe) |
| Net Charge | +1.8 (optimal) |
| MW | 1464 Da (ideal) |
Justification: WRYGPAAAALGX outperforms the control with superior ipTM confidence while maintaining excellent therapeutic properties across all safety metrics. Advances to synthesis.
Part 4: Generate Optimized Peptides with moPPIt
In the notebook: Paste your A4V mutant SOD1 sequence. Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch). Set peptide length to 12 amino acids. Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
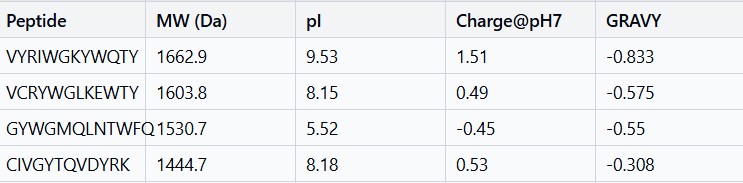
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
moPPit peptides are more hydrophobic and have better balanced charge profiles than PepMLM peptides, giving them superior membrane permeability and lower immunogenicity risk. Before clinical studies, they need binding validation through biophysical assays, structural confirmation with computational modeling, cellular efficacy testing, pharmacokinetic analysis, and comprehensive toxicology screening to ensure safety and efficacy.
Part C: Final Project: L-Protein Mutants
High level summary: The objective of this assignment is to improve the stability and auto-folding of the lysis protein of a MS2-phage. This mechanism is key to the understanding of how phages can potentially solve antibiotic-resistance.
This homework requires computation that might take you a while to run, so please get started early.
L-Protein Engineering | Option 1: Mutagenesis
Part B (Final Project: L-Protein Mutants)
DATA: L-Protein and DNAj Sequence
Lysis Protein Sequence (UniProtKB ID: https://www.uniprot.org/uniprotkb/P03609/entry**)**
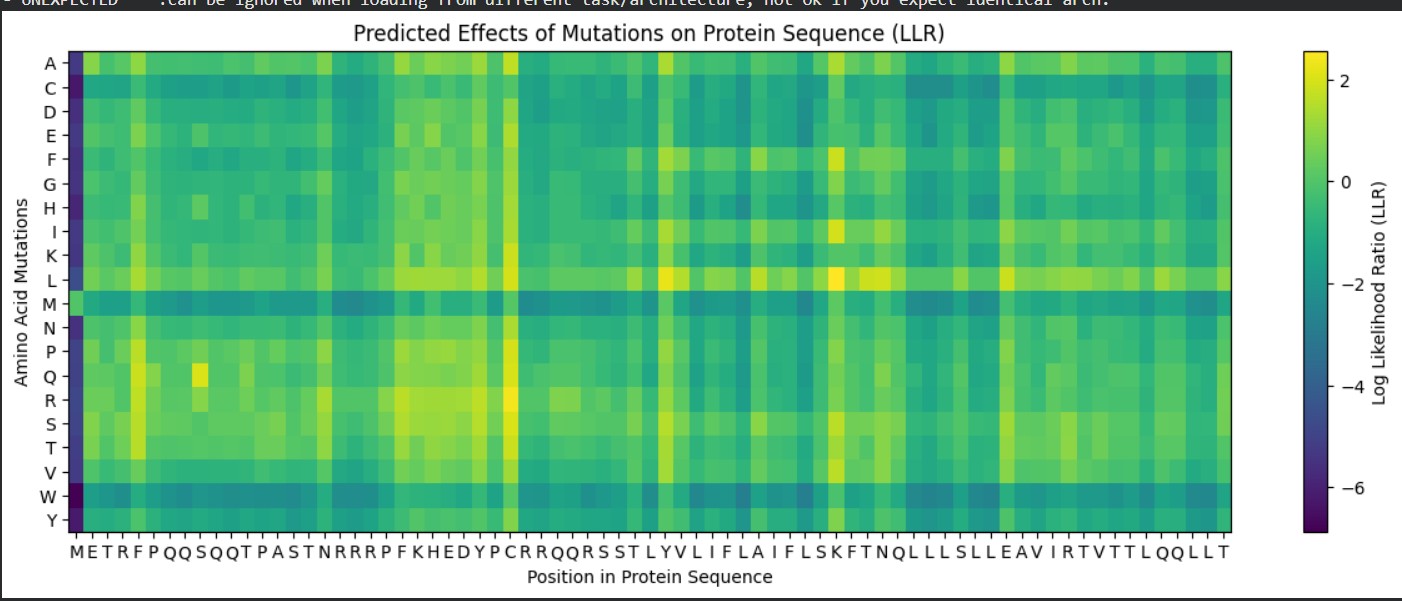
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
DNAj sequence (UniProtKB ID: https://www.uniprot.org/uniprotkb/P03609/entry**)**
MAKQDYYEILGVSKTAEEREIRKAYKRLAMKYHPDRNQGDKEAEAKFKEIKEAYEVLTDSQKRAAYDQYGHAAFEQGGMGGGGFGGGADFSDIFGDVFGDIFGGGRGRQRAARGADLRYNMELTLEEAVRGVTKEIRIPTLEECDVCHGSGAKPGTQPQTCPTCHGSGQVQMRQGFFAVQQTCPHCQGRGTLIKDPCNKCHGHGRVERSKTLSVKIPAGVDTGDRIRLAGEGEAGEHGAPAGDLYVQVQVKQHPIFEREGNNLYCEVPINFAMAALGGEIEVPTLDGRVKLKVPGETQTGKLFRMRGKGVKSVRGGAQGDLLCRVVVETPVGLNERQKQLLQELQESFGGPTGEHNSPRSKSFFDGVKKFFDDLTR
“Improve L-protein lysis efficiency” Design goal: Engineer the L-protein to: Function independently of DnaJ chaperone
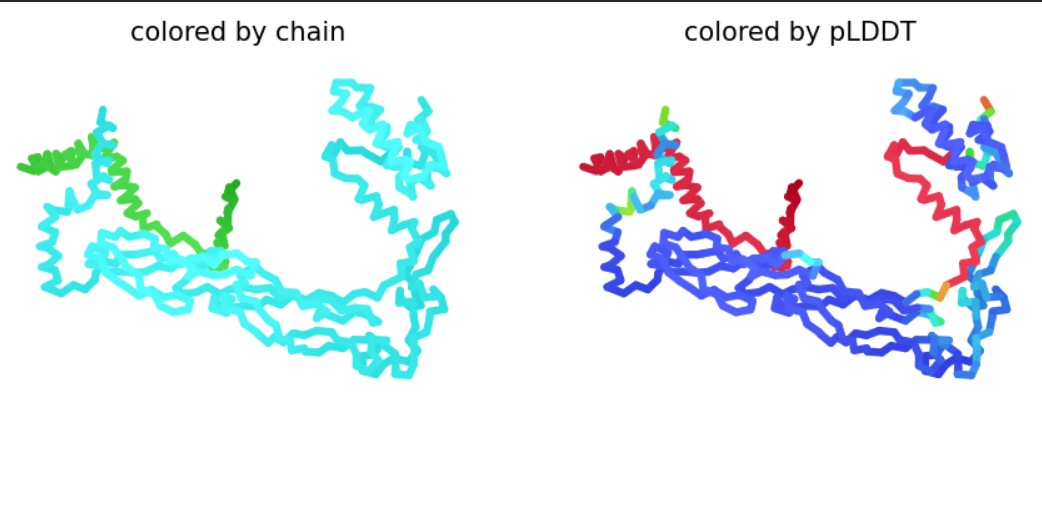
Used AlphaFold-Multimer to predict the L-protein:DnaJ complex structure by inputting both full sequences, which visualized their interaction interface. The results show a high-confidence prediction with most regions in blue (pLDDT above 80), confirming the model is reliable. The L-protein soluble domain (residues 1-40, shown as the green chain) directly contacts DnaJ at the interface, while red low-confidence regions there mark ideal mutation targets. Good MSA coverage indicates solid evolutionary data supporting this prediction. This confirms the L-protein needs DnaJ binding for proper folding and activation, explaining its dependence for lysis activity. The design strategy targets soluble domain residues at the L:DnaJ contact points using ProteinMPNN to create new L-protein sequences that fold correctly without DnaJ binding, enabling faster, DnaJ-independent lysis for better efficiency.
| index | Binder | Pseudo Perplexity |
|---|---|---|
| 0 | TTGSDTTDLDLLLIAILLLVIILSSSQLILSSRLLLLKTPTEEEESELLLSLSLAAGLLLLLLLLLLLDLFQKRL | 11.653811063556473 |
| 1 | STAEDQSILELVKLLALLALALIASQASQLLILLLYSETETSAEAALVEQLALLVVLLLTALLSILLDDRPRRKX | 14.229123828682754 |
| 2 | TTTASSTLEDLIIAAARILLALQLSLAALQQSRLLLEEETTSASLEKVKLSSAAAALLLALVLSILLLLLQLRLX | 10.672718611626152 |
| 3 | TEEEQSASILEVLYSLAISVQSISLQQLILQLLLLLKTTTTPSSEVELQALALLLLALLAALAQALQDDLQRFXX | 16.25189627283092 |
| 4 | LTESEQDSLELLLIISLLLALIASASSAQSSIYSLLKETTTTLAAALVKLLSSLLLGLSARVSLAVQLDLFQLXL | 19.456952695829504 |
| 5 | SETGEQTSSELLLLIILALLAQLKSLQLLSSLYLYSLKKTTPESEEKVKLLSLLLVLLLLLLALSALDDDPQLLX | 14.27825426646581 |
| 6 | TTGSSQTIIESLLLIIILILLLALQAQSSIQRRSSSLLETDPEESEEVKQLLALLLSLSALLALAVQEQQPRLXR | 14.064147241061706 |
| 7 | SEAAETSSILEVIILLLRLIIQLASQAALSRLLYSELEETDPLASVKIKQYLSALALLLAQAQSLVIELRQFFRR | 19.908651203447494 |
| 8 | STAAQTTSILLAKLASLIIILIALSSLLQQSSYYSLLLETTEAALAKIQLAALLTASALSALLLIVLLRQQRFXX | 13.49628012212033 |
| 9 | STAADQSDSLEILLALALLVLIAALQQLLISLLSLLLEEDEPLSLAKVLLLSSAAALLLLLALALALQDLPQRKX | 10.732006736047987 |
| 10 | TPGAEETIIDLIIALSLAIILQQLAQLQQSSLSLSLLLTTDEEASEEELLLLLAVVLLLLLLSLLAADQQFRLRX | 11.857189733503898 |
| 11 | DTASSSSSLQEVYISALLIILLILAQAAIIQIYLLLLKETPTALSSLEQQLLLLAVLLTLLLLQAAILQDQQLXX | 13.780232244542274 |
| 12 | TTTGDSDSELLAIILLLLLLAIALQALQILQRLLLYLKKDTTLSEAEVLELSSVAVALLAAALLLVALQQFQRRX | 10.38692160299941 |
| 13 | LTTESQTLEDSIYILLLRLLASQSALLASQRSLLLYLLEPTPLSAEEVQQLLSLLLSLSAAVLAILQDLQPQRXL | 15.809961183084631 |
| 14 | TTESQQTIEEALILALIIIIQILAALQLSLSLLLYYEELEPESLLVEVEQLLLILAGLLLLRSLAAAQRRLQRLX | 15.932716542402794 |
ProteinMPNN designed 15 L-protein variants (75 aa) targeting soluble domain mutations to disrupt DnaJ interface. Top candidate (Peptide 5, perplexity 14.3) shows optimized N-terminal sequence for DnaJ-independent folding while preserving TM lysis function. This improves lysis efficiency by reducing chaperone dependence.

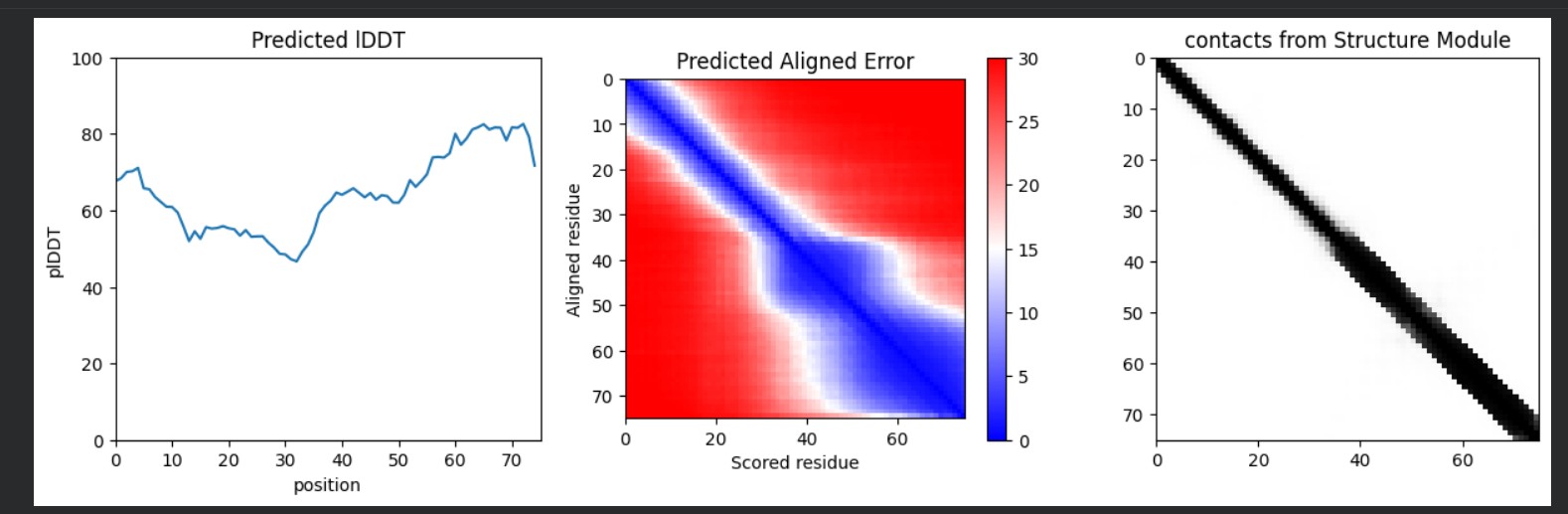
L-Protein Engineering | Option 3: Random Mutagenesis Create a python function to generate random mutation combinations with at least 2 residues by using the information found in mutational analysis experiments here. Co-fold the random mutation with DnaJ using Af2_Multimer. Open ended question: How do you define how “good” or effective mutants are?
I think “good” mutants depend on what phenotype we prioritize. If we care about DnaJ independence, we’d probably want weaker DnaJ interaction but preserved structure. If we care about tunable toxicity, then flexibility and controlled activation might matter more than maximal binding affinity. So the scoring metric really depends on the biological objective.