Week 6 hw: genetic circuits part i
- DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
This process relies on a specialized DNA polymerase with proofreading activity. Unlike standard Taq polymerase, high-fidelity enzymes (such as Phusion DNA Polymerase) possess 3’→5’ exonuclease activity, which allows them to detect and correct errors during DNA synthesis.Phusion High-Fidelity PCR Master Mix
PCR consists of repeated cycles with three main steps:
Denaturation (~98°C)
The double-stranded DNA is separated into single strands.Annealing (~50–72°C)
Primers bind (anneal) to their complementary sequences on the DNA template.Extension (~72°C)
The DNA polymerase synthesizes a new DNA strand by adding nucleotides (dNTPs) to the primers.
During extension, the proofreading activity of high-fidelity polymerases ensures that incorrectly incorporated nucleotides are removed and replaced, resulting in a much lower error rate.
Components of a High-Fidelity PCR Reaction
| Component | Function |
|---|---|
| DNA Polymerase | Synthesizes new DNA strands; proofreading activity increases accuracy |
| dNTPs | Building blocks (A, T, G, C) used to form DNA |
| Reaction Buffer | Maintains optimal pH and ionic conditions for enzyme activity |
| MgCl₂ | Essential cofactor required for polymerase function |
| Primers | Define the start and end points of DNA amplification |
| Template DNA | Target DNA sequence to be amplified |
| Nuclease-free Water | Adjusts final reaction volume |
| Additives (e.g., DMSO) | Improve amplification of difficult or GC-rich templates |
What are some factors that determine primer annealing temperature during PCR?
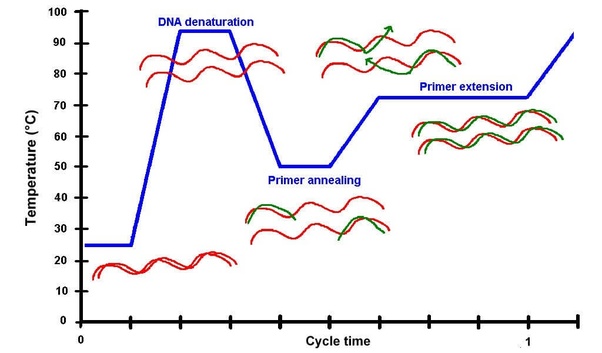
1. DNA Denaturation (~94–98°C)
At high temperature, the double-stranded DNA separates into single strands, making the template accessible.
2. Primer Annealing (~50–65°C)
The temperature is lowered to allow primers to bind (anneal) to their complementary sequences on the DNA template.
The annealing temperature is critical and depends on several factors:
- Primer melting temperature (Tm) → determines optimal binding temperature
- GC content → higher GC increases binding strength and Tm
- Primer length → longer primers bind more stably
- Sequence specificity → affects how well primers match the template
- Salt concentration (Mg²⁺) → stabilizes primer-template interaction
- Additives (e.g., DMSO) → can reduce binding stability
3. Primer Extension (~72°C)
DNA polymerase extends the primers, synthesizing new DNA strands by adding dNTPs.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR vs Restriction Enzyme Digestion
| PCR | Restriction Enzyme Digestion | |
|---|---|---|
| Method | DNA amplification 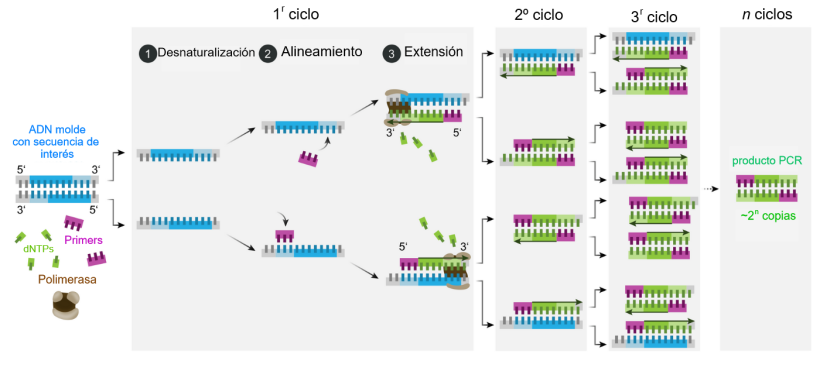
| DNA cleavage 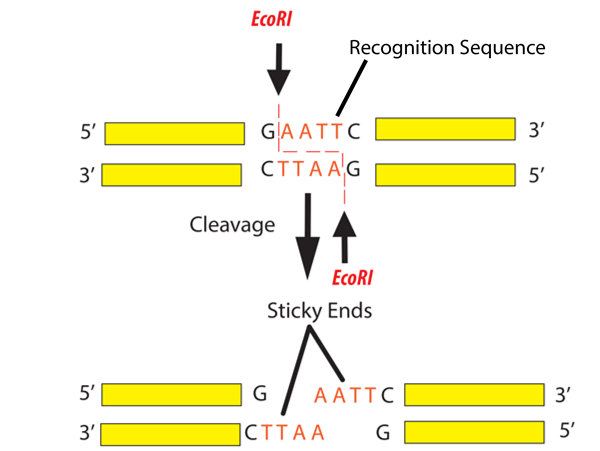
|
| Mechanism | Uses primers and DNA polymerase to copy DNA | Uses restriction enzymes to cut DNA at specific sites |
| Requirements | Template DNA, primers, polymerase, dNTPs | DNA, restriction enzyme(s), buffer |
| Process | Thermal cycling (denaturation, annealing, extension) | Incubation at constant temperature |
| Equipment | Thermocycler | Incubator or water bath |
| Output | Specific amplified DNA fragment | DNA fragments based on restriction sites |
| DNA Quantity | Can amplify small amounts of DNA | Does not amplify DNA |
| Specificity | Determined by primer design | Determined by enzyme recognition sequence |
| Flexibility | High (can modify sequence, add tags) | Limited (depends on existing sites) |
| Typical Use | Amplifying or modifying DNA | Cutting DNA for cloning or analysis |
When to Use Each Method
Use PCR when:
- You need to amplify a specific DNA region
- DNA quantity is low
- You want to modify the sequence (e.g., mutations, tags)
Use Restriction Digestion when:
- You need to cut DNA at known sequences
- You are working with plasmids or cloning workflows
- You want sticky ends for ligation
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To ensure that DNA fragments generated by PCR or restriction enzyme digestion are appropriate for Gibson Assembly, it is essential that they contain overlapping sequences at their ends. These overlapping regions, typically between 20 and 40 base pairs, allow adjacent DNA fragments to anneal to each other during the assembly process. These overlaps are most commonly introduced through primer design during PCR, where additional sequences are added to the 5’ ends of primers to match neighboring fragments.Addgene Gibson Assembly protocol
In addition to proper overlap design, the DNA fragments must be accurately generated and free of mutations. This is especially important when fragments are produced by PCR, where the use of high-fidelity polymerases helps minimize errors in the amplified sequence. Correct fragment orientation and order must also be ensured, since Gibson Assembly joins fragments based on sequence homology, meaning that incorrect design will result in failed or incorrect constructs.
Furthermore, DNA purity and concentration play a critical role in the efficiency of the assembly. Contaminants such as salts or residual enzymes can inhibit the reaction, while imbalanced fragment concentrations may reduce assembly success. According to the protocol, optimal results are achieved when DNA fragments are present in equimolar amounts.
Finally, it is important to note that Gibson Assembly does not require compatible sticky ends, unlike traditional restriction enzyme cloning. Instead, an exonuclease generates single-stranded overhangs, allowing fragments with complementary overlaps to anneal. A DNA polymerase then fills in the gaps, and a ligase seals the nicks, resulting in a continuous double-stranded DNA molecule.
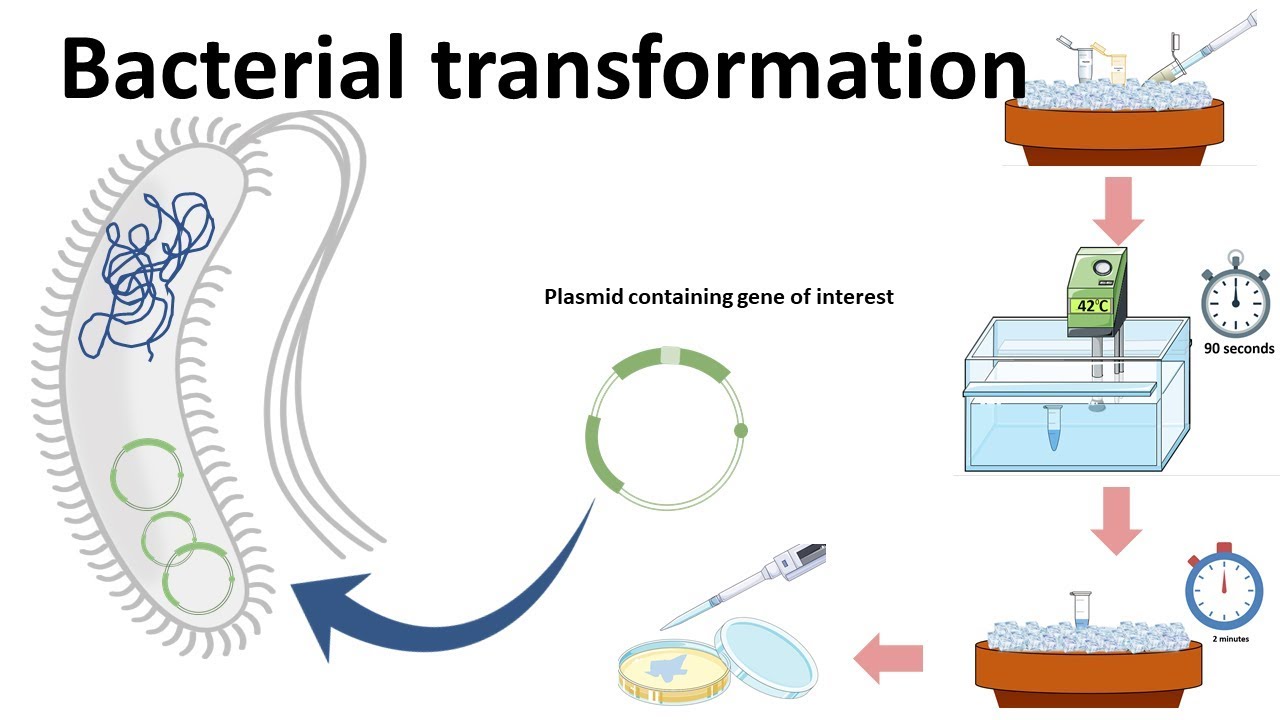
How does the plasmid DNA enter the E. coli cells during transformation?
During bacterial transformation, plasmid DNA enters E. coli cells through a process that temporarily increases the permeability of the cell membrane. This is typically achieved using chemical transformation or electroporation, both of which facilitate the uptake of foreign DNA into the bacterial cell.
In chemical transformation, E. coli cells are first treated with calcium chloride or similar salts, which help neutralize the negative charges on both the DNA and the cell membrane. This allows the plasmid DNA to come closer to the cell surface. A subsequent heat shock (usually at 42°C for a short period) creates a thermal imbalance that induces the formation of temporary pores in the membrane, through which the plasmid DNA can enter the cell.
In electroporation, cells are exposed to a brief, high-voltage electric pulse that disrupts the cell membrane and creates transient pores. These pores allow plasmid DNA to pass directly into the cytoplasm. After the pulse, the membrane reseals, trapping the DNA inside the cell.
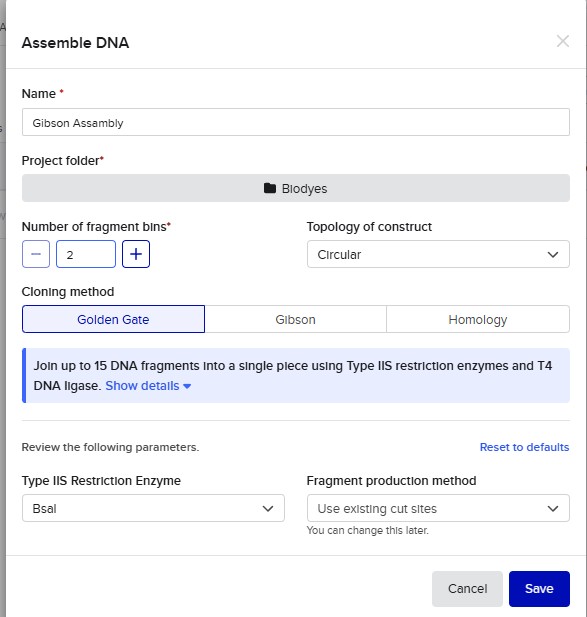
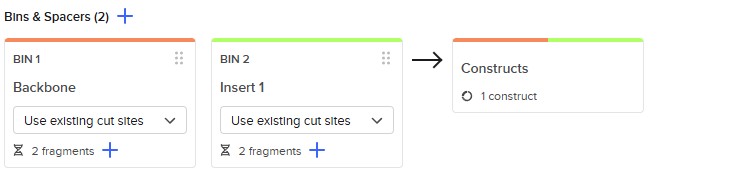
Describe another assembly method in detail (such as Golden Gate Assembly)
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Golden Gate Assembly
Golden Gate Assembly is a molecular cloning method that allows the precise and simultaneous assembly of multiple DNA fragments in a single reaction. It uses Type IIS restriction enzymes, such as BsaI, which cut DNA outside of their recognition sequence, generating custom overhangs. These overhangs can be designed to be unique and complementary, ensuring that DNA fragments assemble in a specific order. During the reaction, the restriction enzyme and DNA ligase work together in a cycling process of digestion and ligation, enabling seamless assembly without leaving extra sequences (scarless cloning). Unlike traditional restriction cloning, this method allows the assembly of multiple fragments in a one-pot reaction with high efficiency. Golden Gate Assembly is widely used in synthetic biology due to its precision, speed, and ability to assemble complex constructs. Overall, it is a powerful technique for constructing plasmids with multiple inserts in a defined orientation.
Flow:
[Fragment A] → [Fragment B] → [Fragment C]
↓ cut (BsaI)
custom overhangs
↓ ligation
final assembled plasmid
Model this assembly method with Benchling or Asimov Kernel!
Asimov Kernel
Create a Repository for your work
Create a blank Notebook entry to document the homework and save it to that Repository
Explore the devices in the Bacterial Demos Repo to understand how the parts work together by running the Simulator on various examples, following the instructions for the simulator found in the “Info” panel (click the “i” icon on the right to open the Info panel)
Create a blank Construct and save it to your Repository
Recreate the Repressilator in that empty Construct by using parts from the Characterized Bacterial Parts repository
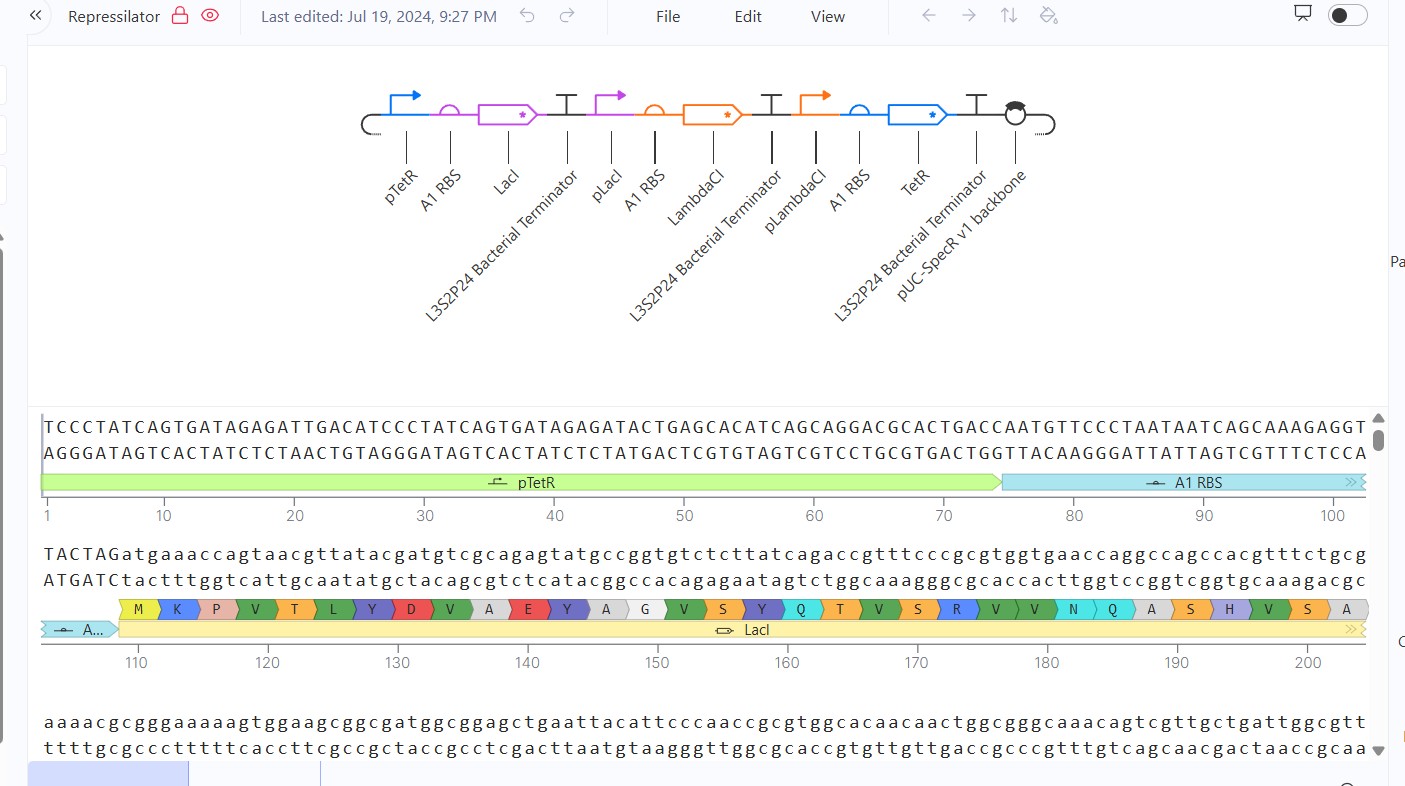
Search the parts using the Search function in the right menu
Drag and drop the parts into the Construct Confirm it works as expected by running the Simulator (“play” button) and compare your results with the Repressilator Construct found in the Bacterial Demos repository
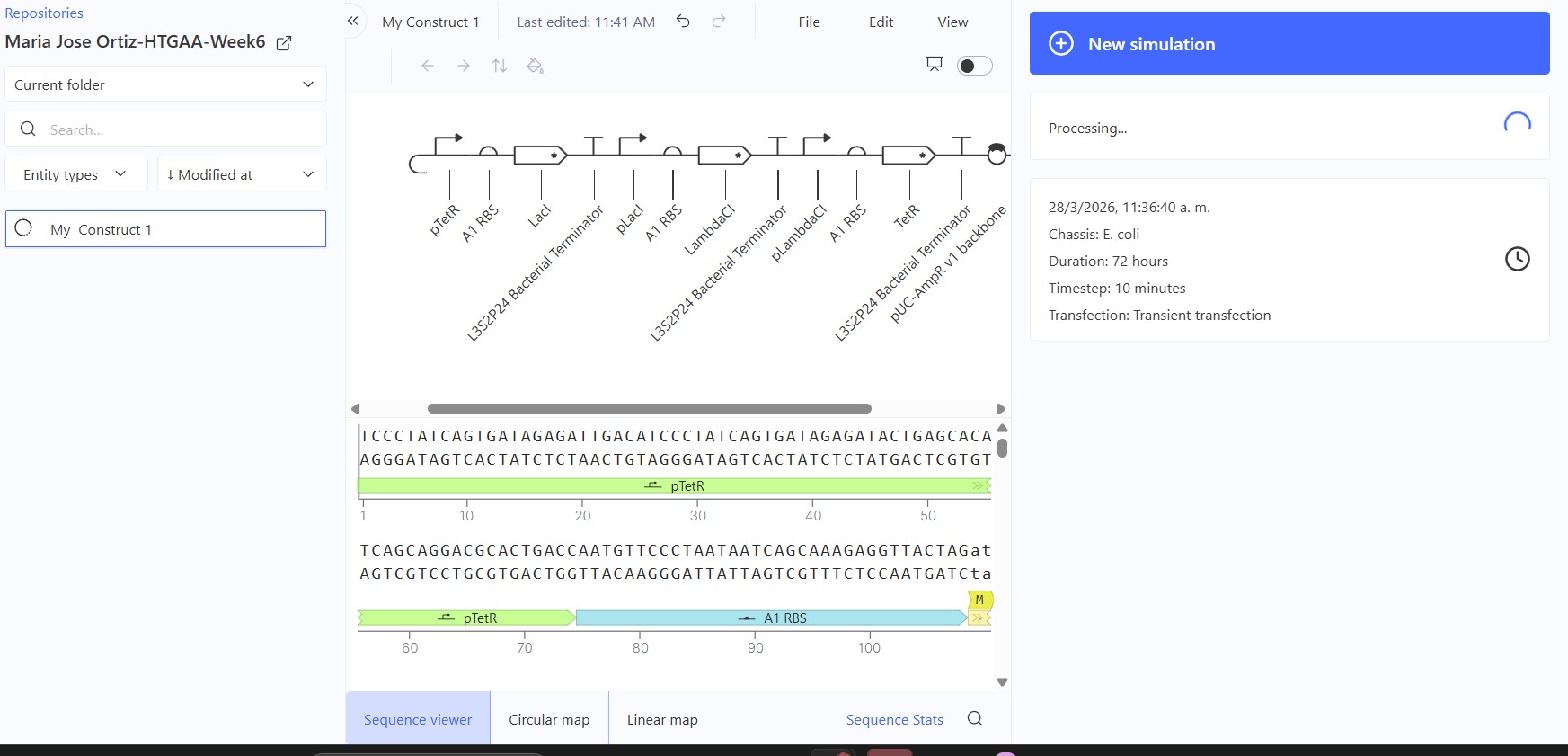
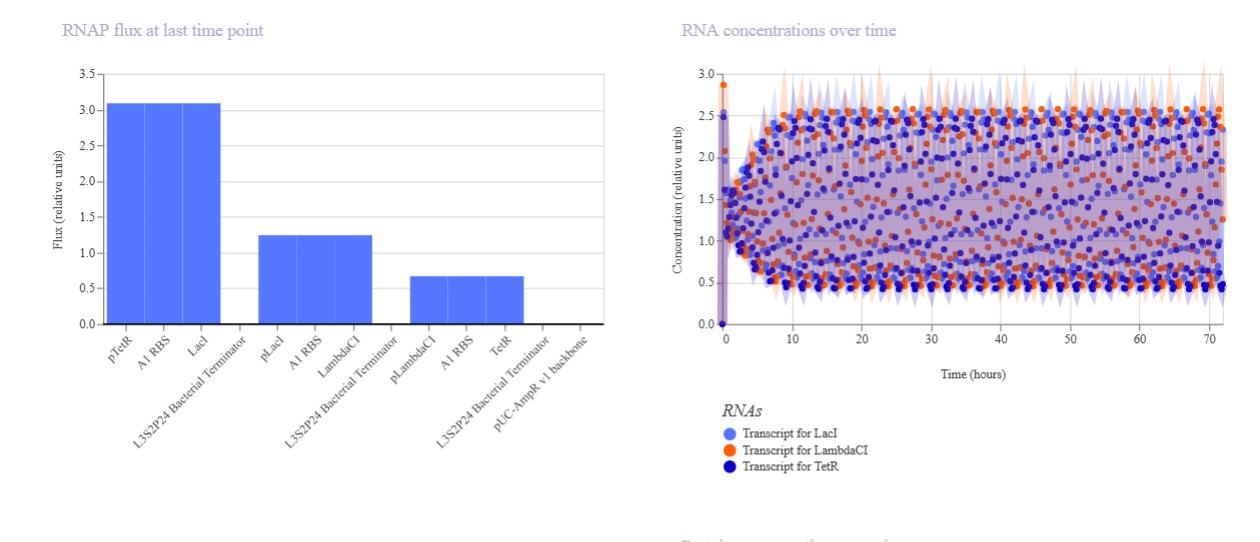
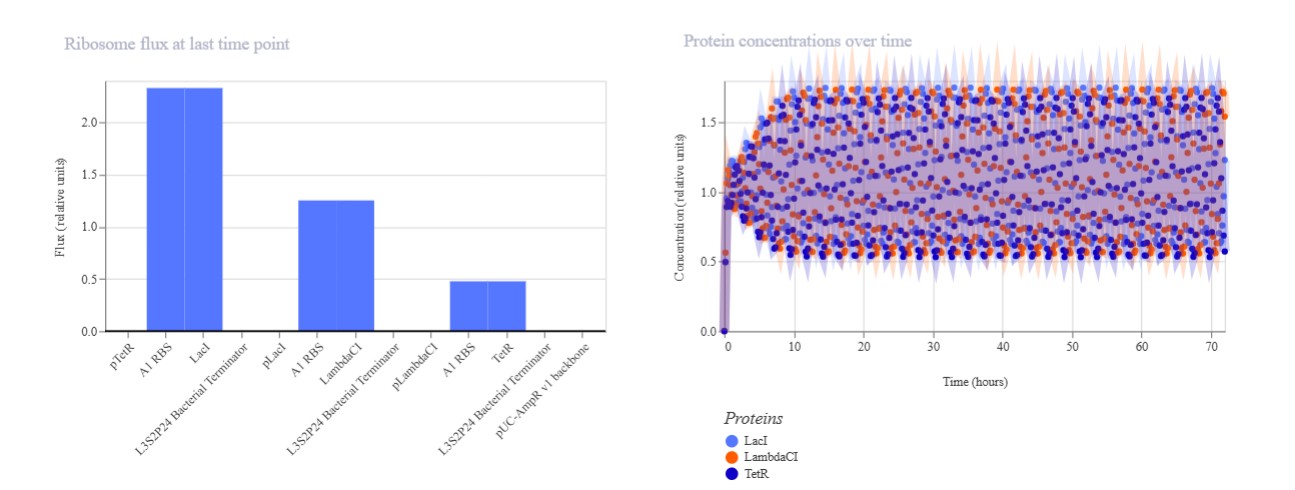
Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo If the results don’t match your expectations, speculate on why and see if you can adjust the simulator settings to get the expected outcome
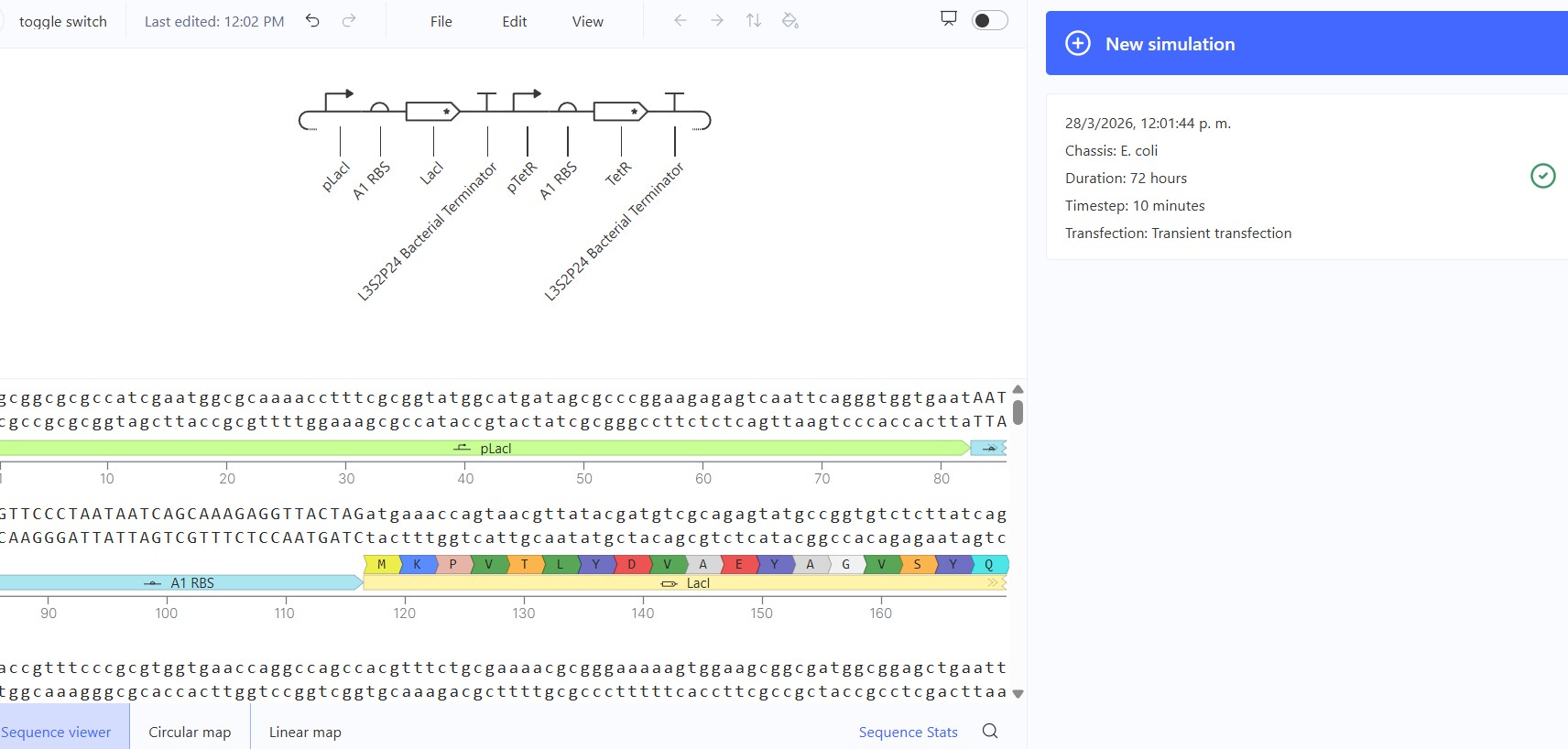
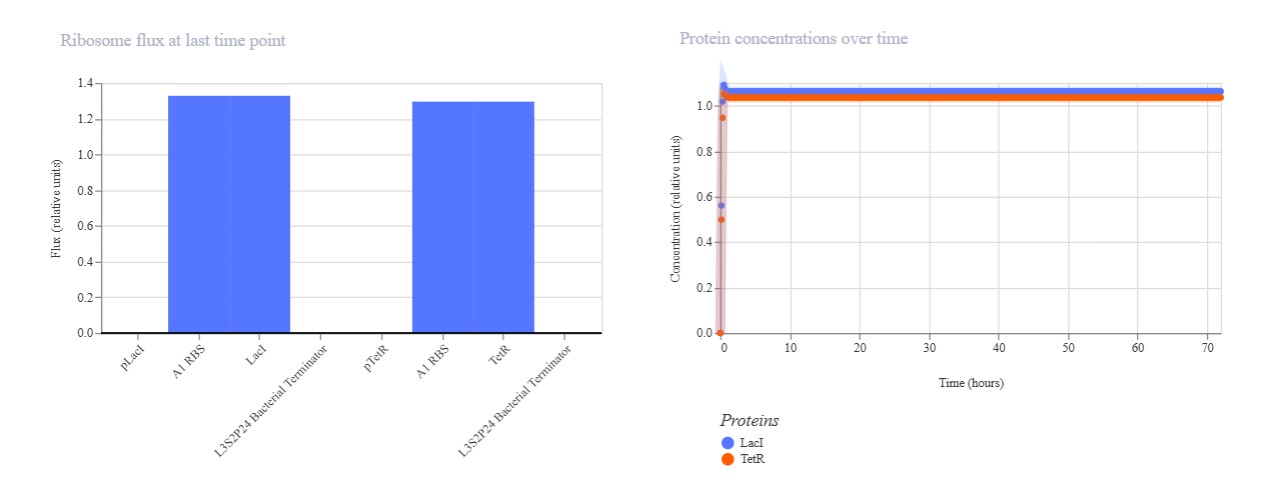
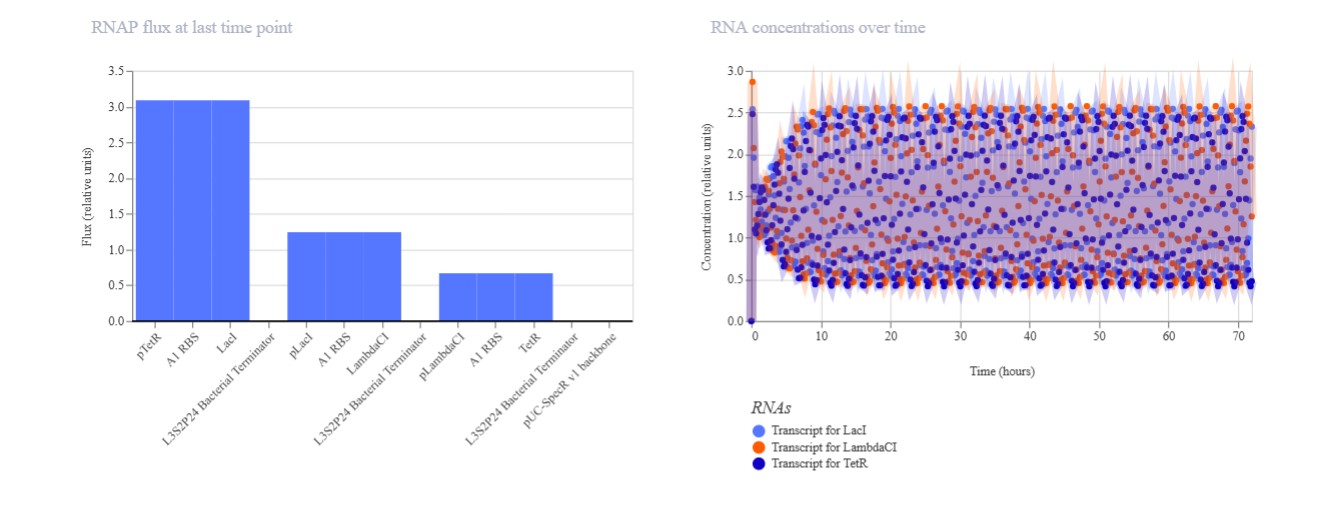
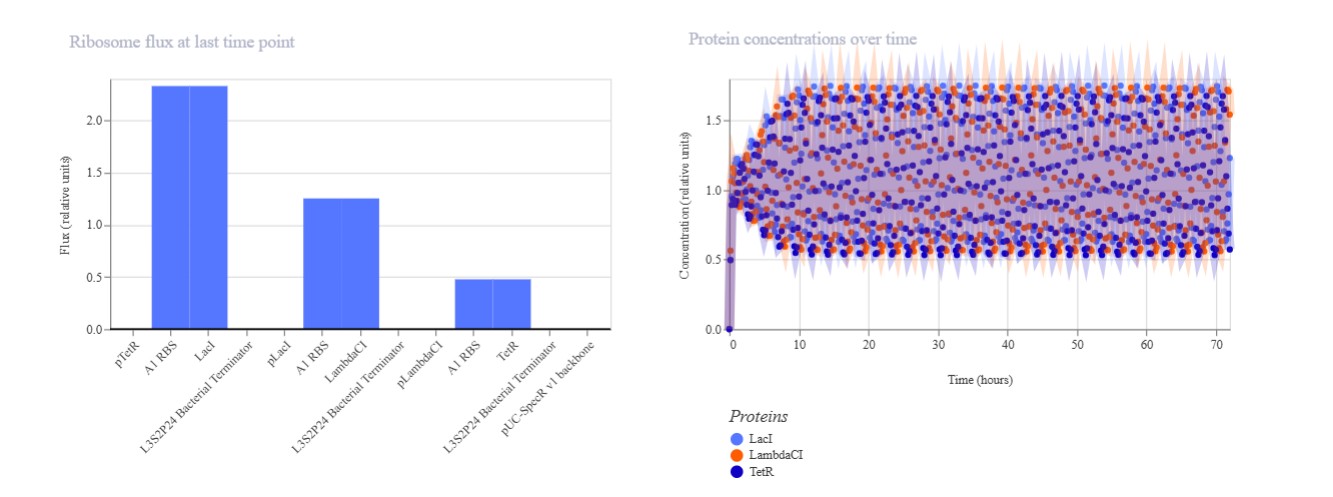
Expected: Bistable behavior with mutual repression - either LacI high/TetR low OR TetR high/LacI low stable states. Simulation should show clear state switch over time. Actual: Achieved toggle functionality. mRNA/protein levels transitioned from LacI-dominant to TetR-dominant state, confirming mutual repression between pLac-LacI and pTet-TetR modules.
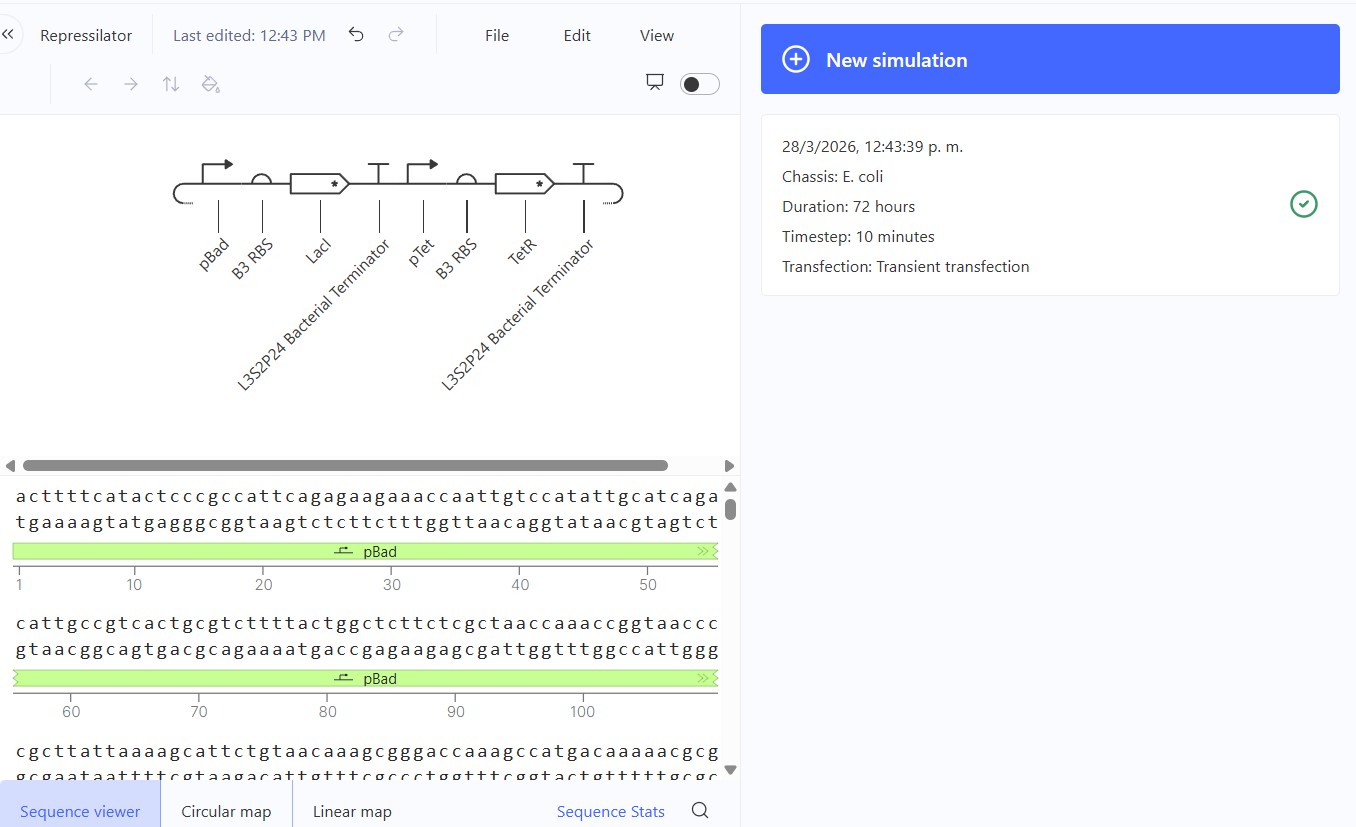
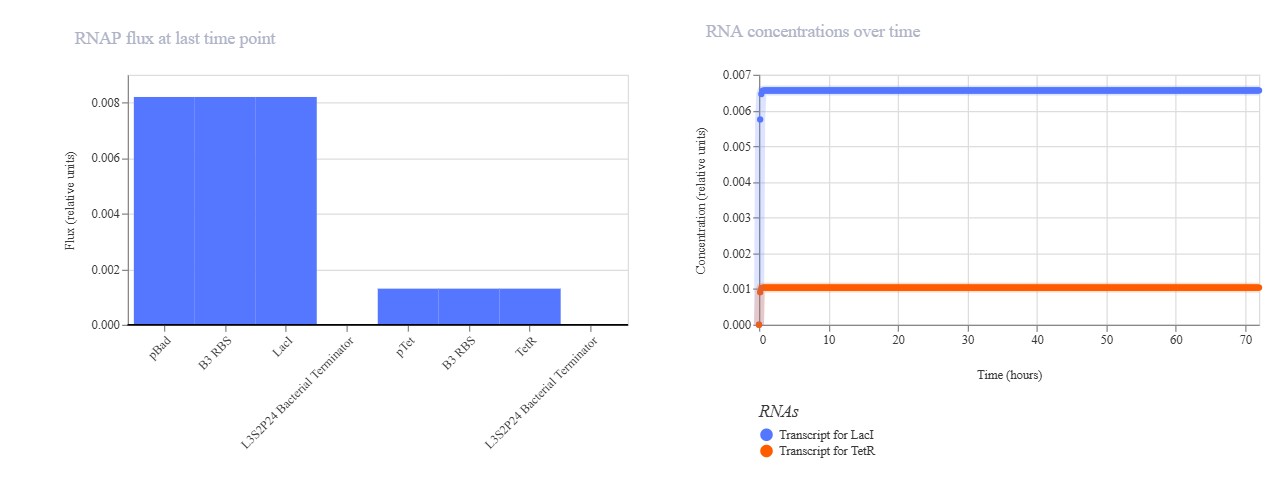
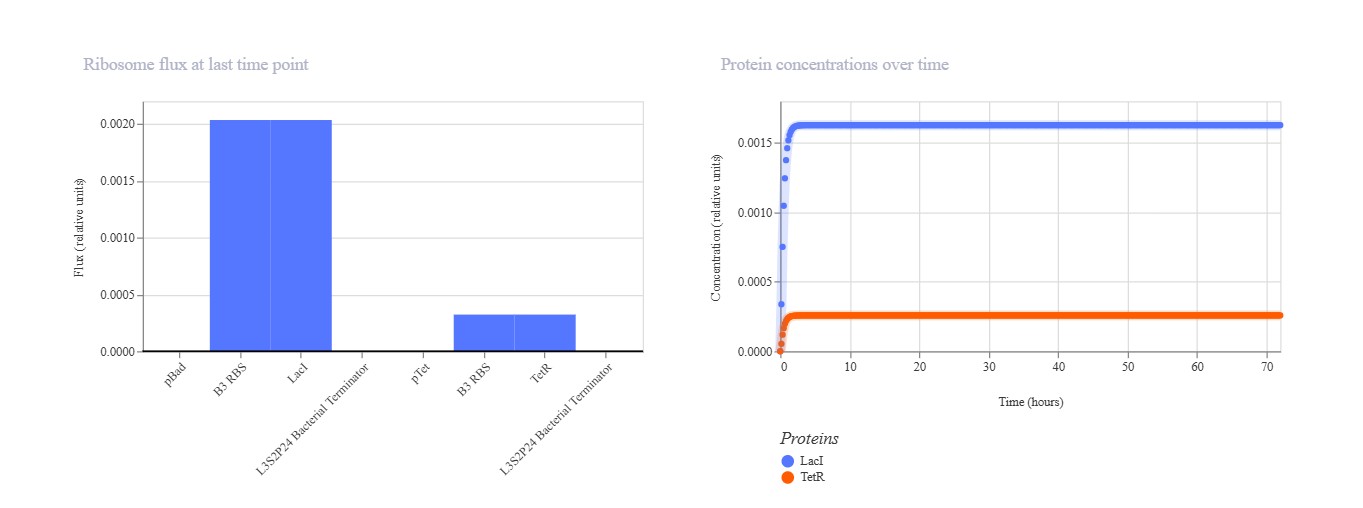
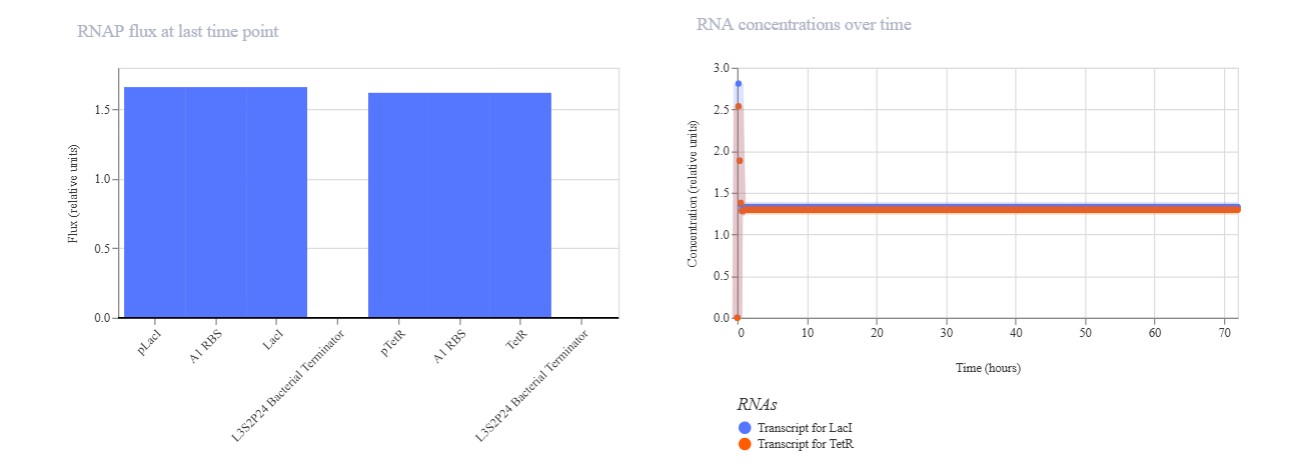
Expected: Similar bistable toggle but with pBAD (arabinose-inducible) replacing pLac for promoter diversity testing. TetR should repress pBAD while LacI represses pTet. Actual: Strong TetR-dominant final state. pBAD mRNA remained low (~0.3) while pTet mRNA high (~1.0), with TetR protein stable high and LacI low. Demonstrates robust toggle with alternative pBAD promoter.
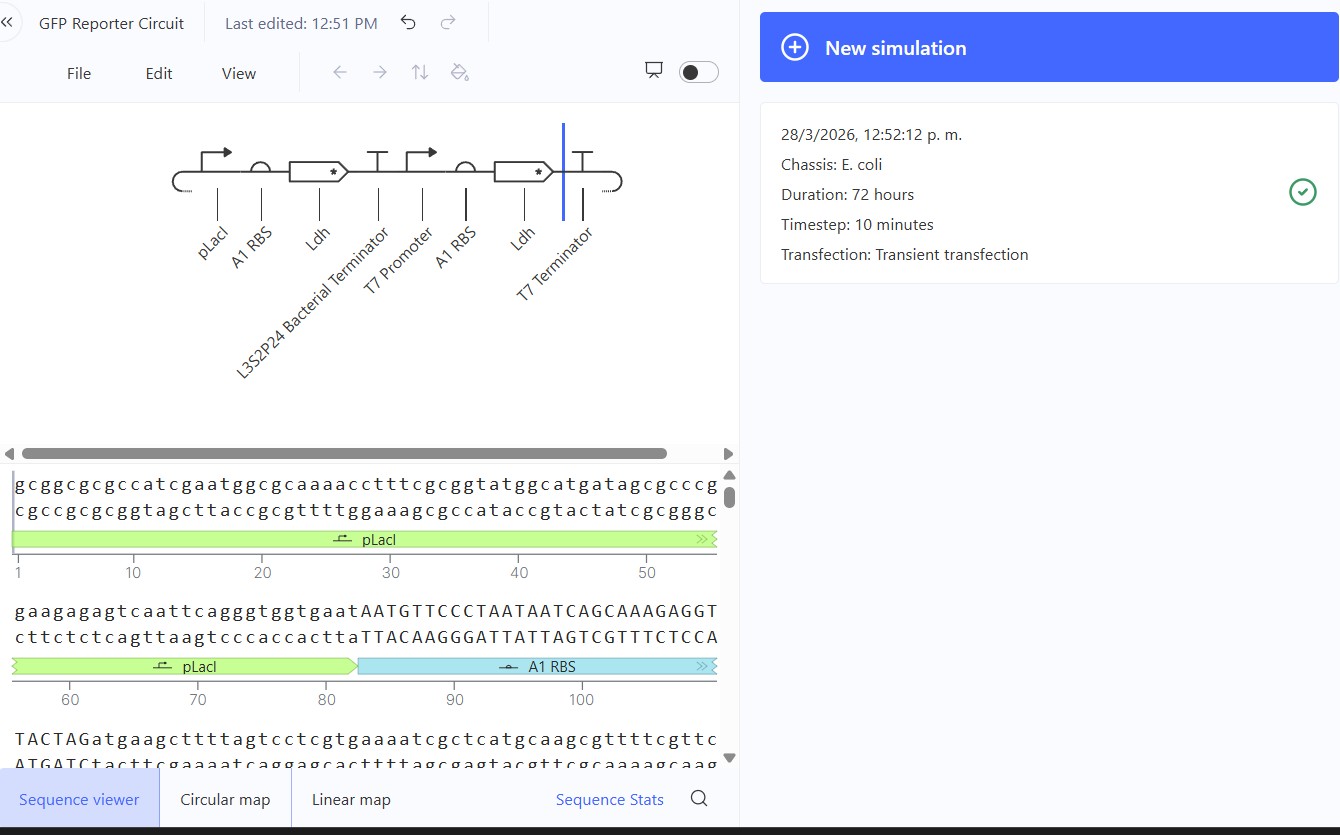
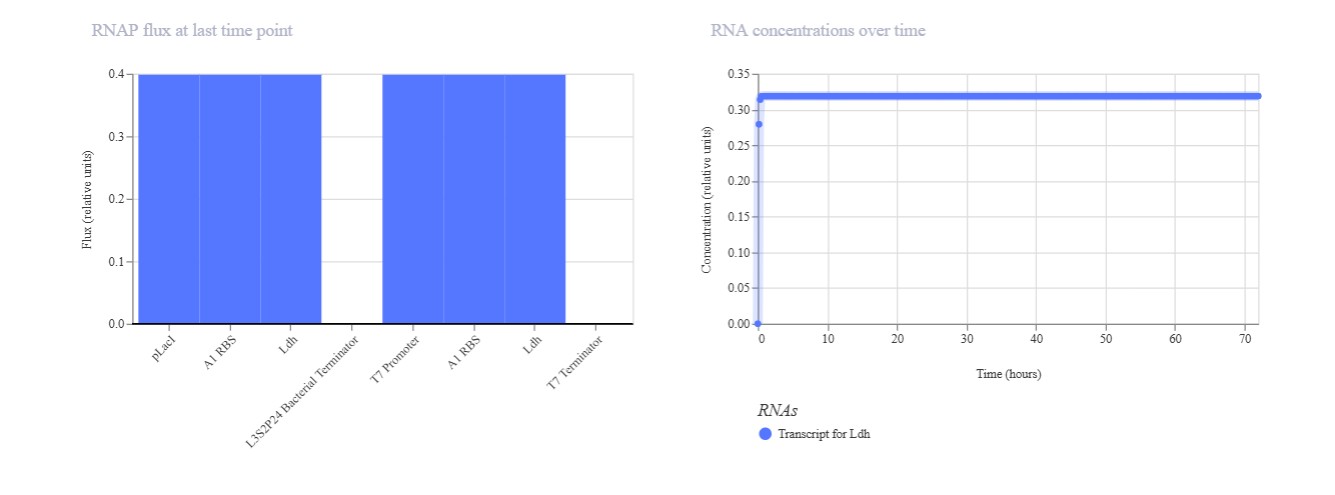
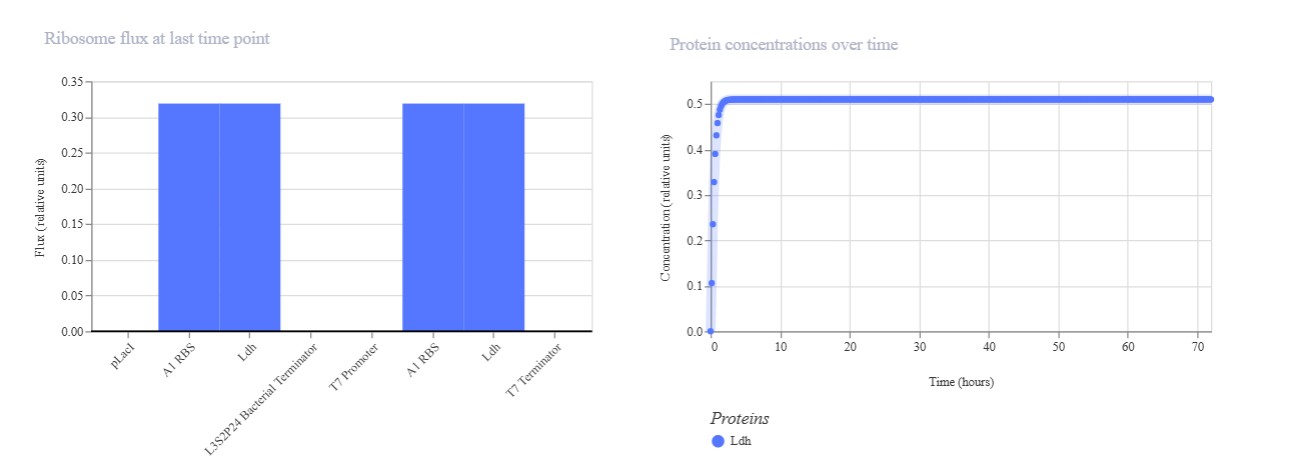
Expected: Comparison of regulated (pLac) vs constitutive (T7) promoters driving identical Ldh reporter. pLac repressed (low), T7 constitutive (high) output. Actual: Perfect promoter strength comparison. T7 promoter dominated (mRNA/protein ~1.0) while pLac remained repressed (~0.2). Confirms T7 » pLac strength hierarchy in E. coli simulation.