Week 2 HW: DNA read / write /edit 🧬
Part I: Benchling & In-silico Gel Art
I created my benchling account and imported the sequence GEN BANK CODE:J02459.1 from the NCBI website. I added the sequence to my benchling proyect folder.
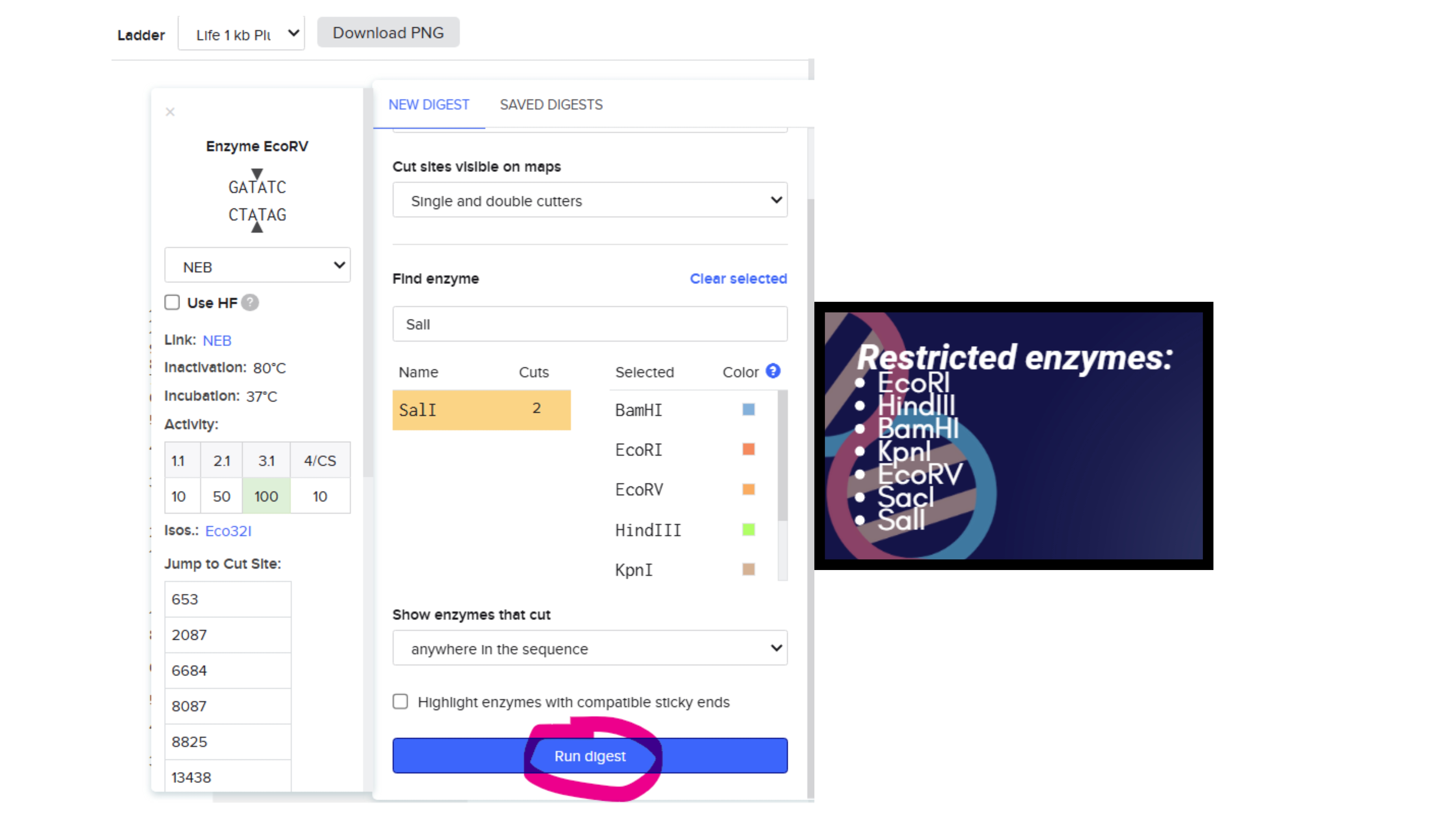
Then I simulated Restriction Enzyme Digestion with adding the following enzymes and then cutted the DNA in the restricted places
Part III: DNA Design Challenge
3.1. Choose your protein:
PROTEIN: Phospholipase A2 Apis mellifera - PA2 APIME
I’ve chosen phospholipase A2 originating from Apis mellifera due to being a key enzymatic factor of honeybee venom and for their study as potential neuroprotective and immunomodulatory agents. Bee venom phospholipase A2 has been reported as having properties that reduce neuroinflammation and protect dopaminergic neurons in animal models of Parkinson’s Disease.
I obtained the protein sequence from Uniprot
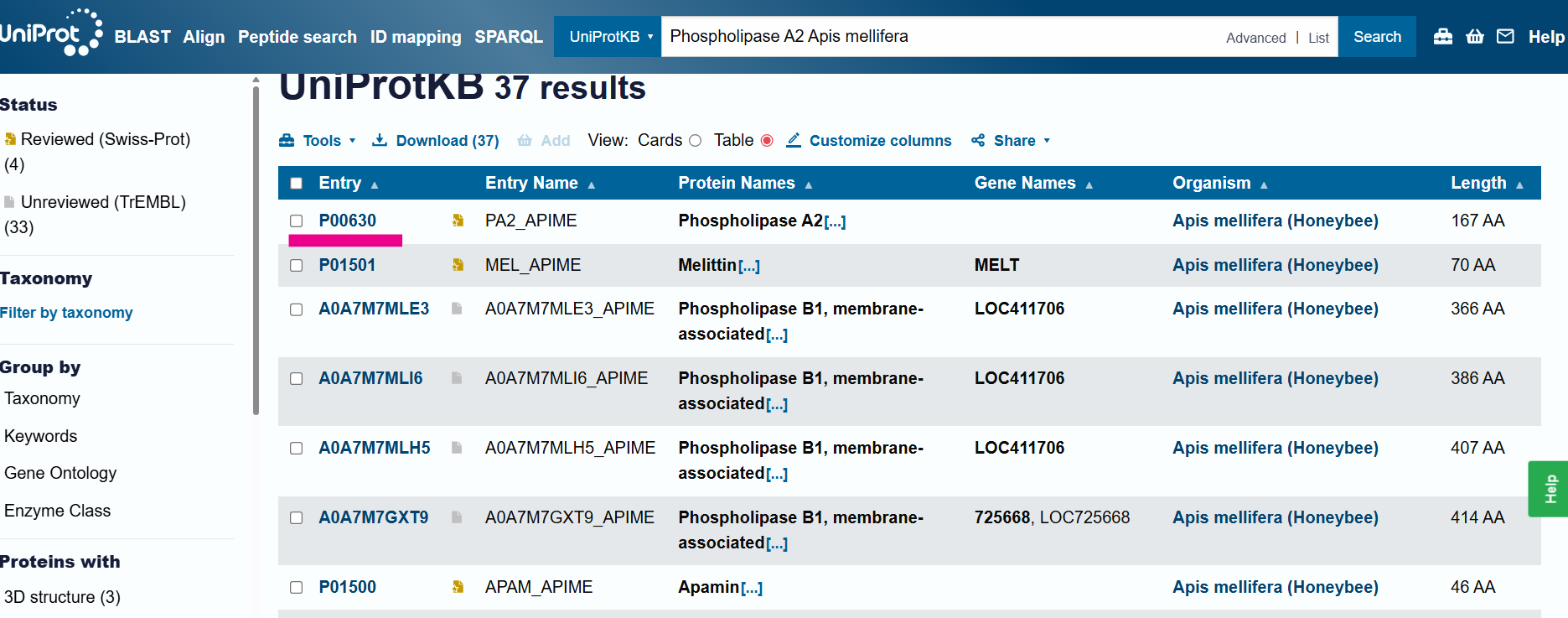
sp|P00630|PA2_APIME Phospholipase A2 OS=Apis mellifera OX=7460 PE=1 SV=3 MQVVLGSLFLLLLSTSHGWQIRDRIGDNELEERIIYPGTLWCGHGNKSSGPNELGRFKHT DACCRTHDMCPDVMSAGESKHGLTNTASHTRLSCDCDDKFYDCLKNSADTISSYFVGKMY FNLIDTKCYKLEHPVTGCGERTEGRCLHYTVDKSKPKVYQWFDLRKY
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
Using a reverse translation tool, I converted my amino acid sequence into a nucleotide sequence based on the standard genetic code. Because multiple codons can encode the same amino acid, the resulting sequence represents one of several possible valid sequences that could produce this protein. I obtained the nucleotides using the following tool available on internet: https://www.bioinformatics.org/sms2/rev_trans.html
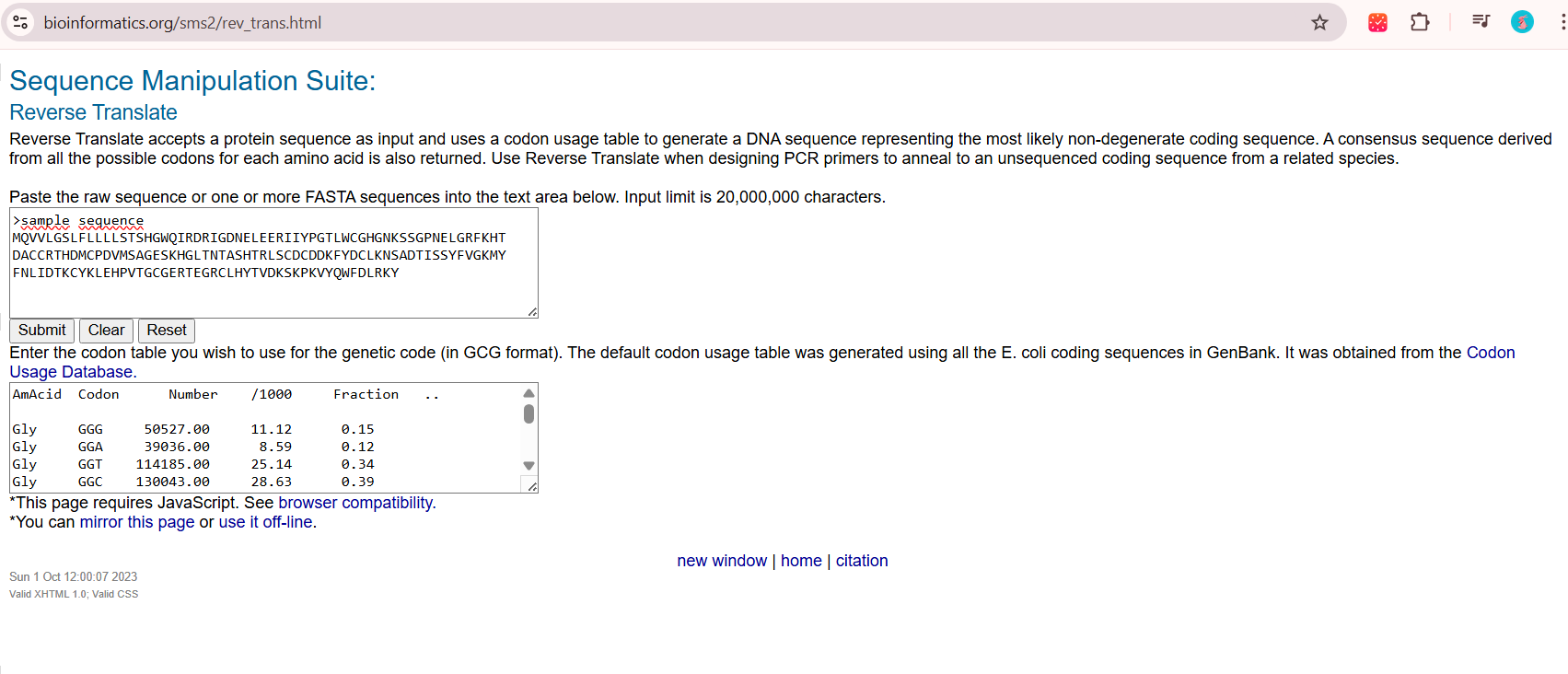
reverse translation of sample sequence to a 501 base sequence of most likely codons. atgcaggtggtgctgggcagcctgtttctgctgctgctgagcaccagccatggctggcag attcgcgatcgcattggcgataacgaactggaagaacgcattatttatccgggcaccctg tggtgcggccatggcaacaaaagcagcggcccgaacgaactgggccgctttaaacatacc gatgcgtgctgccgcacccatgatatgtgcccggatgtgatgagcgcgggcgaaagcaaa catggcctgaccaacaccgcgagccatacccgcctgagctgcgattgcgatgataaattt tatgattgcctgaaaaacagcgcggataccattagcagctattttgtgggcaaaatgtat tttaacctgattgataccaaatgctataaactggaacatccggtgaccggctgcggcgaa cgcaccgaaggccgctgcctgcattataccgtggataaaagcaaaccgaaagtgtatcag tggtttgatctgcgcaaatat
3.3. Codon optimization.
I need to optimize my codons so that the chosen organism can express them accurately.
Chosen organism: E. coli
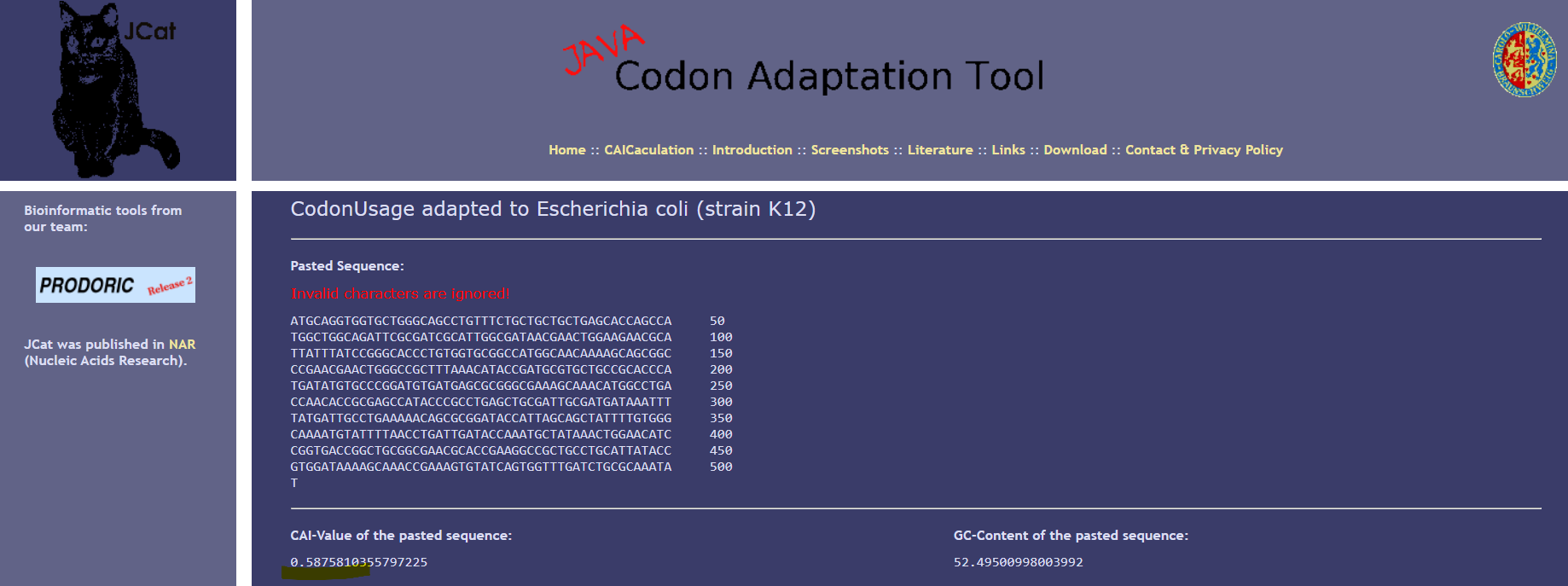
I optimized my sequence for expression in E. coli K12 usign the following tool: JCat I realized that the Codon Adaptation Index improve from a value of 0.58 to 1.0, indicating optimal codon usage for the host organism. The GC content of the optimized sequence (51.3%) is consistent with the genomic GC content of E. coli.

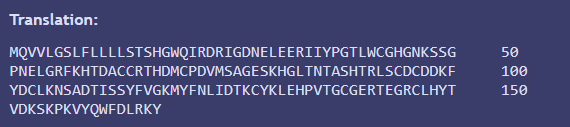
Translation of the optimized sequence confirmed that the amino acid sequence remained unchanged because it matches with the previous protein equence I obtained in part 3.1 from Uniprot.
3.4. You have a sequence! Now what?
Using an expression vector, the recombinant DNA sequence (which has an optimized codon) can be cloned into a host organism. The host cell transcribes this new DNA sequence into mRNA, which is then translated into protein. Protein production can be done using either cell-based or cell-free expression systems; however, toxic proteins should generally be produced using a cell-free expression system to avoid harming the host organism.
Part V: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? I want to analyze eDNA (environmental DNA for short) from the bees’ hives for tracking viruses and monitoring the local flora. It’s important to identify the bacterial/fungal populations and the presence of viruses within the hives to combat Colony Collapse Disorder and to maintain healthy populations of pollinators capable of producing the Phospholipase A2 I’m researching. (ii) What technology or technologies would you use to perform sequencing on your DNA and why? I would use Oxford Nanopore Sequencing because of its portability and ability to produce long reads, which are essential for identifying complex microbial communities in the field *GENERATION: Third generation sequencing system allows sequencing of individual DNA molecules without PCR amplification. *INPUT AND PREPARATION: Input consists of high molecular weight genomic DNA. Library preparation requires breaking genomic DNA into smaller pieces (if needed) and then inserting a sequencing adapter at both ends of each piece. The sequencing adapter contains a “motor-protein” that controls the passage of the DNA through the nanopore. *ESSENTIAL STEPS AND BASE CALLING: During sequencing, the DNA molecule passes through a protein nanopore in an electrically charged membrane. Movement of the DNA through the nanopore causes electrical current changes. The base calling algorithm uses a neural network to translate the electrical current changes into a nucleotide A, T, G or C. *OUTPUT: The final output is a FASTQ file for the output of a long read sequence and quality score.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? I would want to synthesize the expression cassette for the honeybee Phospholipase A2 (PA2_APIME) that I designed in Part 4. The goal is to produce this protein in a cell-free system to study its potential neuroprotective properties for Parkinson’s Disease research without the toxicity issues associated with cellular expression. (ii) What technology or technologies would you use to perform this DNA synthesis and why? I would use Silicon-based DNA synthesis (Twist Bioscience). This technology allows for high-throughput, accurate synthesis of gene fragments and clonal genes at a significantly lower cost than traditional methods *ESSENTIAL STEPS: The process starts with a silicon chip where thousands of oligonucleotides are synthesized in parallel using a phosphoramidite-based chemical process. These small fragments are then harvested, assembled into longer genes (using methods like Gibson Assembly), and verified for accuracy through NGS *LIMITATIONS: While highly scalable, limitations include complexity constraints; sequences with high GC content or long repetitive regions are difficult to synthesize accurately and may take longer to produce or fail the “complexity check.
5.3 DNA Edit
(i) What DNA would you want to edit and why? I would like to edit the genome of E. coli host strains to improve their tolerance to the expression of toxic proteins like Phospholipase A2. By editing specific metabolic pathways or membrane transporters, we could create “super-strains” optimized for the bioproduction of therapeutic venom proteins (ii) What technology or technologies would you use to perform these DNA edits and why? I would use CRISPR/Cas9 because of its high precision, ease of programming via a guide RNA (gRNA), and ability to perform multiple edits simultaneously (multiplexing) *HOW IT EDITS: The Cas9 nuclease acts like molecular scissors. Directed by a gRNA, it binds to a specific target sequence and creates a double-strand break (DSB). The cell then repairs this break through Non-Homologous End Joining (NHEJ) to knock out a gene, or Homology-Directed Repair (HDR) to insert a specific sequence if a template is provided. *PREPARATION AND INPUT: The required inputs include the Cas9 enzyme (or a plasmid encoding it), a custom-designed gRNA targeting the site of interest, and a repair template (DNA) if a specific insertion or change is desired *LIMITATIONS: The main limitations are off-target effects (unintended edits at similar sequences) and varying efficiency depending on the cell type and the specific genomic location being targeted.