Subsections of Homework
Week 1 HW: Principles and Practices
Important: I use ChatGPT and Gemini to help me organize my ideas, translate some concepts and reunite everything!!
Chagas disease is a major public health problem in Latin America, especially in underserved regions. In Argentina, this disease mainly affects the northern part of the country, where high temperatures, rural housing conditions, poverty and limited access to healthcare favor the presence and spread of the insect vector vinchuca or “kissing bug” (scientifically called Triatoma Infestans). Many people living in these areas are diagnosed late or not diagnosed at all.
Early diagnosis is extremely important, since treatment is much more effective in the early stages of infection. However, current diagnostic methods often require multiple tests, specialized equipment, and trained personnel, which are not available at all in low-resource settings. In addition to this, several provinces such as Santiago del Estero, Formosa, Chaco, Tucuman and Jujuy include regions that are extremely difficult to access. Areas like El Impenetrable in Chaco or the rural forested regions are home of communities that lack basic services such as water, electricity and reliable transportation. These create favorable conditions for the presence and reproduction of vinchucas.
Given this, I propose the development of a nanobiosensor for the early diagnosis of Trypanosoma cruzi infection. The idea is to create a portable, low-cost diagnostic tool that could detect parasite-specific biomarkers such as Tc24 and SAPA proteins in simple biological samples like blood, allowing the fast and early diagnosis outside of centralized laboratories.
Governance/Policy goals
The main governance goal of this project is to ensure that the nanobiosensor is developed and used in a safe, ethical and socially responsible way, and it’s accessible to everyone, in order to maximize its public health benefits and minimizing potential harms.
Prevent harm and ensure safe use (non-malfeasance): this technology should not cause any harm due to inaccurate results, misuse, or unsafe handling. It should be easy to use, handled by a primary care doctor.
Promote equity and fair access: this nanobiosensor should be accessible to everyone, especially to those most affected rural communities. The goal is the implementation of “Open Science” licensing for sensor’s design to allow local manufacturing in Argentina, reducing dependency on expensive imports, to avoid creating a technology that only benefits massive healthcare systems.
Build trust and responsible data use: people must trust the diagnostic tool and the institutions using it. This includes clear communication, informed consent, responsible use of data, and education in healthcare systems and machinery.
Potential Governance Actions
a. Field-based validation and approval for point-of-care diagnostics
Purpose: nowadays, the diagnosis of Trypanosoma cruzi infection relies mainly on laboratory-based methods such as ELISA and PCR, which require specialized infrastructure and trained personnel, limiting their use in rural and low-resource endemic areas. At the same time, recent studies have explored biosensor-based diagnostics for other parasitic diseases, including malaria and leishmaniasis, demonstrating the potential of nanobiosensors for rapid and point-of-care detection. However, similar technologies for Chagas disease remain underdeveloped and poorly implemented.
Design: this biosensor would be tested in real environmental conditions (high temperature, limited infrastructure, low-income facilities) in collaboration with local primary care centers and rural doctors. National health authorities (ANMAT) would review the results before approving large-scale use.
Assumptions: it is expected that the validation and approval would be fast and without obstacles. We could also assume that communities and healthcare professionals will easily adopt this technology but we have to think about a potential resistance from large pharmaceutical companies or major laboratories that may see their market threatened.
Risks of failure and success:
- Risks of failure: if major corporations oppose the technology, it could lead to delays in approval and, consequently, limited availability. This sensor could also not be well-received due to a lack of local infrastructure or training.
- Risks of success: If the sensor is successfully adopted, it could create a demand that outpaces local production capacity, potentially causing shortages or uneven distribution.
b. Incentives for affordable and open-access design
Purpose: to reduce inequality in access, this action focuses on keeping the technology affordable and widely available.
Design: public funding or academic grants could require that the nanobiosensor design be shared under open or non-exclusive licenses. Partnerships with public institutions, important researchers and local manufacturers could help reduce costs and support regional production.
Assumptions: this assumes that researchers and developers are willing to share designs, data and manufacturing processes under open or non-exclusive licenses. It also assumes that open access or publicly funded models can remain economically sustainable while maintaining quality standards.
Risks of failure and success:
- Risks of failure: if public funding is insufficient or inconsistent, production quality could decline, resulting in unreliable diagnostic devices. Private companies may also be discouraged from participating due to limited commercial incentives.
- Risks of success: if open-access designs are widely adopted, a lack of clear coordination and quality control could lead to fragmented manufacturing, variable device performance or misuse.
c. Training healthcare workers and engaging communities
Purpose: a diagnostic tool is only useful if used correctly and understood by both patients and healthcare workers.
Design: basic training programs would be implemented for healthcare workers on how to use the sensor, interpret results and explain them to patients. Community education could help reduce fear, stigma or misinformation related to Chagas disease, and also help prevent the infections.
Assumptions: this assumes that training programs can be effectively delivered, that healthcare workers have sufficient time and institutional support to participate and that communities are open to engaging with new diagnostic technologies and to receiving education.
Risks of failure and success:
- Risks of failure: without adequate training or follow-up, healthcare workers may misinterpret results or use this tool incorrectly, leading to inaccurate diagnoses or loss of trust.
- Risks of success: if training and community engagement are highly effective, diagnosis rates may increase rapidly. This could potentially overwhelm healthcare systems that are not fully prepared to provide treatment, monitoring, or long-term care.
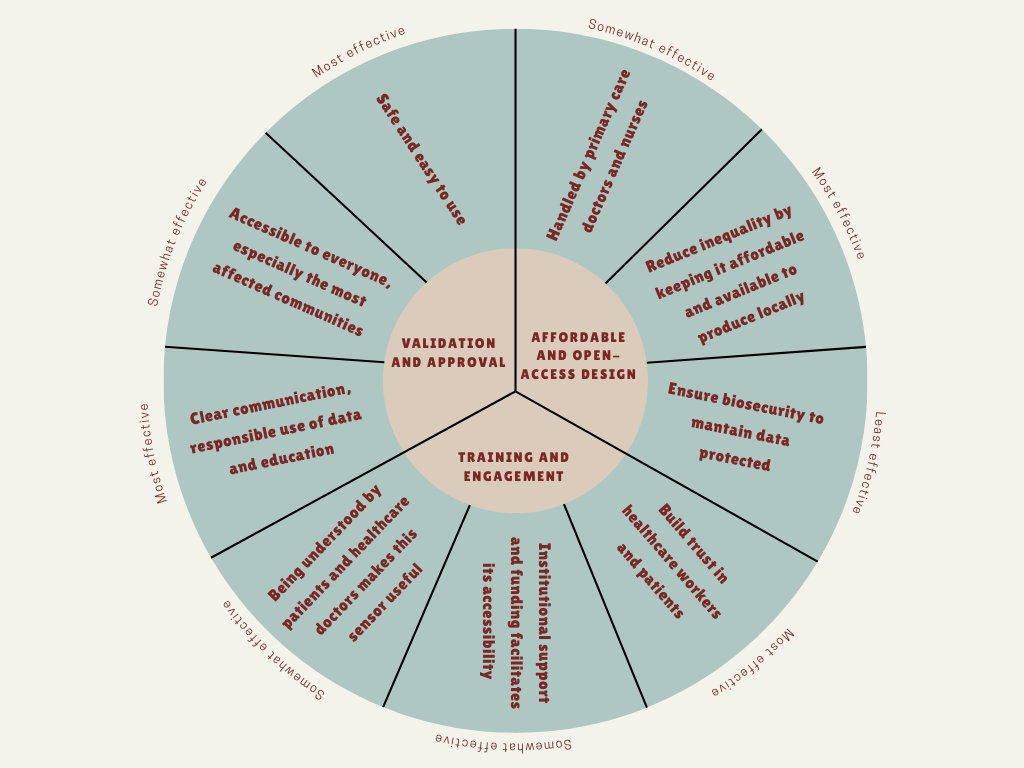
Based on the previous analysis, I would recommend a combination of Field validation and approval and Incentives for local manufacturing. I strongly believe that these actions are not sufficient on their own. For example, sharing the sensor’s “blueprints” ensures this technology is accessible and easy to develop, but it does not guarantee the quality control that only formal regulation can provide. Consequently, I propose that ANMAT as a regulatory agency and the Ministry of Health of Argentina should provide funding and legal approval exclusively to projects that commit to manufacturing this nanobiosensors, and keeping prices accessible not only for the public health system, but also for researchers and scientific institutes.
What we win and what we risk:
- To make the sensor low-cost and locally made, we must take on more responsibility in supervising the process. We will be choosing the “harder” path of managing our own production instead of the “easier” but more expensive path of importing finished technology from abroad. It’s the only way to become independent.
- Adjusting and testing the devices in the extreme conditions where most of the damned patients live will delay the official launch. However, this is a necessary sacrifice. Otherwise, we risk the sensors failing due to environmental conditions, leading to false negatives and destroying the community’s trust in the program.
Assumptions and Uncertainties:
- Political will: this plan assumes that the National Health System will keep Chagas disease as a priority and will not cut the budget needed for its treatment.
- Pathogen evolution: it is well known that pathogens tend to mutate in order to adapt and survive new environmental conditions, so it is uncertain if the protein used in this sensor will continue to function in a future. This is why this device will need periodic updates, as well as the studies on T. cruzi.
- Digital infrastructure: we assume that, even in remote areas, there will be a basic way to perform this analysis and measurements, and there will always be a healthcare worker around to do it.
Subsections of Week 1 HW: Principles and Practices
Week 2 Lecture Prep Assignments
Professor Jacobson:
Biological DNA polymerase is an enzyme that couples a 5’-3’ polymerization domain with a 3’-5’ exonuclease proofreading domain. As this enzyme moves along the template DNA strand, it adds deoxynucleoside-triphosphates (dNTPs) complementary to the exposed base, forming a phosphodiester bond at the primer’s 3’-OH.
This enzyme has an error rate of 1:106 (one error for every million base additions). If an incorrect nucleotide is incorporated, the resulting mismatch destabilizes the nascent strand, the polymerase pauses, and the mismatched base is transferred into an exonuclease pocket, where the 3’-5’ exonuclease clives it off, inserting then the correct base.
The human genome contains about 3.2 billion base pairs, so without further correction, a single replication of the genome would result in approximately 3200 errors. To deal with this discrepancy, biology uses error-correcting mechanisms to mitigate this mismatch:
Polymerase proofreading that removes misincorporated nucleotides,
Post-replication mismatch repair that scans the newly synthesized strand for remaining errors, such as the MutS Repair System, and
Redundancy from having two homologous chromosome sets, allowing cellular quality-control checkpoints to detect and eliminate damaged cells.
An average human protein is encoded by about 1036 bp of coding DNA (≈345 amino acids). Since the genetic code is degenerate, 62 codons specify the same 20 amino acids, where each amino acid is encoded by 2 to 6 different codons.
When synthesizing or expressing these proteins, only a small fraction of these sequences are usable because the DNA and its transcript prevent synonymous codes from being equally effective through several factors like:
Secondary structure interference, where certain DNA or mRNA sequences may fold into stable minimum free energy “hairpins”, blocking the cellular machinery from translating the code,
Codon-bias and tRNA availability: cells preferentially use codons that match abundant tRNAs,
Regulatory motifs: accidental creation of splice sites, ribosome-binding sites or motifs recognized by cellular enzymes which target the mRNA for destruction,
GC-content and stability: extreme GC-rich or AT-rich regions affect DNA stability, replication and transcription efficiency,
and many more factors are the reasons why all of these different codes don’t work for a single protein of interest.
Dr. LeProust
The most used method for oligo synthesis currently is the phosphoramidite method, a chemical process that involves a four-step cycle repeated for each nucleotide added: coupling, capping, oxidation and deblocking.
Direct synthesis of oligonucleotides longer than 200 nt is difficult due to the accumulation of chemical errors and truncated/mutated sequences with each cycle, which significantly reduces the yield of full-length sequences.
Due to the inefficiencies mentioned above, it is not possible to make a 2000 bp gene via direct oligo synthesis, because the yield for a single strand of that length would be effectively zero. Because of the 1:100 error rate, this long sequence would likely contain at least 20 error, making it biologically non-functional. Besides, direct chemical synthesis is generally limited to around 200-300 nt. Instead, genes of this length are created through assembly, using techniques like PCR assembly or Gibson assembly, in order to assemble shorter oligos.
Dr. Church
- The 10 amino acids generally considered essential for animals are:
- Arginine
- Histidine
- Isoleucine
- Leucine
- Lysine
- Methionine
- Phenylalanine
- Threonine
- Valine
- Threonine
The “Lysine Contingency” in Jurassic Park movies was a genetic alteration developed by Dr. Henry Wu where the dinosaurs were engineered to be unable to produce the essential amino acid Lysine. The idea was that the animals would die if they escaped the park because they wouldn’t have access to the lysine supplements provided by their carers.
Knowing that Lysine is already an essential amino acid this breaks the logic of this contingency because animals (including dinosaurs) generally can not produce lysine naturally, so the genetic modification to “remove” this ability was redundant, because they already had it. Therefore, all animals obtain lysine by eating plants or other animals, like red or white meat, cheese, eggs, soy, etc. If the dinosaurs escaped, they would not die from a lack of supplements, they would simply survive by eating standard protein-rich food sources found in the wild.
Week 2 HW: DNA Read, Write and Edit

–> This image shows DNA fragments separated by agarose gel electrophoresis and stained with a fluorescent dye, performed during my Genetic Engineering course.
Part 1: Benchling and In-silico Gel Art
According to the instructions, this is the Gel I designed for p53 human protein (tried my best to make it look like a butterfly i’m sorry!!)
Part 3: DNA Design Challenge
For this assignment I have chosen the human protein p53, often called “guardian of the genome” is a critical tumor suppressor that maintains genomic stability by regulating cycle cell processes, promoting DNA repairing, and inducing apoptosis or senescence in response to cellular stress. It acts as a transcription factor, binding to DNA to stop damaged cells proliferation, potentially cancerous genetic material. I find this protein interesting because of its crucial role in maintaining cellular integrity. I was drawn to it during my medical genetics course last year due to its key function in controlling cell proliferation and its ability to trigger cell cycle arrest or apoptosis when DNA damage is detected. This protein is mutated in nearly 50% of known cancers in humans, and it is also involved in processes like aging, metabolism and DNA repair. This wide range of actions makes p53 an incredibly interesting protein to study in synbio.
After doing the reverse translation on my protein sequence to obtain the DNA sequence, I have performed the codon optimization. This is a fundamental part of genetic engineering because different organisms prefer different codons to code for the same amino acid. By optimizing the codons, you align the sequence with the organism’s natural tRNA abundance, which improves translation efficiency. Without doing this, even if the gene is correct, it might not be efficiently expressed, which can lead to low protein yields or even no expression at all.
I have chosen the optimization for human organisms and I would prefer to express them on HEK293 cells because they are human-derived, making them ideal for expressing human proteins. There are easy to transfect, grow quickly, and they support post-translational modifications, which are crucial for many human proteins.
Now I have the optimized sequence, to produce the protein from the DNA, it is possible to amplify the sequence using PCR, designing primers that include specific restriction sites. Then, I amplify my gene and insert it into an expression vector such as a pEASY or pUC plasmid. This vector has to contain a strong promoter, like the CMV promoter, which is essential for expression in eukaryotic cells. After that, I have to linearize the plasmid using the same restriction enzymes, and transform the HEK293 cells by electroporation. Then, the cells are incubated in fresh culture medium to allow recovery and expression of the introduced DNA. The plasmid is transcribed into mRNA and then translated into the target protein by the cellular machinery. It is also important to have a selectable marker, such as antibiotic selection, to know which cells are expressing the gene. Finally, protein expression can be confirmed using techniques such as Western Blot, ELISA, or fluorescent detection if a GFP was added.
3.5 (Optional)
A single gene can code for multiple proteins at the transcriptional level primarily through alternative splicing, a process where the pre-mRNA, transcribed from the DNA, is spliced in different ways to include or exclude exons (coding regions).
Alignment of the coding DNA and RNA sequence with its translated amino acid sequence for the p53 protein.
Part 4: Prepare a Twist DNA Synthesis order
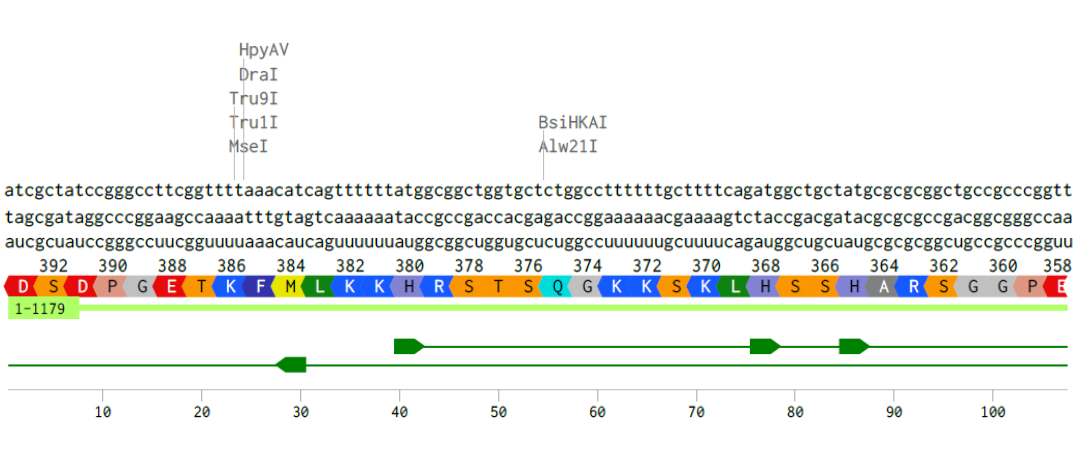
I have built the DNA insert sequence according to the instructions on Benchling and this is what I have obtained:

Link to my E. coli sequence: https://benchling.com/s/seq-aEUjDIoXsdjPsD14jzXd?m=slm-BT3BayyvXI3H27cDW11c
Since I could not have access to the Twist Bioscience software (it says I have to contact a distributor), I was not able to continue with this assignment.
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) I would like to sequence the DNA of the parasite Tritrichomonas foetus, as it is the microorganism I plan to study for my final project. Specifically, I am interested in identifying the genes encoding excretion-secretion antigens, which are believed to play a key role in host-parasite interactions and reproductive impairment, and I want to probe that. Sequencing its DNA would allow me to better understand the molecular basis of its pathogenicity and how these antigens may affect the reproductive capacity of BALB/c mice. Additionally, obtaining genomic information could help identify potential targets for diagnostics, treatments or preventive strategies against infections caused by this parasite.
(ii) I would use NGS for whole-genome sequencing (WGS), as it provides comprehensive coverage of all genes, including my antigens of interest.
This method belongs to second-generation sequencing technologies because it relies on massively parallel sequencing of millions of DNA fragments simultaneously.
The input would be purified genomic DNA extracted from the parasite, which would then be fragmented, followed by adapter ligation and PCR amplification to generate a sequencing library. During sequencing-by-synthesis (e.g. Illumina platform), fluorescently labeled nucleotides are incorporated into the growing DNA strands, and each base is identified by detecting the emitted fluorescence.
The output consists of millions of short sequence reads that can be assembled bioinformatically to reconstruct the genome and identify genes related to pathogenicity and reproductive effects.
5.2 DNA Write
(i) I would like to synthesize a genetic circuit designed as a biosensor to detect excretion-secretion antigens from T. foetus as mentioned above. Early detection in infected bulls is critical because they can act as asymptomatic carriers and spread the parasite to females and other males, causing infertility, significant economic losses in the cattle industry and environmental consequences, as affected animals contribute to the spread of the parasite in pasturelands and water systems. The synthetic DNA construct would include a sensing module responsive to the parasite antigens already mentioned, a regulatory element, and a reporter gene (such as GFP). This biosensor could potentially be used as a rapid diagnostic tool to identify infected animals before the infection spreads within the herd.
(ii) to synthesize the designed genetic circuit, I would use commercial gene synthesis technology, which allows the production of custom DNA sequences with high accuracy, without the need to assemble fragments manually. The essential steps include designing the desired DNA sequence in silico, chemical synthesis of short oligonucleotides, assembly of these fragments into the full-length construct, and cloning into a plasmid vector for delivery. The resulting DNA can be then amplified and used for downstream applications such as transformation into host cells. This method is scalable and precise, however, limitations may include restrictions on how long the DNA sequence can be, the time it takes to receibe the synthesized construct, and possible difficulties with certain sequences, such as those with very high GC content or many repeated regions.
5.3 DNA Edit
(i) I would be interested in editing the human genome using CRISPR-Cas technology to study numerical abnormalities such as trisomy, specifically those causing early miscarriages, preventing babies from reaching full term, or resulting in reduced life expectancy, like Edwards syndrome (trisomy 18) and Patau syndrome (trisomy 13). The goal would be to explore whether removing the extra chromosome in early embryonic stages could restore normal gene dosage and improve developmental outcomes. This type of research could help us better understand the genetic mechanisms underlying severe developmental disorders.
(ii) to perform these DNA edits, I would use CRISPR-Cas9 genome editing technology because it allows precise and targeted modification of specific DNA sequences. This system works by using a guide RNA (sgRNA) designed to match the target DNA region, which directs the Cas9 enzyme to create a double-strand break at that site. The cell’s natural DNA repair mechanisms then repair the break, either by non-homologous end joining or homology-directed repair (which is expected to remove or modify genetic material).
Preparation involves designing specific sgRNAs targeting sequences on the extra chromosome, and delivering the complex Cas9-sgRNAs into cells or embryos, typically using vectors.
Limitations of this method include possible off-target effects, incomplete editing efficiency and mosaicism, where not all cells are edited in the same way.
Week 3 HW: Lab Automation
Week 3: Lab Automation

Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Olsen J.V. et al. Fully Automated Workflow for Integrated Sample Digestion and Evotip Loading Enabling High-Throughput Clinical Proteomics (2024) Mol Cell Proteomics 23(7), 100790. DOI 10.1016/j.mcpro.2024.100790
This article describes a fully automated workflow for preparing clinical proteomics samples, from protein digestion to loading peptides into Evotips (disposable, tip-based C18 reversed-phase trap columns), ready for LC-MS/MS analysis.
They use the Opentrons OT-2 liquid-handling robot, which controls all preparation steps without manual intervention after the initial loading of reagents. The process combines protein capture through aggregation on magnetic beads with enzymatic digestion and, without centrifugation steps, directly transfers the peptides to Evotips using positive pressure, all programmed through downloadable scripts from the Evosep website.
Using this method, up to 192 samples can be processed in parallel in approximately 6 h, which equals to 100 samples/day and eliminates human variability.
In tests with HeLa lysates, the workflow identified ~8.000 protein groups and ~130.000 peptides using an 11.5-min gradient on the Orbitrap Astral, demonstrating high sensitivity and reproducibility. It was also applied to 192 plasma samples from patients with metastatic melanoma, revealing clinically relevant protein changes.
- Write a description about what you intend to do with automation tools for your final project.
For my final project, I want to design a nanobiosensor using metallic nanoparticles (such as gold or silver, maybe (if possible) carbon-based materials) to detect excretion-secretion antigens from the parasite Tritrichomonas foetus, which produces a disease called bovine trichomonosis.
Automation tools will help make the experiments more reproducible, faster, and less dependent on manual work. I would use a liquid-handling robot (for example the Opentrons OT-2) to automate repetitive lab tasks, such as preparing nanoparticle solutions, mixing reagents, functionalizing nanoparticles with antibodies or aptamers, performing washing steps, preparing assay plates, etc. this would allow testing many conditions at the same time (for example, different nanoparticle types or concentrations) to find the best sensor design.
Automation could also be used to test the biosensor performance, maybe by adding samples and controls to plates, preparing serial dilutions of the target antigen, running multiple detection assays in parallel, measuring signals, etc. that would help evaluate sensitivity and specificity of the sensor more efficiently.
I also plan to design simple 3D-printed holders to organize tubes, microplates or sensor chips on the robot deck.
If available, the Ginkgo Nebula platform could be used to screen different antibodies or binding molecules to find the one that recognizes the parasite protein with the highest specificity, improving the performance of the biosensor.
Week 4 HW: Protein Design Part I
Part A. Conceptual questions
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
- Meat has an average 20% of protein, so 500 grams of meat would have 100 grams of protein. An average amino acid mass is 100 Da (100g/mol). So, according to
Avogadro’s number, in a 500 g piece of meat, we are consuming approximately 6.62x1023 molecules of amino acids.
- Why do humans eat beef but do not become a cow, eat fish but do not become fish?
- Dietary proteins are digested into individual amino acids and absorbed and reused to build proteins used for the human metabolism, processes that determine
our identity, not the food we eat.
- Why are there only 20 natural amino acids?
- There are 20 natural amino acids (22 in some organisms) because the standard genetic code evolved to use this 20 structures, maybe because they were
efficient for metabolism, providing sufficient chemical diversity or they were optimal in terms of evolution.
- Can you make other non-natural amino acids? Design some new amino acids.
- Using synbio it is possible to create new amino acids according to what you need or want to do, for example maybe metal-binding amino acids to coordinate
metal ions, or photo-crosslinking amino acids to form bonds when exposed to light, or perhaps adding electronegative elements like Fluor to alter their
electronic properties.
- If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
- L-amino acids form right-handed helices, while D-amino acids would form a left-handed helix.
- Can you discover additional helices in proteins?
- Synbio help us explore and find new structural possibilities since protein folding is certainly a complex issue that, at present, is not fully understood.
Even though they are rare, it is possible to find -helix, 310 helix, foldamers (artificial oligomers), and maybe there are more out there to be discovered.
- Why are most molecular helices right-handed?
- Because biological amino acids are L-chiral, making right-handed helices more sterically favorable and stable energetically.
- Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
- β-sheets are made of protein strands that lie next to each other and are connected by many hydrogen bonds, forming a very flat and extended surface with
many places where it can stick to another sheet, so they can easily attach and stack together. Some amino acids in proteins are hydrophobic and when the β-
sheets form, these parts can be exposed. To avoid water, they stick to each other hiding from water. Aggregation happens because it is energetically
favorable, so the driving forces are the hydrophobic effect, the hydrogen bonds to increase stabilization and sticking together to lower the free energy.
- Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
- In amyloid diseases (like Alzheimer’s) some proteins misfold, losing their normal shape and refolding into β-sheet structures. Since these structures are
very stable, it is hard to go back from there. These misfolded proteins then stick to each other, form long fibers (amyloid fibrils), accumulate in tissues
and damage cells.
Since these structures are very strong, stable and able to self-assemble, it is possible to use them as materials. Scientist are studying them for creating
nanofibers, biomaterials, tissue scaffolds, and drug delivery systems.
Part B. Protein Analysis and Visualization
For this assignment I chose the p53 protein (P04637), which is a multifunctional transcription factor that induces cell cycle arrest, DNA repairs or apoptosis upon binding to its target DNA sequence in human organisms. Acts as a tumor suppressor in many tumor types; induces growth arrest or apoptosis depending on the physiological circumstances and cell type. I particularly find its activity fascinating since I am interested in cancer diseases and trying to understand how they work and how the human body is prepared to regulate these processes.
i. This protein has 393 amino acids, being P (proline) the most frequent one (11.45%)
ii. Running a BLAST analysis, I have found this protein has 250 sequence homologs, going from the organism Pan paniscus (Pygmy chimpanzee) with a 100% homology, to Suricata suricata (Meerkat) with a 79.6%.
iii. This protein belongs to the proteins p53 family, also known as family p53/p63/p73 because they all share a similar structure with conserved domains and related functions.
i. The first high resolution structure solved of the oligomerization domain was deposited in 1994 and released in 1995 using a multi-dimensional NMR. (https://www.rcsb.org/structure/1OLG#entity-1)
ii. This protein is often linked with DNA apart from the protein since it is a transcription factor, so it binds to the DNA to regulate genes. It also has antibody fragments used for crystallization, regulating proteins and peptides like an 11-residue recruitment peptide in a complex with CDK2/CyclinA. Zinc ions are also present, used as a cofactor (binds 1 zinc ion per subunit).
iii. p53 has 6 different domains, and each domain belongs to a different structure classification family:
• 2AC0 A:96-289 (SCOP ID 8024487): Family: p53 DNA-binding domain-like
• 1AIE A:326-356 (SCOP ID 8025247): Family: p53 tetramerization domain
• 2AC0 A:96-289 (SCOP ID 8036866): Family: p53-like transcription factors
• 1AIE A:326-356 (SCOP ID 8037626): Family: p53 tetramerization domain
• 3DAC P:17-28 (SCOP ID 8050972): Family: p53 transactivation domain (TAD)
• 3DAC P:17-28 (SCOP ID 8093389): Family: p53 TAD
- ii.
 Protein visualized as “cartoon”
Protein visualized as “cartoon”
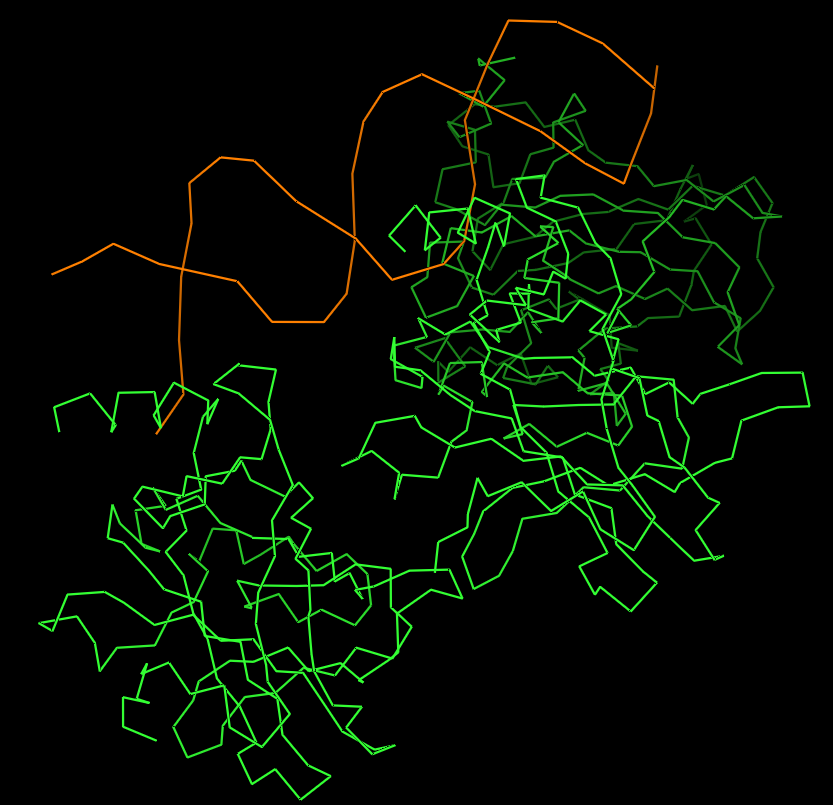 Protein visualized as “ribbon”
Protein visualized as “ribbon”
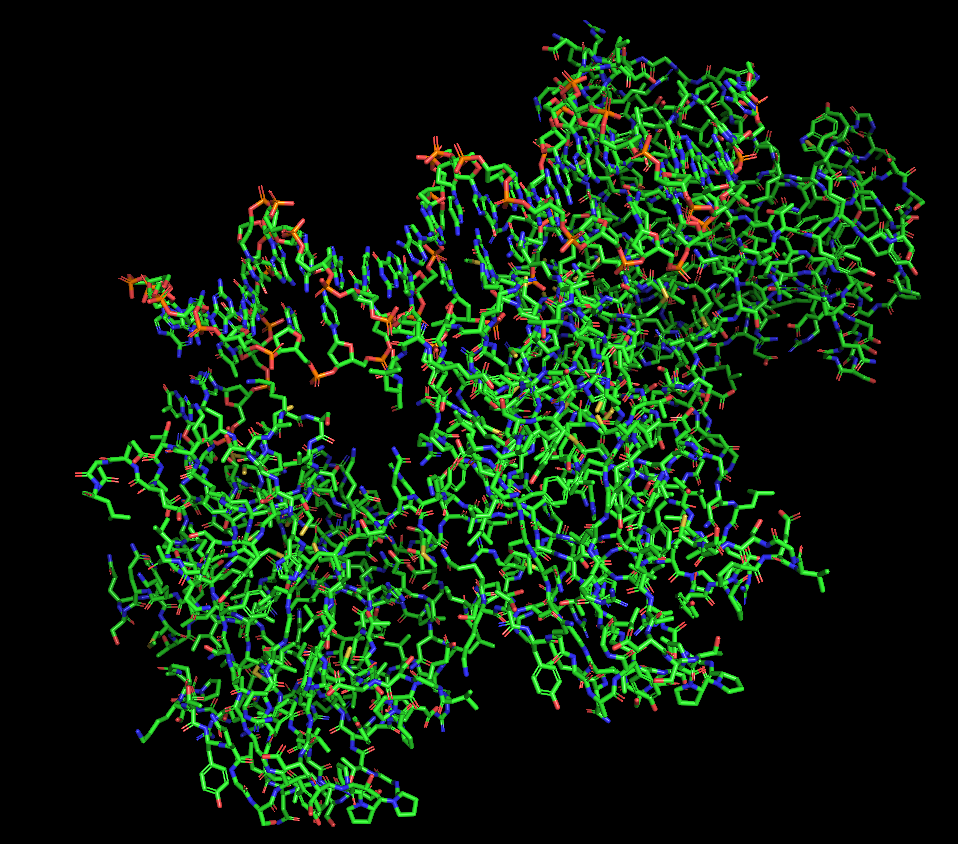 Protein visualized as “sticks”
Protein visualized as “sticks”
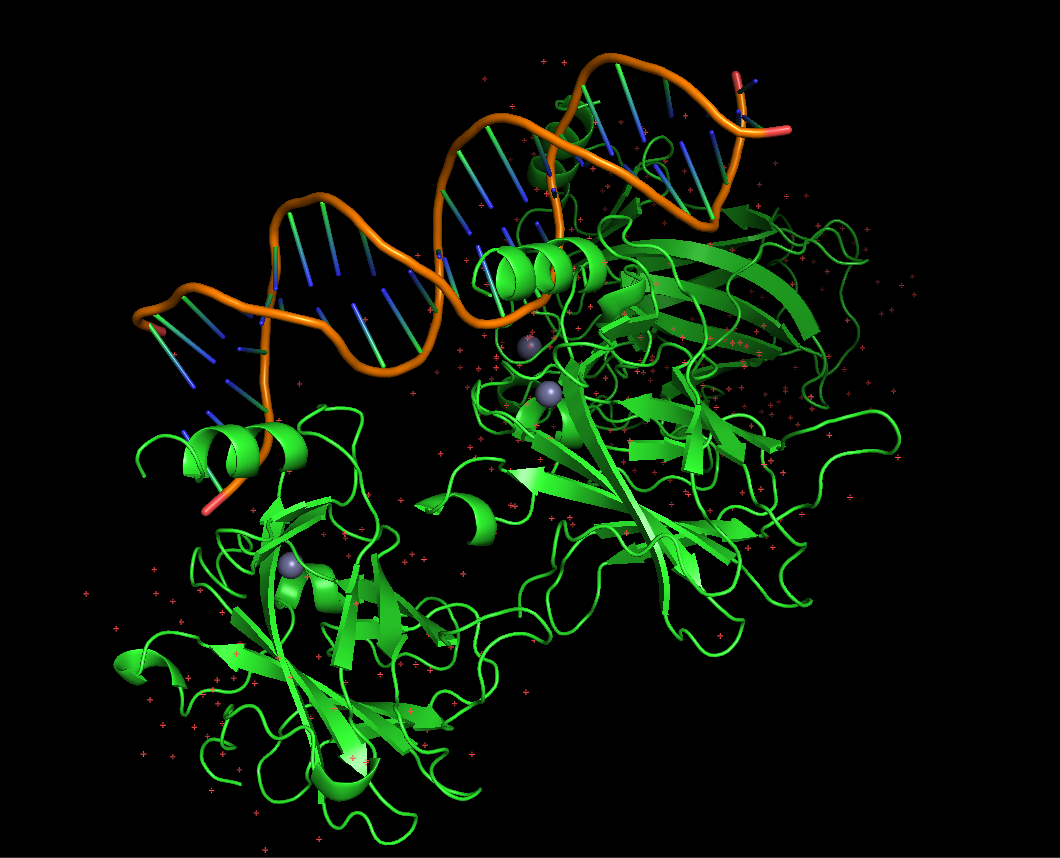
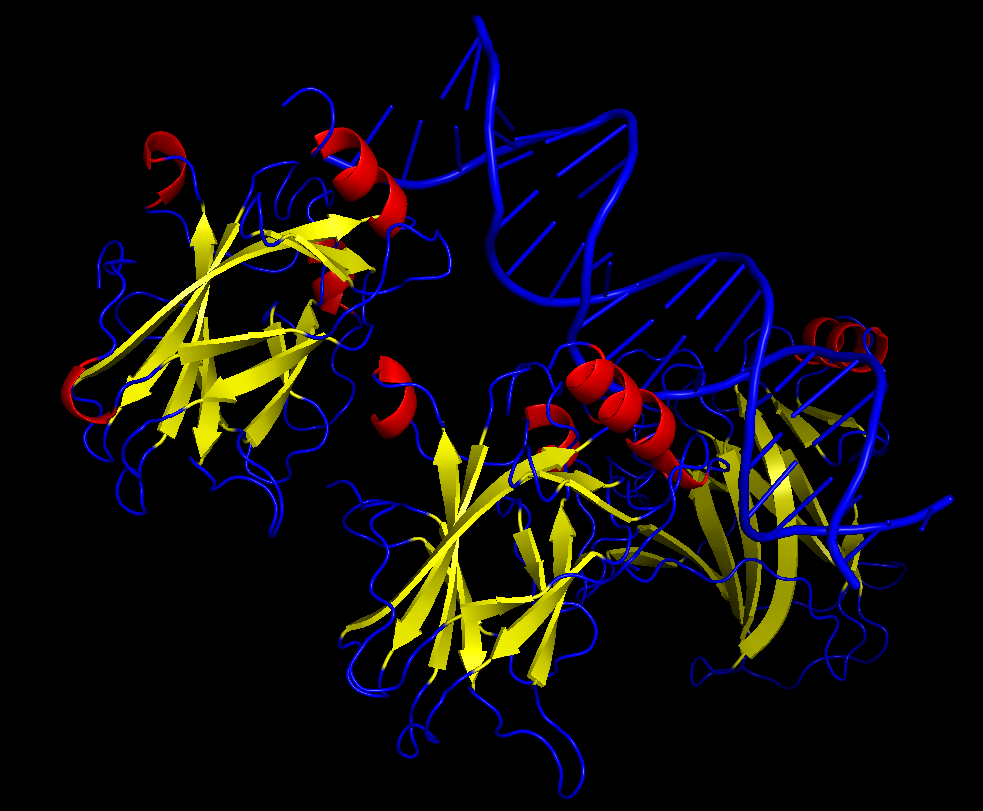
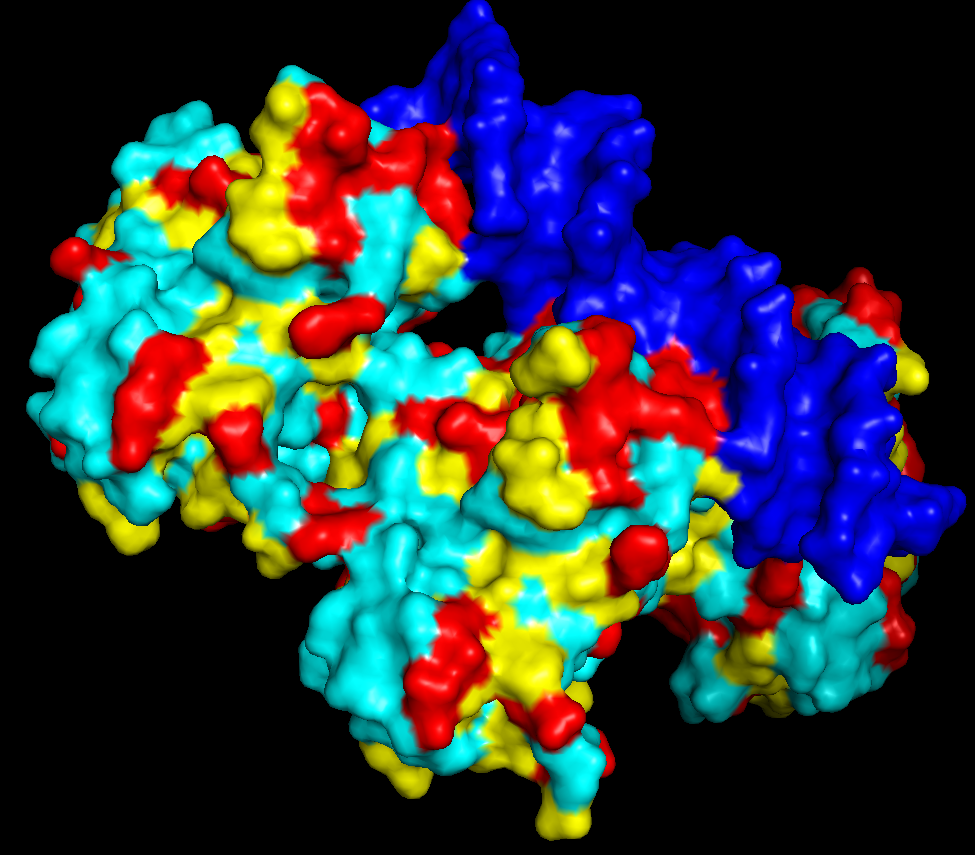
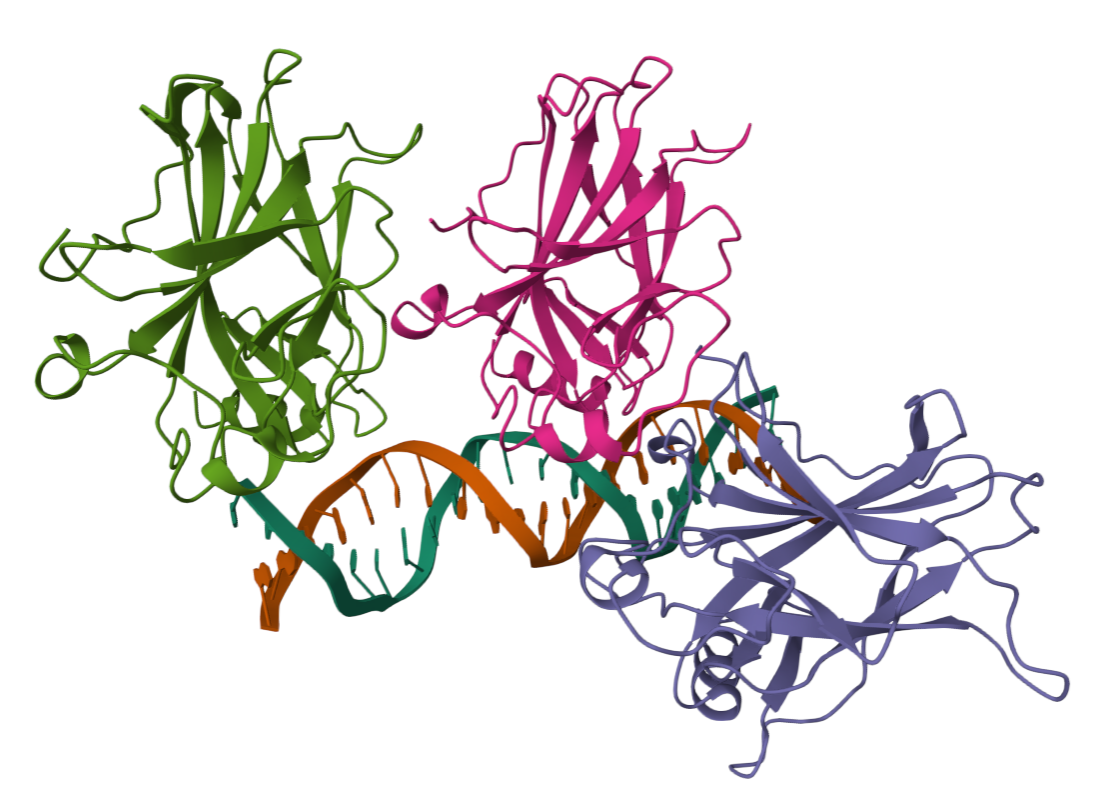
iii. The protein is colored by secondary structure: alpha-helices in red, beta-sheets in yellow, loops in blue. The structure also shows p53 bound to DNA.
iv. Hydrophobic residues (yellow) are mainly located in the interior of the protein forming a stable core. Hydrophilic residues (blue) are most exposed on the surface where they can interact with water or DNA. Charged residues are colored in red.
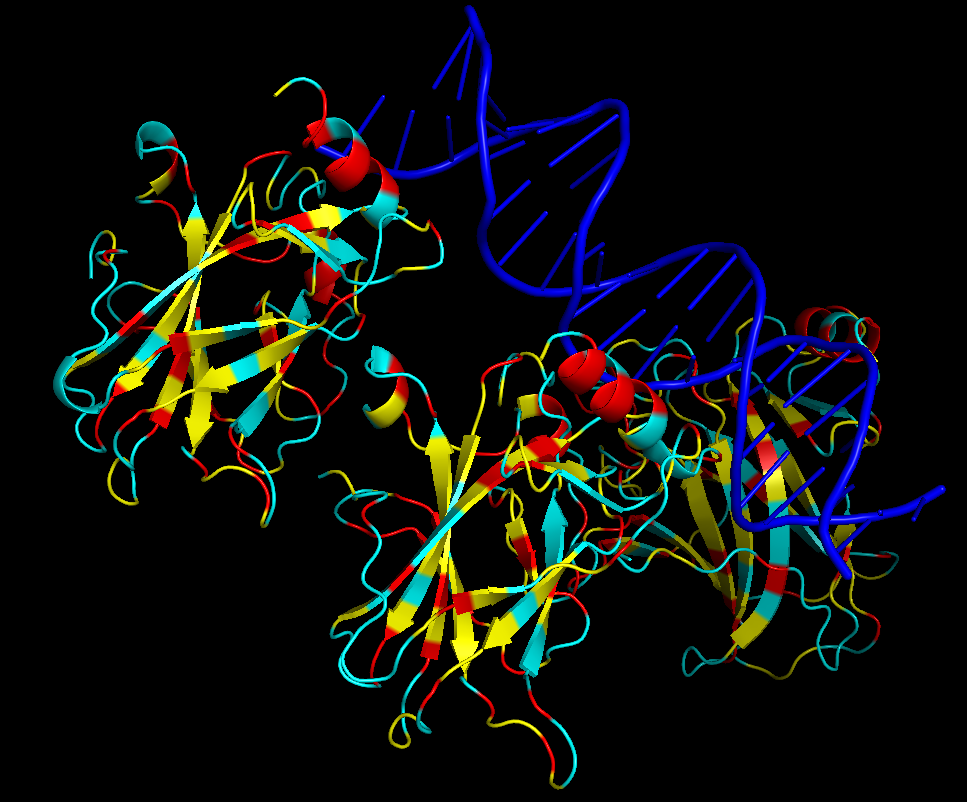
v. The protein surface shows “holes” or binding pockets where other molecules, like DNA or peptides, can bind. Such pockets are important for p53’s biological function.
PART C
C1. Protein Language Modeling
- Deep Mutational Scans
b. After generating the deep mutational scan of protein p53, I have found that most mutations are tolerated in some regions showed in yellow and green colors, but other certain positions display strongly unfavorable mutations across many amino acid substitutions, colored in blue (unfavorable) and dark violet (VERY unfavorable). These positions likely correspond to functionally important residues, such as those involved in DNA binding or structural stability.
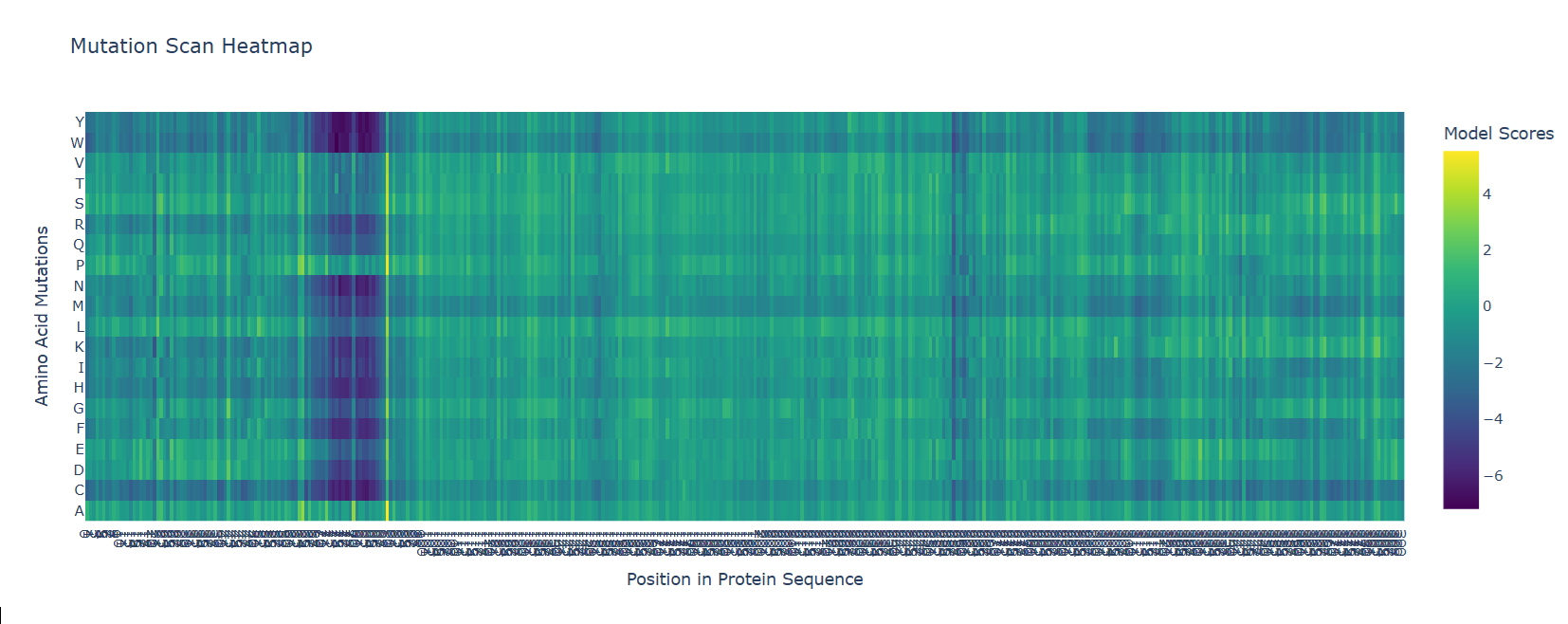 Mutational scan for p53
Mutational scan for p53
A clear pattern is observed at positions where nearly all mutations are highly unfavorable (violet and blue). One mutation that stands out is the substitution at position 74 to tryptophan (W), that shows a strongly negative score (-7.186), suggesting that this mutation is highly unfavorable and likely disrupts the protein’s structure or function.
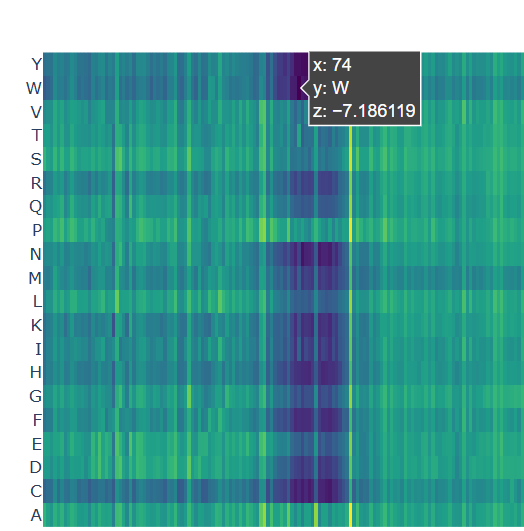 Chosen mutation for p53 (very dark violet)
Chosen mutation for p53 (very dark violet)
- Latent Space Analysis
b. When analyzing the points in the t-SNE 3D plot for the Paramecium caudatum hemoglobin as provided, proteins here are located near a cluster of similar proteins, suggesting functional or structural similarity with its neighbors. For example, I have found a cluster including matches for Clostridium botulinum and the tetanus neurotoxin from C. botulinum.
c. I placed my selected protein in the resulting 3D map, where it sits within a dense cluster of the latent space. Its position suggests that p53 shares common structural features or evolutionary patterns with many other proteins in the dataset.

C2. Protein Folding
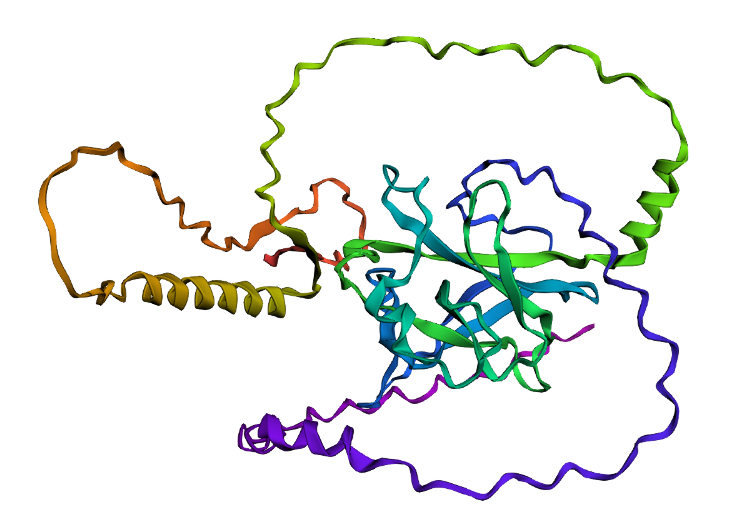
- I folded the p53 protein using ESMFold and compared the predicted structure with the experimental structure from PyMOL (PDB ID: 1TUP). They match partially, because beta-sheets and alpha-helices that form the core of the protein in the ESMFold prediction align closely with the ones in PyMOL. According to the structural differences, PyMOL structure includes p53 bound to DNA as a complex, whereas the ESMFold prediction shows a single isolated chain (monomer).
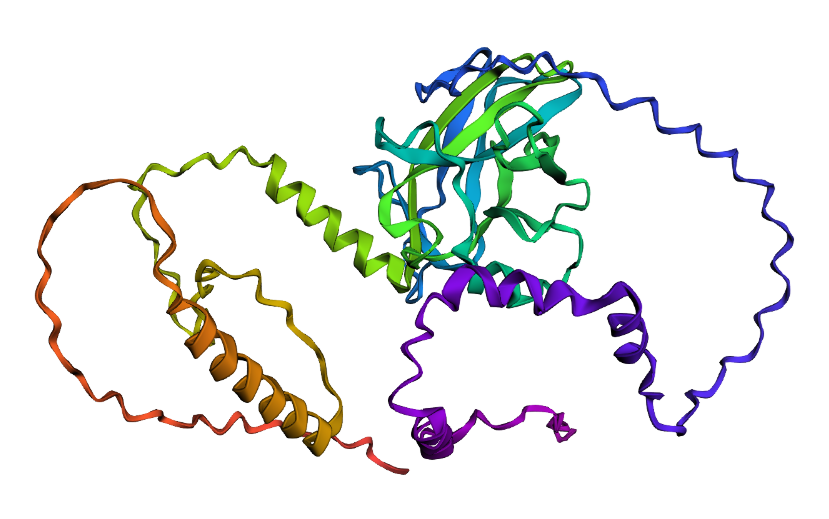 Folded protein structure predicted by ESMFold.
Folded protein structure predicted by ESMFold.
 Folded protein structure predicted by PDB.
Folded protein structure predicted by PDB.
- First I changed only one amino acid and the structure remained stable. Then I changed 5 more amino acids and the folding was a bit different but still possible. Next, changed 20 amino acids but still showing, even though had a different folding structure. Lastly, I deleted 15 amino acids and the protein was still showing with some other structural changes (including the previous changes I made on the sequence). Given the differences, it is possible to say that the core domain maintains its architecture, indicating that the protein exhibits structural robustness.
C3. Protein generation
- The predicted sequence for my protein (PDB: 1TUP) is:
GPPVPPTARDPGAYGFTLGFEATGTGASVTSTYSPALNTIYAKLNAAVPVRLLTTAPPPAGTRVRFRLVYADEAYRTTVVRRSPKAAAADDSDGRRPPDFVLSILDDPDAEYVRDPETGWLSVTVPYRPPPPGATATTYLLAFNETTTAKGGLDGNKVLLVVELLDADGALLGRDSAYVRVVANPGAAAAAAEAAK
And the original sequence is:
MEEPQSDPSVEPPLSQETFSDLWKLLPENNVLSPLPSQAMDDLMLSPDDIEQWFTEDPGPDEAPRMPEAAPPVAPAPAAPTPAAPAPAPSWPLSSSVPSQKTYQGSYGFRLGFLHSGTAKSVTCTYSPALNKMFCQLAKTCPVQLWVDSTPPPGTRVRAMAIYKQSQHMTEVVRRCPHHERCSDSDGLAPPQHLIRVEGNLRVEYLDDRNTFRHSVVVPYEPPEVGSDCTTIHYNYMCNSSCMGGMNRRPILTIITLEDSSGNLLGRNSFEVRVCACPGRDRRTEEENLRKKGEPHHELPPGSTKRALPNNTSSSPQPKKKPLDGEYFTLQIRGRERFEMFRELNEALELKDAQAGKEPGGSRAHSSHLKSKKGQSTSRHKKLMFKTEGPDSD
 Even though I kept getting an error while trying to run the ESMFold with this new sequence (and could not complete the task in my Google Colab), I compared them by myself and I can clearly see that the new predicted sequence is way shorter and different than the original one.
I asked Gemini to compare them and found that there is a very low level of sequence identity between the two of them. The original sequence is 393 amino acids long and includes flexible, disordered regions, while the predicted sequence is shorter (approximately 190 amino acids) and focuses on the DNA-binding core domain. The probabilities from ProteinMPNN show that the model prioritized stability. It replaced many of the original amino acids with new ones that better fit the 3D backbone.
So, even though the letters are different, the predicted 3D structure matches the original p53 backbone almost perfectly (according to Gemini), proving that the model succesfuly redesigned the protein, maintaining its functional shape while using a different primary sequence.
Even though I kept getting an error while trying to run the ESMFold with this new sequence (and could not complete the task in my Google Colab), I compared them by myself and I can clearly see that the new predicted sequence is way shorter and different than the original one.
I asked Gemini to compare them and found that there is a very low level of sequence identity between the two of them. The original sequence is 393 amino acids long and includes flexible, disordered regions, while the predicted sequence is shorter (approximately 190 amino acids) and focuses on the DNA-binding core domain. The probabilities from ProteinMPNN show that the model prioritized stability. It replaced many of the original amino acids with new ones that better fit the 3D backbone.
So, even though the letters are different, the predicted 3D structure matches the original p53 backbone almost perfectly (according to Gemini), proving that the model succesfuly redesigned the protein, maintaining its functional shape while using a different primary sequence.
Part D: Brainstorm on Bacteriohage Engineering
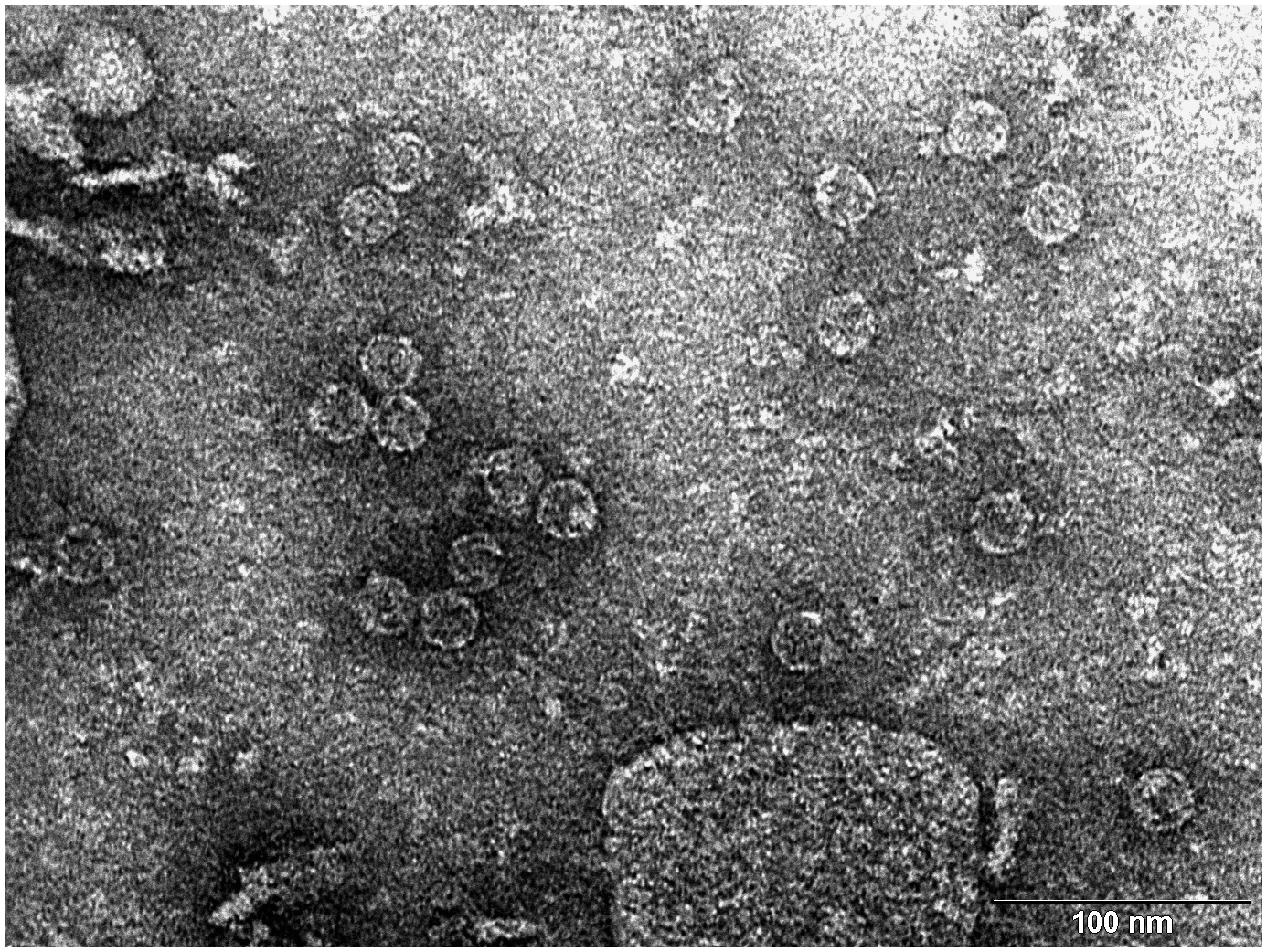 Transmission electron microscopy (TEM) photograph of the intact MS2 phage-like particles (MS2 PLP) present in the supernatant after ultrasonic disruption of E. coli production cells. (Mikel P, Vasickova P, Tesarik R, Malenovska H, Kulich P, Vesely T and Kralik P (2016) Preparation of MS2 Phage-Like Particles and Their Use As Potential Process Control Viruses for Detection and Quantification of Enteric RNA Viruses in Different Matrices. Front. Microbiol. 7:1911. doi: 10.3389/fmicb.2016.01911)
Transmission electron microscopy (TEM) photograph of the intact MS2 phage-like particles (MS2 PLP) present in the supernatant after ultrasonic disruption of E. coli production cells. (Mikel P, Vasickova P, Tesarik R, Malenovska H, Kulich P, Vesely T and Kralik P (2016) Preparation of MS2 Phage-Like Particles and Their Use As Potential Process Control Viruses for Detection and Quantification of Enteric RNA Viruses in Different Matrices. Front. Microbiol. 7:1911. doi: 10.3389/fmicb.2016.01911)
(Important: I couldn’t find a group in this opportunity so I was my own group!)
Proposal: Engineering a DnaJ independent MS2 lysis protein for enhanced Phage Therapy
The L- protein of MS2 phages needs DnaJ because this is a chaperone from the Hsp40 family that helps the full-length L protein fold correctly. In the host cell, DnaJ forms a complex with the highly basic N-terminal domain of L. This complex allows L to adopt a conformation that can interact with its target (still unknown) and cause cell lysis.
When the chaperone is mutated or removed, the lysis process is delayed or completely blocked at certain conditions, even though L accumulates normally, showing that the lack of interaction with DnaJ prevents a step happening after folding, not the synthesis of the toxic protein itself.
According to this, the main goal is to engineer a Dna-J independent version of the MS2 L protein. By removing this dependency and stabilizing the C-terminal lytic core, I aim to create a protein that triggers bacterial lysis faster and more reliably across different bacterial strains without needing additional co-factors.
Using the tools practiced in this weeks’ recitation, this is the proposed bioinformatics pipeline:
Identify the region that really matters: mutagenesis experiments showed that the 67 loss-of-function alleles are concentrated in the C-terminal half of L around the LS motif. The N-terminal domain (residues 1-42) acts as a regulatory break because it creates a strict dependency on the host chaperone DnaJ for proper holding. There, the first 36 to 42 amino acids (N-terminal domain) are nonessential for the killing mechanism itself and removing them speeds up lysis. In addition, the Lytic Core corresponds to the last 30 amino acids, which include the LS motif and the transmembrane helix. So, I will keep the LS motif (Leu48-Ser49) and the Lys50 residue as they are essential for membrane interaction.
Search for homology and keep the essentials:
- Use BLAST against UniProt to obtain L-like sequences from other leviviruses (similar to MS2). Using ESM2 (Protein Language Model), I will perform an in silico Deep Mutational Scan to rank possible mutations, helping me find specific substitutions in the membrane helix that increase stability without breaking the essential LS motif.
Model the structure through Computational Tools:
After finding the best mutation candidates, I will upload the core sequence to ESMFold and visualize it (and compare with PyMOL) to confirm that the transmembrane helix is correctly inserted and capable of membrane insertion.
Once the structure is confirmed, could be useful to use ProteinMPNN to generate a new (and much more robust) sequence for the protein, making it more stable for biotechnology applications.
Finally, I will perform a Latent Space Analysis using t-SNE to validate the engineered designs. This map acts as a functional “sanity check” by clustering the artificial sequence with known active and natural variants of the original protein. If my candidate falls within the functional cluster and stays far away from known loss-of-function mutants (like those affecting the LS motif) it confirms that the protein is likely to be active in the lab, maintaining the original properties necessary to interact with its target.
Potential pitfalls:
Unknown target: since the host membrane target protein is unknown, it is not possible to predict (and confirm) the exact binding interface.
Lysis/assembly balance: if lysis happens to fast, it might kill the bacteria before enough phage progeny are assembled.
Week 5 HW: Protein Design: Part II
PART A: SOD1 Binder Peptide Design
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
SOD1 sequence: MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
SOD1 Mutated (A4V) sequence: MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Part 1: Generate Binders with PepMLM
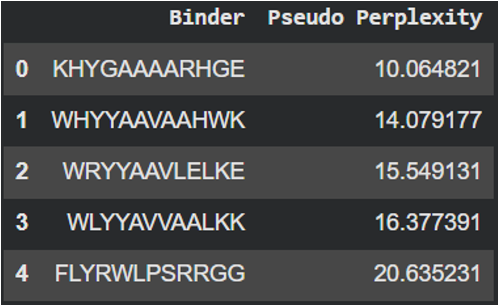
The pseudo-perplexity score reflects how likely a peptide-sequence is according to the patterns learned by this protein language model. Lower values indicate sequences that are more consistent with common protein sequence patterns. Analyzing the designed peptides, KHYGAAAARHGE showed the lowest score (10.06) suggesting that it is the most probable peptide according to the model. In contrast, the reference peptide (FLYRWLPSRRGG) showed the highest value (20.63) indicating that its amino acid composition is less typical compared to the sequences learned by this model.
Part 2: Evaluate Binders with AlphaFold3
- Protein-peptide KHYGAAAARHGE: the prediction produced an ipTM score of 0.5 suggesting a weak or uncertain interaction between the mutant SOD1 protein and the designed peptide. In the graphic, the peptide is spatially separated from the prtein surface and does not clearly localize near the N-terminal region where the A4V mutation is located. The PAE map also shows higher uncertainty between both of them, indicating that AlphaFold does not predict a stable binding interaction in this configuration. Although this PepMLM-designed peptide showed the lowest perplexity score, the prediction not showing a clear interaction with the mutant suggests that, while the peptide sequence is statistically possible, it may not form a strong binding interaction with SOD1.
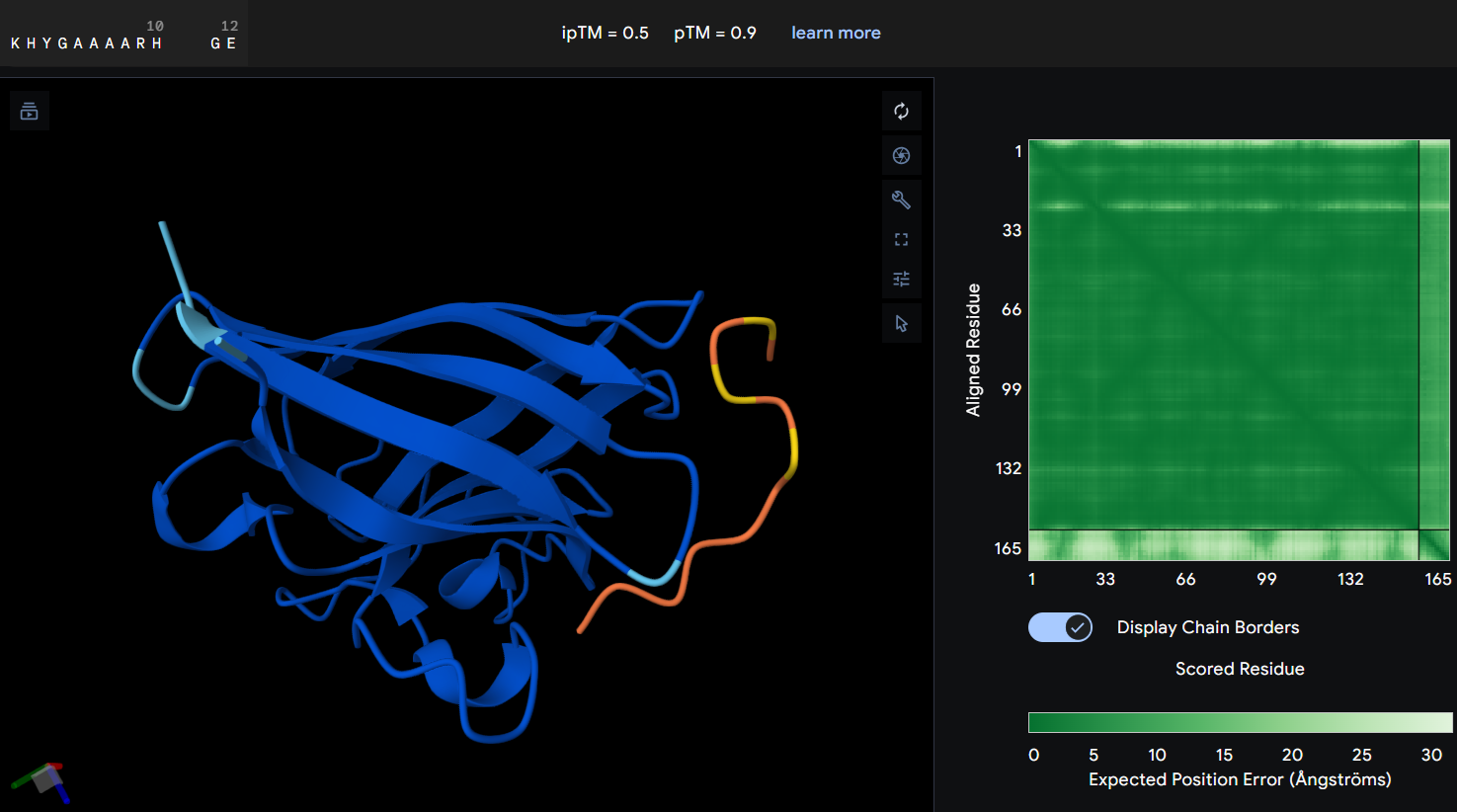
- Protein-peptide WHYYAAVAAHWK: this second designed peptide showed an ipTM score of 0.4 indicating low confidence in the predicted protein-peptide interaction. In this graphic, the peptide does not form a clearly defined binding interface. The PAE map also suggest uncertainty in the relative postioning between the peptide and the protein. This model does not support a strong or stable interaction between this peptide and the mutant SOD1 structure.
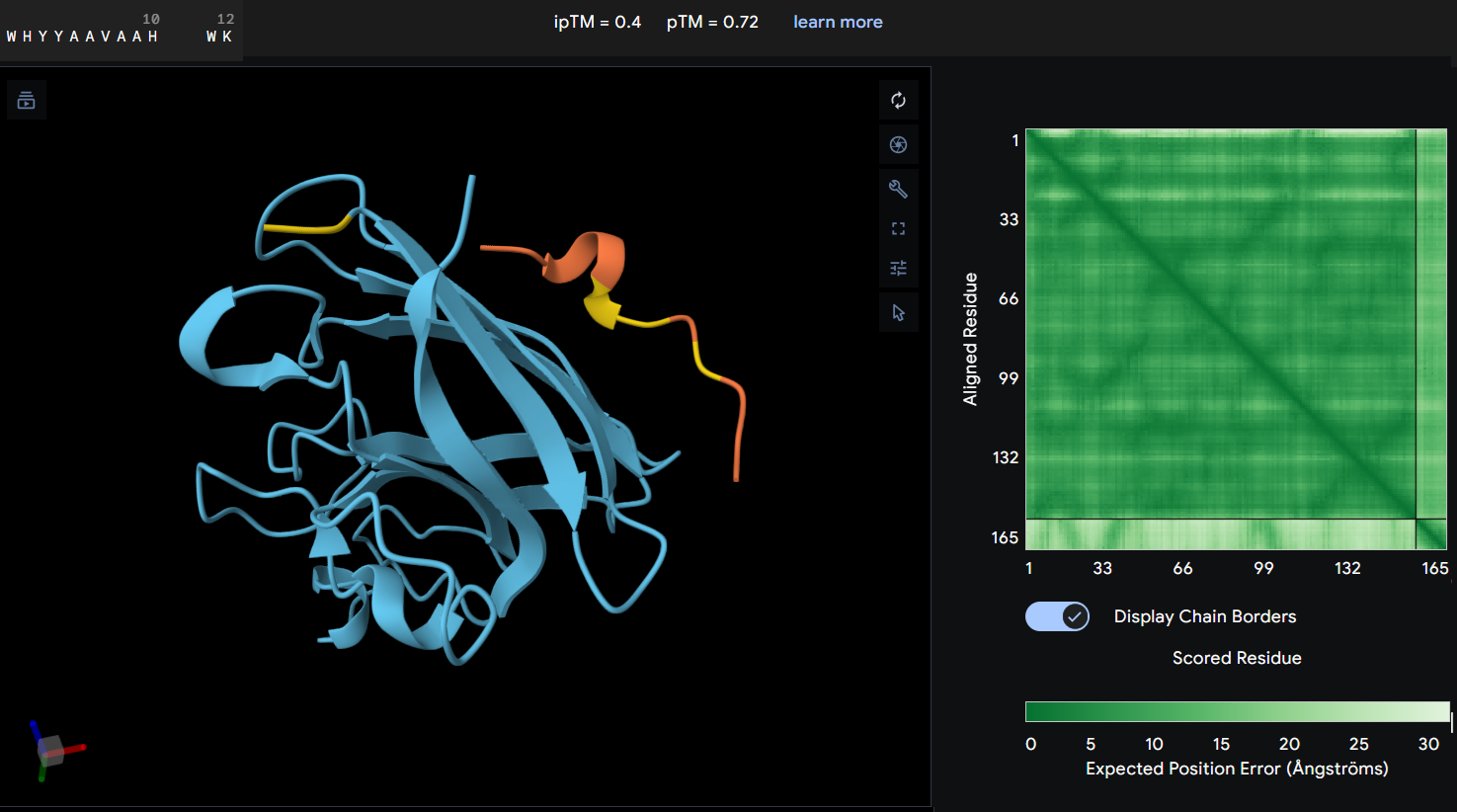
- Proteine-peptide WRYYAAVLELKE: this peptide produced an ipTM score of 0.22 indicating a very low confidence in the predicted protein-peptide interaction. In the structural model, the peptide appears clearly separated from the SOD1 surface and does not form any binding interface. The PAE map also shows high uncertainty between the peptide and the protein. These results suggest that this peptide is unlikely to form a stable interaction with the mutant SOD1 potein.
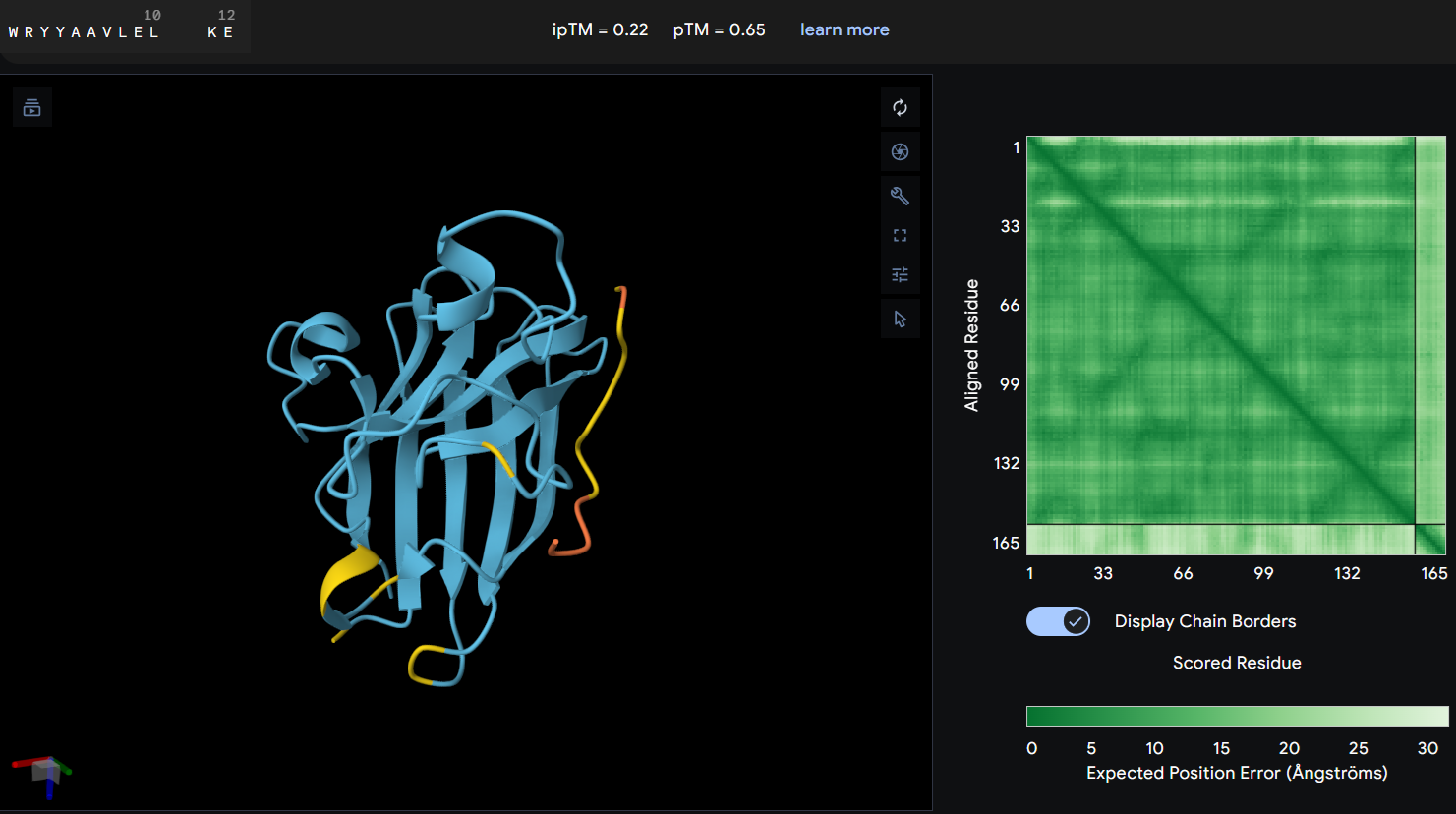
- Protein-peptide WLYYAVVAALKK: this peptide showed an ipTM score of 0.36 reflecting a lack of predicted interaction with the protein. The predicted structure shows the peptide distant from the protein’s core, and the PAE map confirms this with a high error values between the two chains.
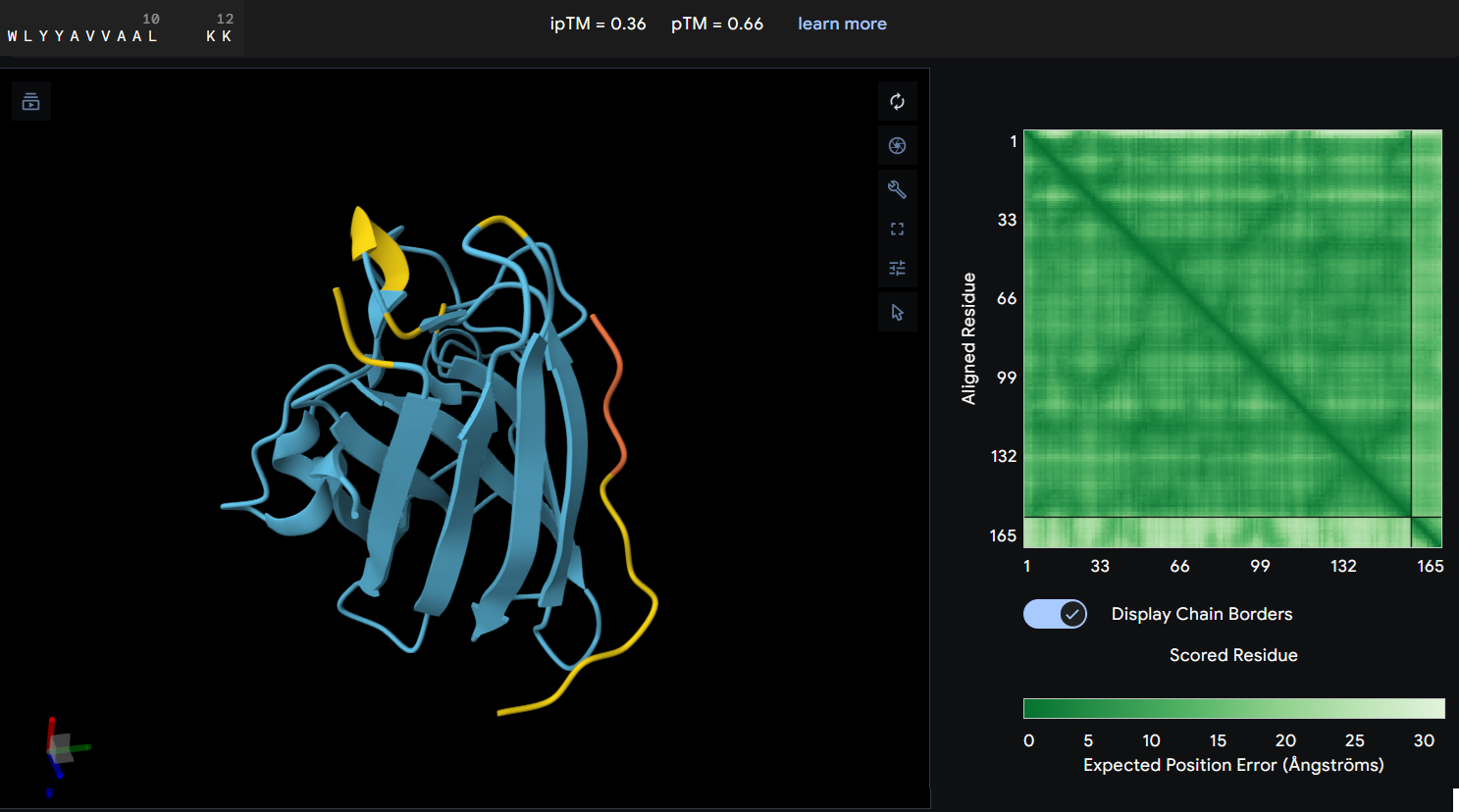
- Protein-peptide FLYRWLPSRRGG: the given peptide for this activity resulted in the lowest docking confidence with an ipTM of 0.35. In the predicted structure, there is a complete lack of interaction as the peptide remains spatially dissociated from the protein. This observation is supported by the PAE map, which shows maximum error values for the chain distances, indicating that the model cannot predict a stable interface.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
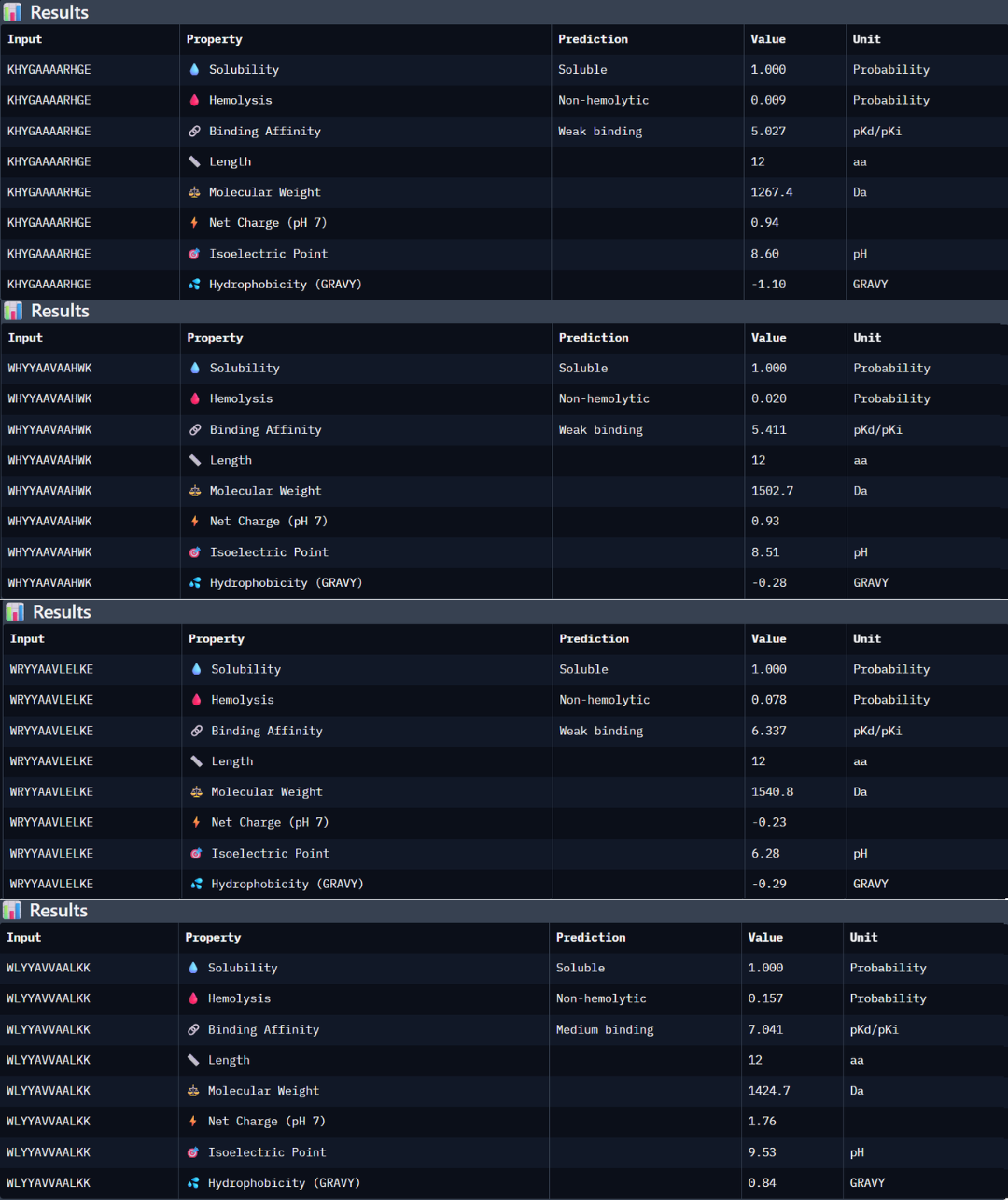
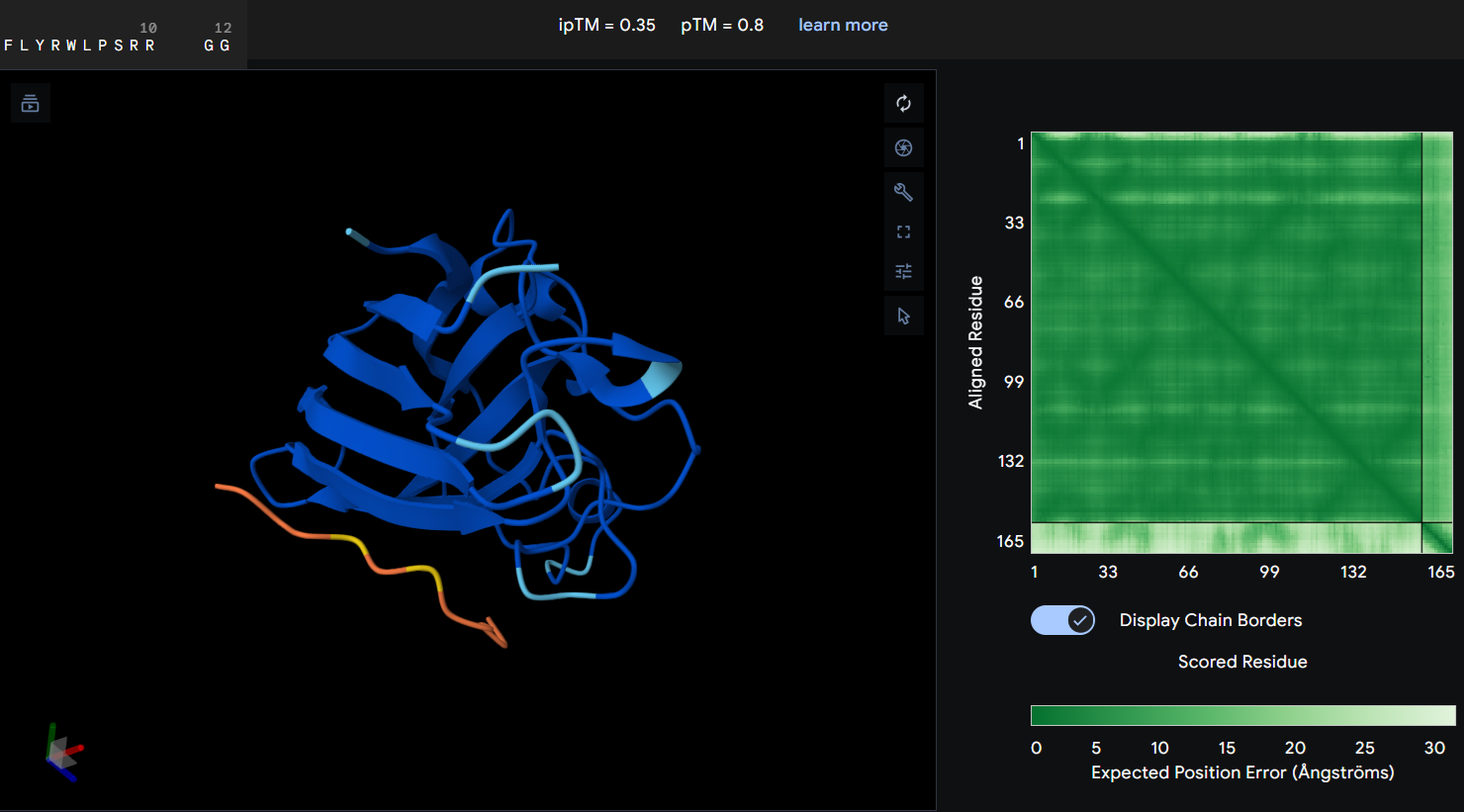
Peptide 1: the peptide (KHYGAAAARHGE) stands out as the most promising candidate from the given peptides. Structurally, it has the highest ipTM score of 0.5, but even though the 3D model is not showing a binding site with the protein, the PAE matrix shows certain dark green shadows indicating a more trusting prediction. While PeptiVerse predicts a weak binding affinity (5.027), this is consistent with the early-stage design of these peptides. This sequence maintains an excellent therapeutic profile, being fully soluble (1000 probability) and non-hemolytic (0.009 probability). The balance between its structural stability and its safe properties makes it the main choice over the other peptides with high perplexity scores.
Peptide 2: the peptide WHYYAAVAAHWK shows a higher predicted binding affinity in PeptiVerse (5.411) compared to the previous one, but its structural confidence in AlphaFold is lower with an ipTM of 0.4 suggesting that the predicted affinity does not always translate into a stable structural complex. This peptide is also highly soluble (1.000 probability) and non-hemolytic (0.020 probability).
Peptide 3: this third peptide WRYYAAVLELKE shows a discrepancy between the prediction and structural reality. Although PeptiVerse predicts the highest binding affinity (6.337), AF returns a very low ipTM of 0.22. additionally, while the peptide remains soluble, its hemolysis probability (0.078) is higher.
Peptide 4: WLYYAVVAALKK achieved the highest predicted affinity in PeptiVerse (7.041), its structural docking in AF remains poor with an ipTM of 0.36. the peptide fails to localize to the A4V site or the dimer interface. Furthermore, this sequence carries the highest hemolysis probability (0.157) of all peptides, making it the least favorable candidate from a therapeutic point of view. These results demonstrate that a high affinity score is insufficient if not accompanied by structural stability and a safe profile.
According to the previous analysis, I have chosen the KHYGAAAARHGE peptide as the lead candidate because it demonstrates the most robust balance between structural viability and therapeutic safety. Although other peptides like WLYYAVVAALKK showed higher predicted chemical affinity in PeptiVerse, they failed to produce a stable interaction in AF an presented significantly higher hemolysis risks. The chosen peptide possess the most favorable pharmacological profile, confirming that the lowest perplexity score from PepMLM was the most reliable indicator of a biologically possible and safe binder.
Part 4: Generate Optimized Peptides with moPPIt
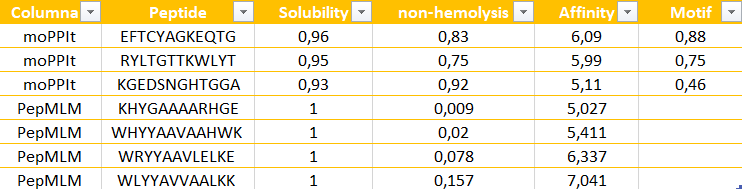
The peptides generated by moPPit differ significantly from the PepMLM ones in their precision and optimization. While PepMLM produced “possible” sequences based on general “likelihood”, moPPit allows for a controlled design, letting us pick exactly where we want the peptide to bind, such as the A4V mutation side as I chose (residues 2-6). This ‘multi-objective’ approach optimizes binding strength (affinity), solubility and safety all at the same time during the creation of the peptide, rather than just checking them at the end. As a result, peptides like EFTCYAGKEQTG show much stronger predicted binding and better focus on the target area.
Evaluation before clinical studies:
To make sure these peptides are safe and effective before testing them in humans, I would follow these steps:
- Structural check using AlphaFold to see if the peptide actually stays attached to the protein in a 3D simulation.
- Lab Binding Tests, like an ELISA to see if my protein and peptide show a colorimetric signal.
- Safety tests, performing a hemolysis assay in the lab to confirm the peptide does not damage any red blood cells.
- Effectiveness, to see if the peptide successfully stops the mutant aggregation in a test tube.
PART C: Final Project: L-Protein Mutants
Stage 1: I performed a two-part analysis to understand the MS2 lysis (L) protein
Evolutionary conservation (pBLAST &ClustalOmega): after performing the alignment between the similar protein sequences in other phages and the original L-protein sequence, I identified those conserved residues which have not changed over evolution and are likey essential for function, and variable residues (shown as blank spaces), which have changed and might tolerate engineering.
Experimental mutation data: analysis of the given laboratory data listing various L-protein mutations and whether they successfully caused lysis in E. coli.
Conclusions:
- Conserved and essential regions: the L-protein has two critical domains:
- Soluble N-terminal domain (residues 1-40): interacts with DnaJ. Residues 25-38 are extremely conserved and likely form the core DnaJ binding site.
- Transmembrane domain (residues 41-75): forming the lysis pore. The start of this domain (residues 41-49) is very conserved and necessary for membrane insertion.
- Experimental fragility: the experimental data revealed a crucial fact: the very beginning of the protein (residues 1-15) is extremely sensitive. Almost all changes here prevented the protein from even being produced, resulting in zero lysis. It is mandatory to avoid these positions.
- Safe positions to mutate: based on the integrated approach, it has been concluded that we must avoid mutating conserved sites, avoid the critical DnaJ core (25-38) and avoid the experimentally fragile N-terminus (1-15). According to this, the safest and most promising areas for engineering are:
The soluble loop (residues 16-24), positions between the N-terminus and the conserved binding core. Changing them might alter the interaction to become independent of the specific DnaJ mutation without destroying the protein itself.
The transmembrane domain (residues 50-75): this region seems less sensitive to total expression failure and is the key to improving lysis speed and efficiency.
 Possible mutations analysis on Google Colab
Possible mutations analysis on Google Colab
To design an improved version of the MS2 L-protein that is actually independent of the DnaJ chaperone or to increase killing efficiency to bypass bacterial resistance, this is the followed strategy:
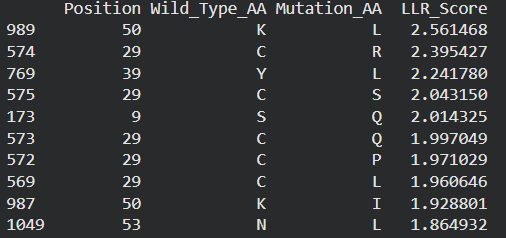
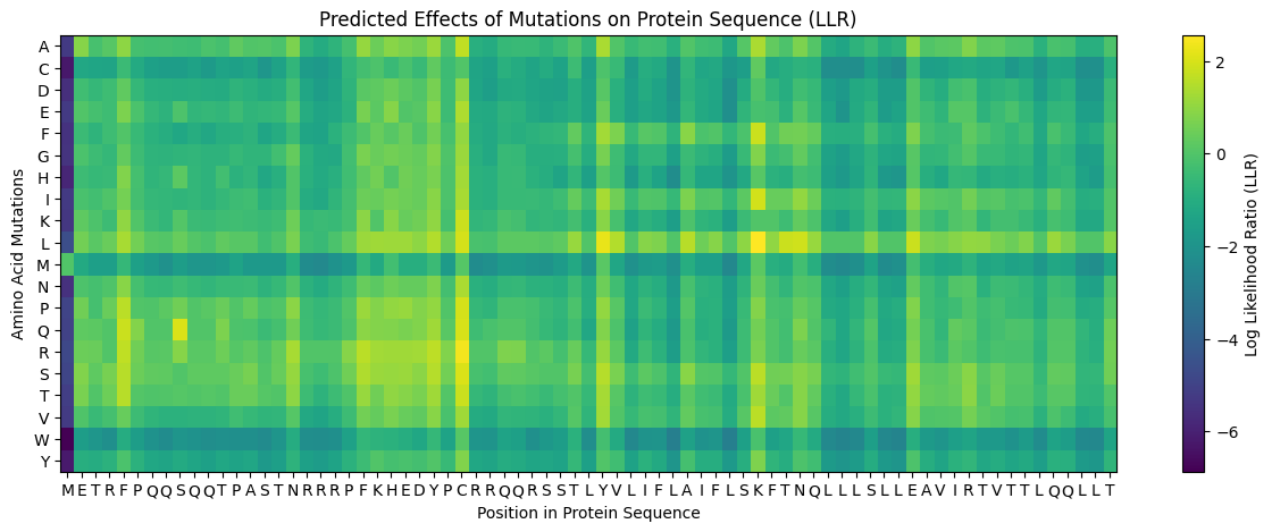
Predict the stability and functional impact of every possible mutation using ESM-1v. the results are visualized in the heatmap, where the rows represent the 75 positions of the L-protein, the columns represent the 20 different amino acids we could use for mutations, the bright yellow/clear cells indicate high log-likelihood scores, meaning the mutation is predicted to be stable and safe (dark purple indicate negative scores, warning that the mutation might break the protein).
Correlation between AI scores vs. Experimental data: after cross-referencing the Colab scores with the given database for the L-protein mutants, I found a strong correlation between the experimental data and the predicted scores. While the laboratory data (L-protein mutants spreadsheet) shows that mutations in the N-terminus (positions 1-5) result in zero lysis, these positions are completely absent from the ‘Top Mutations’ list generated by the Colab, which only includes stable changes with positive scores. This proves the ESM captures the protein’ s fragility perfectly.
These are the mutations I choose after doing the actual analysis, by strictly filtering the Top Mutations table generated in the Colab, prioritizing the highest LLR scores to ensure structural stability.
- Position 53 (L): score 1.86, Transmembrane region
- Position 50 (L): score 2.56, Transmembrane region
- Position 39 (L): score 2.24, Soluble region
- Position 40 (L): score 1.47, Soluble region
- Position 52 (L): score 1.81, Transmembrane region
Positions 39 and 40 (Soluble region) aim to maintain protein expression while potentially altering host chaperone interactions.
Positions 50, 53 and 52 (Transmembrane region) are designed to enhance or stabilize the multimeric assembly required for efficient bacterial lysis.
Multimeric Assembly
Sequences for AlphaFold:
- Variant 1 (Y39L) METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYLLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
- Variant 2 (V40L) METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLLFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
- Variant 3 (F50L) METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAILLSKFTNQLLLSLLEAVIRTVTTLQQLLT
- Variant 4 (S53L) METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLLKFTNQLLLSLLEAVIRTVTTLQQLLT
- Variant 5 (Double Y39L + F50L) METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYLLIFLAILLSKFTNQLLLSLLEAVIRTVTTLQQLLT
After generating the multimeric assembly for Variant 3 (because it had the highest LLR score) using AF2, I compared it against the WT structure. The results show that the mutant successfully maintains its octameric symmetry, forming a stable ring-like structure with a clear central pore. Although the pLDDT scores remain low in the disordered N-terminal and C-terminal tails, the core transmembrane assembly is preserved showing a clearly defined central pore. This suggests that substituting Phenylalanine for a more flexible Leucine at this position stabilizes the transmembrane helix without disrupting the quaternary assembly required for bacterial membrane perforation.
To conclude, I designed this new version to beat the bacteria’s defenses. By making the lysis pore stronger without changing the most important parts of the protein, we can kill the bacteria more quickly. This gives the E. coli less time to protect itself using its chaperones, making it much harder for it to become resistant.
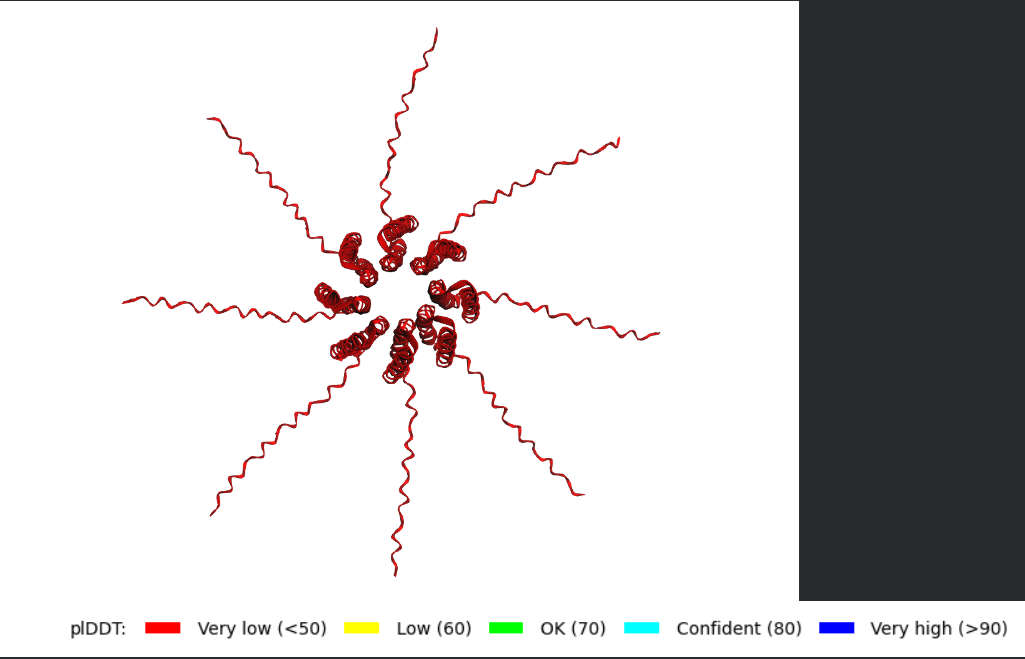 Predicted 3D structure on AF2 Multimer where it is easy to see the expected octameric structure.
Predicted 3D structure on AF2 Multimer where it is easy to see the expected octameric structure.
 Predicted 3D structure on AF2 Multimer for the F50L octamer confirming the mutation preserves the structural integrity of the protein.
Predicted 3D structure on AF2 Multimer for the F50L octamer confirming the mutation preserves the structural integrity of the protein.
Week 6 HW: Genetic Circuits: Part I
- What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
- This master mix contains a Phusion high-fidelity DNA polymerase, an enzyme that synthesizes new DNA strands during PCR and has proofreading activity that reduces the error rate compared to standard polymerases. The enzyme needs Mg+2 ions as a cofactor required for the polymerase activity. This mix also contains dNTPs (deoxynucleotide triphosphates), which are the building blocks used by the polymerase to synthesize new DNA. Another key component is the reaction buffer, which maintains the optimal pH and salt conditions required for the enzyme to function properly. Finally, water is used as the solvent.
- What are some factors that determine primer annealing temperature during PCR?
- The annealing temperature mainly depends on the melting temperature (Tm) of the primers, which is influenced by:
- Primer length: longer primers have higher melting temperatures
- G-C content: G-C base pairs form three hydrogen bonds increasing primer stability (but more energy is required to hydrolyze them) compared to A-T pairs.
- Sequence composition and presence of secondary structures (like hairpins or dimers)
- Salt concentration and reaction conditions
Usually the annealing temperature is chosen to be a few degrees lower than the calculated Tm of the primers to allow efficient binding while maintaining specificity.
- There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
- PCR and restriction enzyme digestion are both methods to generate linear DNA fragments, but working differently.
PCR amplifies a specific DNA region using primers and a DNA polymerase. The primers determine the exact boundaries of the fragment being amplified, which makes PCR very flexible. This technique is useful when we want to amplify a specific gene or add sequences such as overlaps or tags to the ends of the DNA.
Restriction enzyme digestion uses these enzymes that cut DNA at specific recognition sequences. This method requires that the restriction sites already exist in the sequence. The protocol usually involves incubating the DNA with the enzyme under optimal buffer and temperature conditions.
In terms of when to use each method, PCR is preferable when we need custom DNA fragments, sequence modification, or large amplification of DNA. Restriction digestion is often sued when we want to cut plasmids or DNA molecular at defined natural restriction sites, especially when preparing vectors for cloning.
- How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
- To ensure this fragments are compatible, they must contain overlapping homologous regions at their ends. Typically, these overlaps are about 20-40 base pairs long and are designed to match the adjacent DNA fragment. It is possible to design these PCR primers that include the necessary overlap sequences at their 5’ ends. After amplification, the primers will contain the overlaps needed for assembly. It is also important to check that the fragments are correctly sized and free of unwanted sequences using gel electrophoresis.
- How does the plasmid DNA enter the E. coli cells during transformation?
- During transformation plasmid DNA enters E. coli cells when the bacterial membrane becomes temporarily permeable. In chemical transformation, cells are first treated with calcium chloride, which helps neutralize the negative charges of both the DNA and the cell membrane. Then a heat shock step is applied, that creates a sudden temperature change that helps DNA molecules pass through the membrane and enter the cell.
There are other methods such electroporation, a short electrical pulse applied to the cells in order to create temporary pores in the membrane, allowing plasmid DNA to enter the cytoplasm.
- Describe another assembly method in detail (such as Golden Gate Assembly)
a. Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
b. Model this assembly method with Benchling or Asimov Kernel!
a) Another DNA assembly method is Gateway cloning, which is used in systems such as the CloneMiner™ kit, provided by Invitrogen. This method is based on site-specific recombination using att sites (attB, attP, attL and attR) derived from phage lambda. In this approach, PCR products are first generated with attB adapters, which allow the DNA fragment to combine with a donor vector through the action of recombination enzymes. Unlike Gibson or Golden Gate Assembly, this method does not rely on ligation or overlapping sequences, but instead on highly specific enzymatic recombination. This results in the formation of a continuous and functional DNA molecule. Gateway cloning is especially useful for generating cDNA libraries because it allows efficient and directional insertion of many different DNA fragments into vectors.
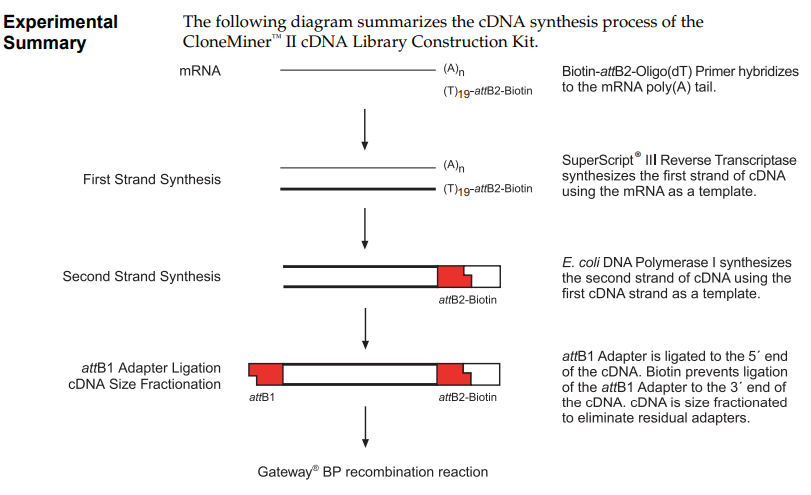
CloneMiner™ II cDNA Library Construction Kit. High-quality cDNA libraries without the use of restriction enzyme cloning techniques. Provided by Invitrogen. Obtained from their Catalog Number A11180.
b) To create this plasmid, I simulated a Gateway BP reaction using Benchling. First, I added attB sites to the Bos taurus Calpain (CAPN1) gene sequence. Then, I combined this gene with the pDONR™221 vector. During this simulation, the gene replaces the original vector’s center, and the sites transform into attL1 and attL2. The final result is this Entry Clone, which is now ready to be moved into an expression vector.
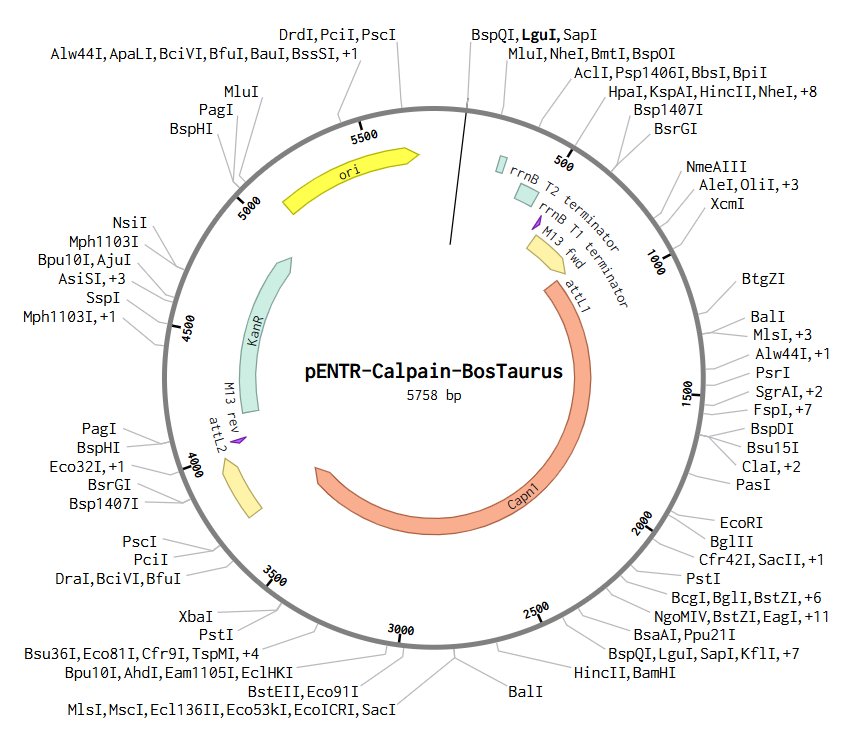
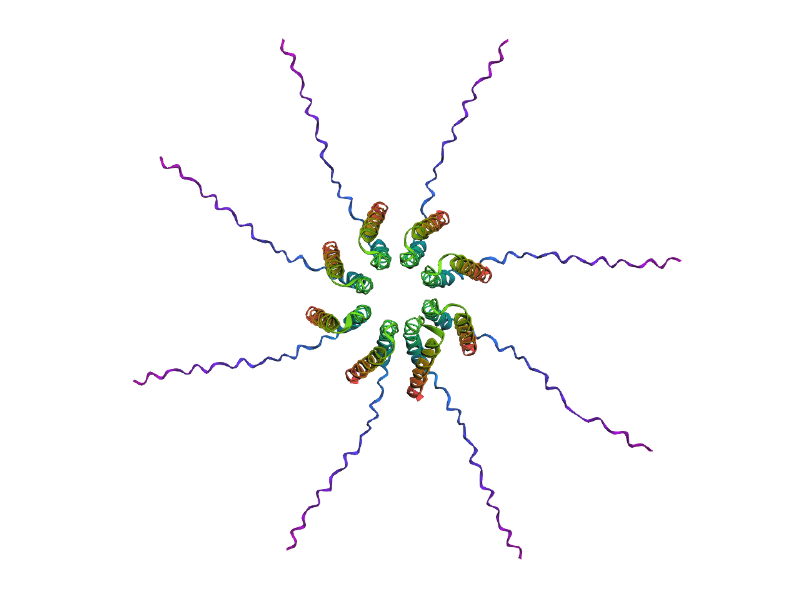
Asimov Kernel
- For this assignment I have simulated the Repressilator as expected, and comparing it to the Repressilator Construct Demo given in Asimov, I have found they work exactly the same, as seen in all the plots.
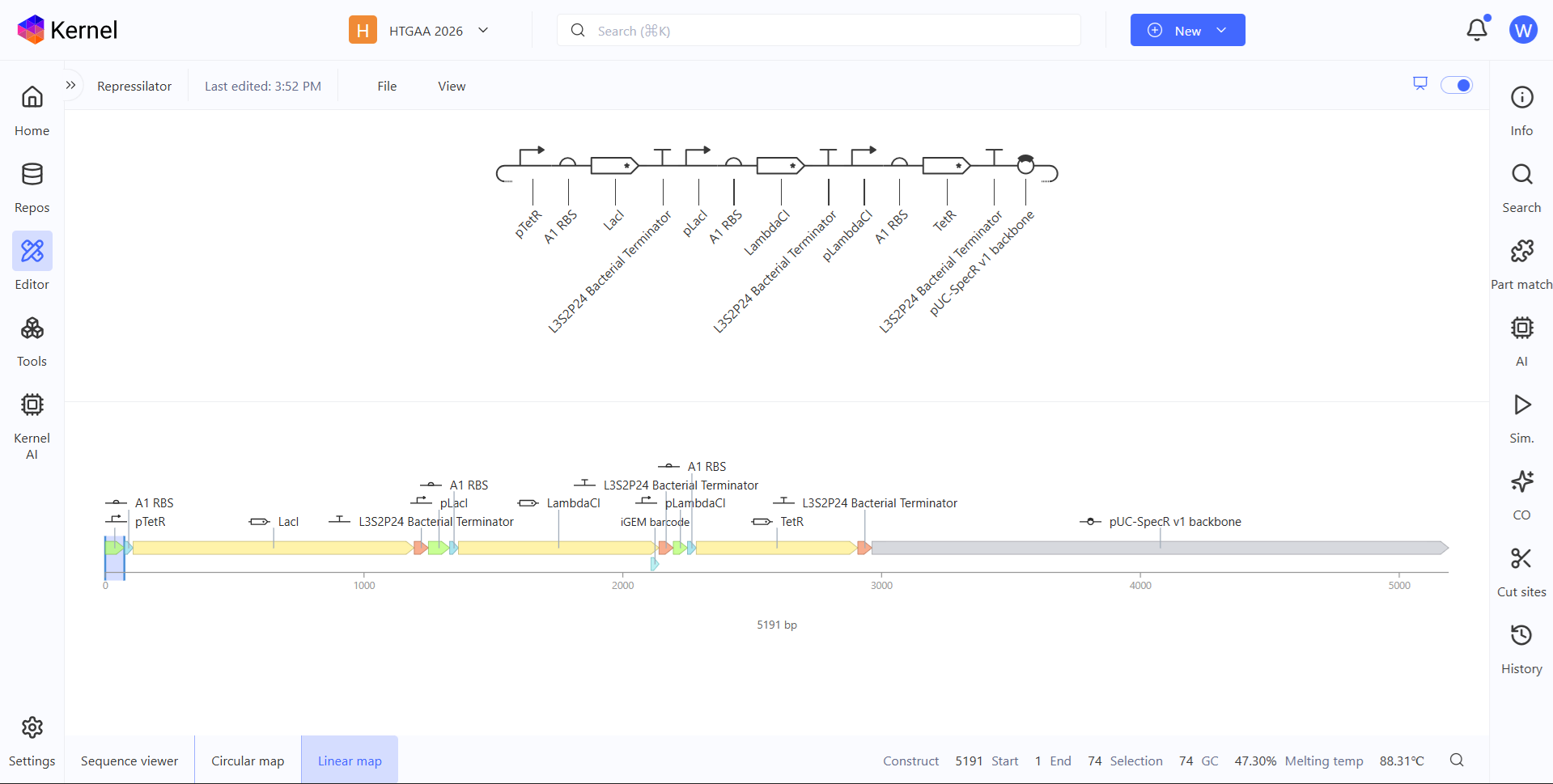
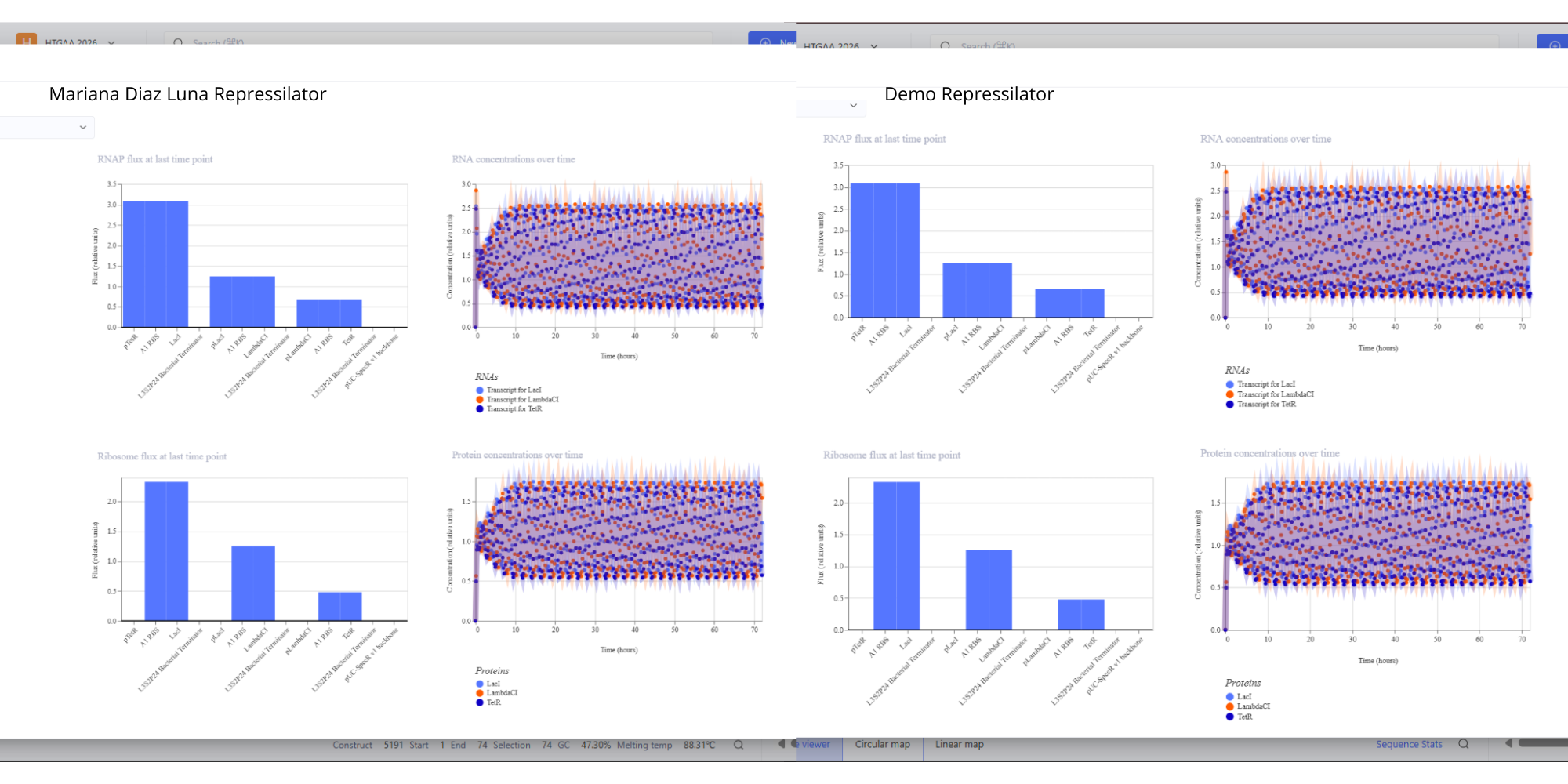 Image showing the comparison between the Repressilator I have made and the Demo.
Image showing the comparison between the Repressilator I have made and the Demo.
- See 3 constructs below:
I. 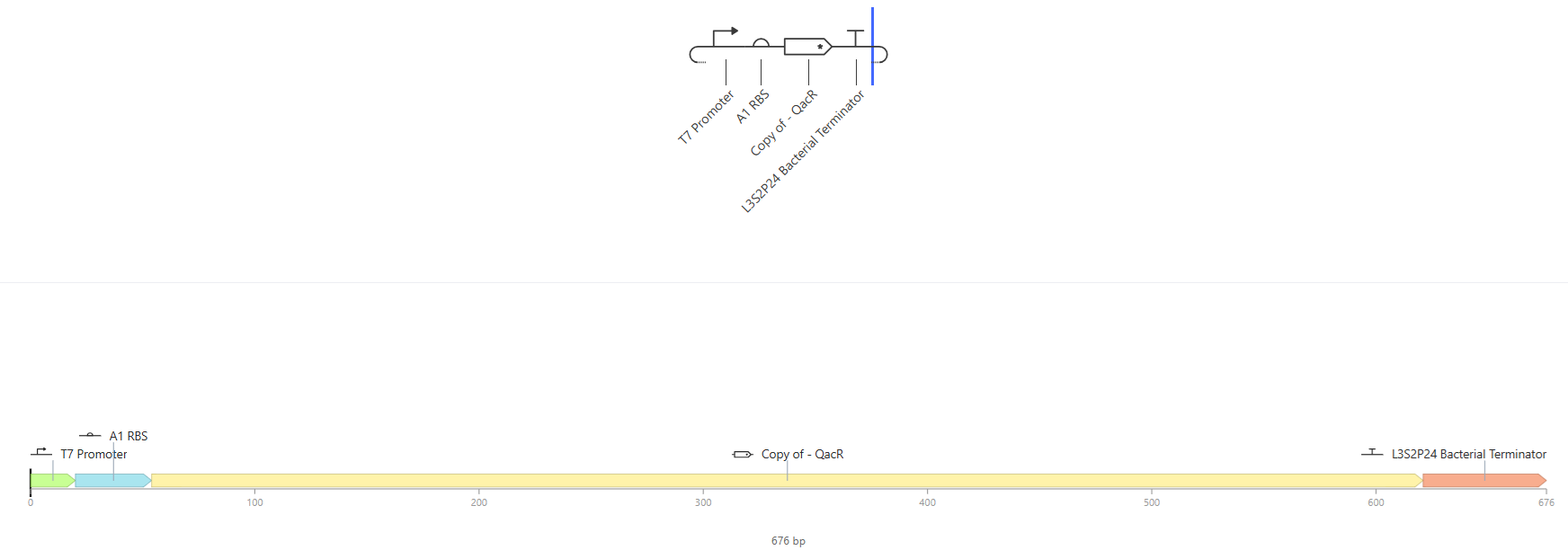
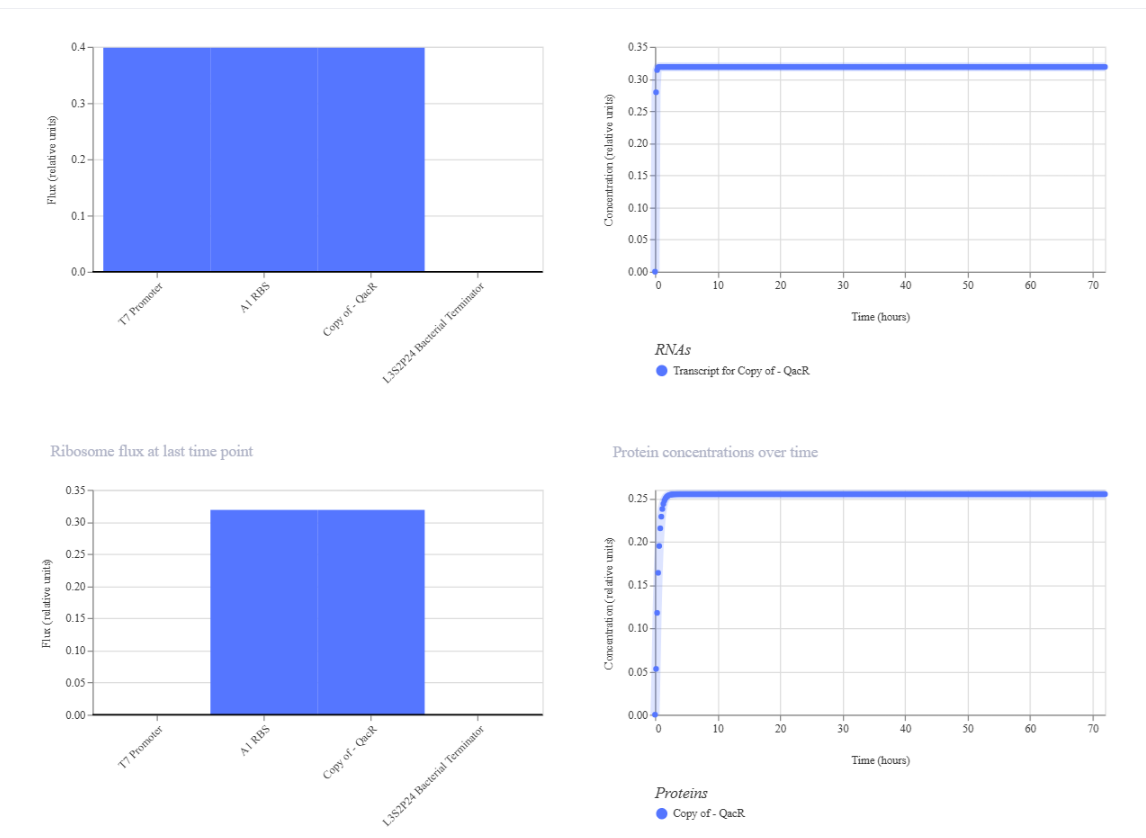
Results where as expected, with high expression levels in the construct when simulated, with a drop-off after Terminator.
II. 
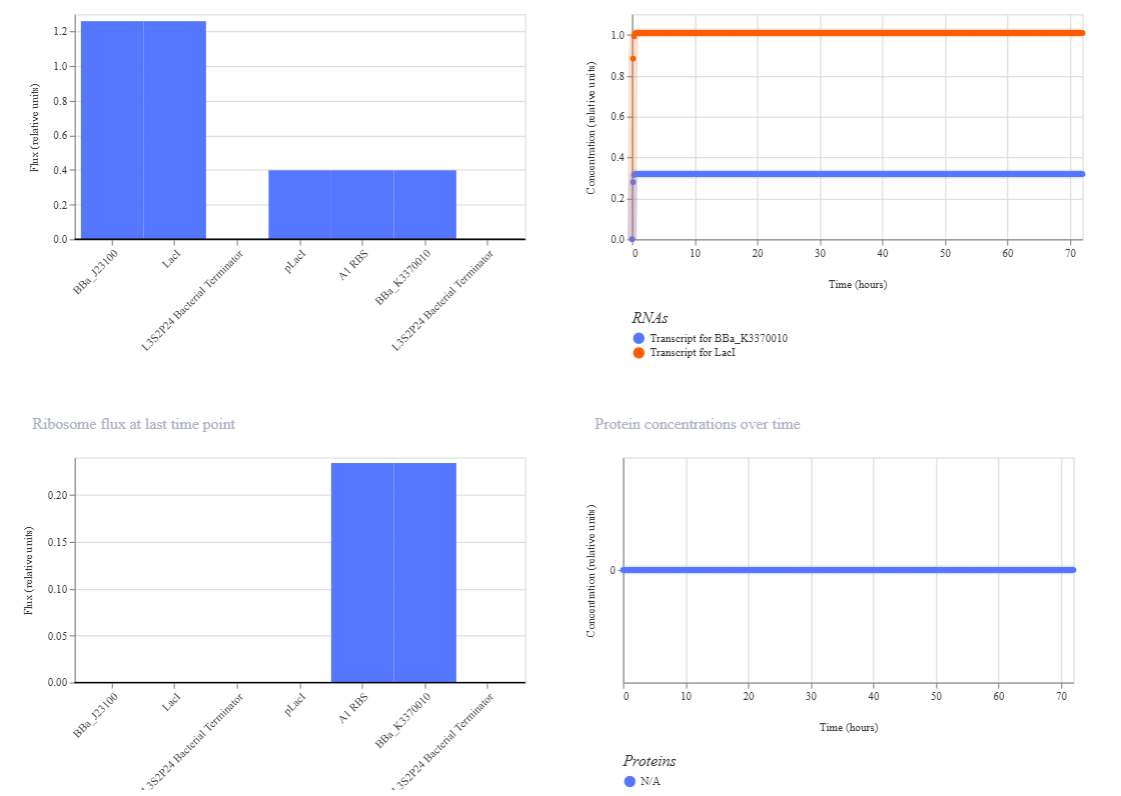 In this circuit, the expression of GFP is controlled by a lac-derived promoter regulated by LacI. The system responds to the presence or absence of IPTG. In absence of IPTG LacI binds to the lac operator region within the promoter, blocking GFP gene. As a result, GFP levels should be very low. When IPTG is added it is expected to bind to LacI and inactivate it, so GFP is now transcribed.
When running the simulation, the expected difference between conditions with and without IPTG is not observed, maybe because the GFP used is not properly recognized as a coding sequence (even though I have tried several of them) or is not expressed in my system.
In this circuit, the expression of GFP is controlled by a lac-derived promoter regulated by LacI. The system responds to the presence or absence of IPTG. In absence of IPTG LacI binds to the lac operator region within the promoter, blocking GFP gene. As a result, GFP levels should be very low. When IPTG is added it is expected to bind to LacI and inactivate it, so GFP is now transcribed.
When running the simulation, the expected difference between conditions with and without IPTG is not observed, maybe because the GFP used is not properly recognized as a coding sequence (even though I have tried several of them) or is not expressed in my system.
III.
Week 7 HW: Genetic Circuits: Part II
Part 1: Intracellular Artificial Neural Networks (IANNs):
- What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
These IANNs have several advantages compared to traditional genetic circuits:
- Continuous responses instead of binary outputs, which makes them more similar to real biological systems, where gene expression is not just “on or off” but varies in intensity.
- Better handling of noisy biological environments, because IANNs can integrate multiple inputs and average signals, making them more robust to fluctuations caused by these noisy systems.
- Ability to learn complex patterns compared to Boolean circuits that are limited to simple logic, while IANNs can approximate complex nonlinear functions allowing more sophisticated decision-making.
- This neural-like architectures can be extended to multiple layers, enabling hierarchical processing.
- IANNs are more biologically realistic, since gene regulatory networks in cells already behave more like analog systems.
- Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
Application: Smart cancer cell detection and response system
- An IANN could be engineered in mammalian cells to detect a specific combination of cancer biomarkers and trigger a therapeutic response.
Input:
- Expression level of oncogene A
- Expression level of oncogene B
- Hypoxia signal
Processing: each input contributes with a weight, similar to neural networks. The system integrates all those signals and applies a threshold-like function to decide if the combined patterns whether matches or not a cancer profile. If it does, the output is activated.
Output:
i. Expression of a pro-apoptotic protein (inducing cell death) OR
ii. Expression of a fluorescent reporter for diagnosis
Limitations: biological noise and variability in this cell systems may affect accuracy, as well as cross-talk with endogenous pathways causing unintended interactions. It may also be hard to precisely control gene expression levels and, since cancerous cells mutate all the time, this instability could break the system over time.
- Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
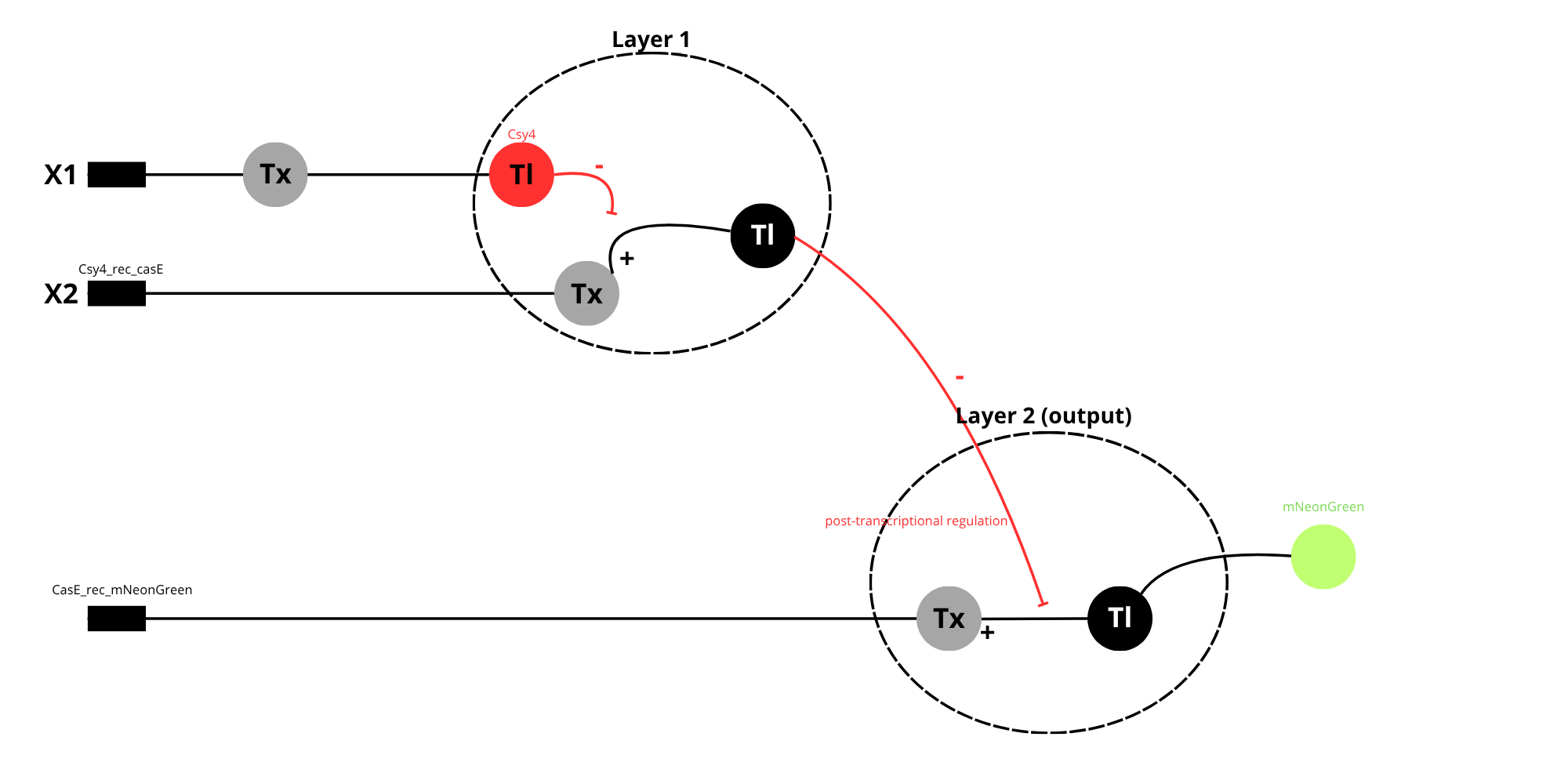
Diagram showing a two-layer intracellular perceptron.
In layer 1, the input X1 is transcribed and translated to produce the endoribonuclease Csy4, acting as the output of the first layer. Csy4 the regulates Layer 2 as the post-transcriptional level by cleaving the mRNA produced from input X”, which contains a Csy4 recognition site.
In Layer 2, X2 is transcribed and translated to produce a fluorescent protein (Y). However, when Csy4 is present, it reduces mRNA stability, leading to lower protein expression.
This system is considered a multiplayer perceptron because the output of the first layer controls the behavior of the second layer, allowing more complex signal processing.
Part 2: Fungal Materials
- What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
- Some examples of fungal materials include biodegradable packing, eco-leather, and construction materials such as ecological bricks. Fungal packaging is typically produced using mycelium grown on agricultural waste, where it acts as a natural binder, forming a lightweight, foam-like composite material. This is used as a sustainable alternative to petroleum-based plastics and Styrofoam in protective packaging, as it can absorb impacts and be molded into specific shapes during growth.
Mycelium-based leather is developed by controlling fungal growth to prodce dense, sheet-like structures. These materials are then processed (compressed, dried and sometimes chemically treated) to achieve mechanical properties similar to animal leather. They are used in fashion and textile industry for products such as shoes, bags and clothing.
Fungal bricks are created by growing mycelium through lignocellulosic substrates, where it binds the particles into a solid composite. Once growth is complete, the material is dried to stop further biological activity. These bricks are lightweight, biodegradable and can provide thermal insulation.
The main advantages of fungal materials include biodegradability, low environmental impact and the ability to use renewable feedstocks such as agricultural residues. Additionally, their production generally requires less energy compared to traditional materials.
As limitations, their mechanical strength and long-term durability are often lower than those of conventional materials like plastics, concrete or treated leather. They can also be sensitive to moisture, biological degradation, and environmental variability, which may restrict their use in certain conditions or require additional processing to improve stability.
- What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
- One interesting application would be to engineer fungi to produce “self-healing” construction materials. These modified fungi could be able to respond to mechanical damage; such as cracks in a fungal brick. When damage occurs, the fungus could be activated to regrow its mycelial network and repair the structure. This could be achieved by engineering gene circuits that are activated by stress signals or exposure to oxygen and moisture.
Additionally, fungi could be modified to produce extracellular polymers or bidngind proteins that improve the mechanical strength of the material during the repair process.
As an advantage is possible to mention the fact that this would extend the lifetime of sustainable building materials and reduce the need for maintenance or replacement, making construction more environmentally friendly and cost-effective. Using fungi for this purpose is especially advantageous because their natural growth as filamentous networks allows them to penetrate and reconnect damaged areas, something that is difficult to achieve with bacteria. However, challenges include controlling fungal growth to prevent overproliferation, ensuring long-term stability, and designing genetic systems that respond reliably to environmental signals.
Week 9 HW: Cell-free Systems
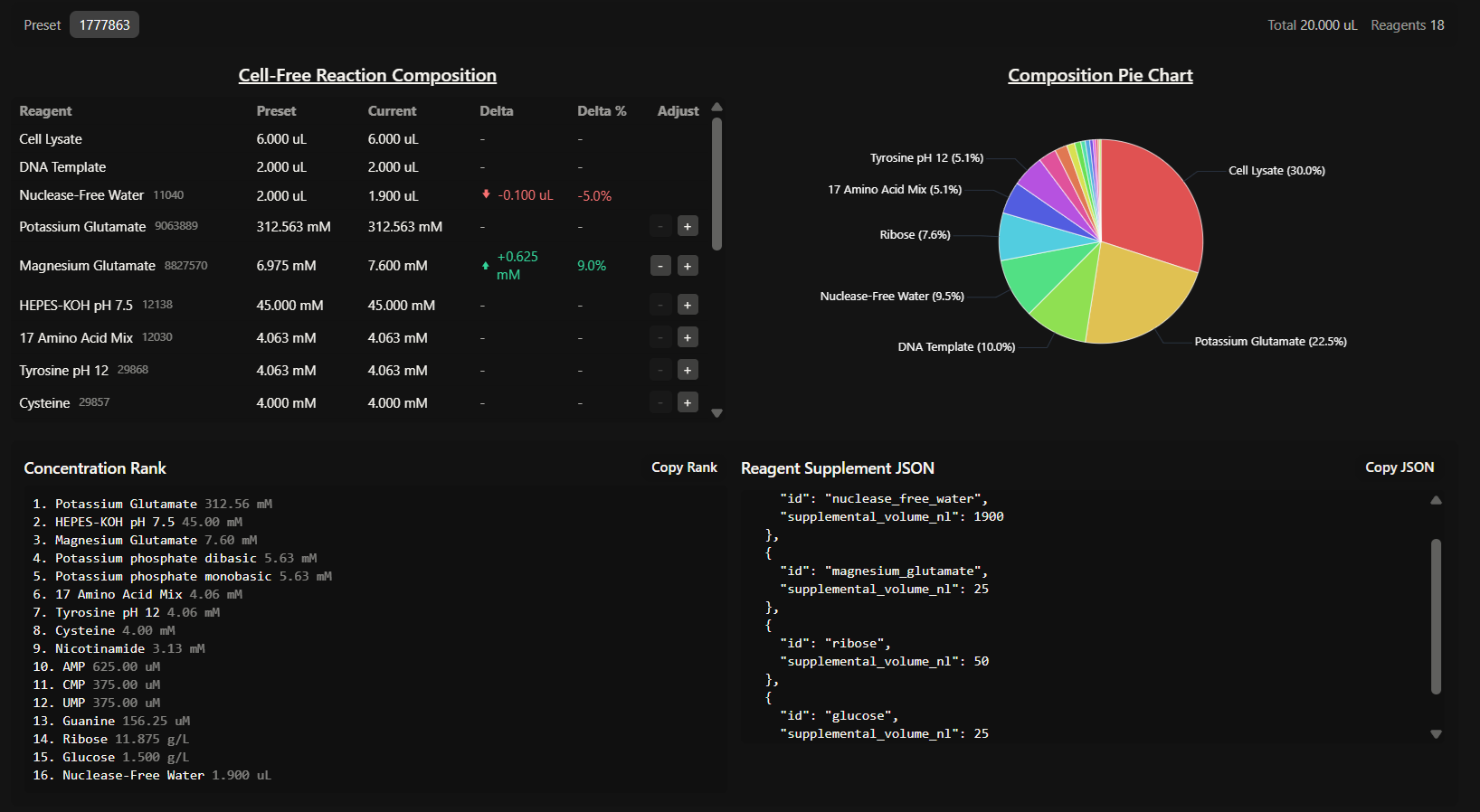
GENERAL HW QUESTIONS
- Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
CFPS transitioned from a “black box” scenario to an open controllable system, so flexibility and control are the main advantages here. This allows us to precisely manipulate the concentrations of amino acids, salts, and templates. It also allows for the addition of non-canonical amino acids or cytotoxic agents that would otherwise kill a living host.
Cases where CFPS is more beneficial:
- Production of cytotoxic proteins, because proteins that disrupt membrane integrity or interfere with vital cellular processes (like certain toxins or antimicrobial peptides) cannot be produced in vivo.
- Rapid prototyping: CFPS bypasses the time-consuming steps of cloning, transformation and cell cultivation, reducing the cycle from days to hours.
- Describe the main components of a cell-free expression system and explain the role of each component.
A standard CFPS system consists of:
- Whole cell extract (lysate): provides the essential molecular machinery, like ribosomes, enzymes, initiation/elongation factors, etc.
- Energy solution: containing an energy source (like glucose or phosphoenolpyruvate) and a buffer system to maintain an optimal pH and ionic strength.
- Reaction mix: includes the DNA template (plasmid or PCR product), RNA polymerase, nucleotides and amino acids.
- Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy provision is critical because protein synthesis is metabolically expensive: for every peptide bond formed, multiple high-energy phosphate bonds are hydrolyzed. Without regeneration, the accumulation of inorganic phosphate inhibits the reaction, and the system reaches thermodynamic equilibrium quickly, ceasing production.
A method to ensure continuous ATP supply could be a semi-continuous or continuous-exchange bioreactor, which uses a dialysis membrane to facilitate the constant diffusion of fresh substrates like ATP and NTPs into the reaction chamber, while simultaneously removing inhibitory metabolic byproducts, increasing the system’s productivity.
- Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic systems (e.g. E. coli) are simpler and faster, being highly efficient for expressing smaller, robust proteins like GFP because they lack complex compartmentalization, allowing for rapid coupled transcription and translation. However, they do not have post-translational modifications or are very limited, so it is not possible to produce human or therapeutic proteins that require them. Instead, eukaryotic systems are preferred. Although they generally offer lower yields and involve more complex preparation, they provide the necessary machinery for folding and glycosylation.
In a prokaryotic system, I would choose to produce a GFP because it is a robust, non-glycosylated protein commonly used as a reporter. The high yield of E. coli lysates makes it ideal for quick quantification.
My eukaryotic choice would be Human Erythropoietin because it requires specific glycosylation patterns to be biologically active and stable in the bloodstream that only this eukaryotic cells can provide.
- How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Expressing membrane proteins in these systems could be challenging due to their hydrophobic nature, which leads to aggregation and precipitation in aqueous lysates. To overcome this, may be possible to add synthetic lipids or detergents to the reaction, which will provide a hydrophobic scaffold for the protein to insert into during translation. We could also use lysates enriched with specific chaperones (like DnaJ/DnaK) to assist in proper insertion.
- Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
a. Template degradation: checking the purity of the DNA and ensuring the environment is RNase-free by adding RNase inhibitors to the mix and avoiding contamination.
b. Magnesium imbalance: ribosome stability and polymerase activity are extremely sensitive to magnesium concentration, so it is possible to perform a Mg+2 titration to ensure there is no difference.
c. Codon bias: if expressing a human gene in E. coli lysate, the “rare codons” could ruin translation. A solution would be to supplement the reaction with tRNAs for rare codons or use a codon-optimized sequence.
HW QUESTIONS FROM KATE ADAMALA
I would design a synthetic minimal cell (SMC) for pathogen detection and response to function as a “sentinel” to expand the therapeutic capacity against hospital-acquired infections, specifically targeting Pseudomonas aeruginosa. The input would be a Quorum Sensing molecule produced by P. aeruginosa called 3-oxo-C12-HSL, and the therapeutic output a Bacteriophage T4 Lysozyme (a potent antibacterial enzyme).
A fundamental aspect of this design is the encapsulation of the cell-free machinery within a phospholipid bilayer. This function cannot be realized by cell-free transcription and translation alone. Without the liposome membrane, both the genetic circuit amd the synthesized enzymes would be immediately diluted, preventing the high macromolecular crowding required for efficient reaction kinetics. The membrane acts as a diffusion barrier and a protective shield, preserving the interal system from exogenous proteases and nucleases commonly found in clinical environments, which would otherwise degrade the system before it reaches the pathogen.
To implement this the SMC encapsulates an E. coli-derived PURE (Protein synthesis using recombinant elements) system and a specialized genetic circuit, including the lasR gene for constitutive sensor expression and the genes for α-hemolysin (aHL) and T4 lysozyme, both controlled by the PlasI promoter. This setup allows a great communication strategy: while the membrane is naturally permeable to the small 3-oxo-C12-HSL input molecule, the large lysozyme output can only exit the SMC through the aHL pores, which are synthesized only after the pathogen signal is detected. This ensures a localized and triggered response, effectively creating a “smart pill” that remains dormant until it senses a high concentration of the target pathogen.
Finally, the success of this synthetic system will be measured by its ability to inhibit bacterial growth. In an experimental setting, I would monitor the optical density of P. aeruginosa cultures in the presence of the SMCs. A significant reduction in growth, confirmed by fluorescence assays, would demonstrate this system successfully detected the quorum-sensing signal and released a sufficient concentration of lysozyme to neutralize the infection.
Lipids and genes:
- Lipids: POPC, cholesterol
- Enzymes: bacterial cell-free Tx/TI (PURE System)
- Genes: lasR (constitutive expression of the transcription activator)
- Α-hemolysin (aHL) under the PlasI promoter
- T4 Lysozyme under the PlasI promoter
- Target cells: P. aeruginosa (WT)
HW QUESTIONS FROM PETER NGUYEN
I would choose to design a smart athletic textile integrated with these cell-free systems that act as a real-time metabolic sensor, providing a visible colorimetric readout of lactic acid levels through sweat activation. The integration of this sensor addresses a critical need in sports physiology, because lactate serves as a vital biomarker to identify the lactate threshold, the point beyond which metabolic acidosis triggers muscle fatigue and performance decline.
The fabric is manufactured by embedding a cell-free “master mix” directly into the fibers of a synthetic textile. To make it, I would use a genetic circuit where a specific transcription factor (like LldR) senses lactate. In the presence of high lactic acid levels in the user’s sweat, the circuit is activated to express a high-intensity chromoprotein. To ensure the reaction stayls localized, the cell-free components are encapsulated in micro-hydrogels added into the fabric. The user’s sweat acts as the rehydration agent, triggering the on-demand protein synthesis, providing a localized visual map of muscle fatigue directly on the system.
This garment addresses the growing need for non-invasive, real-time physiological monitoring in both sports and physical therapy. Currently, measuring metabolic markers like lactate requires blood draws or expensive electronic sensors. This “living” textile provides a zero-power, lightweight and intuitive way for athletes to optimize their training intensity and for patients in rehabilitation to monitor its health through simple visual feedback.
So, the activation will be through the user’s sweat as the natural trigger. The sensitivity of the circuit is tuned so that a baseline amount of moisture is required, preventing accidental activation by ambient humidity. Following Dr. Nguyen’s protocols, stability will be addressed using the cell-free mix supplemented with lyoprotectans like trehalose and sucrose, which form a glassy state around the proteins and DNA, allowing the garment to be stored at room temperature in a sealed package without losing activity. To address the “one-and-done” nature of cell-free systems, the grment is designed with replaceable bio-patches in high-sweat areas like the lower back or underarms. This hybrid approach combines a durable, washable textile with low-cost, disposable biological insert.
HW QUESTIONS FROM ALLY HUANG
- Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Cosmic radiation is a primary barrier to long-term human spaceflight causing oxidative stress and DNA damage. Even though our own DNA machinery repairs the damage, the amount of radiation exposure accumulates during their time in space making it really challenging to alleviate. Monitoring this risks in real-time is vital for astronaut’s health. Currently, samples must often be returned to Earth for complex analysis. That’s why BioBits offers a portable, cell-free alternative to detect these threats in situ without the metabolic burden of maintaining live cell cultures. Developing a rapid, on-station diagnostic for radiation-induced molecular damage is significant for deep-space missions where immediate medical decision-making is essential for long-term health monitoring.
- Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
Reactive oxygen species (ROS)-responsive promoter regulating expression of a GFP reporter protein.
- Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
ROS are key indicators of oxidative stress, which increases under space conditions. By designing a ROS-responsive genetic circuit in a cell-free system we can directly link oxidative stress levels to measurable GFP fluorescence. This allows real-time detection without requiring living cells. Monitoring ROS levels helps evaluate astronaut health risks. The simplicity and portability of the system makes it ideal for space missions, where traditional laboratory assays are not feasible.
- Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
The hypothesis is that a freeze-dried cell-free expression system incorporating a ROS-responsive promoter can reliably detect oxidative stress by producing a quanfifiable fluorescent signal. Specifically, higher ROS levels will activate the promoter leading to increased GFP expression.
This approach is based on the principle that oxidative stress activates specific regulatory elements in biological systems. By coupling these elements to a reporter gene in a cell-free format we eliminate the need for living cells while preserving biological responsiveness.
The goal of this project is to develop a simple, robust biosensor that can function in space conditions and provide rapid and interpretable results, enabling astronauts to monitor their oxidative stress levels in situ and serve as a model for detecting other environmental or physiological changes during space missions.
- Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
Freeze-dried reactions containing the ROS-responsive GFP construct will be rehydrated with samples exposed to varying ROS levels (eg. Hydrogen peroxide). A negative control with no ROS and a positive control with a known ROS concentration will be included. Reactions will be incubated using the miniPCR device, and fluorescence will be measured with the P51 viewer. Data will be collected as fluorescence intensity over time. Inscreased fluorescence compared to controls will indicate a ppsitive detection. This setup allows quantitative comparison of oxidative stress levels under simulated space conditions.
Week 10 HW: Advanced Imaging and Measurement Technology
Homework: FINAL PROJECT
One of the main aspects to measure in this work is the expression level of the APP since the goal of the CRISPRi system is to reduce its transcription. This could be measured using qPCR (mRNA levels of APP), extracting the RNA from treated and control cells, performing reverse transcription and quantifying gene expression differences.
It is also important to measure whether changes in gene expression translate into protein level changes, using Western Blot to detect APP protein levels or ELISA to quantify amyloid-beta peptides.
Lastly, I need to confirm that these nanoparticles are entering cells and reaching the expected location. Using fluorescence microscopy, it is possible to label those nanoparticles with fluorescent markers and allow visualization inside cells. Confocal microscopy is also useful for a more precise localization.
Waters Part I: Molecular Weight
After analyzing eGFP molecular weight including the His-tag and linker, the theoretical pI/Mw: 5.90 / 28006.60
I have selected two pairs of charge states, one between 900 and 1000: 903.7148 (n+1) and 933.7349 (n).
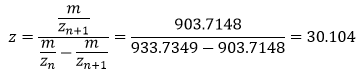
And the other between 800 and 900: 848.9758 (n+1) and 875.4421 (n)

To determine the MW of the protein:

Accuracy of the measurement:
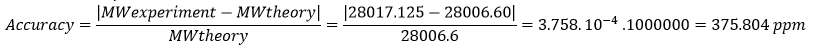
- In large proteins like eGFP the peaks get “crowded” because the charge state is high, so as the charge increases the distance between isotopes becomes very small, and the peaks start to overlap. The instrument’s resolution of 30.000 is high enough to distinguish individual isotopic peaks, so the software has successfully assigned specific m/z values. Techicnally yes, the charge state can be observed even though we cannot distinguish those peaks.
Waters Part II: Secondary/Tertiary structure
Native protein conformations show the natural state of a protein with minimum charges, and denatured conformations are obtained when digesting the protein structure with enzymes into individual highly charged peptides, where each basic residue tends to be protonated.
When a protein unfolds, its internal basic residues become exposed to the solvent. In ESI-MS this leads to a higher number of protons being attached to the protein, consequently showing a distribution of peaks at lower m/z values compared to the native protein. Additionally, the denatured protein exhibits a larger range of charge states due to the increased conformational flexibility of the unfolded polypeptide chain.
There is a lot of noise in that part of the spectrum, so that signal is weak and the labels are likely pointing to noise artifacts or small impurities rather than the actual isotopic distribution of the protein. Those labeled values at ̴2800 m/z are not reliable for determining the charge state.
Waters Part III: Peptide Mapping – primary structure
There are 20 (twenty) Lysines and 6 (six) Arginines in the amino acid structure.
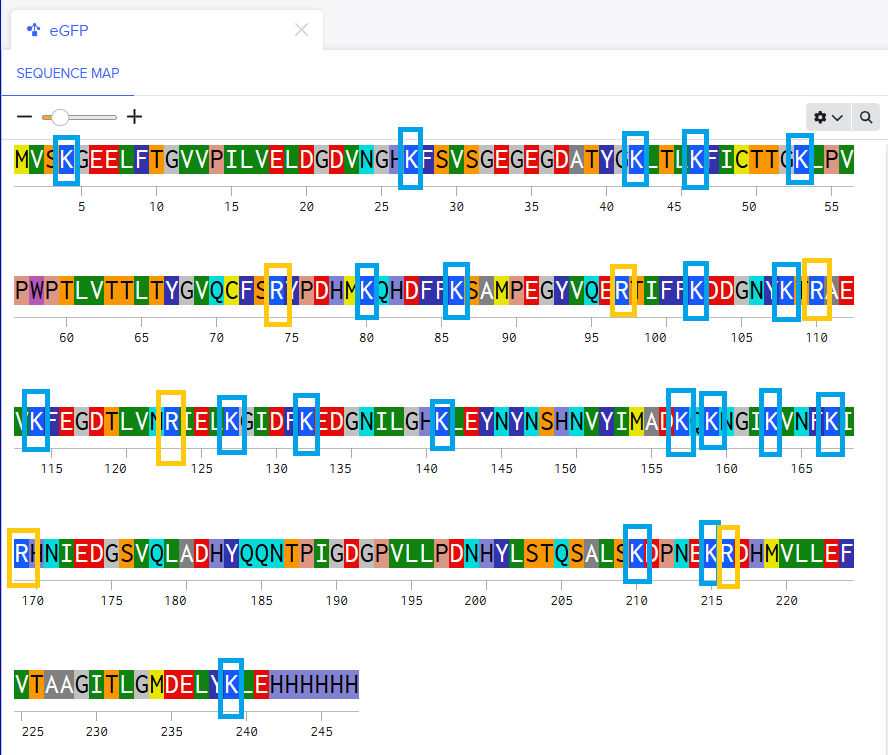
After performing the tryptic digestion with trypsin, will be generated 19 peptides according to Expasy PeptideMass.
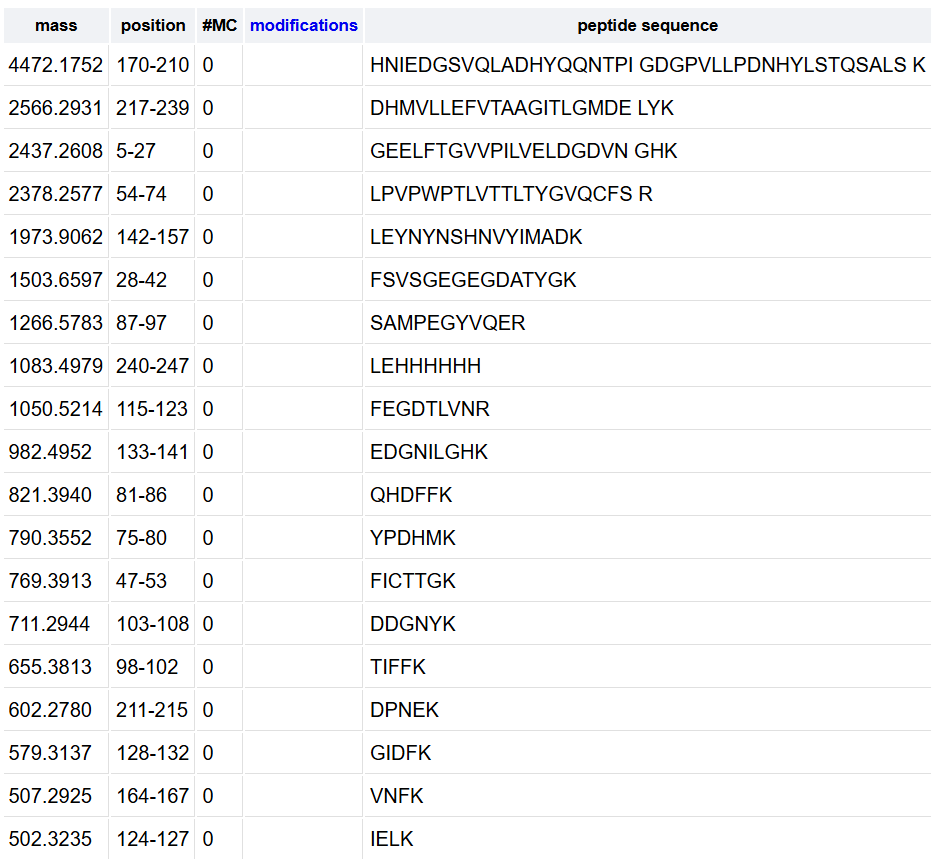
To calculate the 10% threshold I identified the highest peak with the maximum abundance at 4.87 minutes reaching 1.2x10ʌ7counts, and the 10% of this maximum is 1.2x10ʌ6 counts. With this, any peak that goes higher than the 2.10ʌ6 line is above this threshold, counting 17 chromatographic peaks between 0.5 and 6 minutes.
There are fewer peaks in the chromatogram than the number of peptides predicted. This occurs because some peptides may co-elute, meaning they exit the column at the same time and appear as a single peak. Some peptides may also have low inonization efficiency, resulting in peaks with a relative abundance below the 10% threshold used for counting.
The most abundant peak for the peptide eluting at 2.78 minutes is m/z = 525.76712.
Isotope separation:
- Peak 1: 525.76712
- Peak 2: 526.25918
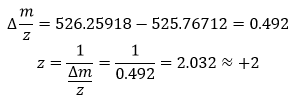
To calculate the mass of the singly charged form ([M+H]+):

- Based on the PeptideMass analysis and the previous calculation:
- Experimental [M+H]+: 1050.52697
- Theoretical: 1050.5214, sequence FEGDTLVNR (position 115-123)
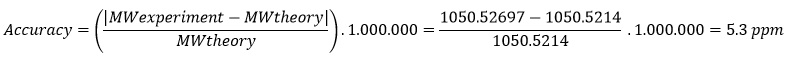 An error of 5.30 ppm indicates high mass accuracy, according to the high-resolution instrument used which generally provides accuracy <10 ppm.
An error of 5.30 ppm indicates high mass accuracy, according to the high-resolution instrument used which generally provides accuracy <10 ppm.
According to the peptide identification data shown on the image, the percentage of the sequence confirmed by peptide mapping is 88% meaning that 88% of the total amino acids in the protein were successfully identified as peptides during the LC-MS analysis. For my own analysis, the 90.7% of the eGFP sequence was covered.
Using my results from Question 2 and the previous selected peptide FEGDTLVNR, I have obtained the next Fragment Ion Table for the +1 charge state, which confirms the identity of the pepdtide eluting at 2.78 minutes. The calculated y-ion series matches the experimental peaks in Figure 5c.
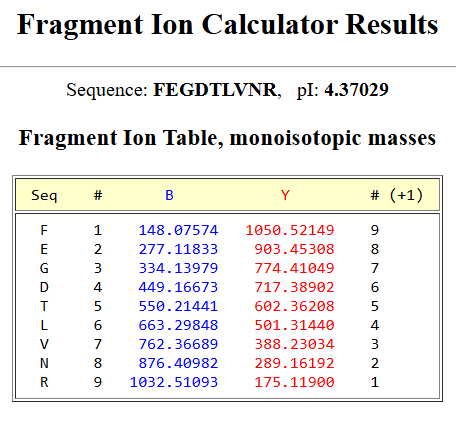
- According to the previous results, the experimental data and the theoretical one, the data makes sense because the experimental coverage (88%) is very close to the theoretical maximum obtained on PeptideMass (90.7%). The small difference of 2.7% is expected in LC-MS experiments as some predicted peptides may have low ionization efficiency or may not meet the 10% relative abundance threshold required for positive identification. With this, the results strongly indicate that the sample is the eGFP standard.
Waters Part IV - Oligomers
To identify the peaks, first I need to convert the units from kDa to MDa:

Waters Part V - Did I make GFP?
According to the MW of the protein calculated on Part I, the mass error of 375 ppm is quite normal, because for a complete protein the charge is so high the isotopes are very close together. Even with the high resolution, the machine still struggles to find the exact center of the peak, so the error is multiplied by 30 when calculating the final mass.
This is why we did the Peptide Mapping analyzing one single peptide (FEGDTLVNR) so the machine could see every single isotope clearly. By choosing one specific peak to calculate the mass and obtaining an accuracy of 5.3 ppm I could confirm that this was the exact sequence of my protein. Yes, I did make GFP

Week 11 HW: Bioproduction & Cloudlabs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
In the initial artwork (before the complete design was changed :()) I made that little flower using mRFP, sfGFP and mKO2.
I actually loved this project because we had the opportunity to contribute to the HTGAA effort. I would also like to say that Ronan’s page is easy to understand and use, thanks Ronan!
One thing I didn’t like was the fact that when this project was launched and we got the email, we started making our own figures but then (close to the deadline) some other students changed everything on the original design (can’t blame them though I know it was colaborative!). Maybe for next year the deadline could be shorter so it stays the original design or there’s not much time to re-design it?
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
- Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
a. E. coli Lysate
• BL21 (DE3) Star Lysate (includes T7 RNA Polymerase): bacterial cell extract containing all the molecular machinery needed for protein synthesis
b. Salts/Buffer
• Potassium Glutamate: maintains the ionic strength of the solution
• HEPES-KOH pH 7.5: buffer that keeps pH stable at 7.5 (close to physiological conditions)
• Magnesium Glutamate: provides Mg+2 ions essential for ribosome assembly and enzymatic activity
• Potassium phosphate monobasic
• Potassium phosphate dibasic: these two forms together act a buffer providing phosphate groups
c. Energy / Nucleotide System
• Ribose: backbone for building nucleotides
• Glucose: energy source
• AMP
• CMP
• GMP
• UMP: these four nucleoside monophosphates are necessary for RNA synthesis
• Guanine: can be converted into GMP and then GTP
d. Translation Mix (Amino Acids)
• 17 Amino Acid Mix: provides 17 of the 20 standard amino acids for protein building
• Tyrosine
• Cysteine: tyrosine and cysteine are added separately because are prone to oxidation and poorly soluble at neutral pH
e. Additives
• Nicotinamide: precursor to NAD+, necessary coenzyme for metabolic reactions
f. Backfill
• Nuclease Free Water: to bring the reaction volume to the final desired amount, plus it has no enzymes that could degrade RNA or DNA in the reaction.
- Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences).
The main difference is in the energy/nucleotide system: the 1-hour PEP-NTP mis provides energy through PEP-Mono and Maltodextrin and fully built NTPs ready to use immediately, while the 20-hour NMP-Ribose-Glucose system uses Ribose + Glucose and NMPs as simpler precursors that the enzymes convert into usable NTPs over time.
The 1-hour system also includes several additives absent in the 20-hour version: spermidine to stabilize ribosomes, DMSO to help with solubility, cAMP as a gene expression regulator, and folinic acid that supports amino acid synthesis.
- Bonus question: How can transcription occur if GMP is not included but Guanine is?
Enzymes present in the E. coli lysate can take the free guanine and attach it to a ribose-phosphate group to synthesize GMP on its way. That GMP can be phosphorylated to GDP and then GTP.
Part C: Planning the Global Experiment – Cell-Free Master Mix Design
- Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each).
• sfGFP: this Green fluorescent protein is very important because it has an exceptional folding robustness, so it was engineered so that its folding is not affected in any conditions.
• mRFP1: the biggest limitation is its low brightness, so it is needed a lot of protein to get a decent readout. In addition, some molecules get stuck during maturation, which makes the signal even lower.
• mKO2: this protein matures very slowly: it takes about 2 hours just to reach half of its final brightness, so it takes much more time to analyze the final results
• mTurquoise2: it is one of the most reliable fluorescent proteins to use as a reporter in cell-free systems because it basically converts almost every proton it absorbs into emitted fluorescence, so even small amounts of expressed protein will produce a strong signal.
• mScarlet_I: this protein has been engineered to have a marked maturation acceleration in cells, so compared to its sister protein mScarlet, this one is faster to light up in a cell-free reaction
• Electra2: this blue fluorescent protein tends to aggregate forming non-fluorescent aggregates instead of properly folding. It requires molecular oxygen for chromophore maturation, which must be present in sufficient quantity in the reaction.
- Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
My hypothesis is that increasing the concentrations of Ribose, Glucose and Magnesium Glutamate in the NMP-Ribose mastermix will maximize mKO2 fluorescence over a 36-hour incubation.
In a long-term reaction like this, the main issues are the energy depletion and byproduct inhibition, specifically the accumulation of inorganic phosphate which can inhibit key enzymes and slow down the reaction. By increasing ribose and glucose we provide a larger and longer-lasting carbon and energy soiurce, preventing early termination of transcription and translation. Additionally, increasing magnesium glutamate is crucial because Mg+2 ions are essential cofactors for ribosome stability and RNA polymerase activity, and in extended incubation periods free Mg+2 becomes sequestered by those accumulated phosphate groups, reducing its availability in the reaction.
mKO2 is particularly useful for rthis extended format because of its slow maturation half-time and its long-lived non-fluorescent intermediate state, meaning a significant fraction of the protein will still be completing chromophore formation past the 20-hour mark. The expected effect is a slow-burn metabolic rate that keeps the lysate active long enough for mKO2 to fully mature, resulting in a higher cumulative fluorescent yield at 36 hours compared to the standard 20-hour mix.
- he second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24). You can begin composing master mix compositions here.
I tested and changed the reagent master mix compositions according to my hypothesis, increasing concentrations of Glucose, Ribose and Magnesium Glutamate as shown in the image: