Week 2 HW: DNA Read, Write and Edit

–> This image shows DNA fragments separated by agarose gel electrophoresis and stained with a fluorescent dye, performed during my Genetic Engineering course.
Part 1: Benchling and In-silico Gel Art
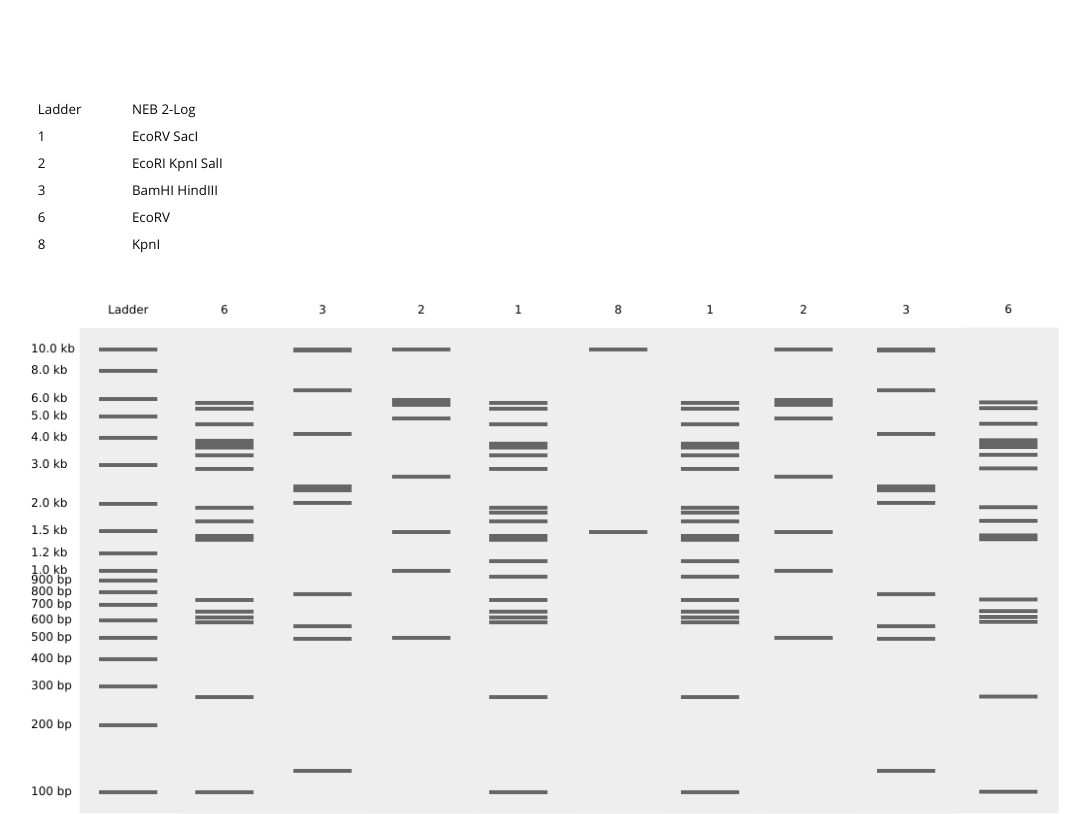
According to the instructions, this is the Gel I designed for p53 human protein (tried my best to make it look like a butterfly i’m sorry!!)
Part 3: DNA Design Challenge
For this assignment I have chosen the human protein p53, often called “guardian of the genome” is a critical tumor suppressor that maintains genomic stability by regulating cycle cell processes, promoting DNA repairing, and inducing apoptosis or senescence in response to cellular stress. It acts as a transcription factor, binding to DNA to stop damaged cells proliferation, potentially cancerous genetic material. I find this protein interesting because of its crucial role in maintaining cellular integrity. I was drawn to it during my medical genetics course last year due to its key function in controlling cell proliferation and its ability to trigger cell cycle arrest or apoptosis when DNA damage is detected. This protein is mutated in nearly 50% of known cancers in humans, and it is also involved in processes like aging, metabolism and DNA repair. This wide range of actions makes p53 an incredibly interesting protein to study in synbio.
After doing the reverse translation on my protein sequence to obtain the DNA sequence, I have performed the codon optimization. This is a fundamental part of genetic engineering because different organisms prefer different codons to code for the same amino acid. By optimizing the codons, you align the sequence with the organism’s natural tRNA abundance, which improves translation efficiency. Without doing this, even if the gene is correct, it might not be efficiently expressed, which can lead to low protein yields or even no expression at all. I have chosen the optimization for human organisms and I would prefer to express them on HEK293 cells because they are human-derived, making them ideal for expressing human proteins. There are easy to transfect, grow quickly, and they support post-translational modifications, which are crucial for many human proteins.
Now I have the optimized sequence, to produce the protein from the DNA, it is possible to amplify the sequence using PCR, designing primers that include specific restriction sites. Then, I amplify my gene and insert it into an expression vector such as a pEASY or pUC plasmid. This vector has to contain a strong promoter, like the CMV promoter, which is essential for expression in eukaryotic cells. After that, I have to linearize the plasmid using the same restriction enzymes, and transform the HEK293 cells by electroporation. Then, the cells are incubated in fresh culture medium to allow recovery and expression of the introduced DNA. The plasmid is transcribed into mRNA and then translated into the target protein by the cellular machinery. It is also important to have a selectable marker, such as antibiotic selection, to know which cells are expressing the gene. Finally, protein expression can be confirmed using techniques such as Western Blot, ELISA, or fluorescent detection if a GFP was added.
3.5 (Optional)
A single gene can code for multiple proteins at the transcriptional level primarily through alternative splicing, a process where the pre-mRNA, transcribed from the DNA, is spliced in different ways to include or exclude exons (coding regions).
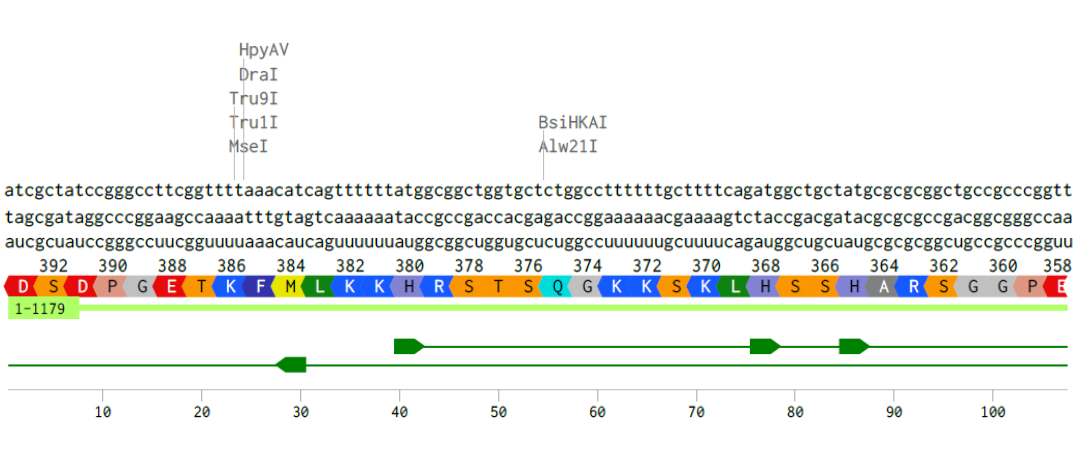
Alignment of the coding DNA and RNA sequence with its translated amino acid sequence for the p53 protein.
Part 4: Prepare a Twist DNA Synthesis order
I have built the DNA insert sequence according to the instructions on Benchling and this is what I have obtained:

Link to my E. coli sequence: https://benchling.com/s/seq-aEUjDIoXsdjPsD14jzXd?m=slm-BT3BayyvXI3H27cDW11c
Since I could not have access to the Twist Bioscience software (it says I have to contact a distributor), I was not able to continue with this assignment.
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) I would like to sequence the DNA of the parasite Tritrichomonas foetus, as it is the microorganism I plan to study for my final project. Specifically, I am interested in identifying the genes encoding excretion-secretion antigens, which are believed to play a key role in host-parasite interactions and reproductive impairment, and I want to probe that. Sequencing its DNA would allow me to better understand the molecular basis of its pathogenicity and how these antigens may affect the reproductive capacity of BALB/c mice. Additionally, obtaining genomic information could help identify potential targets for diagnostics, treatments or preventive strategies against infections caused by this parasite.
(ii) I would use NGS for whole-genome sequencing (WGS), as it provides comprehensive coverage of all genes, including my antigens of interest.
This method belongs to second-generation sequencing technologies because it relies on massively parallel sequencing of millions of DNA fragments simultaneously.
The input would be purified genomic DNA extracted from the parasite, which would then be fragmented, followed by adapter ligation and PCR amplification to generate a sequencing library. During sequencing-by-synthesis (e.g. Illumina platform), fluorescently labeled nucleotides are incorporated into the growing DNA strands, and each base is identified by detecting the emitted fluorescence.
The output consists of millions of short sequence reads that can be assembled bioinformatically to reconstruct the genome and identify genes related to pathogenicity and reproductive effects.
5.2 DNA Write
(i) I would like to synthesize a genetic circuit designed as a biosensor to detect excretion-secretion antigens from T. foetus as mentioned above. Early detection in infected bulls is critical because they can act as asymptomatic carriers and spread the parasite to females and other males, causing infertility, significant economic losses in the cattle industry and environmental consequences, as affected animals contribute to the spread of the parasite in pasturelands and water systems. The synthetic DNA construct would include a sensing module responsive to the parasite antigens already mentioned, a regulatory element, and a reporter gene (such as GFP). This biosensor could potentially be used as a rapid diagnostic tool to identify infected animals before the infection spreads within the herd.
(ii) to synthesize the designed genetic circuit, I would use commercial gene synthesis technology, which allows the production of custom DNA sequences with high accuracy, without the need to assemble fragments manually. The essential steps include designing the desired DNA sequence in silico, chemical synthesis of short oligonucleotides, assembly of these fragments into the full-length construct, and cloning into a plasmid vector for delivery. The resulting DNA can be then amplified and used for downstream applications such as transformation into host cells. This method is scalable and precise, however, limitations may include restrictions on how long the DNA sequence can be, the time it takes to receibe the synthesized construct, and possible difficulties with certain sequences, such as those with very high GC content or many repeated regions.
5.3 DNA Edit
(i) I would be interested in editing the human genome using CRISPR-Cas technology to study numerical abnormalities such as trisomy, specifically those causing early miscarriages, preventing babies from reaching full term, or resulting in reduced life expectancy, like Edwards syndrome (trisomy 18) and Patau syndrome (trisomy 13). The goal would be to explore whether removing the extra chromosome in early embryonic stages could restore normal gene dosage and improve developmental outcomes. This type of research could help us better understand the genetic mechanisms underlying severe developmental disorders.
(ii) to perform these DNA edits, I would use CRISPR-Cas9 genome editing technology because it allows precise and targeted modification of specific DNA sequences. This system works by using a guide RNA (sgRNA) designed to match the target DNA region, which directs the Cas9 enzyme to create a double-strand break at that site. The cell’s natural DNA repair mechanisms then repair the break, either by non-homologous end joining or homology-directed repair (which is expected to remove or modify genetic material).
Preparation involves designing specific sgRNAs targeting sequences on the extra chromosome, and delivering the complex Cas9-sgRNAs into cells or embryos, typically using vectors.
Limitations of this method include possible off-target effects, incomplete editing efficiency and mosaicism, where not all cells are edited in the same way.