Week 4 HW: Protein Design Part I
Part A. Conceptual questions
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
- Meat has an average 20% of protein, so 500 grams of meat would have 100 grams of protein. An average amino acid mass is 100 Da (100g/mol). So, according to Avogadro’s number, in a 500 g piece of meat, we are consuming approximately 6.62x1023 molecules of amino acids.
- Why do humans eat beef but do not become a cow, eat fish but do not become fish?
- Dietary proteins are digested into individual amino acids and absorbed and reused to build proteins used for the human metabolism, processes that determine our identity, not the food we eat.
- Why are there only 20 natural amino acids?
- There are 20 natural amino acids (22 in some organisms) because the standard genetic code evolved to use this 20 structures, maybe because they were efficient for metabolism, providing sufficient chemical diversity or they were optimal in terms of evolution.
- Can you make other non-natural amino acids? Design some new amino acids.
- Using synbio it is possible to create new amino acids according to what you need or want to do, for example maybe metal-binding amino acids to coordinate metal ions, or photo-crosslinking amino acids to form bonds when exposed to light, or perhaps adding electronegative elements like Fluor to alter their electronic properties.
- If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
- L-amino acids form right-handed helices, while D-amino acids would form a left-handed helix.
- Can you discover additional helices in proteins?
- Synbio help us explore and find new structural possibilities since protein folding is certainly a complex issue that, at present, is not fully understood. Even though they are rare, it is possible to find -helix, 310 helix, foldamers (artificial oligomers), and maybe there are more out there to be discovered.
- Why are most molecular helices right-handed?
- Because biological amino acids are L-chiral, making right-handed helices more sterically favorable and stable energetically.
- Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
- β-sheets are made of protein strands that lie next to each other and are connected by many hydrogen bonds, forming a very flat and extended surface with many places where it can stick to another sheet, so they can easily attach and stack together. Some amino acids in proteins are hydrophobic and when the β- sheets form, these parts can be exposed. To avoid water, they stick to each other hiding from water. Aggregation happens because it is energetically favorable, so the driving forces are the hydrophobic effect, the hydrogen bonds to increase stabilization and sticking together to lower the free energy.
- Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
- In amyloid diseases (like Alzheimer’s) some proteins misfold, losing their normal shape and refolding into β-sheet structures. Since these structures are very stable, it is hard to go back from there. These misfolded proteins then stick to each other, form long fibers (amyloid fibrils), accumulate in tissues and damage cells. Since these structures are very strong, stable and able to self-assemble, it is possible to use them as materials. Scientist are studying them for creating nanofibers, biomaterials, tissue scaffolds, and drug delivery systems.
Part B. Protein Analysis and Visualization
For this assignment I chose the p53 protein (P04637), which is a multifunctional transcription factor that induces cell cycle arrest, DNA repairs or apoptosis upon binding to its target DNA sequence in human organisms. Acts as a tumor suppressor in many tumor types; induces growth arrest or apoptosis depending on the physiological circumstances and cell type. I particularly find its activity fascinating since I am interested in cancer diseases and trying to understand how they work and how the human body is prepared to regulate these processes.
i. This protein has 393 amino acids, being P (proline) the most frequent one (11.45%)
ii. Running a BLAST analysis, I have found this protein has 250 sequence homologs, going from the organism Pan paniscus (Pygmy chimpanzee) with a 100% homology, to Suricata suricata (Meerkat) with a 79.6%.
iii. This protein belongs to the proteins p53 family, also known as family p53/p63/p73 because they all share a similar structure with conserved domains and related functions.
i. The first high resolution structure solved of the oligomerization domain was deposited in 1994 and released in 1995 using a multi-dimensional NMR. (https://www.rcsb.org/structure/1OLG#entity-1)
ii. This protein is often linked with DNA apart from the protein since it is a transcription factor, so it binds to the DNA to regulate genes. It also has antibody fragments used for crystallization, regulating proteins and peptides like an 11-residue recruitment peptide in a complex with CDK2/CyclinA. Zinc ions are also present, used as a cofactor (binds 1 zinc ion per subunit).
iii. p53 has 6 different domains, and each domain belongs to a different structure classification family:
• 2AC0 A:96-289 (SCOP ID 8024487): Family: p53 DNA-binding domain-like
• 1AIE A:326-356 (SCOP ID 8025247): Family: p53 tetramerization domain
• 2AC0 A:96-289 (SCOP ID 8036866): Family: p53-like transcription factors
• 1AIE A:326-356 (SCOP ID 8037626): Family: p53 tetramerization domain
• 3DAC P:17-28 (SCOP ID 8050972): Family: p53 transactivation domain (TAD)
• 3DAC P:17-28 (SCOP ID 8093389): Family: p53 TAD
- ii.
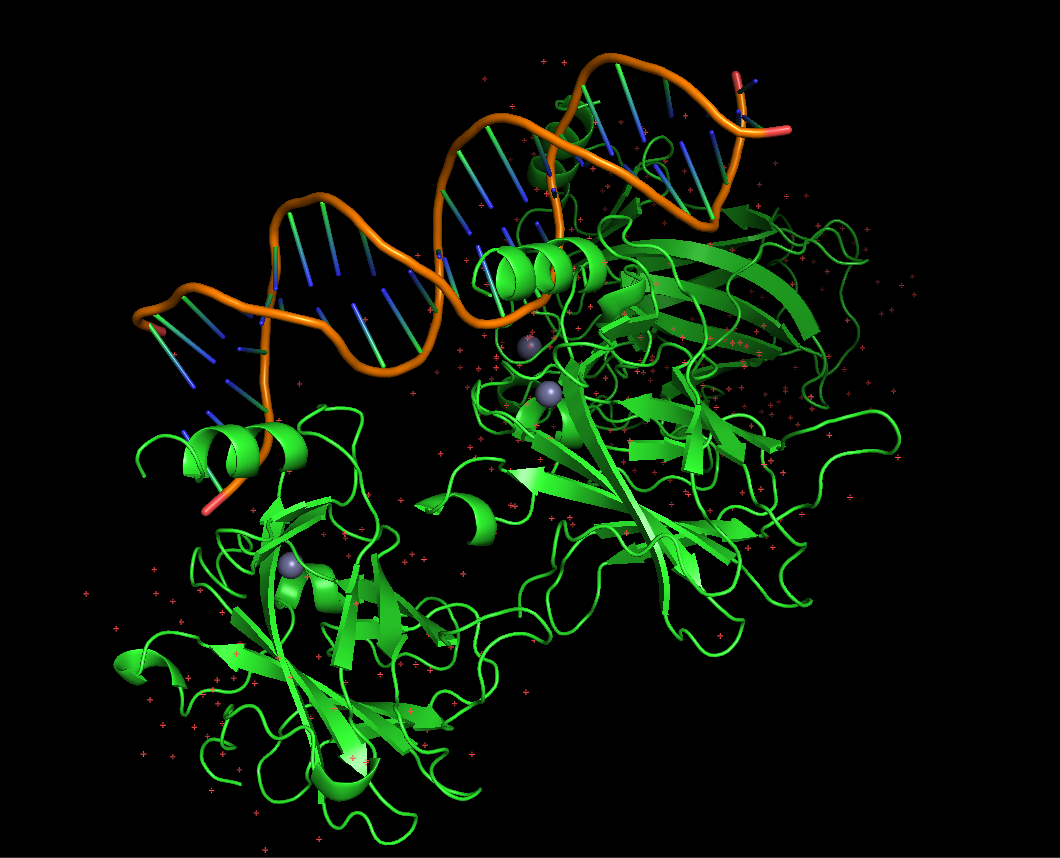
 Protein visualized as “cartoon”
Protein visualized as “cartoon”
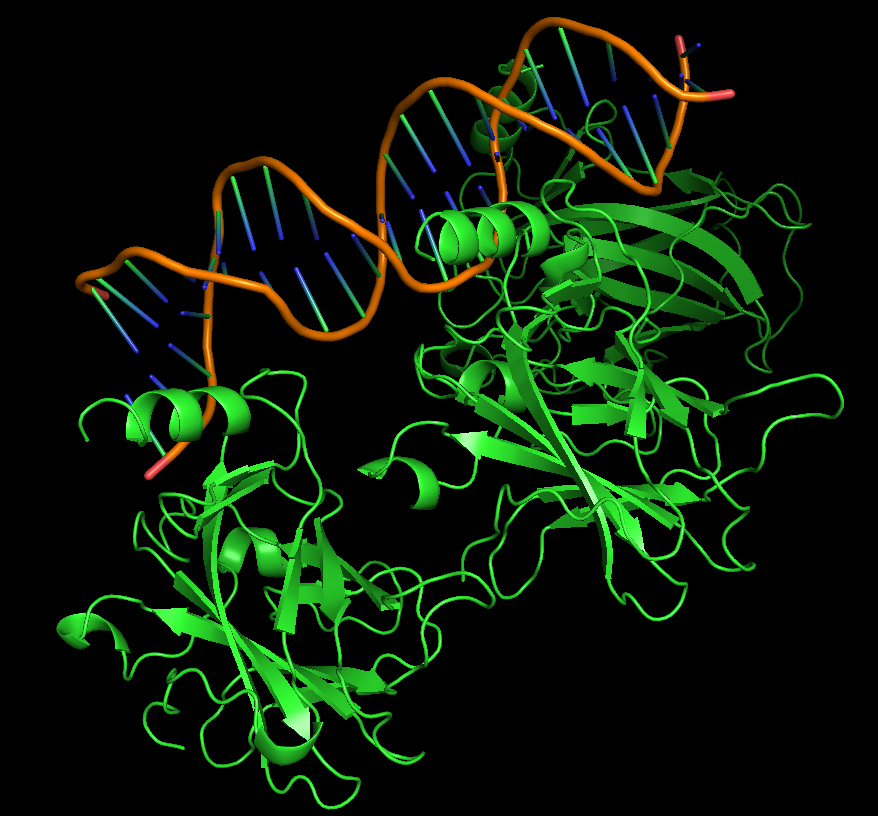
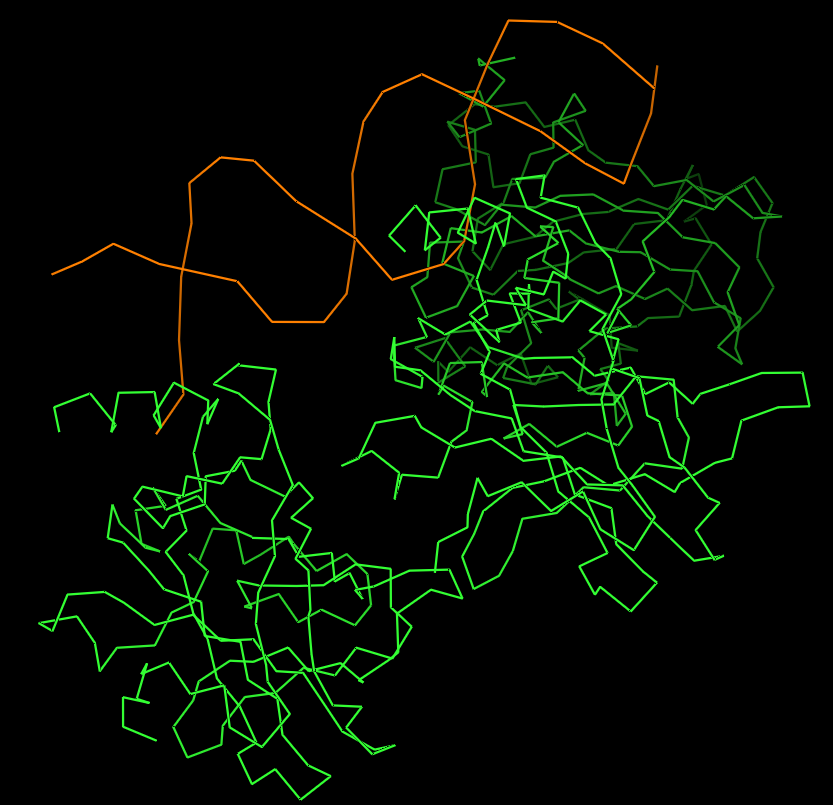 Protein visualized as “ribbon”
Protein visualized as “ribbon”
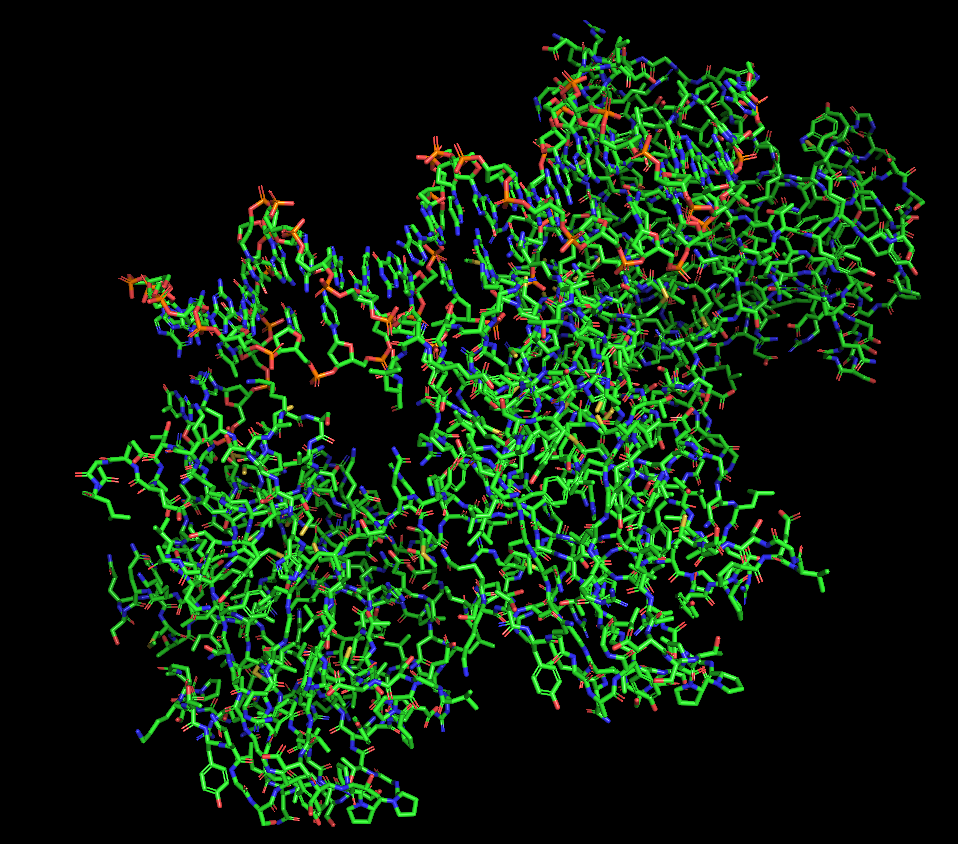 Protein visualized as “sticks”
Protein visualized as “sticks”
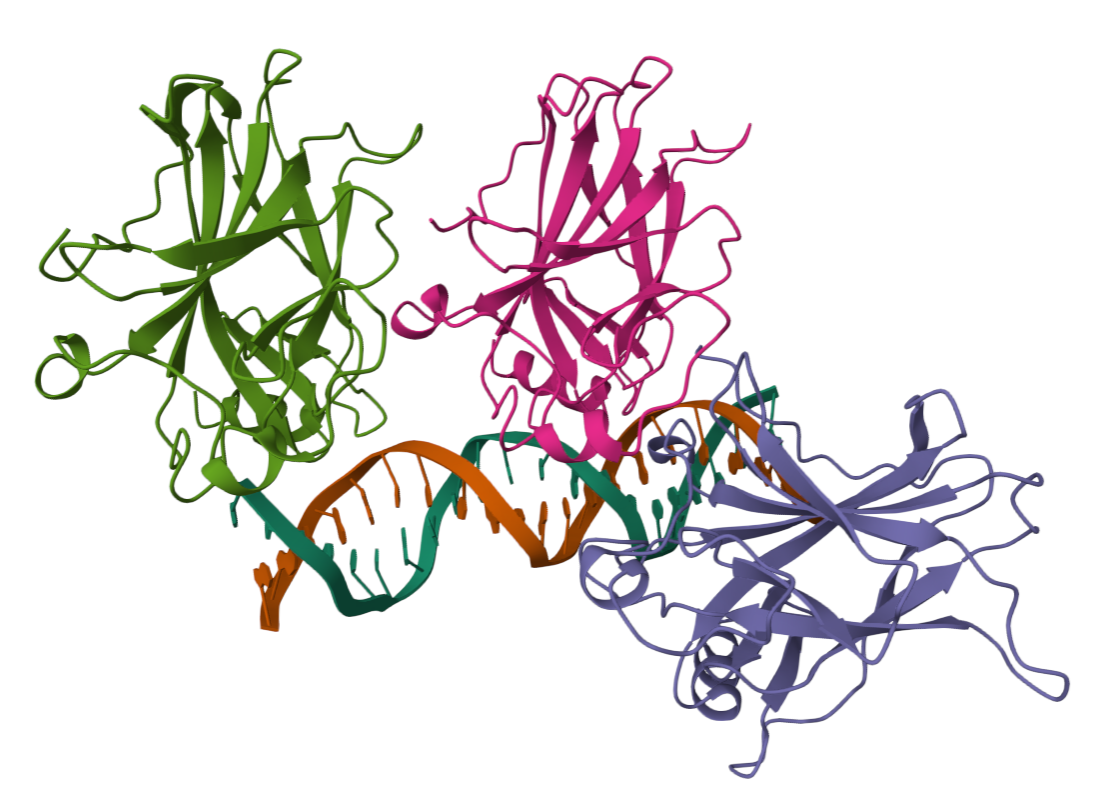
iii. The protein is colored by secondary structure: alpha-helices in red, beta-sheets in yellow, loops in blue. The structure also shows p53 bound to DNA.
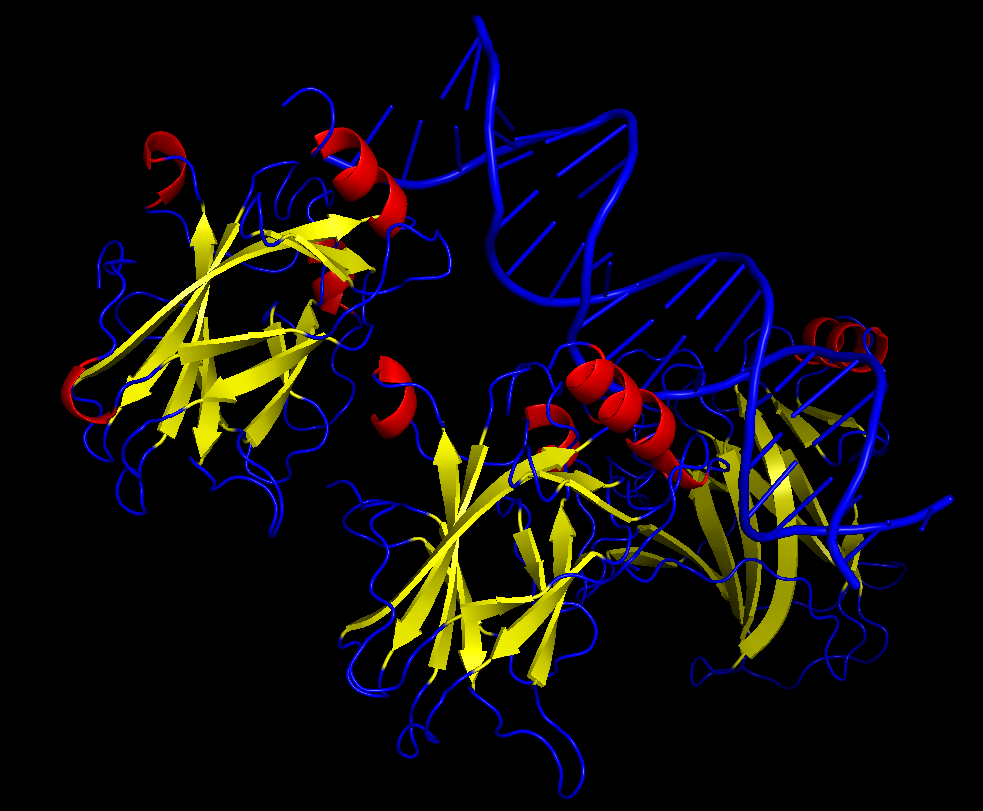
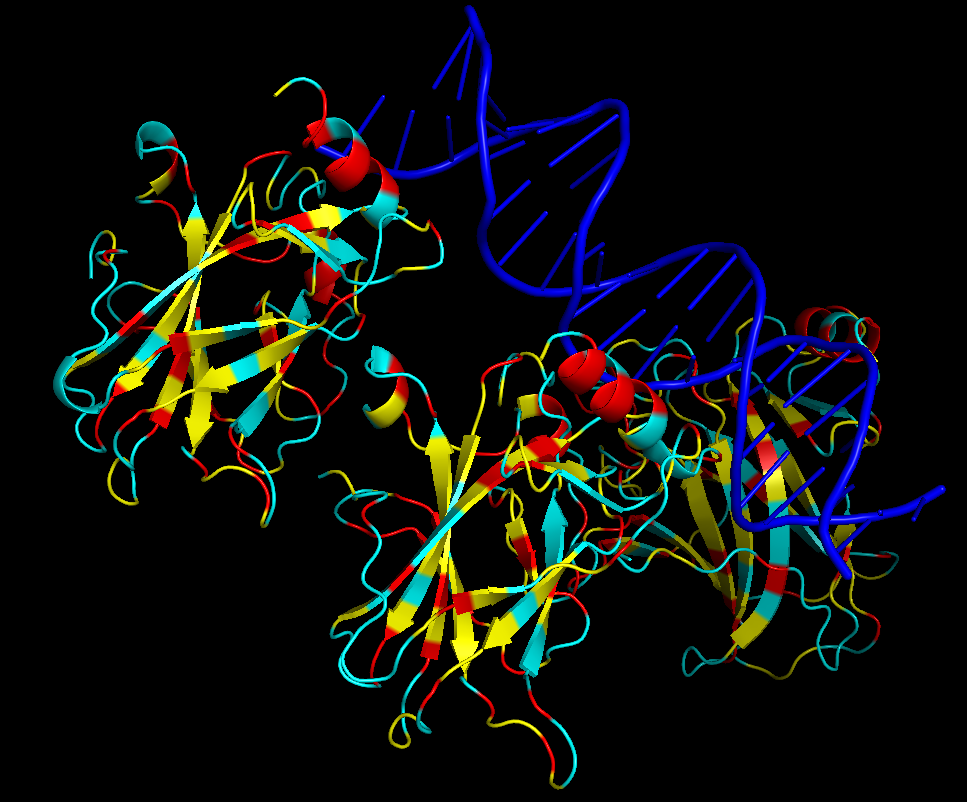
iv. Hydrophobic residues (yellow) are mainly located in the interior of the protein forming a stable core. Hydrophilic residues (blue) are most exposed on the surface where they can interact with water or DNA. Charged residues are colored in red.
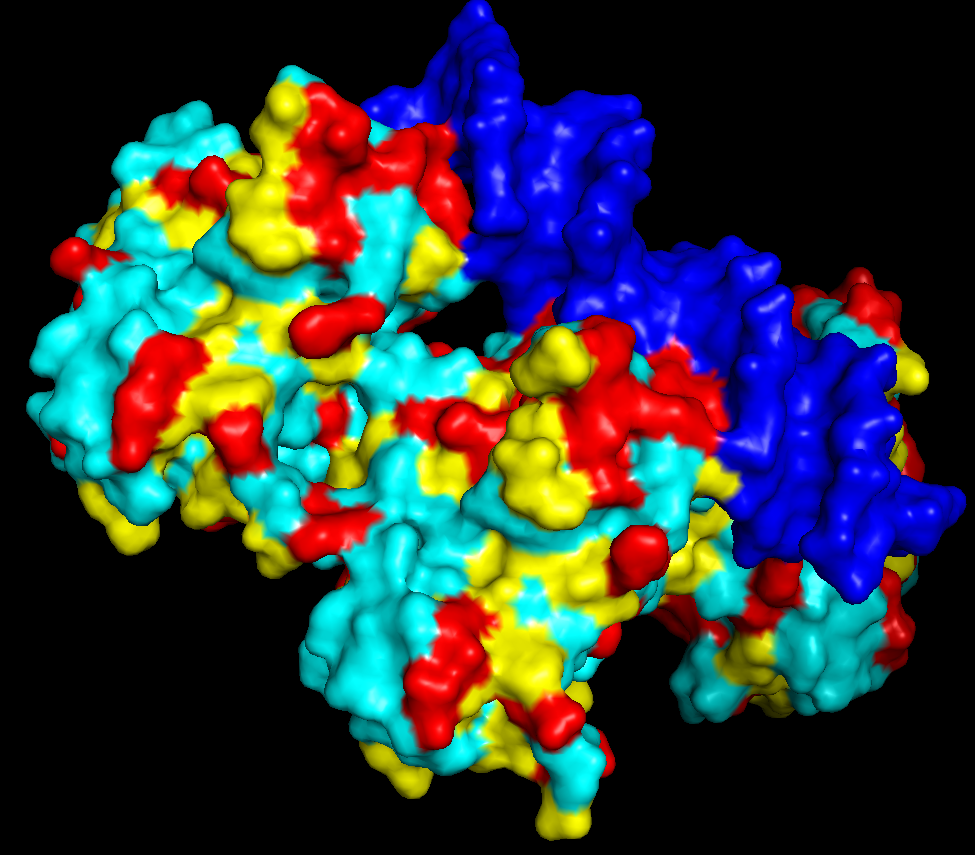
v. The protein surface shows “holes” or binding pockets where other molecules, like DNA or peptides, can bind. Such pockets are important for p53’s biological function.
PART C
C1. Protein Language Modeling
- Deep Mutational Scans
b. After generating the deep mutational scan of protein p53, I have found that most mutations are tolerated in some regions showed in yellow and green colors, but other certain positions display strongly unfavorable mutations across many amino acid substitutions, colored in blue (unfavorable) and dark violet (VERY unfavorable). These positions likely correspond to functionally important residues, such as those involved in DNA binding or structural stability.
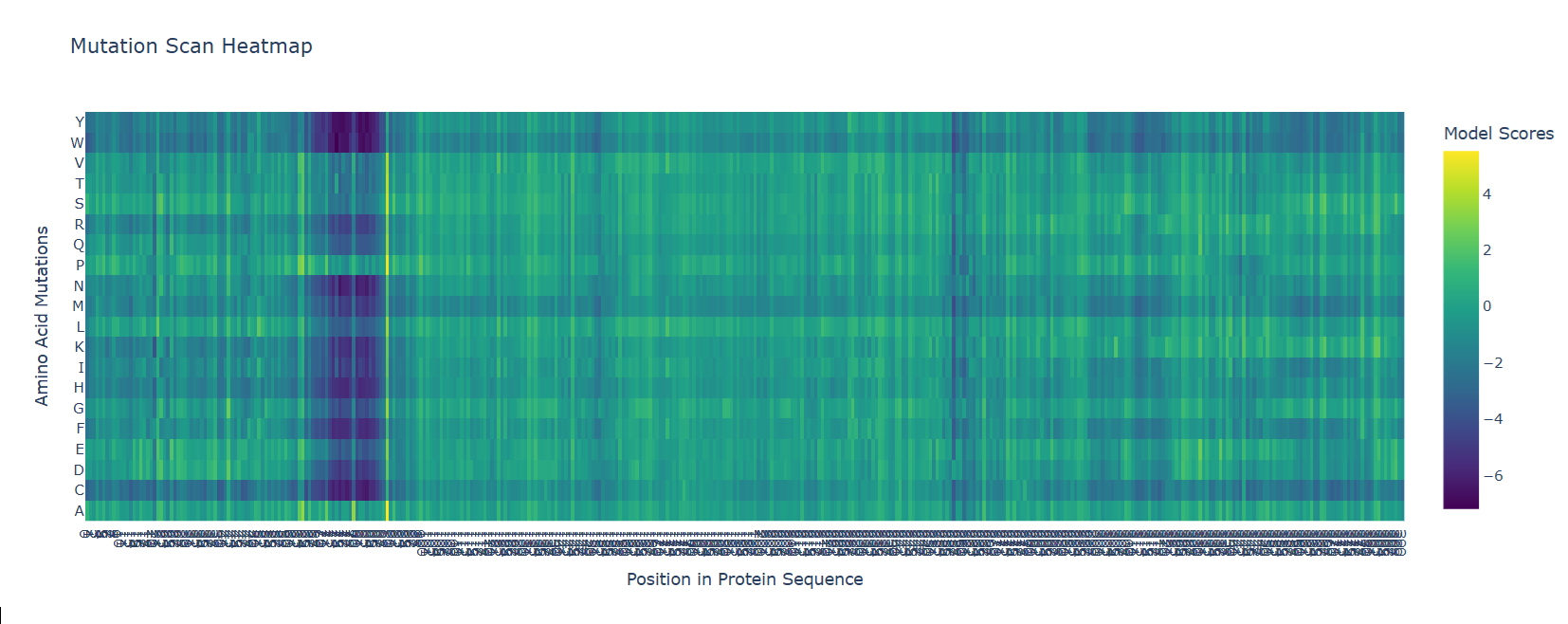 Mutational scan for p53
Mutational scan for p53
A clear pattern is observed at positions where nearly all mutations are highly unfavorable (violet and blue). One mutation that stands out is the substitution at position 74 to tryptophan (W), that shows a strongly negative score (-7.186), suggesting that this mutation is highly unfavorable and likely disrupts the protein’s structure or function.
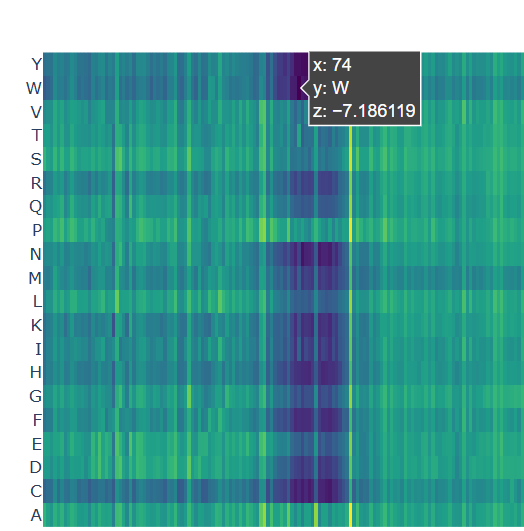 Chosen mutation for p53 (very dark violet)
Chosen mutation for p53 (very dark violet)
- Latent Space Analysis
b. When analyzing the points in the t-SNE 3D plot for the Paramecium caudatum hemoglobin as provided, proteins here are located near a cluster of similar proteins, suggesting functional or structural similarity with its neighbors. For example, I have found a cluster including matches for Clostridium botulinum and the tetanus neurotoxin from C. botulinum.
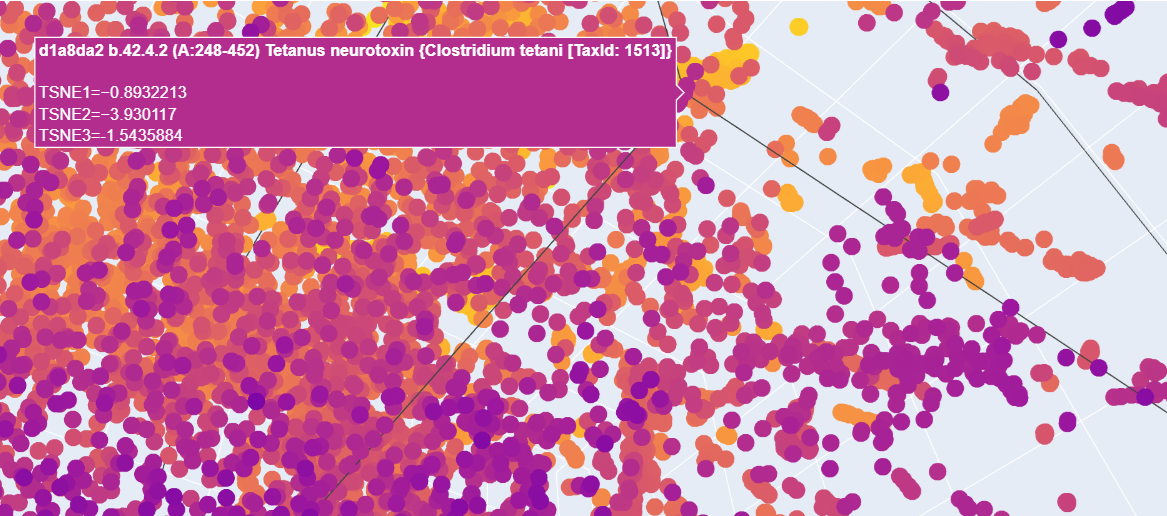
c. I placed my selected protein in the resulting 3D map, where it sits within a dense cluster of the latent space. Its position suggests that p53 shares common structural features or evolutionary patterns with many other proteins in the dataset.

C2. Protein Folding
- I folded the p53 protein using ESMFold and compared the predicted structure with the experimental structure from PyMOL (PDB ID: 1TUP). They match partially, because beta-sheets and alpha-helices that form the core of the protein in the ESMFold prediction align closely with the ones in PyMOL. According to the structural differences, PyMOL structure includes p53 bound to DNA as a complex, whereas the ESMFold prediction shows a single isolated chain (monomer).
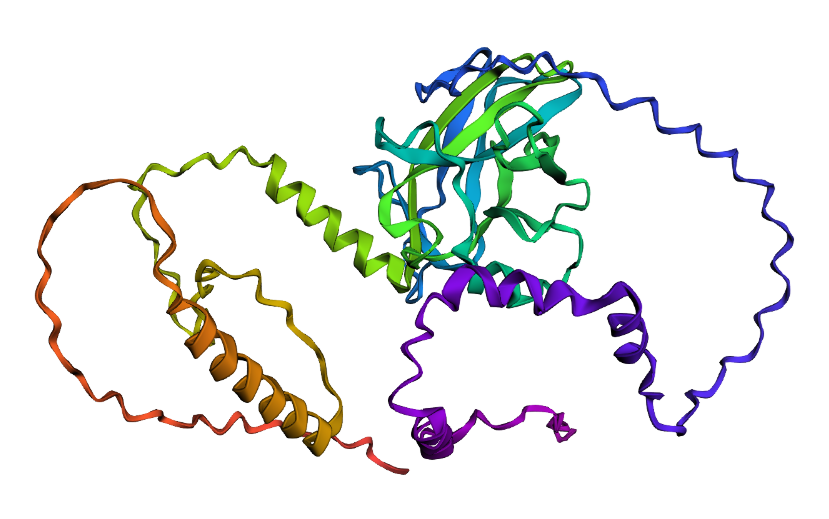 Folded protein structure predicted by ESMFold.
Folded protein structure predicted by ESMFold.
 Folded protein structure predicted by PDB.
Folded protein structure predicted by PDB.
- First I changed only one amino acid and the structure remained stable. Then I changed 5 more amino acids and the folding was a bit different but still possible. Next, changed 20 amino acids but still showing, even though had a different folding structure. Lastly, I deleted 15 amino acids and the protein was still showing with some other structural changes (including the previous changes I made on the sequence). Given the differences, it is possible to say that the core domain maintains its architecture, indicating that the protein exhibits structural robustness.
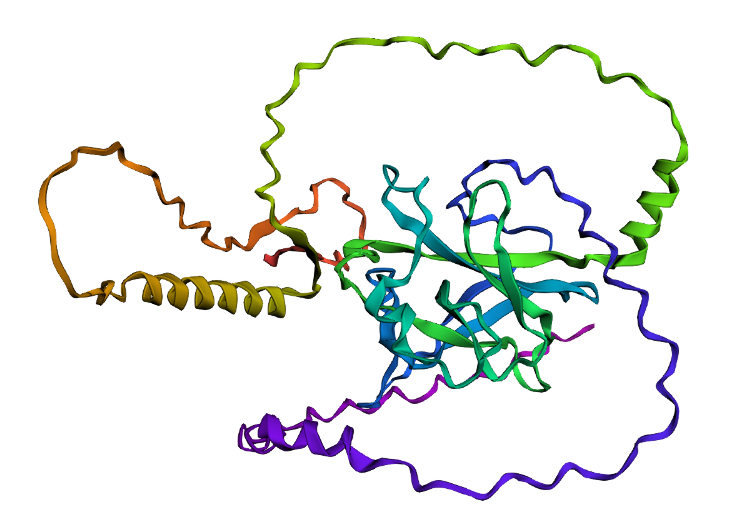
C3. Protein generation
- The predicted sequence for my protein (PDB: 1TUP) is:
GPPVPPTARDPGAYGFTLGFEATGTGASVTSTYSPALNTIYAKLNAAVPVRLLTTAPPPAGTRVRFRLVYADEAYRTTVVRRSPKAAAADDSDGRRPPDFVLSILDDPDAEYVRDPETGWLSVTVPYRPPPPGATATTYLLAFNETTTAKGGLDGNKVLLVVELLDADGALLGRDSAYVRVVANPGAAAAAAEAAK
And the original sequence is:
MEEPQSDPSVEPPLSQETFSDLWKLLPENNVLSPLPSQAMDDLMLSPDDIEQWFTEDPGPDEAPRMPEAAPPVAPAPAAPTPAAPAPAPSWPLSSSVPSQKTYQGSYGFRLGFLHSGTAKSVTCTYSPALNKMFCQLAKTCPVQLWVDSTPPPGTRVRAMAIYKQSQHMTEVVRRCPHHERCSDSDGLAPPQHLIRVEGNLRVEYLDDRNTFRHSVVVPYEPPEVGSDCTTIHYNYMCNSSCMGGMNRRPILTIITLEDSSGNLLGRNSFEVRVCACPGRDRRTEEENLRKKGEPHHELPPGSTKRALPNNTSSSPQPKKKPLDGEYFTLQIRGRERFEMFRELNEALELKDAQAGKEPGGSRAHSSHLKSKKGQSTSRHKKLMFKTEGPDSD
 Even though I kept getting an error while trying to run the ESMFold with this new sequence (and could not complete the task in my Google Colab), I compared them by myself and I can clearly see that the new predicted sequence is way shorter and different than the original one.
I asked Gemini to compare them and found that there is a very low level of sequence identity between the two of them. The original sequence is 393 amino acids long and includes flexible, disordered regions, while the predicted sequence is shorter (approximately 190 amino acids) and focuses on the DNA-binding core domain. The probabilities from ProteinMPNN show that the model prioritized stability. It replaced many of the original amino acids with new ones that better fit the 3D backbone.
So, even though the letters are different, the predicted 3D structure matches the original p53 backbone almost perfectly (according to Gemini), proving that the model succesfuly redesigned the protein, maintaining its functional shape while using a different primary sequence.
Even though I kept getting an error while trying to run the ESMFold with this new sequence (and could not complete the task in my Google Colab), I compared them by myself and I can clearly see that the new predicted sequence is way shorter and different than the original one.
I asked Gemini to compare them and found that there is a very low level of sequence identity between the two of them. The original sequence is 393 amino acids long and includes flexible, disordered regions, while the predicted sequence is shorter (approximately 190 amino acids) and focuses on the DNA-binding core domain. The probabilities from ProteinMPNN show that the model prioritized stability. It replaced many of the original amino acids with new ones that better fit the 3D backbone.
So, even though the letters are different, the predicted 3D structure matches the original p53 backbone almost perfectly (according to Gemini), proving that the model succesfuly redesigned the protein, maintaining its functional shape while using a different primary sequence.
Part D: Brainstorm on Bacteriohage Engineering
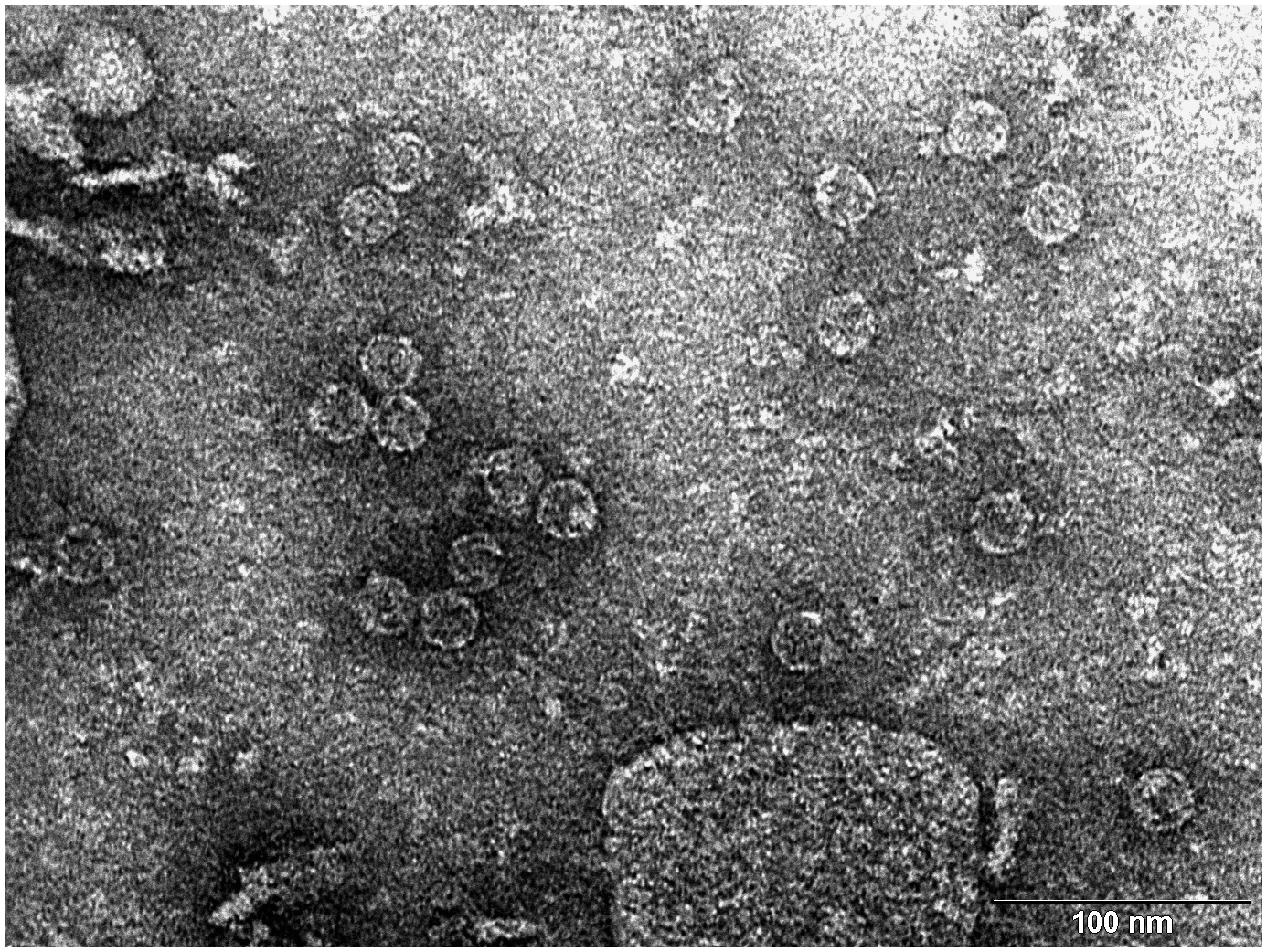 Transmission electron microscopy (TEM) photograph of the intact MS2 phage-like particles (MS2 PLP) present in the supernatant after ultrasonic disruption of E. coli production cells. (Mikel P, Vasickova P, Tesarik R, Malenovska H, Kulich P, Vesely T and Kralik P (2016) Preparation of MS2 Phage-Like Particles and Their Use As Potential Process Control Viruses for Detection and Quantification of Enteric RNA Viruses in Different Matrices. Front. Microbiol. 7:1911. doi: 10.3389/fmicb.2016.01911)
Transmission electron microscopy (TEM) photograph of the intact MS2 phage-like particles (MS2 PLP) present in the supernatant after ultrasonic disruption of E. coli production cells. (Mikel P, Vasickova P, Tesarik R, Malenovska H, Kulich P, Vesely T and Kralik P (2016) Preparation of MS2 Phage-Like Particles and Their Use As Potential Process Control Viruses for Detection and Quantification of Enteric RNA Viruses in Different Matrices. Front. Microbiol. 7:1911. doi: 10.3389/fmicb.2016.01911)
(Important: I couldn’t find a group in this opportunity so I was my own group!)
Proposal: Engineering a DnaJ independent MS2 lysis protein for enhanced Phage Therapy
The L- protein of MS2 phages needs DnaJ because this is a chaperone from the Hsp40 family that helps the full-length L protein fold correctly. In the host cell, DnaJ forms a complex with the highly basic N-terminal domain of L. This complex allows L to adopt a conformation that can interact with its target (still unknown) and cause cell lysis.
When the chaperone is mutated or removed, the lysis process is delayed or completely blocked at certain conditions, even though L accumulates normally, showing that the lack of interaction with DnaJ prevents a step happening after folding, not the synthesis of the toxic protein itself. According to this, the main goal is to engineer a Dna-J independent version of the MS2 L protein. By removing this dependency and stabilizing the C-terminal lytic core, I aim to create a protein that triggers bacterial lysis faster and more reliably across different bacterial strains without needing additional co-factors.
Using the tools practiced in this weeks’ recitation, this is the proposed bioinformatics pipeline:
Identify the region that really matters: mutagenesis experiments showed that the 67 loss-of-function alleles are concentrated in the C-terminal half of L around the LS motif. The N-terminal domain (residues 1-42) acts as a regulatory break because it creates a strict dependency on the host chaperone DnaJ for proper holding. There, the first 36 to 42 amino acids (N-terminal domain) are nonessential for the killing mechanism itself and removing them speeds up lysis. In addition, the Lytic Core corresponds to the last 30 amino acids, which include the LS motif and the transmembrane helix. So, I will keep the LS motif (Leu48-Ser49) and the Lys50 residue as they are essential for membrane interaction.
Search for homology and keep the essentials:
- Use BLAST against UniProt to obtain L-like sequences from other leviviruses (similar to MS2). Using ESM2 (Protein Language Model), I will perform an in silico Deep Mutational Scan to rank possible mutations, helping me find specific substitutions in the membrane helix that increase stability without breaking the essential LS motif.
Model the structure through Computational Tools:
After finding the best mutation candidates, I will upload the core sequence to ESMFold and visualize it (and compare with PyMOL) to confirm that the transmembrane helix is correctly inserted and capable of membrane insertion.
Once the structure is confirmed, could be useful to use ProteinMPNN to generate a new (and much more robust) sequence for the protein, making it more stable for biotechnology applications.
Finally, I will perform a Latent Space Analysis using t-SNE to validate the engineered designs. This map acts as a functional “sanity check” by clustering the artificial sequence with known active and natural variants of the original protein. If my candidate falls within the functional cluster and stays far away from known loss-of-function mutants (like those affecting the LS motif) it confirms that the protein is likely to be active in the lab, maintaining the original properties necessary to interact with its target.
Potential pitfalls:
Unknown target: since the host membrane target protein is unknown, it is not possible to predict (and confirm) the exact binding interface.
Lysis/assembly balance: if lysis happens to fast, it might kill the bacteria before enough phage progeny are assembled.