Week 5 HW: Protein Design: Part II
PART A: SOD1 Binder Peptide Design
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
SOD1 sequence: MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
SOD1 Mutated (A4V) sequence: MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Part 1: Generate Binders with PepMLM
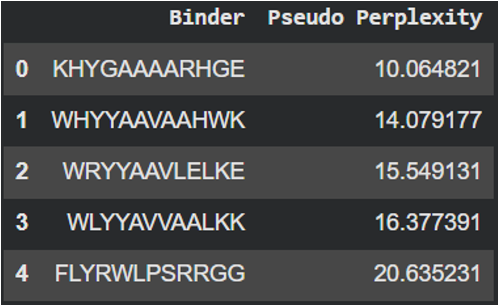
The pseudo-perplexity score reflects how likely a peptide-sequence is according to the patterns learned by this protein language model. Lower values indicate sequences that are more consistent with common protein sequence patterns. Analyzing the designed peptides, KHYGAAAARHGE showed the lowest score (10.06) suggesting that it is the most probable peptide according to the model. In contrast, the reference peptide (FLYRWLPSRRGG) showed the highest value (20.63) indicating that its amino acid composition is less typical compared to the sequences learned by this model.
Part 2: Evaluate Binders with AlphaFold3
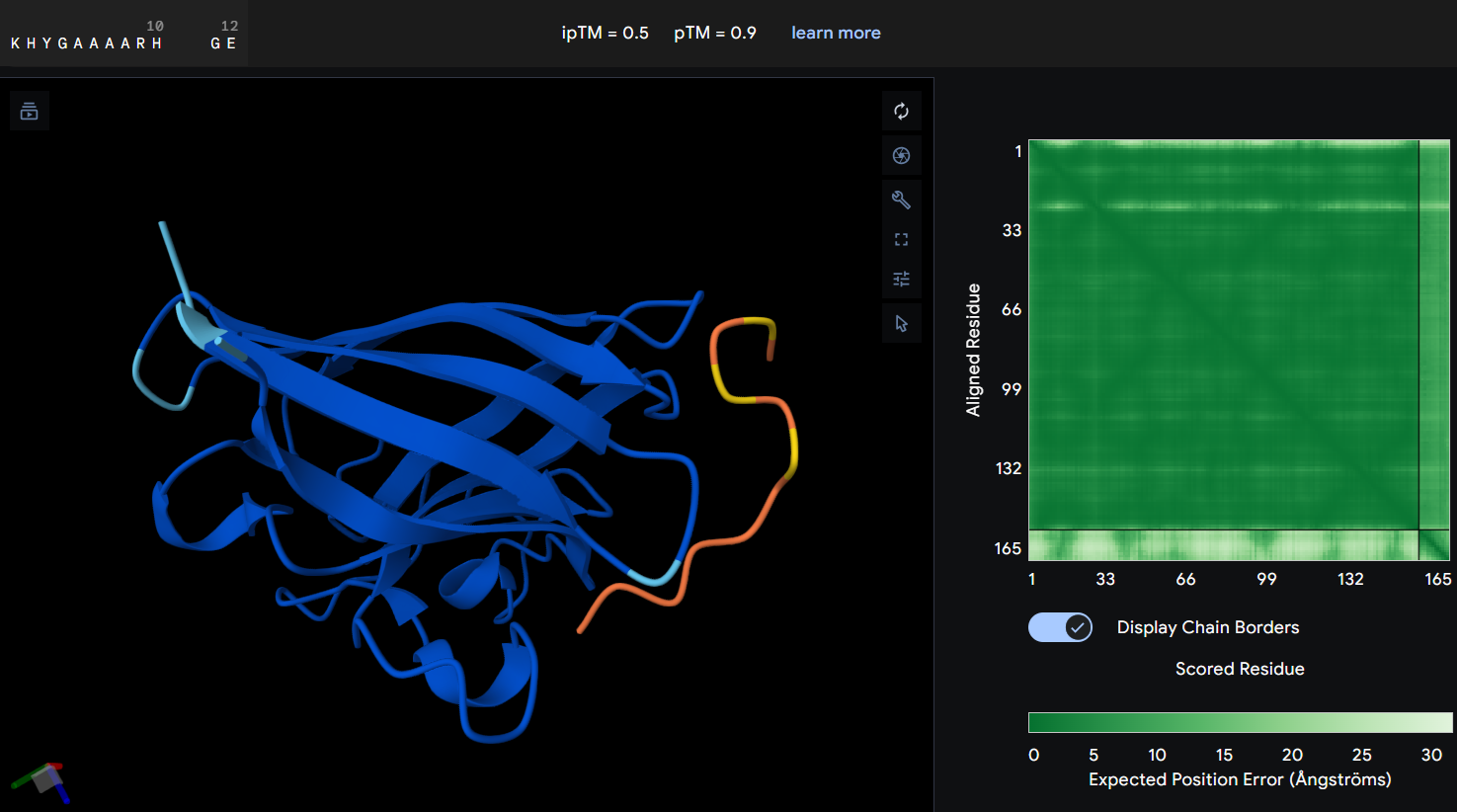
- Protein-peptide KHYGAAAARHGE: the prediction produced an ipTM score of 0.5 suggesting a weak or uncertain interaction between the mutant SOD1 protein and the designed peptide. In the graphic, the peptide is spatially separated from the prtein surface and does not clearly localize near the N-terminal region where the A4V mutation is located. The PAE map also shows higher uncertainty between both of them, indicating that AlphaFold does not predict a stable binding interaction in this configuration. Although this PepMLM-designed peptide showed the lowest perplexity score, the prediction not showing a clear interaction with the mutant suggests that, while the peptide sequence is statistically possible, it may not form a strong binding interaction with SOD1.
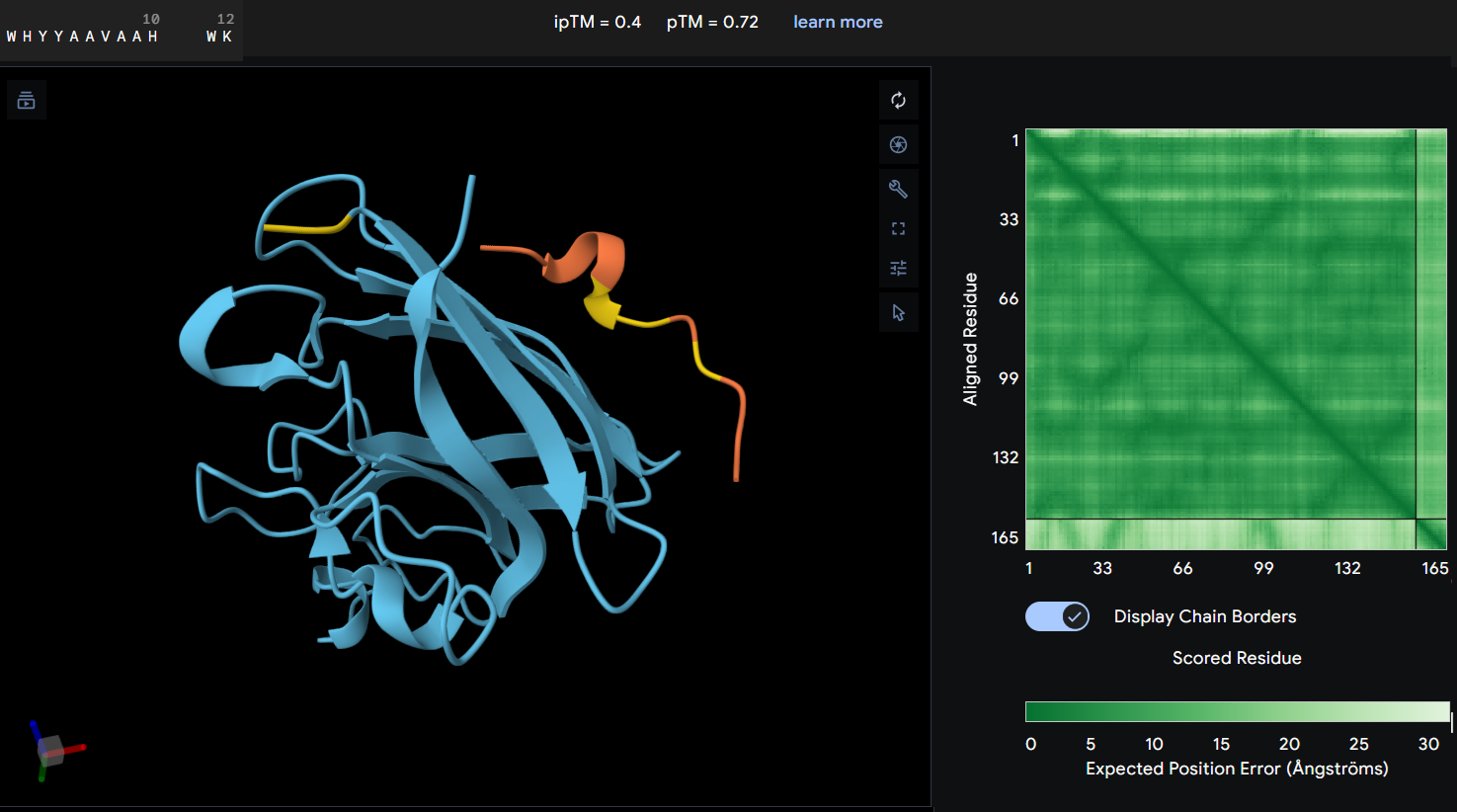
- Protein-peptide WHYYAAVAAHWK: this second designed peptide showed an ipTM score of 0.4 indicating low confidence in the predicted protein-peptide interaction. In this graphic, the peptide does not form a clearly defined binding interface. The PAE map also suggest uncertainty in the relative postioning between the peptide and the protein. This model does not support a strong or stable interaction between this peptide and the mutant SOD1 structure.
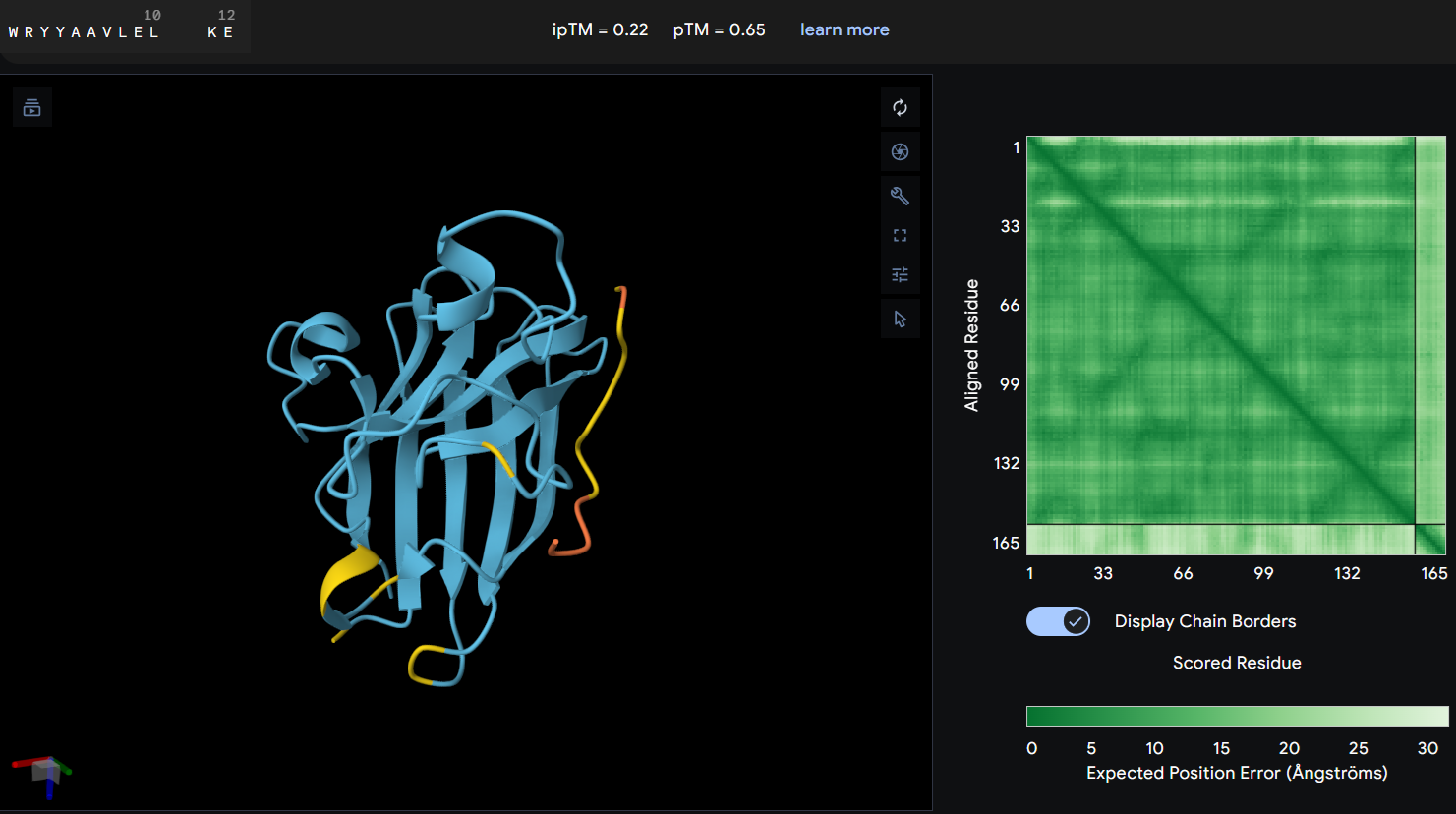
- Proteine-peptide WRYYAAVLELKE: this peptide produced an ipTM score of 0.22 indicating a very low confidence in the predicted protein-peptide interaction. In the structural model, the peptide appears clearly separated from the SOD1 surface and does not form any binding interface. The PAE map also shows high uncertainty between the peptide and the protein. These results suggest that this peptide is unlikely to form a stable interaction with the mutant SOD1 potein.
- Protein-peptide WLYYAVVAALKK: this peptide showed an ipTM score of 0.36 reflecting a lack of predicted interaction with the protein. The predicted structure shows the peptide distant from the protein’s core, and the PAE map confirms this with a high error values between the two chains.
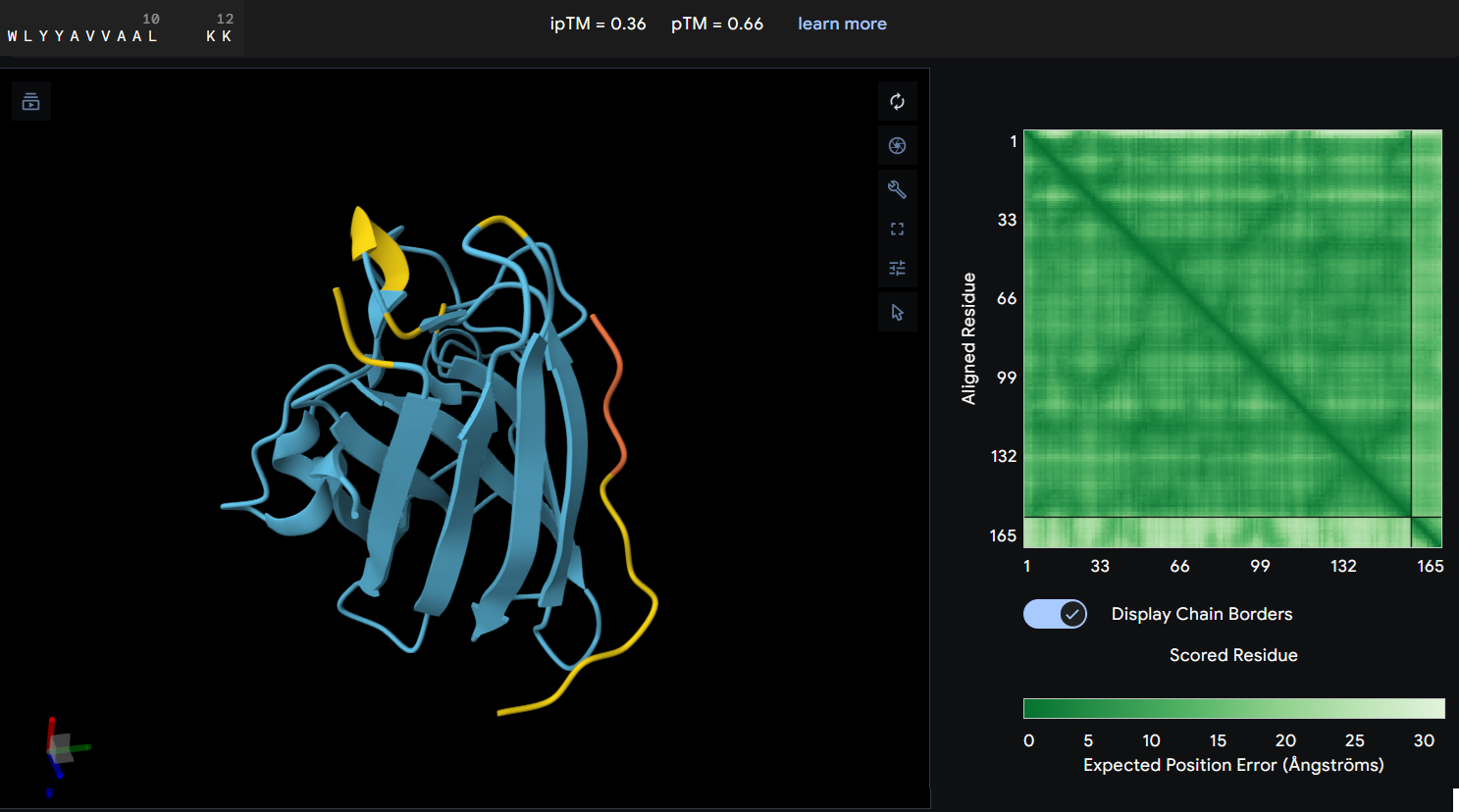
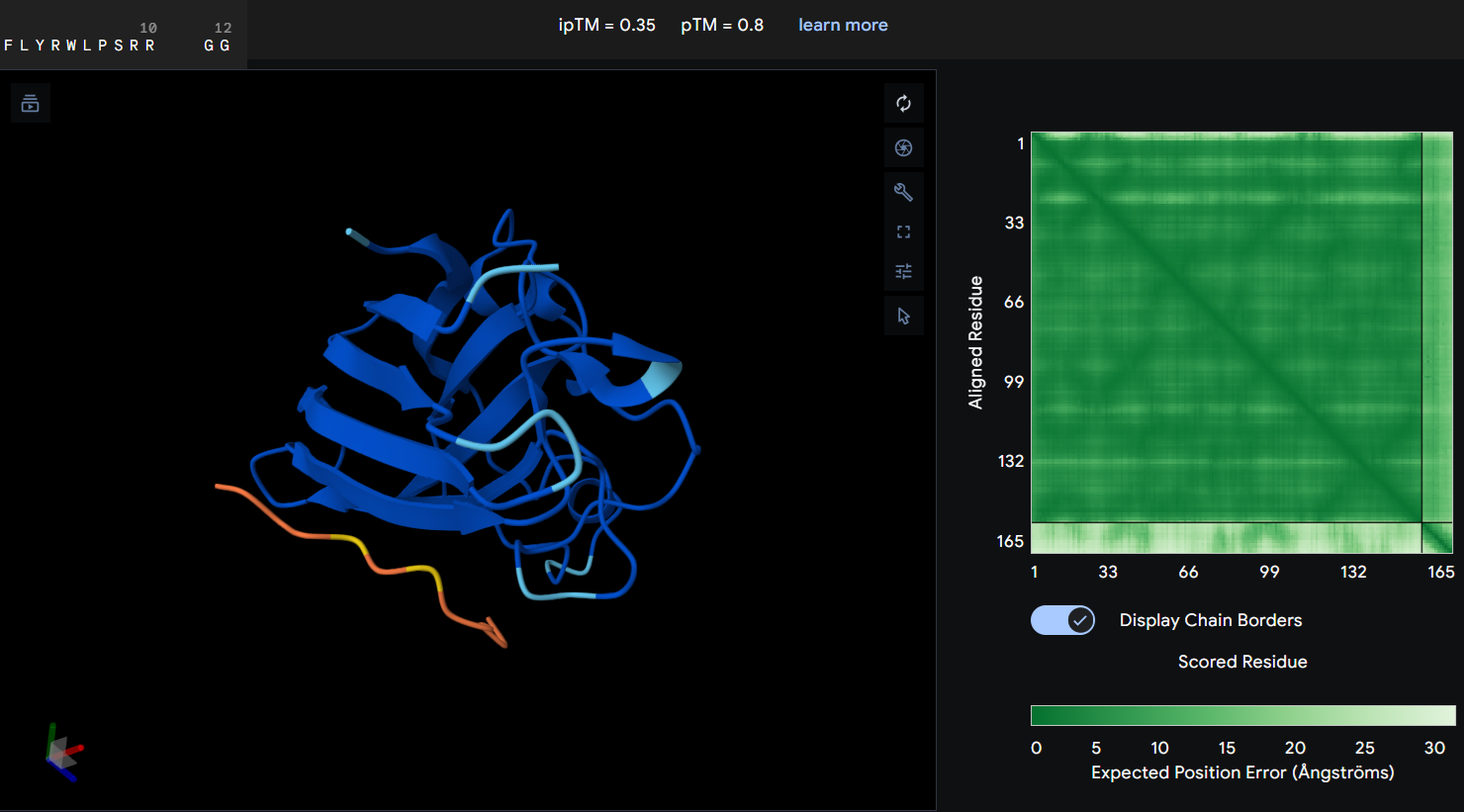
- Protein-peptide FLYRWLPSRRGG: the given peptide for this activity resulted in the lowest docking confidence with an ipTM of 0.35. In the predicted structure, there is a complete lack of interaction as the peptide remains spatially dissociated from the protein. This observation is supported by the PAE map, which shows maximum error values for the chain distances, indicating that the model cannot predict a stable interface.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
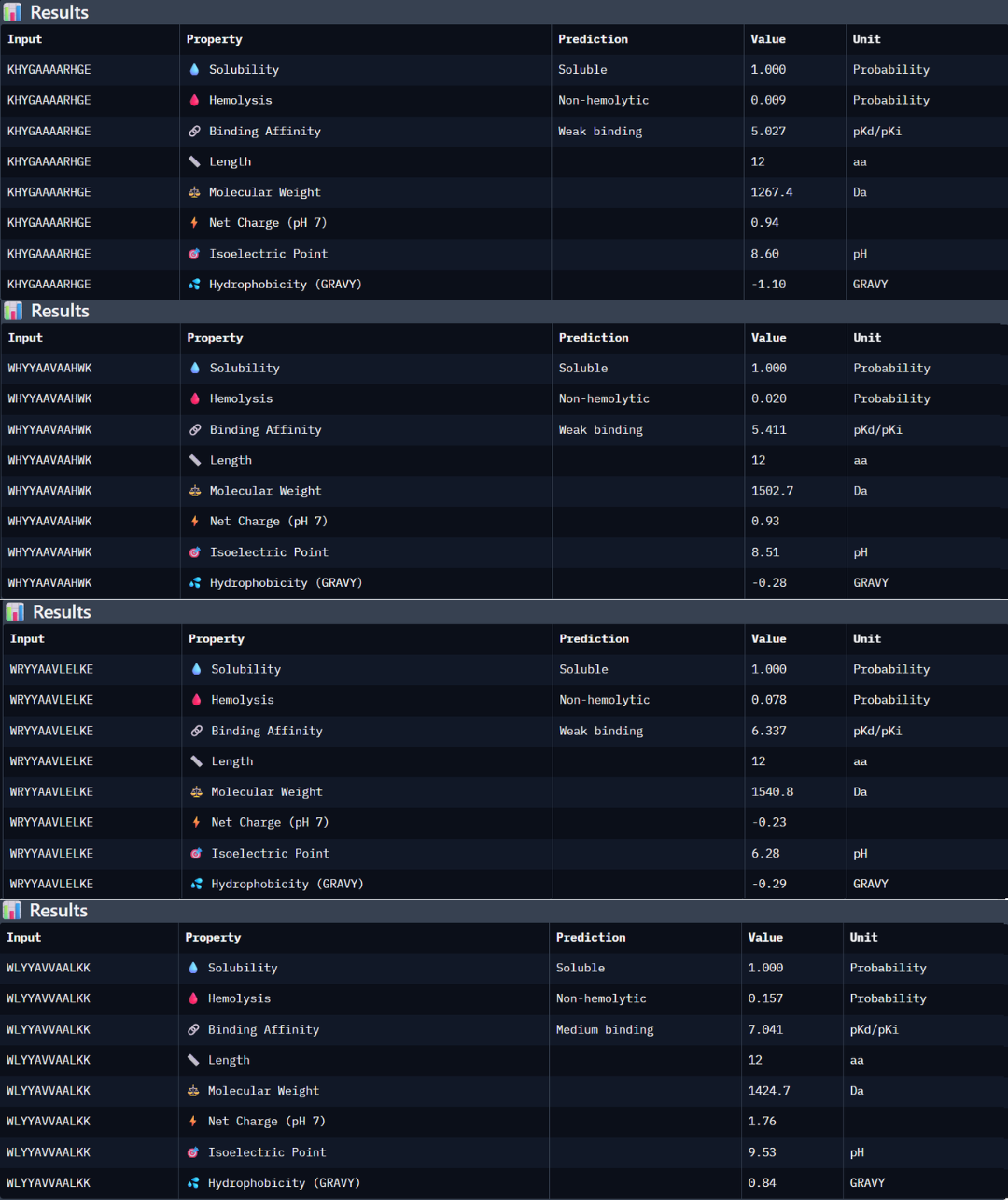
Peptide 1: the peptide (KHYGAAAARHGE) stands out as the most promising candidate from the given peptides. Structurally, it has the highest ipTM score of 0.5, but even though the 3D model is not showing a binding site with the protein, the PAE matrix shows certain dark green shadows indicating a more trusting prediction. While PeptiVerse predicts a weak binding affinity (5.027), this is consistent with the early-stage design of these peptides. This sequence maintains an excellent therapeutic profile, being fully soluble (1000 probability) and non-hemolytic (0.009 probability). The balance between its structural stability and its safe properties makes it the main choice over the other peptides with high perplexity scores.
Peptide 2: the peptide WHYYAAVAAHWK shows a higher predicted binding affinity in PeptiVerse (5.411) compared to the previous one, but its structural confidence in AlphaFold is lower with an ipTM of 0.4 suggesting that the predicted affinity does not always translate into a stable structural complex. This peptide is also highly soluble (1.000 probability) and non-hemolytic (0.020 probability).
Peptide 3: this third peptide WRYYAAVLELKE shows a discrepancy between the prediction and structural reality. Although PeptiVerse predicts the highest binding affinity (6.337), AF returns a very low ipTM of 0.22. additionally, while the peptide remains soluble, its hemolysis probability (0.078) is higher.
Peptide 4: WLYYAVVAALKK achieved the highest predicted affinity in PeptiVerse (7.041), its structural docking in AF remains poor with an ipTM of 0.36. the peptide fails to localize to the A4V site or the dimer interface. Furthermore, this sequence carries the highest hemolysis probability (0.157) of all peptides, making it the least favorable candidate from a therapeutic point of view. These results demonstrate that a high affinity score is insufficient if not accompanied by structural stability and a safe profile.
According to the previous analysis, I have chosen the KHYGAAAARHGE peptide as the lead candidate because it demonstrates the most robust balance between structural viability and therapeutic safety. Although other peptides like WLYYAVVAALKK showed higher predicted chemical affinity in PeptiVerse, they failed to produce a stable interaction in AF an presented significantly higher hemolysis risks. The chosen peptide possess the most favorable pharmacological profile, confirming that the lowest perplexity score from PepMLM was the most reliable indicator of a biologically possible and safe binder.
Part 4: Generate Optimized Peptides with moPPIt
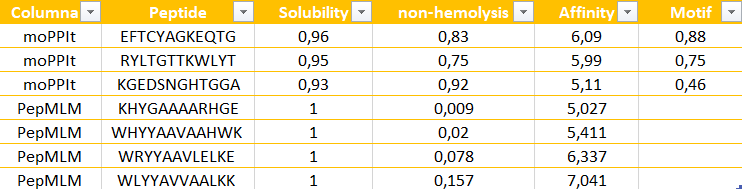
The peptides generated by moPPit differ significantly from the PepMLM ones in their precision and optimization. While PepMLM produced “possible” sequences based on general “likelihood”, moPPit allows for a controlled design, letting us pick exactly where we want the peptide to bind, such as the A4V mutation side as I chose (residues 2-6). This ‘multi-objective’ approach optimizes binding strength (affinity), solubility and safety all at the same time during the creation of the peptide, rather than just checking them at the end. As a result, peptides like EFTCYAGKEQTG show much stronger predicted binding and better focus on the target area.
Evaluation before clinical studies: To make sure these peptides are safe and effective before testing them in humans, I would follow these steps:
- Structural check using AlphaFold to see if the peptide actually stays attached to the protein in a 3D simulation.
- Lab Binding Tests, like an ELISA to see if my protein and peptide show a colorimetric signal.
- Safety tests, performing a hemolysis assay in the lab to confirm the peptide does not damage any red blood cells.
- Effectiveness, to see if the peptide successfully stops the mutant aggregation in a test tube.
PART C: Final Project: L-Protein Mutants
Stage 1: I performed a two-part analysis to understand the MS2 lysis (L) protein
Evolutionary conservation (pBLAST &ClustalOmega): after performing the alignment between the similar protein sequences in other phages and the original L-protein sequence, I identified those conserved residues which have not changed over evolution and are likey essential for function, and variable residues (shown as blank spaces), which have changed and might tolerate engineering.
Experimental mutation data: analysis of the given laboratory data listing various L-protein mutations and whether they successfully caused lysis in E. coli.
Conclusions:
- Conserved and essential regions: the L-protein has two critical domains:
- Soluble N-terminal domain (residues 1-40): interacts with DnaJ. Residues 25-38 are extremely conserved and likely form the core DnaJ binding site.
- Transmembrane domain (residues 41-75): forming the lysis pore. The start of this domain (residues 41-49) is very conserved and necessary for membrane insertion.
- Experimental fragility: the experimental data revealed a crucial fact: the very beginning of the protein (residues 1-15) is extremely sensitive. Almost all changes here prevented the protein from even being produced, resulting in zero lysis. It is mandatory to avoid these positions.
- Safe positions to mutate: based on the integrated approach, it has been concluded that we must avoid mutating conserved sites, avoid the critical DnaJ core (25-38) and avoid the experimentally fragile N-terminus (1-15). According to this, the safest and most promising areas for engineering are:
The soluble loop (residues 16-24), positions between the N-terminus and the conserved binding core. Changing them might alter the interaction to become independent of the specific DnaJ mutation without destroying the protein itself.
The transmembrane domain (residues 50-75): this region seems less sensitive to total expression failure and is the key to improving lysis speed and efficiency.
 Possible mutations analysis on Google Colab
Possible mutations analysis on Google Colab
To design an improved version of the MS2 L-protein that is actually independent of the DnaJ chaperone or to increase killing efficiency to bypass bacterial resistance, this is the followed strategy:
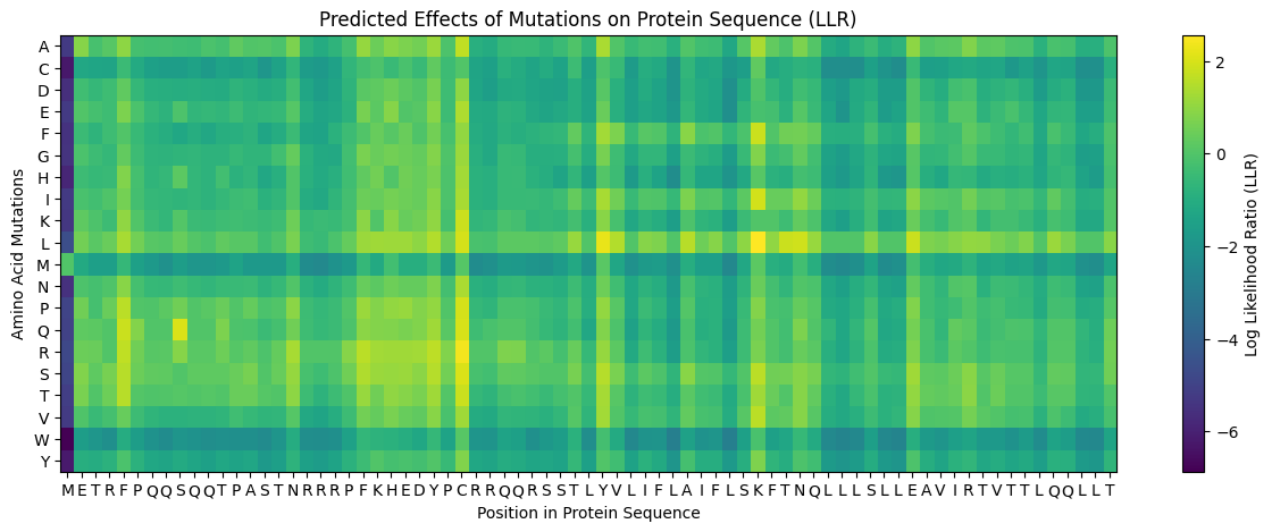
Predict the stability and functional impact of every possible mutation using ESM-1v. the results are visualized in the heatmap, where the rows represent the 75 positions of the L-protein, the columns represent the 20 different amino acids we could use for mutations, the bright yellow/clear cells indicate high log-likelihood scores, meaning the mutation is predicted to be stable and safe (dark purple indicate negative scores, warning that the mutation might break the protein).
Correlation between AI scores vs. Experimental data: after cross-referencing the Colab scores with the given database for the L-protein mutants, I found a strong correlation between the experimental data and the predicted scores. While the laboratory data (L-protein mutants spreadsheet) shows that mutations in the N-terminus (positions 1-5) result in zero lysis, these positions are completely absent from the ‘Top Mutations’ list generated by the Colab, which only includes stable changes with positive scores. This proves the ESM captures the protein’ s fragility perfectly.
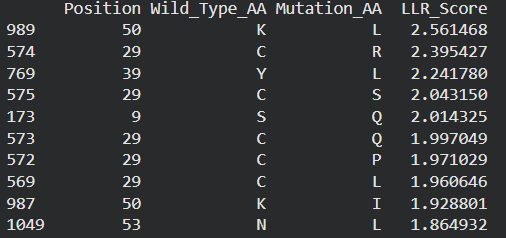
These are the mutations I choose after doing the actual analysis, by strictly filtering the Top Mutations table generated in the Colab, prioritizing the highest LLR scores to ensure structural stability.
- Position 53 (L): score 1.86, Transmembrane region
- Position 50 (L): score 2.56, Transmembrane region
- Position 39 (L): score 2.24, Soluble region
- Position 40 (L): score 1.47, Soluble region
- Position 52 (L): score 1.81, Transmembrane region
Positions 39 and 40 (Soluble region) aim to maintain protein expression while potentially altering host chaperone interactions. Positions 50, 53 and 52 (Transmembrane region) are designed to enhance or stabilize the multimeric assembly required for efficient bacterial lysis.
Multimeric Assembly
Sequences for AlphaFold:
- Variant 1 (Y39L) METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYLLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
- Variant 2 (V40L) METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLLFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
- Variant 3 (F50L) METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAILLSKFTNQLLLSLLEAVIRTVTTLQQLLT
- Variant 4 (S53L) METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLLKFTNQLLLSLLEAVIRTVTTLQQLLT
- Variant 5 (Double Y39L + F50L) METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYLLIFLAILLSKFTNQLLLSLLEAVIRTVTTLQQLLT
After generating the multimeric assembly for Variant 3 (because it had the highest LLR score) using AF2, I compared it against the WT structure. The results show that the mutant successfully maintains its octameric symmetry, forming a stable ring-like structure with a clear central pore. Although the pLDDT scores remain low in the disordered N-terminal and C-terminal tails, the core transmembrane assembly is preserved showing a clearly defined central pore. This suggests that substituting Phenylalanine for a more flexible Leucine at this position stabilizes the transmembrane helix without disrupting the quaternary assembly required for bacterial membrane perforation. To conclude, I designed this new version to beat the bacteria’s defenses. By making the lysis pore stronger without changing the most important parts of the protein, we can kill the bacteria more quickly. This gives the E. coli less time to protect itself using its chaperones, making it much harder for it to become resistant.
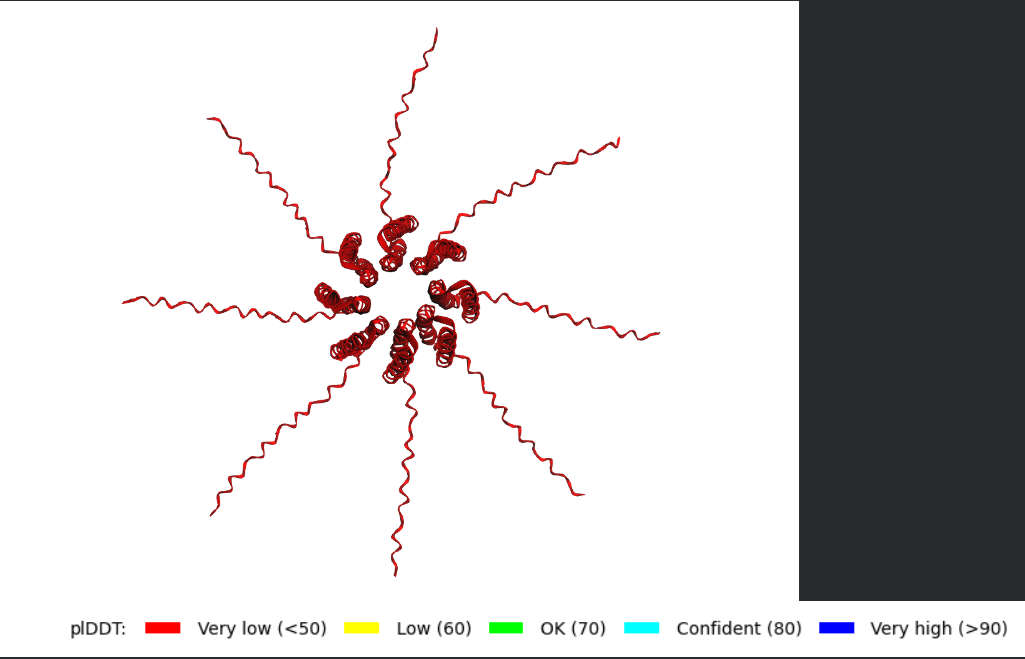 Predicted 3D structure on AF2 Multimer where it is easy to see the expected octameric structure.
Predicted 3D structure on AF2 Multimer where it is easy to see the expected octameric structure.
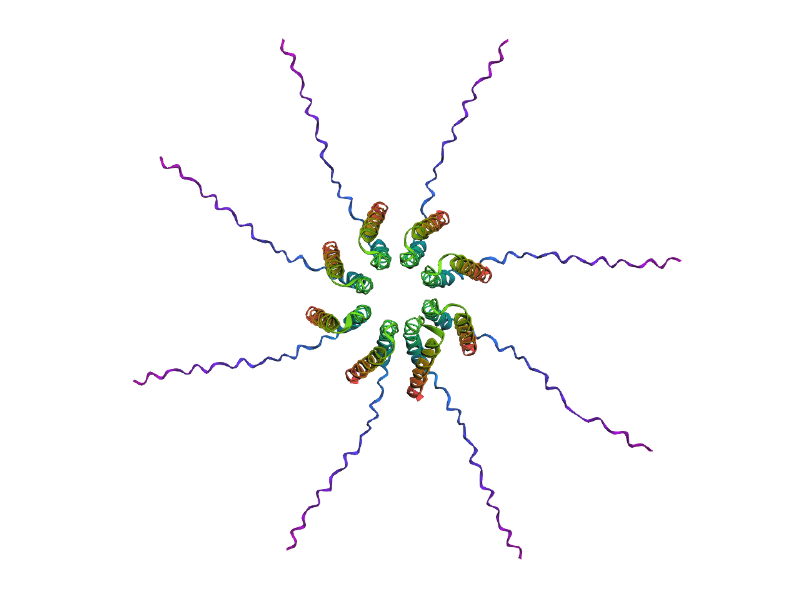 Predicted 3D structure on AF2 Multimer for the F50L octamer confirming the mutation preserves the structural integrity of the protein.
Predicted 3D structure on AF2 Multimer for the F50L octamer confirming the mutation preserves the structural integrity of the protein.