Week 6 HW: Genetic Circuits: Part I
- What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
- This master mix contains a Phusion high-fidelity DNA polymerase, an enzyme that synthesizes new DNA strands during PCR and has proofreading activity that reduces the error rate compared to standard polymerases. The enzyme needs Mg+2 ions as a cofactor required for the polymerase activity. This mix also contains dNTPs (deoxynucleotide triphosphates), which are the building blocks used by the polymerase to synthesize new DNA. Another key component is the reaction buffer, which maintains the optimal pH and salt conditions required for the enzyme to function properly. Finally, water is used as the solvent.
- What are some factors that determine primer annealing temperature during PCR?
- The annealing temperature mainly depends on the melting temperature (Tm) of the primers, which is influenced by:
- Primer length: longer primers have higher melting temperatures
- G-C content: G-C base pairs form three hydrogen bonds increasing primer stability (but more energy is required to hydrolyze them) compared to A-T pairs.
- Sequence composition and presence of secondary structures (like hairpins or dimers)
- Salt concentration and reaction conditions Usually the annealing temperature is chosen to be a few degrees lower than the calculated Tm of the primers to allow efficient binding while maintaining specificity.
- There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
- PCR and restriction enzyme digestion are both methods to generate linear DNA fragments, but working differently. PCR amplifies a specific DNA region using primers and a DNA polymerase. The primers determine the exact boundaries of the fragment being amplified, which makes PCR very flexible. This technique is useful when we want to amplify a specific gene or add sequences such as overlaps or tags to the ends of the DNA. Restriction enzyme digestion uses these enzymes that cut DNA at specific recognition sequences. This method requires that the restriction sites already exist in the sequence. The protocol usually involves incubating the DNA with the enzyme under optimal buffer and temperature conditions. In terms of when to use each method, PCR is preferable when we need custom DNA fragments, sequence modification, or large amplification of DNA. Restriction digestion is often sued when we want to cut plasmids or DNA molecular at defined natural restriction sites, especially when preparing vectors for cloning.
- How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
- To ensure this fragments are compatible, they must contain overlapping homologous regions at their ends. Typically, these overlaps are about 20-40 base pairs long and are designed to match the adjacent DNA fragment. It is possible to design these PCR primers that include the necessary overlap sequences at their 5’ ends. After amplification, the primers will contain the overlaps needed for assembly. It is also important to check that the fragments are correctly sized and free of unwanted sequences using gel electrophoresis.
- How does the plasmid DNA enter the E. coli cells during transformation?
- During transformation plasmid DNA enters E. coli cells when the bacterial membrane becomes temporarily permeable. In chemical transformation, cells are first treated with calcium chloride, which helps neutralize the negative charges of both the DNA and the cell membrane. Then a heat shock step is applied, that creates a sudden temperature change that helps DNA molecules pass through the membrane and enter the cell. There are other methods such electroporation, a short electrical pulse applied to the cells in order to create temporary pores in the membrane, allowing plasmid DNA to enter the cytoplasm.
- Describe another assembly method in detail (such as Golden Gate Assembly)
a. Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
b. Model this assembly method with Benchling or Asimov Kernel!
a) Another DNA assembly method is Gateway cloning, which is used in systems such as the CloneMiner™ kit, provided by Invitrogen. This method is based on site-specific recombination using att sites (attB, attP, attL and attR) derived from phage lambda. In this approach, PCR products are first generated with attB adapters, which allow the DNA fragment to combine with a donor vector through the action of recombination enzymes. Unlike Gibson or Golden Gate Assembly, this method does not rely on ligation or overlapping sequences, but instead on highly specific enzymatic recombination. This results in the formation of a continuous and functional DNA molecule. Gateway cloning is especially useful for generating cDNA libraries because it allows efficient and directional insertion of many different DNA fragments into vectors.
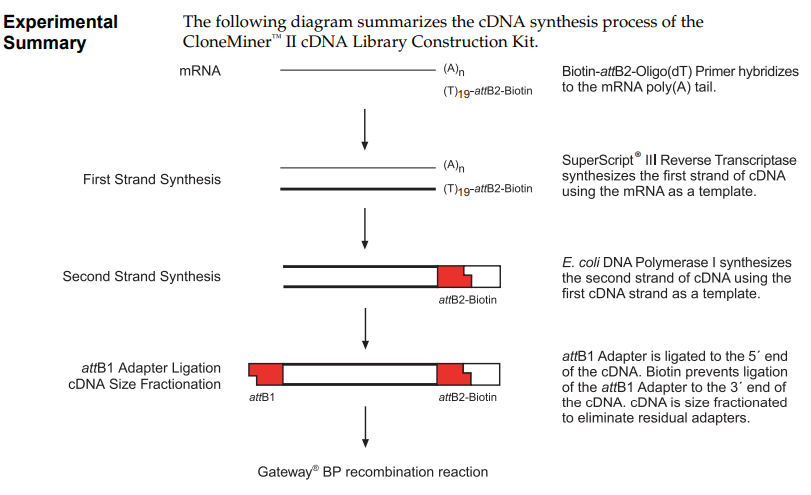
CloneMiner™ II cDNA Library Construction Kit. High-quality cDNA libraries without the use of restriction enzyme cloning techniques. Provided by Invitrogen. Obtained from their Catalog Number A11180.
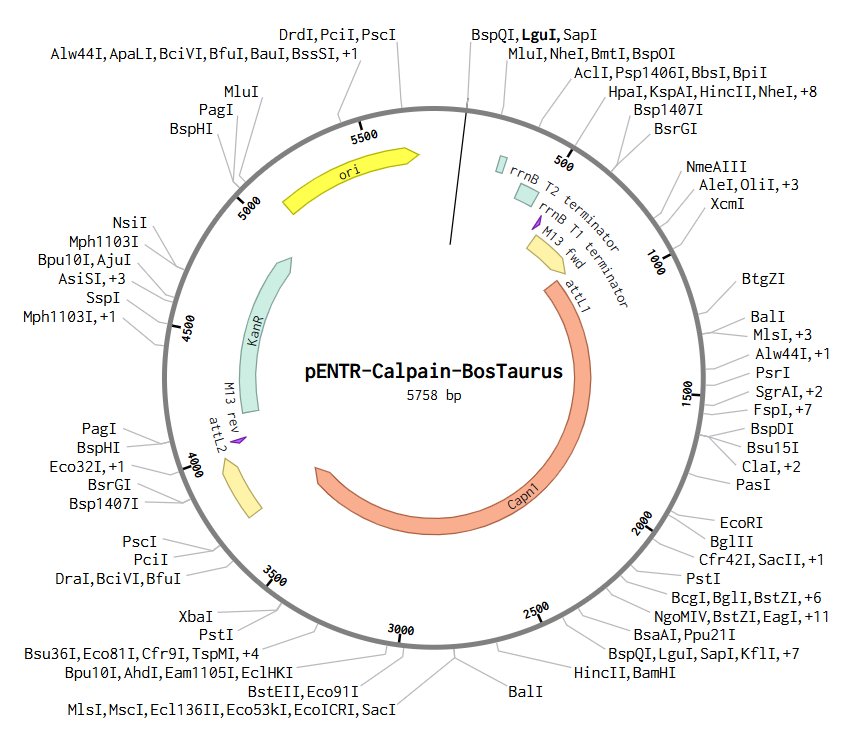
b) To create this plasmid, I simulated a Gateway BP reaction using Benchling. First, I added attB sites to the Bos taurus Calpain (CAPN1) gene sequence. Then, I combined this gene with the pDONR™221 vector. During this simulation, the gene replaces the original vector’s center, and the sites transform into attL1 and attL2. The final result is this Entry Clone, which is now ready to be moved into an expression vector.
Asimov Kernel
- For this assignment I have simulated the Repressilator as expected, and comparing it to the Repressilator Construct Demo given in Asimov, I have found they work exactly the same, as seen in all the plots.
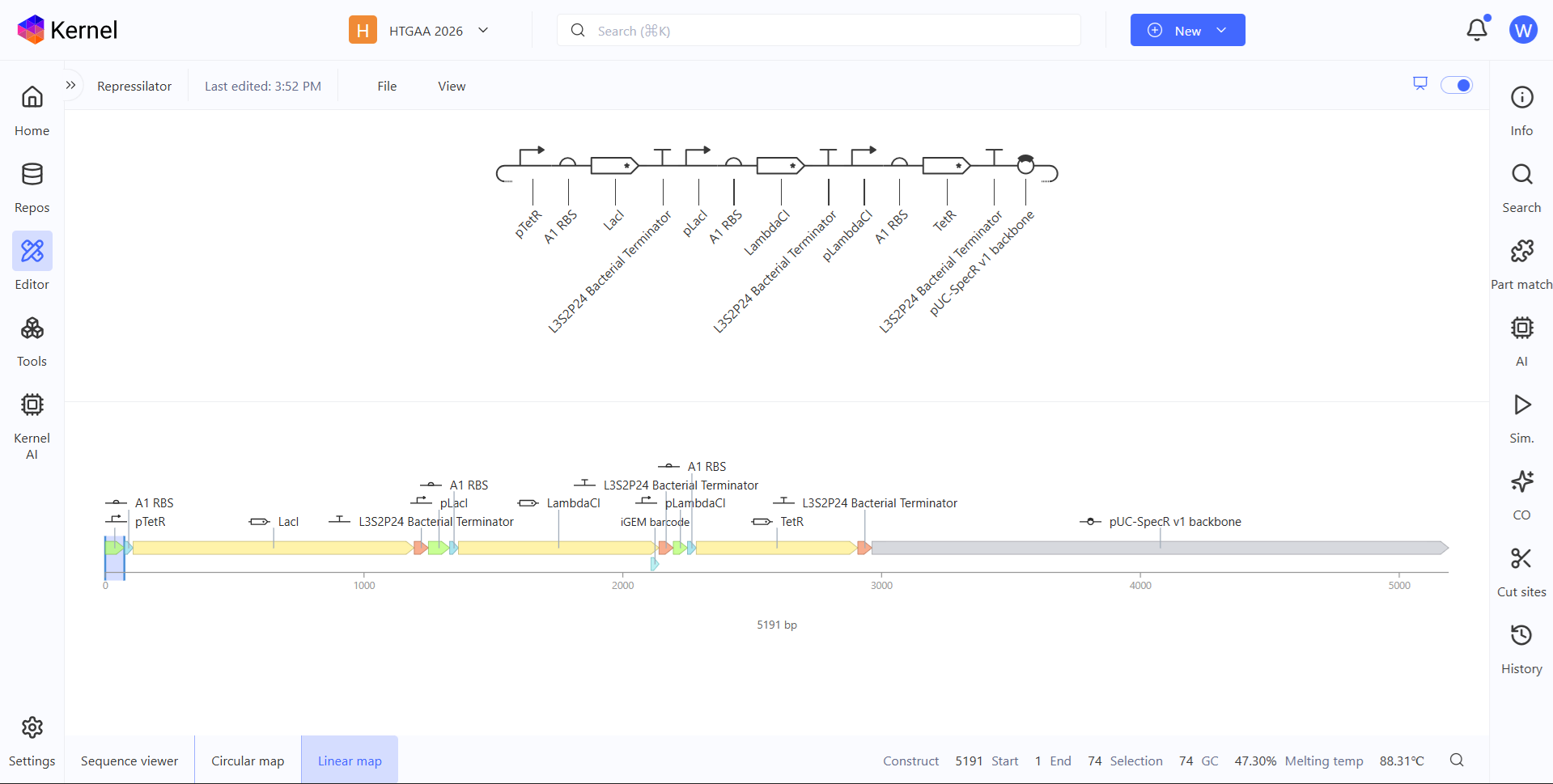
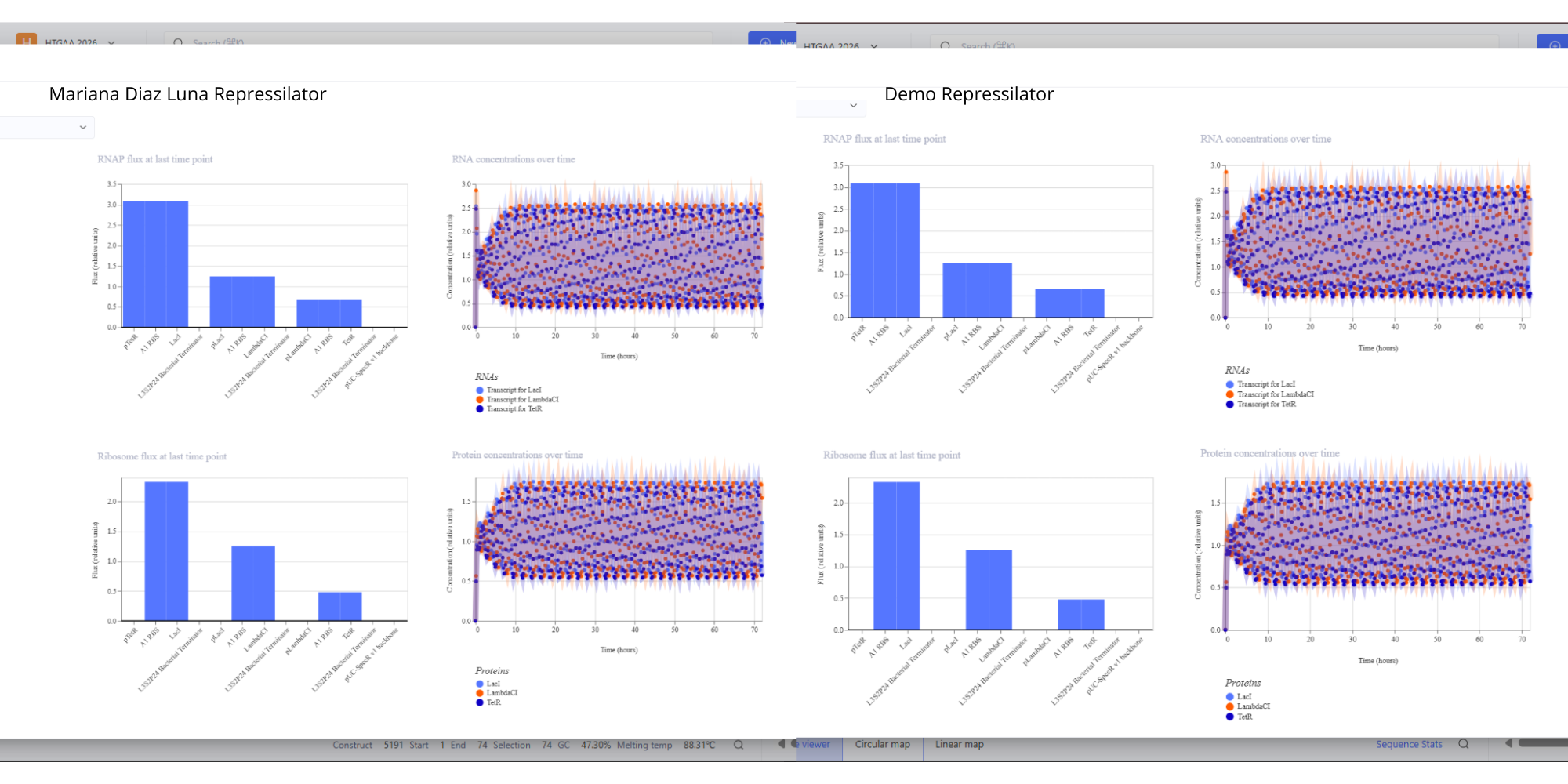 Image showing the comparison between the Repressilator I have made and the Demo.
Image showing the comparison between the Repressilator I have made and the Demo.
- See 3 constructs below:
I. 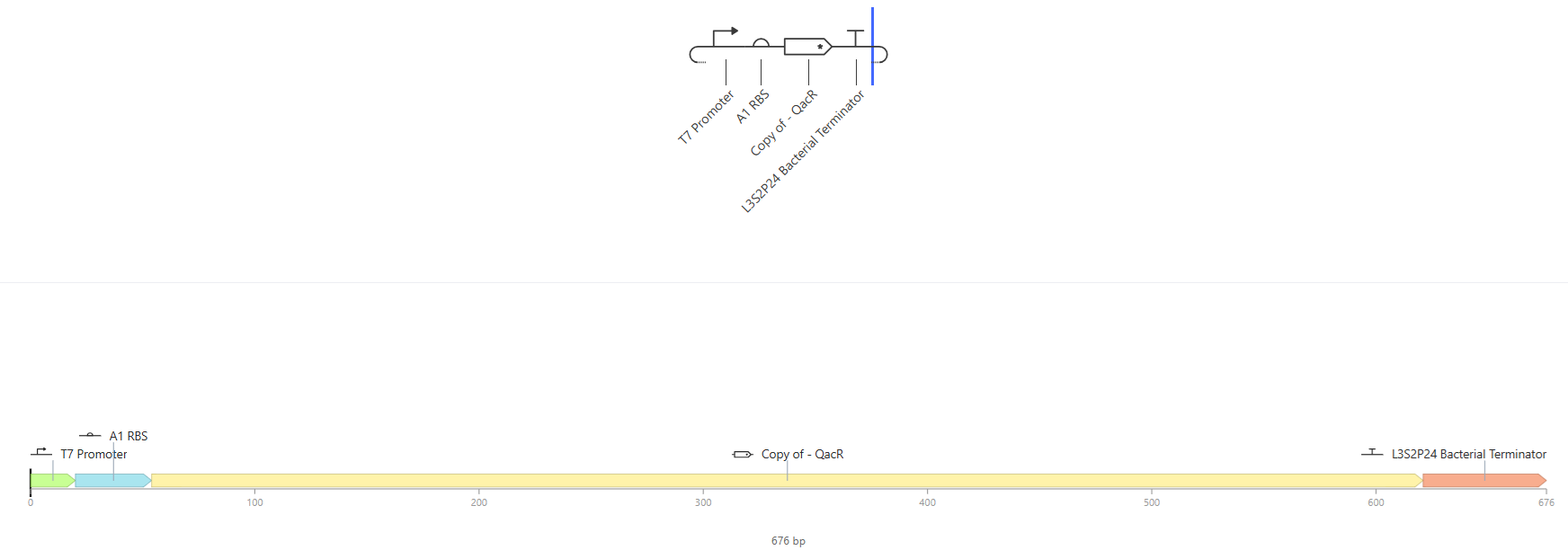
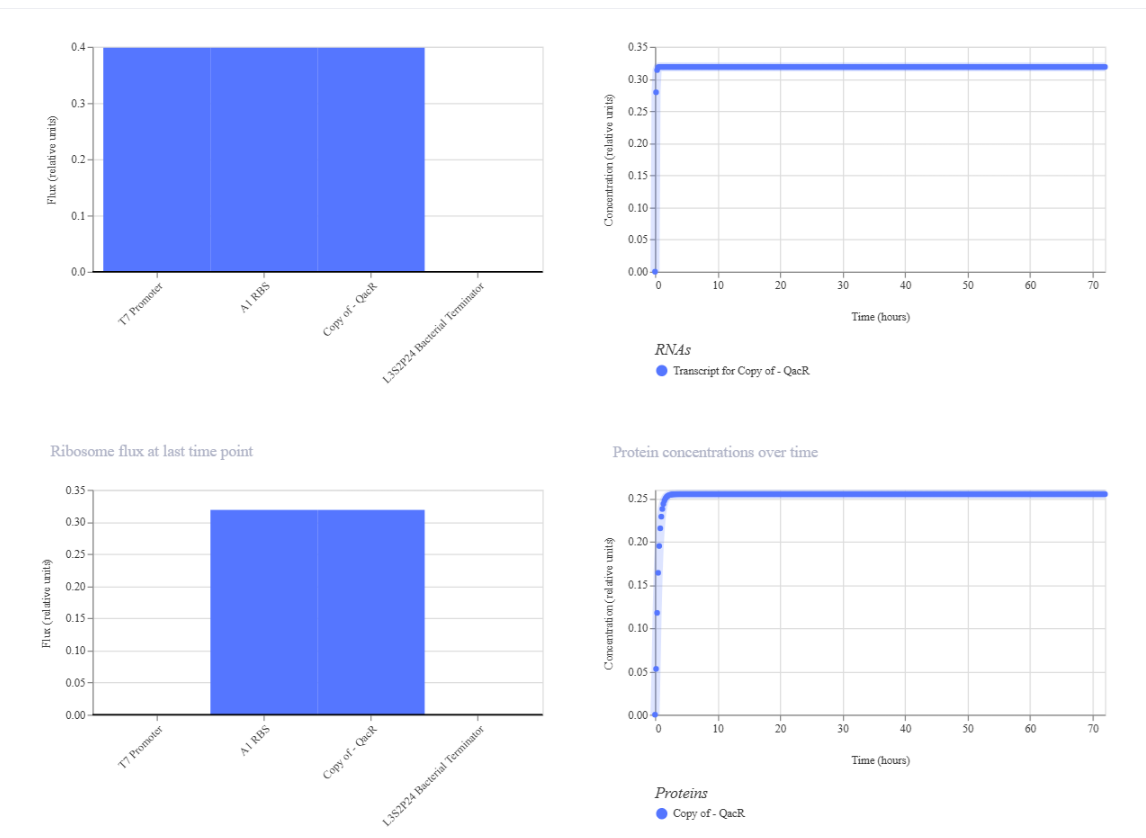
Results where as expected, with high expression levels in the construct when simulated, with a drop-off after Terminator.
II. 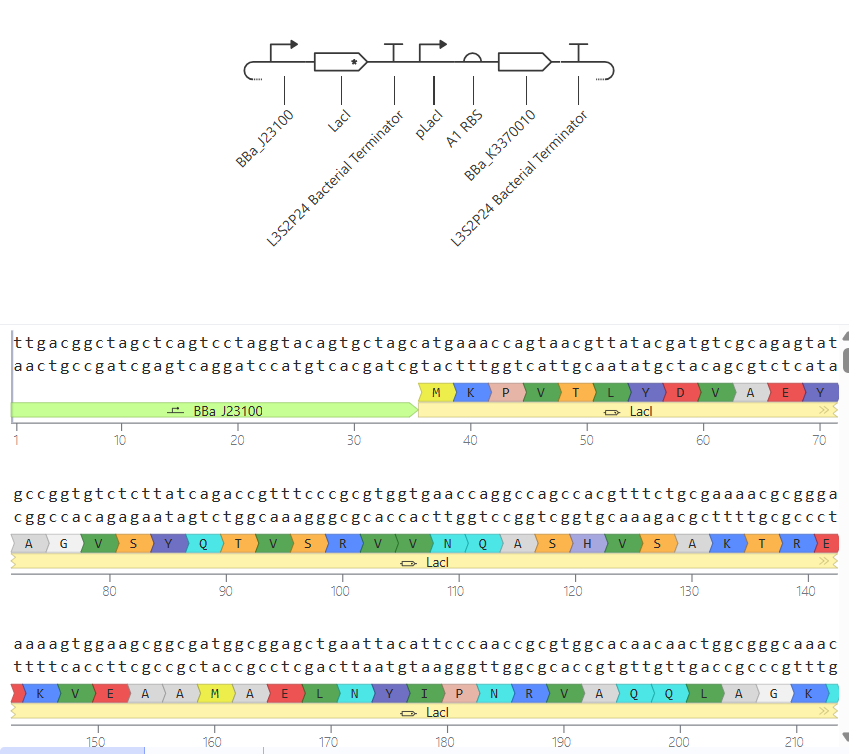
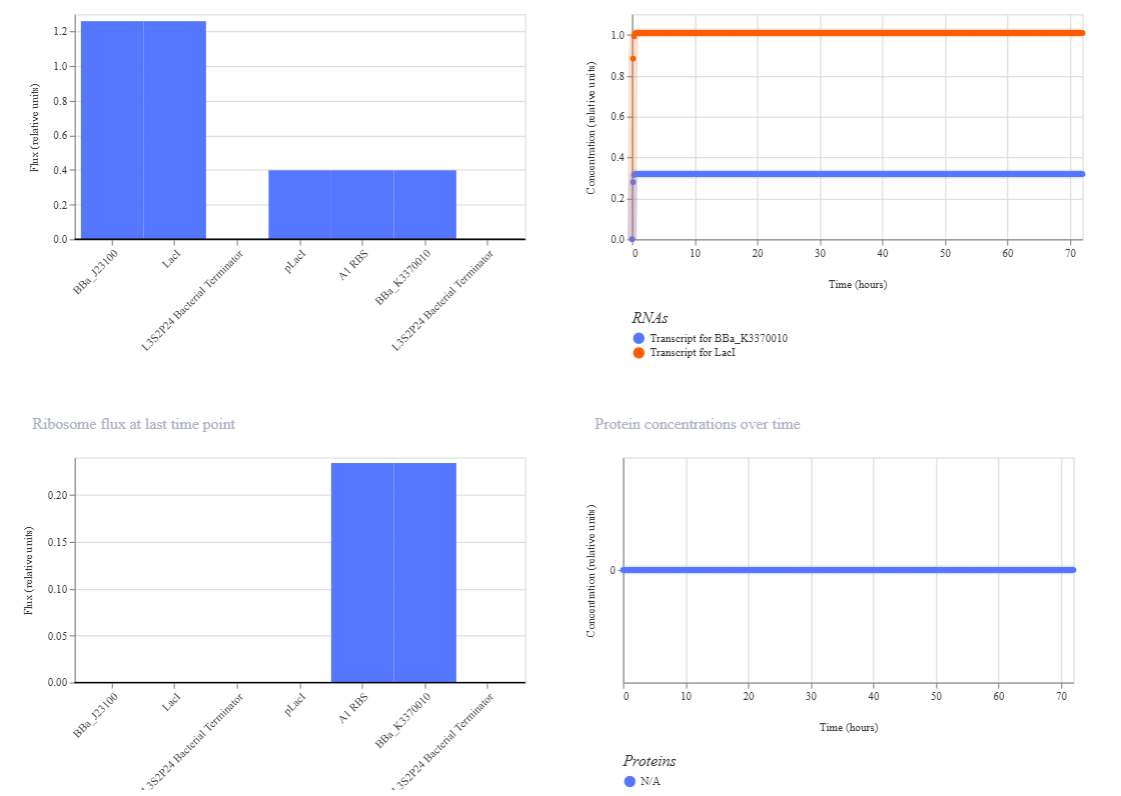 In this circuit, the expression of GFP is controlled by a lac-derived promoter regulated by LacI. The system responds to the presence or absence of IPTG. In absence of IPTG LacI binds to the lac operator region within the promoter, blocking GFP gene. As a result, GFP levels should be very low. When IPTG is added it is expected to bind to LacI and inactivate it, so GFP is now transcribed.
When running the simulation, the expected difference between conditions with and without IPTG is not observed, maybe because the GFP used is not properly recognized as a coding sequence (even though I have tried several of them) or is not expressed in my system.
In this circuit, the expression of GFP is controlled by a lac-derived promoter regulated by LacI. The system responds to the presence or absence of IPTG. In absence of IPTG LacI binds to the lac operator region within the promoter, blocking GFP gene. As a result, GFP levels should be very low. When IPTG is added it is expected to bind to LacI and inactivate it, so GFP is now transcribed.
When running the simulation, the expected difference between conditions with and without IPTG is not observed, maybe because the GFP used is not properly recognized as a coding sequence (even though I have tried several of them) or is not expressed in my system.
III.