Week 10 HW: Advanced Imaging and Measurement Technology
Homework: FINAL PROJECT
One of the main aspects to measure in this work is the expression level of the APP since the goal of the CRISPRi system is to reduce its transcription. This could be measured using qPCR (mRNA levels of APP), extracting the RNA from treated and control cells, performing reverse transcription and quantifying gene expression differences.
It is also important to measure whether changes in gene expression translate into protein level changes, using Western Blot to detect APP protein levels or ELISA to quantify amyloid-beta peptides.
Lastly, I need to confirm that these nanoparticles are entering cells and reaching the expected location. Using fluorescence microscopy, it is possible to label those nanoparticles with fluorescent markers and allow visualization inside cells. Confocal microscopy is also useful for a more precise localization.
Waters Part I: Molecular Weight
After analyzing eGFP molecular weight including the His-tag and linker, the theoretical pI/Mw: 5.90 / 28006.60
I have selected two pairs of charge states, one between 900 and 1000: 903.7148 (n+1) and 933.7349 (n).
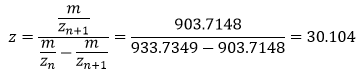
And the other between 800 and 900: 848.9758 (n+1) and 875.4421 (n)

To determine the MW of the protein:

Accuracy of the measurement:
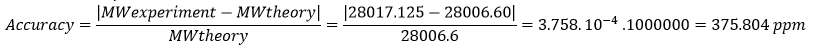
- In large proteins like eGFP the peaks get “crowded” because the charge state is high, so as the charge increases the distance between isotopes becomes very small, and the peaks start to overlap. The instrument’s resolution of 30.000 is high enough to distinguish individual isotopic peaks, so the software has successfully assigned specific m/z values. Techicnally yes, the charge state can be observed even though we cannot distinguish those peaks.
Waters Part II: Secondary/Tertiary structure
Native protein conformations show the natural state of a protein with minimum charges, and denatured conformations are obtained when digesting the protein structure with enzymes into individual highly charged peptides, where each basic residue tends to be protonated. When a protein unfolds, its internal basic residues become exposed to the solvent. In ESI-MS this leads to a higher number of protons being attached to the protein, consequently showing a distribution of peaks at lower m/z values compared to the native protein. Additionally, the denatured protein exhibits a larger range of charge states due to the increased conformational flexibility of the unfolded polypeptide chain.
There is a lot of noise in that part of the spectrum, so that signal is weak and the labels are likely pointing to noise artifacts or small impurities rather than the actual isotopic distribution of the protein. Those labeled values at ̴2800 m/z are not reliable for determining the charge state.
Waters Part III: Peptide Mapping – primary structure
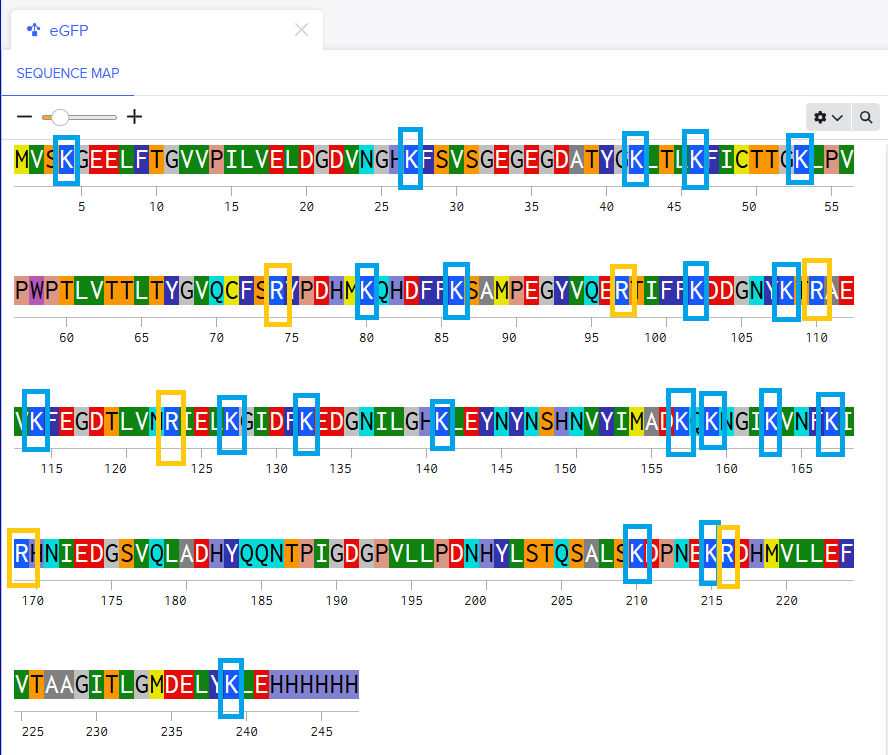
There are 20 (twenty) Lysines and 6 (six) Arginines in the amino acid structure.
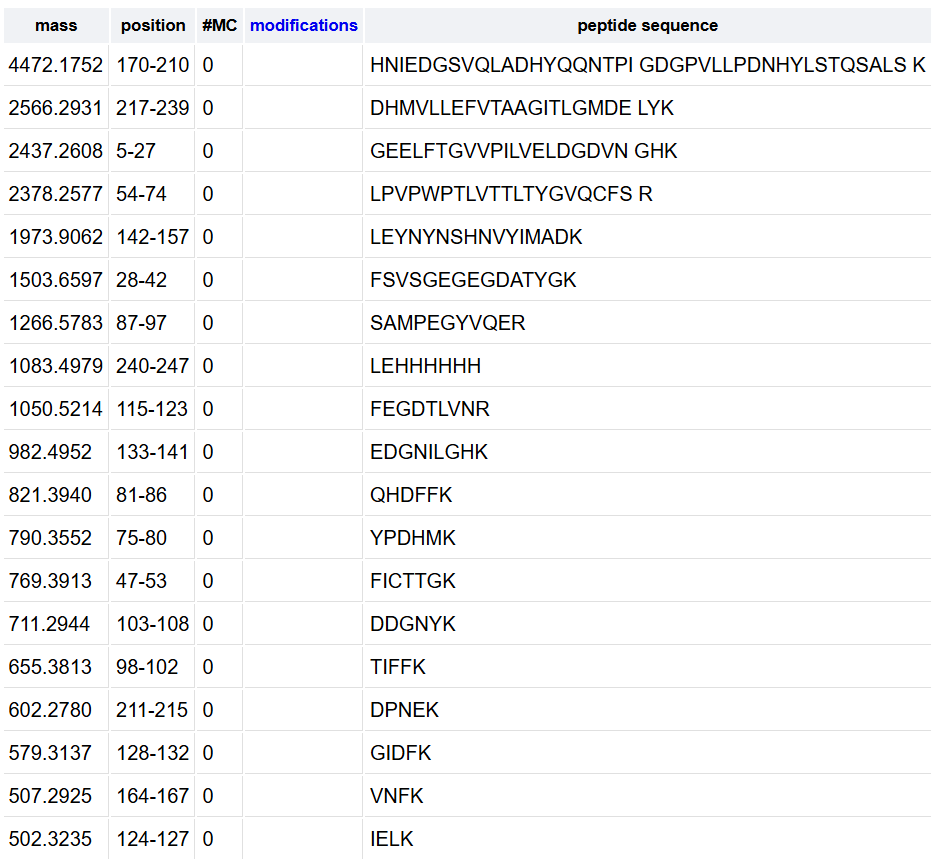
After performing the tryptic digestion with trypsin, will be generated 19 peptides according to Expasy PeptideMass.
To calculate the 10% threshold I identified the highest peak with the maximum abundance at 4.87 minutes reaching 1.2x10ʌ7counts, and the 10% of this maximum is 1.2x10ʌ6 counts. With this, any peak that goes higher than the 2.10ʌ6 line is above this threshold, counting 17 chromatographic peaks between 0.5 and 6 minutes.
There are fewer peaks in the chromatogram than the number of peptides predicted. This occurs because some peptides may co-elute, meaning they exit the column at the same time and appear as a single peak. Some peptides may also have low inonization efficiency, resulting in peaks with a relative abundance below the 10% threshold used for counting.
The most abundant peak for the peptide eluting at 2.78 minutes is m/z = 525.76712. Isotope separation:
- Peak 1: 525.76712
- Peak 2: 526.25918
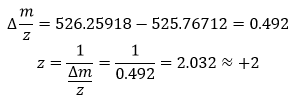
To calculate the mass of the singly charged form ([M+H]+):

- Based on the PeptideMass analysis and the previous calculation:
- Experimental [M+H]+: 1050.52697
- Theoretical: 1050.5214, sequence FEGDTLVNR (position 115-123)
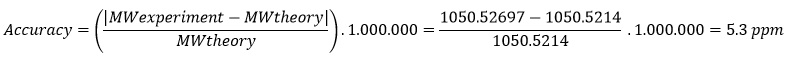 An error of 5.30 ppm indicates high mass accuracy, according to the high-resolution instrument used which generally provides accuracy <10 ppm.
An error of 5.30 ppm indicates high mass accuracy, according to the high-resolution instrument used which generally provides accuracy <10 ppm.
According to the peptide identification data shown on the image, the percentage of the sequence confirmed by peptide mapping is 88% meaning that 88% of the total amino acids in the protein were successfully identified as peptides during the LC-MS analysis. For my own analysis, the 90.7% of the eGFP sequence was covered.
Using my results from Question 2 and the previous selected peptide FEGDTLVNR, I have obtained the next Fragment Ion Table for the +1 charge state, which confirms the identity of the pepdtide eluting at 2.78 minutes. The calculated y-ion series matches the experimental peaks in Figure 5c.
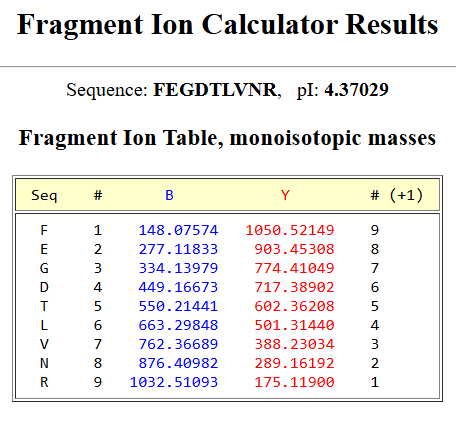
- According to the previous results, the experimental data and the theoretical one, the data makes sense because the experimental coverage (88%) is very close to the theoretical maximum obtained on PeptideMass (90.7%). The small difference of 2.7% is expected in LC-MS experiments as some predicted peptides may have low ionization efficiency or may not meet the 10% relative abundance threshold required for positive identification. With this, the results strongly indicate that the sample is the eGFP standard.
Waters Part IV - Oligomers
To identify the peaks, first I need to convert the units from kDa to MDa:

Waters Part V - Did I make GFP?
According to the MW of the protein calculated on Part I, the mass error of 375 ppm is quite normal, because for a complete protein the charge is so high the isotopes are very close together. Even with the high resolution, the machine still struggles to find the exact center of the peak, so the error is multiplied by 30 when calculating the final mass. This is why we did the Peptide Mapping analyzing one single peptide (FEGDTLVNR) so the machine could see every single isotope clearly. By choosing one specific peak to calculate the mass and obtaining an accuracy of 5.3 ppm I could confirm that this was the exact sequence of my protein. Yes, I did make GFP
