Group Final Project
Week 4 Part D: Brainstorm on Bacteriohage Engineering
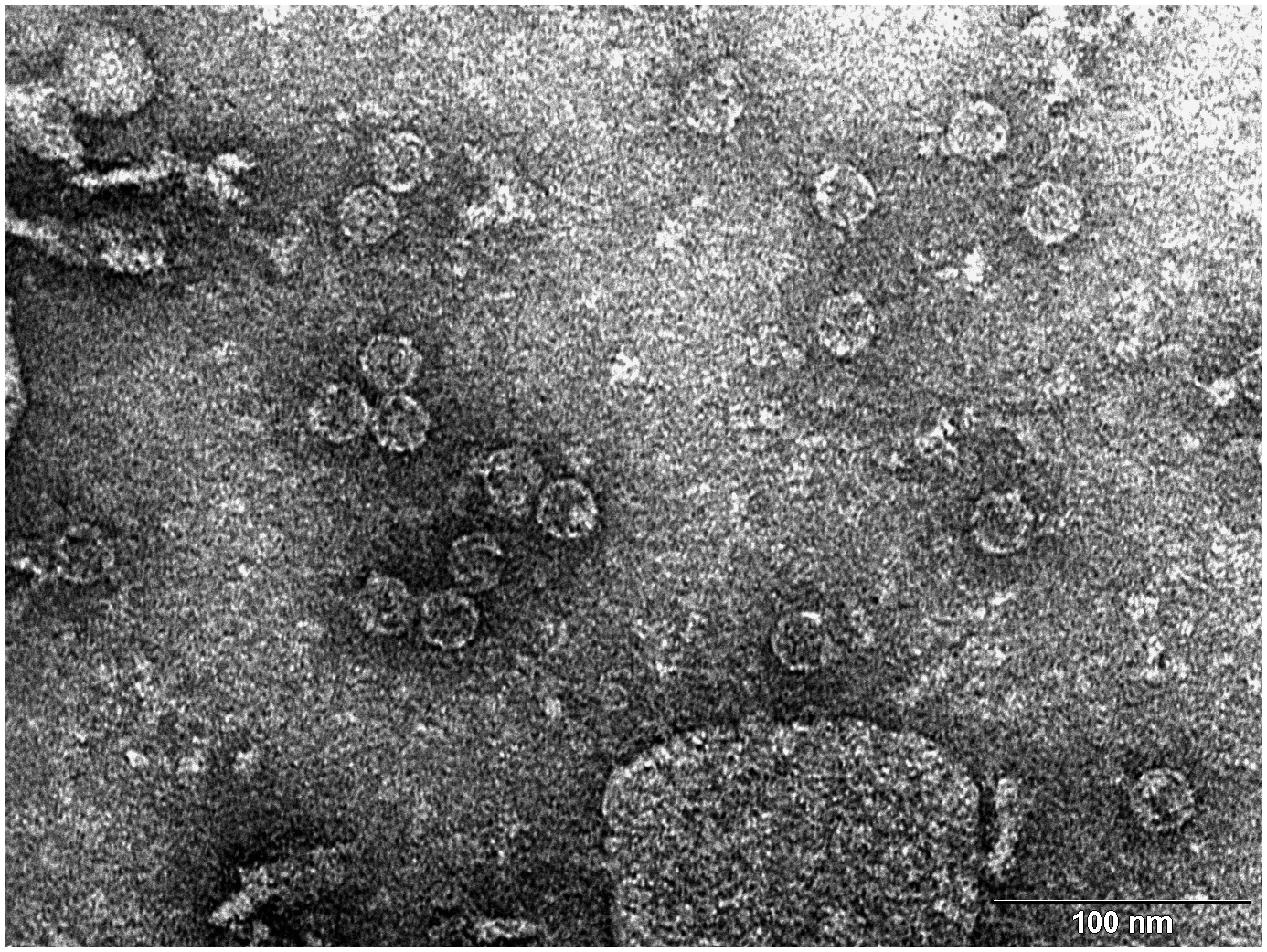 Transmission electron microscopy (TEM) photograph of the intact MS2 phage-like particles (MS2 PLP) present in the supernatant after ultrasonic disruption of E. coli production cells. (Mikel P, Vasickova P, Tesarik R, Malenovska H, Kulich P, Vesely T and Kralik P (2016) Preparation of MS2 Phage-Like Particles and Their Use As Potential Process Control Viruses for Detection and Quantification of Enteric RNA Viruses in Different Matrices. Front. Microbiol. 7:1911. doi: 10.3389/fmicb.2016.01911)
Transmission electron microscopy (TEM) photograph of the intact MS2 phage-like particles (MS2 PLP) present in the supernatant after ultrasonic disruption of E. coli production cells. (Mikel P, Vasickova P, Tesarik R, Malenovska H, Kulich P, Vesely T and Kralik P (2016) Preparation of MS2 Phage-Like Particles and Their Use As Potential Process Control Viruses for Detection and Quantification of Enteric RNA Viruses in Different Matrices. Front. Microbiol. 7:1911. doi: 10.3389/fmicb.2016.01911)
(Important: I couldn’t find a group in this opportunity so I was my own group!)
Proposal: Engineering a DnaJ independent MS2 lysis protein for enhanced Phage Therapy
The L- protein of MS2 phages needs DnaJ because this is a chaperone from the Hsp40 family that helps the full-length L protein fold correctly. In the host cell, DnaJ forms a complex with the highly basic N-terminal domain of L. This complex allows L to adopt a conformation that can interact with its target (still unknown) and cause cell lysis.
When the chaperone is mutated or removed, the lysis process is delayed or completely blocked at certain conditions, even though L accumulates normally, showing that the lack of interaction with DnaJ prevents a step happening after folding, not the synthesis of the toxic protein itself. According to this, the main goal is to engineer a Dna-J independent version of the MS2 L protein. By removing this dependency and stabilizing the C-terminal lytic core, I aim to create a protein that triggers bacterial lysis faster and more reliably across different bacterial strains without needing additional co-factors.
Using the tools practiced in this weeks’ recitation, this is the proposed bioinformatics pipeline:
Identify the region that really matters: mutagenesis experiments showed that the 67 loss-of-function alleles are concentrated in the C-terminal half of L around the LS motif. The N-terminal domain (residues 1-42) acts as a regulatory break because it creates a strict dependency on the host chaperone DnaJ for proper holding. There, the first 36 to 42 amino acids (N-terminal domain) are nonessential for the killing mechanism itself and removing them speeds up lysis. In addition, the Lytic Core corresponds to the last 30 amino acids, which include the LS motif and the transmembrane helix. So, I will keep the LS motif (Leu48-Ser49) and the Lys50 residue as they are essential for membrane interaction.
Search for homology and keep the essentials:
- Use BLAST against UniProt to obtain L-like sequences from other leviviruses (similar to MS2). Using ESM2 (Protein Language Model), I will perform an in silico Deep Mutational Scan to rank possible mutations, helping me find specific substitutions in the membrane helix that increase stability without breaking the essential LS motif.
Model the structure through Computational Tools:
After finding the best mutation candidates, I will upload the core sequence to ESMFold and visualize it (and compare with PyMOL) to confirm that the transmembrane helix is correctly inserted and capable of membrane insertion.
Once the structure is confirmed, could be useful to use ProteinMPNN to generate a new (and much more robust) sequence for the protein, making it more stable for biotechnology applications.
Finally, I will perform a Latent Space Analysis using t-SNE to validate the engineered designs. This map acts as a functional “sanity check” by clustering the artificial sequence with known active and natural variants of the original protein. If my candidate falls within the functional cluster and stays far away from known loss-of-function mutants (like those affecting the LS motif) it confirms that the protein is likely to be active in the lab, maintaining the original properties necessary to interact with its target.
Potential pitfalls:
Unknown target: since the host membrane target protein is unknown, it is not possible to predict (and confirm) the exact binding interface.
Lysis/assembly balance: if lysis happens to fast, it might kill the bacteria before enough phage progeny are assembled.
Week 5 PART C: Final Project: L-Protein Mutants
Stage 1: I performed a two-part analysis to understand the MS2 lysis (L) protein
Evolutionary conservation (pBLAST &ClustalOmega): after performing the alignment between the similar protein sequences in other phages and the original L-protein sequence, I identified those conserved residues which have not changed over evolution and are likey essential for function, and variable residues (shown as blank spaces), which have changed and might tolerate engineering.
Experimental mutation data: analysis of the given laboratory data listing various L-protein mutations and whether they successfully caused lysis in E. coli.
Conclusions:
- Conserved and essential regions: the L-protein has two critical domains:
- Soluble N-terminal domain (residues 1-40): interacts with DnaJ. Residues 25-38 are extremely conserved and likely form the core DnaJ binding site.
- Transmembrane domain (residues 41-75): forming the lysis pore. The start of this domain (residues 41-49) is very conserved and necessary for membrane insertion.
- Experimental fragility: the experimental data revealed a crucial fact: the very beginning of the protein (residues 1-15) is extremely sensitive. Almost all changes here prevented the protein from even being produced, resulting in zero lysis. It is mandatory to avoid these positions.
- Safe positions to mutate: based on the integrated approach, it has been concluded that we must avoid mutating conserved sites, avoid the critical DnaJ core (25-38) and avoid the experimentally fragile N-terminus (1-15). According to this, the safest and most promising areas for engineering are:
The soluble loop (residues 16-24), positions between the N-terminus and the conserved binding core. Changing them might alter the interaction to become independent of the specific DnaJ mutation without destroying the protein itself.
The transmembrane domain (residues 50-75): this region seems less sensitive to total expression failure and is the key to improving lysis speed and efficiency.
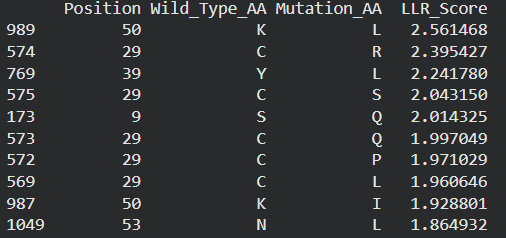 Possible mutations analysis on Google Colab
Possible mutations analysis on Google Colab
To design an improved version of the MS2 L-protein that is actually independent of the DnaJ chaperone or to increase killing efficiency to bypass bacterial resistance, this is the followed strategy:
Predict the stability and functional impact of every possible mutation using ESM-1v. the results are visualized in the heatmap, where the rows represent the 75 positions of the L-protein, the columns represent the 20 different amino acids we could use for mutations, the bright yellow/clear cells indicate high log-likelihood scores, meaning the mutation is predicted to be stable and safe (dark purple indicate negative scores, warning that the mutation might break the protein).
Correlation between AI scores vs. Experimental data: after cross-referencing the Colab scores with the given database for the L-protein mutants, I found a strong correlation between the experimental data and the predicted scores. While the laboratory data (L-protein mutants spreadsheet) shows that mutations in the N-terminus (positions 1-5) result in zero lysis, these positions are completely absent from the ‘Top Mutations’ list generated by the Colab, which only includes stable changes with positive scores. This proves the ESM captures the protein’ s fragility perfectly.
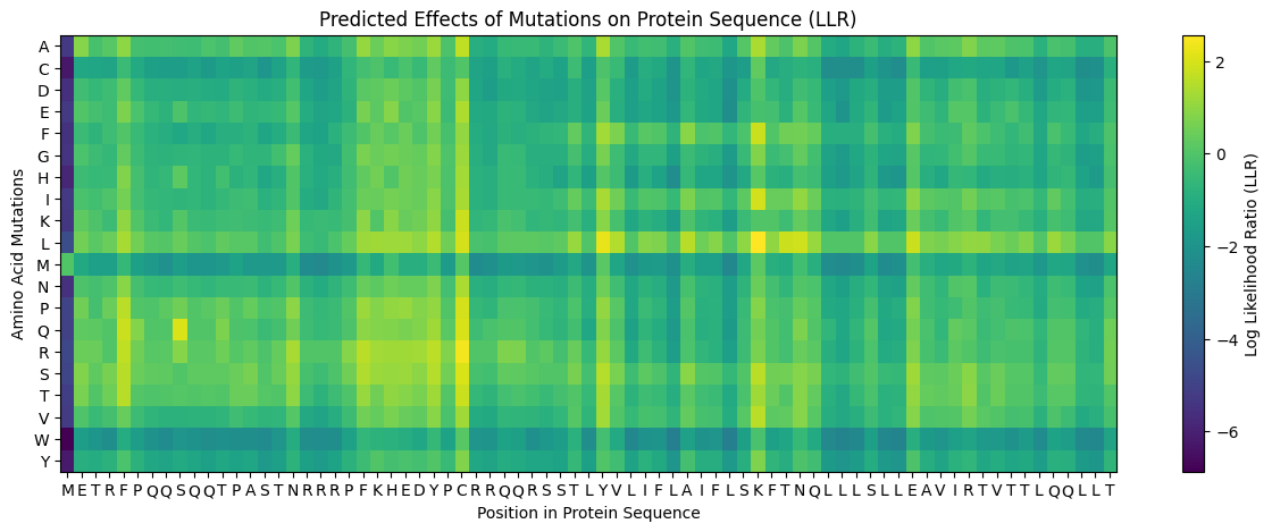
These are the mutations I choose after doing the actual analysis, by strictly filtering the Top Mutations table generated in the Colab, prioritizing the highest LLR scores to ensure structural stability.
- Position 53 (L): score 1.86, Transmembrane region
- Position 50 (L): score 2.56, Transmembrane region
- Position 39 (L): score 2.24, Soluble region
- Position 40 (L): score 1.47, Soluble region
- Position 52 (L): score 1.81, Transmembrane region
Positions 39 and 40 (Soluble region) aim to maintain protein expression while potentially altering host chaperone interactions. Positions 50, 53 and 52 (Transmembrane region) are designed to enhance or stabilize the multimeric assembly required for efficient bacterial lysis.
Multimeric Assembly
Sequences for AlphaFold:
- Variant 1 (Y39L) METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYLLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
- Variant 2 (V40L) METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLLFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
- Variant 3 (F50L) METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAILLSKFTNQLLLSLLEAVIRTVTTLQQLLT
- Variant 4 (S53L) METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLLKFTNQLLLSLLEAVIRTVTTLQQLLT
- Variant 5 (Double Y39L + F50L) METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYLLIFLAILLSKFTNQLLLSLLEAVIRTVTTLQQLLT
After generating the multimeric assembly for Variant 3 (because it had the highest LLR score) using AF2, I compared it against the WT structure. The results show that the mutant successfully maintains its octameric symmetry, forming a stable ring-like structure with a clear central pore. Although the pLDDT scores remain low in the disordered N-terminal and C-terminal tails, the core transmembrane assembly is preserved showing a clearly defined central pore. This suggests that substituting Phenylalanine for a more flexible Leucine at this position stabilizes the transmembrane helix without disrupting the quaternary assembly required for bacterial membrane perforation. To conclude, I designed this new version to beat the bacteria’s defenses. By making the lysis pore stronger without changing the most important parts of the protein, we can kill the bacteria more quickly. This gives the E. coli less time to protect itself using its chaperones, making it much harder for it to become resistant.
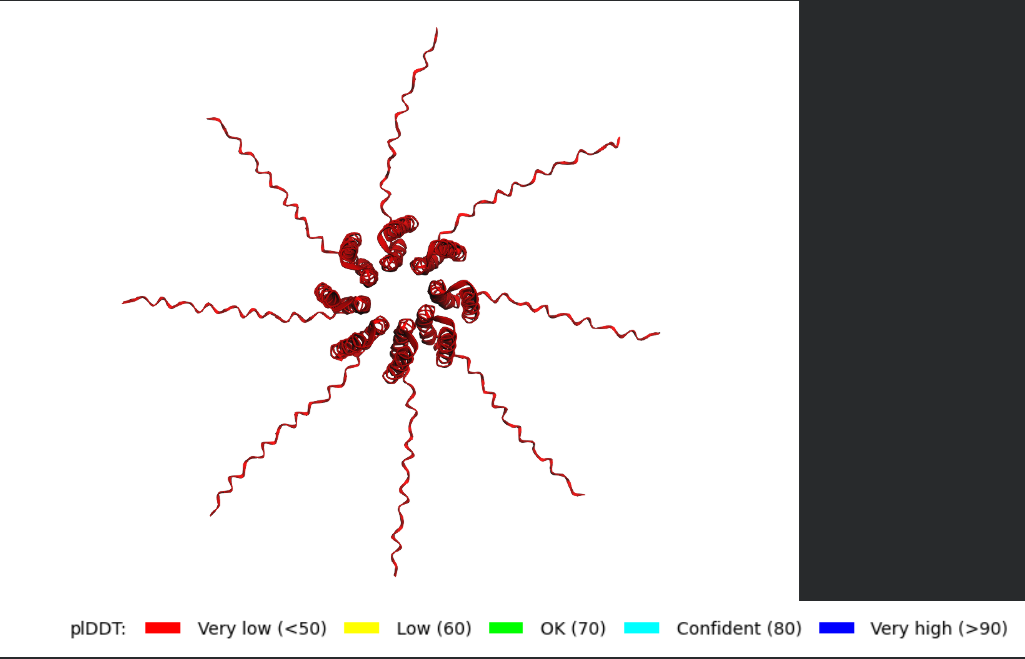 Predicted 3D structure on AF2 Multimer where it is easy to see the expected octameric structure.
Predicted 3D structure on AF2 Multimer where it is easy to see the expected octameric structure.
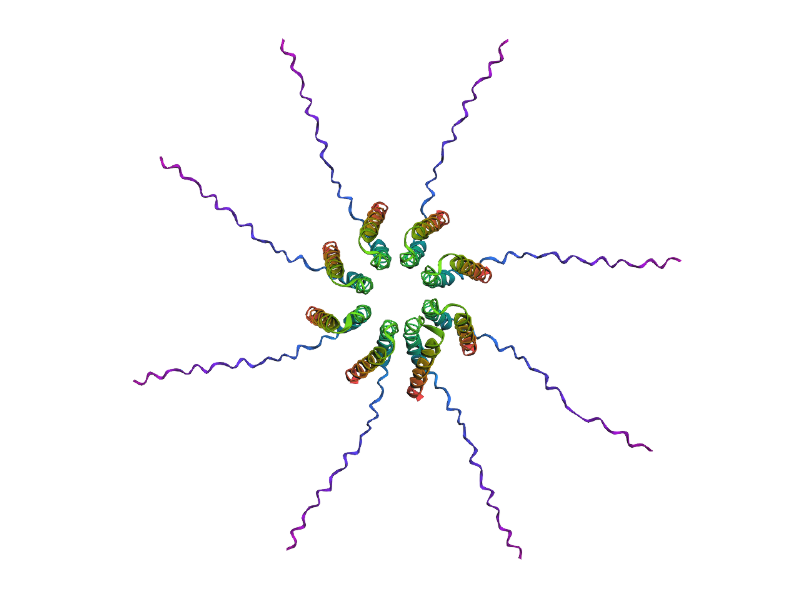 Predicted 3D structure on AF2 Multimer for the F50L octamer confirming the mutation preserves the structural integrity of the protein.
Predicted 3D structure on AF2 Multimer for the F50L octamer confirming the mutation preserves the structural integrity of the protein.