Week 4 HW: Protein Design Part 1
Homework: Protein Design I
Part A. Conceptual Questions
1) How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
~ 21% of meat is protein content (Smith et al. 2022) therefore, 500g meet contains about 105g of protein.
Using the approximation of average amino acid ≈ 100 Da ≈ 100 g/mol for ~100 g protein: 100/100=1.00 mol
Avogadro’s number: 1 mole = 6.02214076×10²³ 1.00 mol × 6.022×10²³ ≈ 6.02×10²³ amino-acid molecules
2) Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Beef/fish supplies raw materials and energy, but it doesn’t transfer “cow/fish identity”. What we eat is digested first meaning the proteins, fats, and carbohydrates are broken down into small building blocks (amino acids, fatty acids, sugars), absorbed, and then reassembled into human molecules under human genetic and hormonal control.
3) Why are there only 20 natural amino acids?
Doig (2017) hypothesizes that the canonical set of 20 standard amino acids is best understood as an evolved “alphabet” that became fixed early because this set is sufficient and practical for building stable, soluble proteins. This set enables soluble folded structures with close-packed hydrophobic cores and ordered binding pockets, rather than being selected because each amino acid was needed for catalysis (since RNA catalysts were already effective enough). Once early life standardized a working translation system around this set, changing the alphabet would have been costly, so it became effectively locked in (“frozen”). Other references, such as Freeland et al. (2000), suggest that 20 is a good number for minimizing damage from errors (mutation/mistranslation).
4) Where did amino acids come from before enzymes that make them, and before life started?
Amino acids could plausibly have come from abiotic chemistry on early Earth. Proposed routes include cyanosulfidic protometabolism and amino-acid formation from electrical discharges in simple “primitive Earth” gas mixtures (the classic Miller experiment).
5) Can you discover additional helices in proteins?
Beyond the α-helix, proteins commonly contain 3₁₀ helices and π helices (less frequent helical variants), as well as polyproline II helices (common in Pro-rich/disordered regions) and the specialized collagen triple helix.
6) Why are most molecular helices right-handed?
Right-handed helices dominate because natural biomolecules are made from single-handed monomers, and the right-handed twist is the lowest-energy way to repeat their geometry without clashes.
7) Why do β-sheets tend to aggregate?
β-sheet aggregation buries exposed hydrophobic side chains and releases ordered water from their surfaces, which is strongly favorable, lowering enthalpy.
8) What is the driving force for β-sheet aggregation?
β-sheet aggregation is driven mainly by the hydrophobic effect and stabilized/propagated by intermolecular backbone H-bonding in the cross-β structure (often reinforced by tight steric-zipper packing).
9) Why do many amyloid diseases form β-sheets?
β-sheet architecture is an unusually generic, stable, and self-templating way for polypeptide backbones to stick together when normal folding fails. In a β-sheet, the peptide backbone forms regular hydrogen bonds. This conformation makes amyloid fibrils thermodynamically stable and hard to clear, because once a small β-sheet nucleus forms, it can seed further growth by recruiting more monomers and templating the same β-rich structure.
Part B: Protein Analysis and Visualization
Question 1
I selected poly(3-hydroxyalkanoate) depolymerase (PhaZ) because it is the key enzyme that degrades PHB, which directly controls whether a microbe accumulates bioplastic (useful for biotechnology) or breaks it down (relevant for environmental fate). phaZ inactivation is commonly discussed as a strategy to reduce PHA mobilization and increase polymer retention.
Question 2
MPEPYIFRTVELDDQSIRTAVRPGKPHLTPLLIFNGIGANLELVFPFIEALDPDLEVIAFDVPGVGGSSTPRHPYRFPGLAKLTARMLDYLDYGQVSAIGVSWGGALAQQFAHDYPERCKKLVLAATAAGAVMVPGKPKVLWMMASPRRYVQPSHVIRIAPLIYGGAFRRDPDLAMHHASKVRSGGKLGYYWQLFAGLGWTSIHWLHKIHQPTLVLAGDDDPLIPLVNMRLLAWRIPNAQLHIIDDGHLFLITRAEAVAPIIMKFLQEERQRAVMHPRPASGG
BLAST Result Lenght: 283 aa Most frequent amino acid: Leucine (L), 32/283 = 11.3%
250 hits Reviewed (Swiss-Prot) homologs: 1
It belongs to the PHA depolymerase (PhaZ) family, which is part of the broader α/β-hydrolase enzyme superfamily.
Question 3
AF_AFP26495F1 - COMPUTED STRUCTURE MODEL OF POLY(3-HYDROXYALKANOATE) DEPOLYMERASE
This is not an experimentally solved structure, so there is no X-ray/EM “resolution” value. RCSB explicitly states: “There are no experimental data to verify the accuracy of this computed structure model. See Model Confidence metrics below for all regions of the polypeptide chain.” Instead, quality is reported by AlphaFold confidence. Global pLDDT: 91.95 (very high confidence overall)
RCSB lists 1 unique protein chain (monomer A1) and no ligands/non-protein entities.
Structure classification family: InterPro annotations classify it as Poly(3-hydroxyalkanoate) depolymerase (IPR011942) and an alpha/beta hydrolase fold protein (Alpha/beta hydrolase fold-1 domain, AB hydrolase superfamily).
Question 4
I opened AF-Q9R9W3-F1-model_v6 in PyMOL and visualized it in cartoon, ribbon, and ball-and-stick representations.
Colored by secondary structure, it shows a mixed α/β fold with more helices than β-sheets.
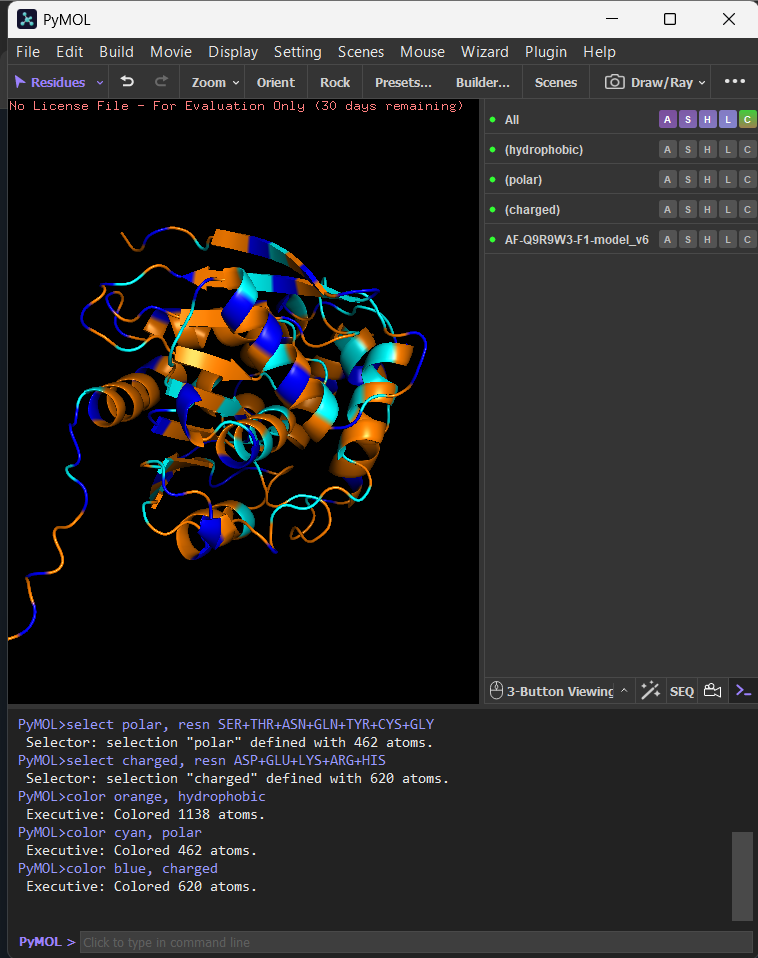
Colored by residue type, hydrophobic residues are enriched in the core (and in a few surface patches), while polar/charged residues are mostly surface-exposed, consistent with solubility.
The surface view shows clear cavities/clefts, consistent with potential binding pockets (e.g., a substrate-binding groove typical of hydrolases).
Part C. Using ML-Based Protein Design Tools
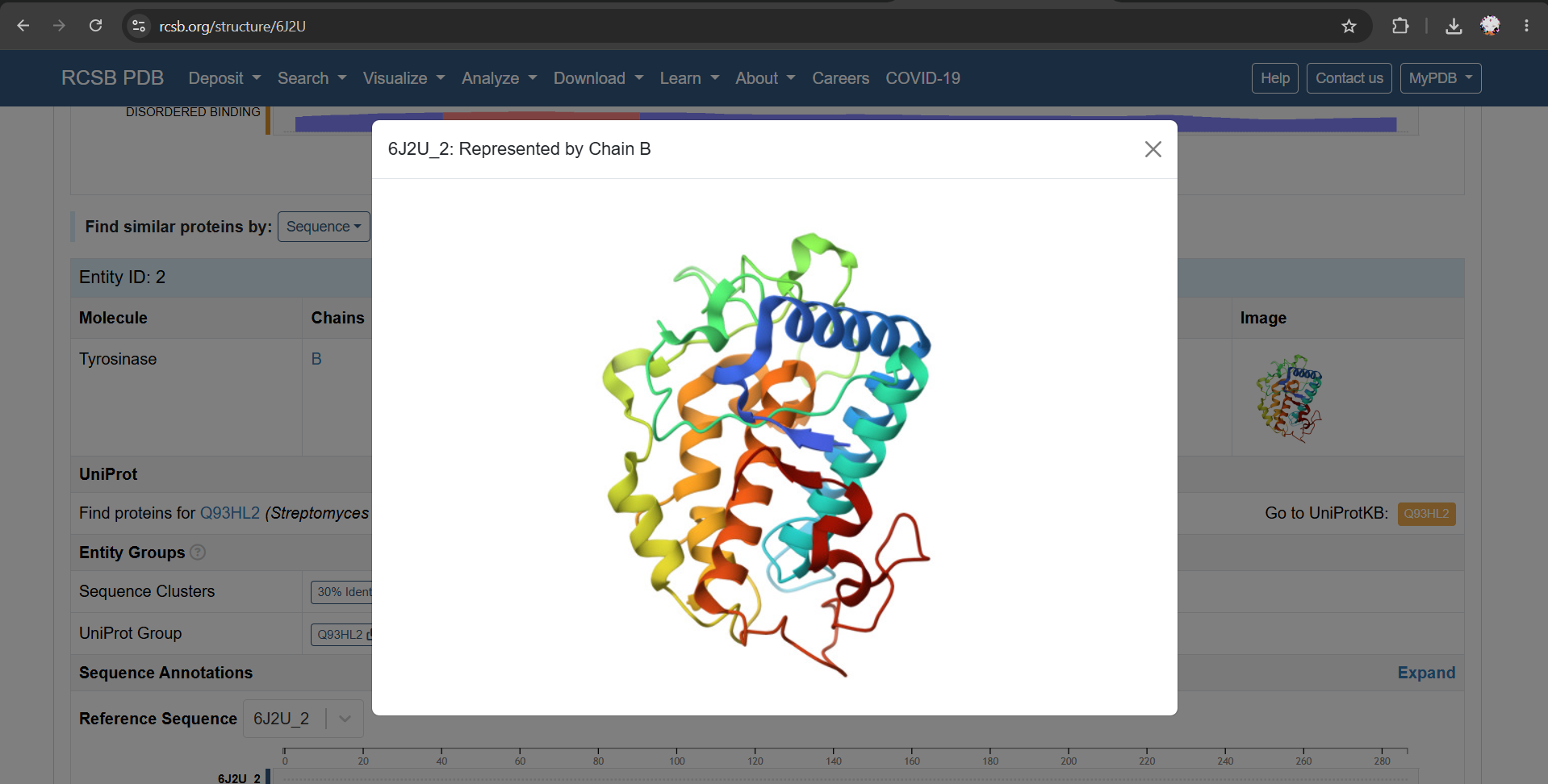
For this section, I chose PDB 6J2U as a structural reference. This entry contains a heterodimeric complex between MelC1, the tyrosinase caddy/cofactor protein, and MelC2, the tyrosinase enzyme from Streptomyces avermitilis. For my analysis, I focused on the MelC2 tyrosinase chain (6J2U_2: Represented by Chain B).
https://colab.research.google.com/drive/1cOMreGB8zHQAy063H0HyahlEamIXSSPf?usp=sharing
C1. Protein Language Modeling
Question 1
a) I used the Chain B sequence from PDB 6J2U, including the N-terminal expression tag present in the deposited sequence.
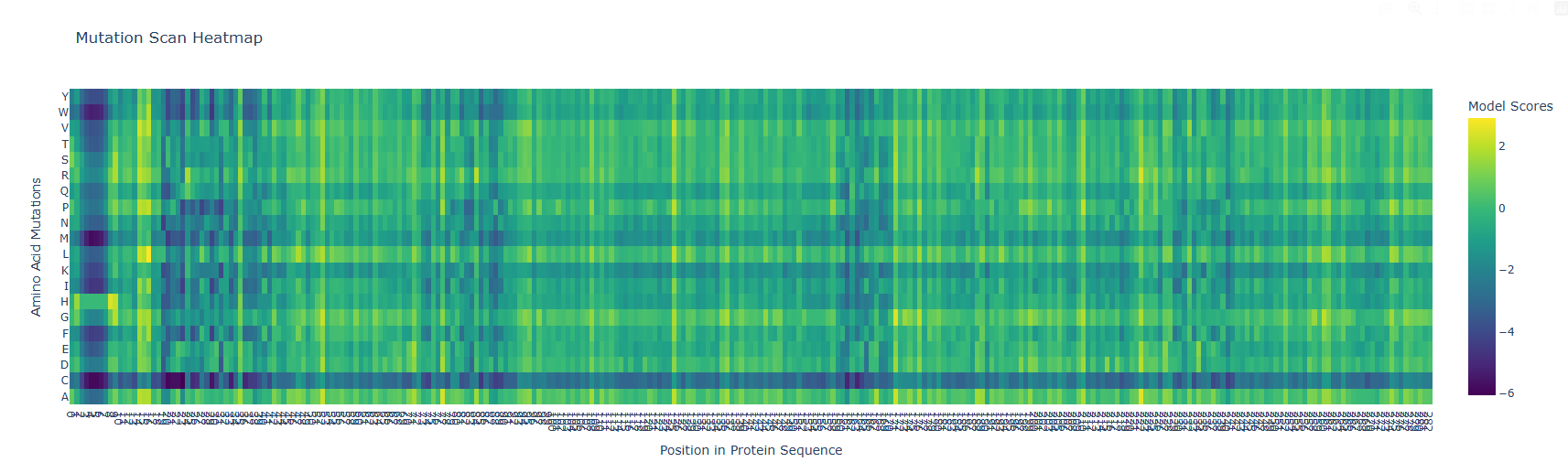
b) The vertical darker columns at certain positions are highly constrained residues where most substitutions are penalized. That usually indicates structural importance (core packing, tight turns, or residues critical for fold stability). Positions with mostly neutral colors across many substitutions are likely surface-exposed or in flexible loops, where the model predicts more tolerance.
After generating the ESM2 mutational scan heatmap, I found it difficult to confidently interpret specific patterns only by visual inspection, because the plot contains many residues and mutations compressed into a dense matrix. To make the interpretation more objective, I used ChatGPT to help me write small analysis snippets to quantify the heatmap directly. I run a script to calculate the average ESM2 score for each mutant amino acid across all positions and found out that substitutions to cysteine are broadly disfavored across the MelC2 sequence.
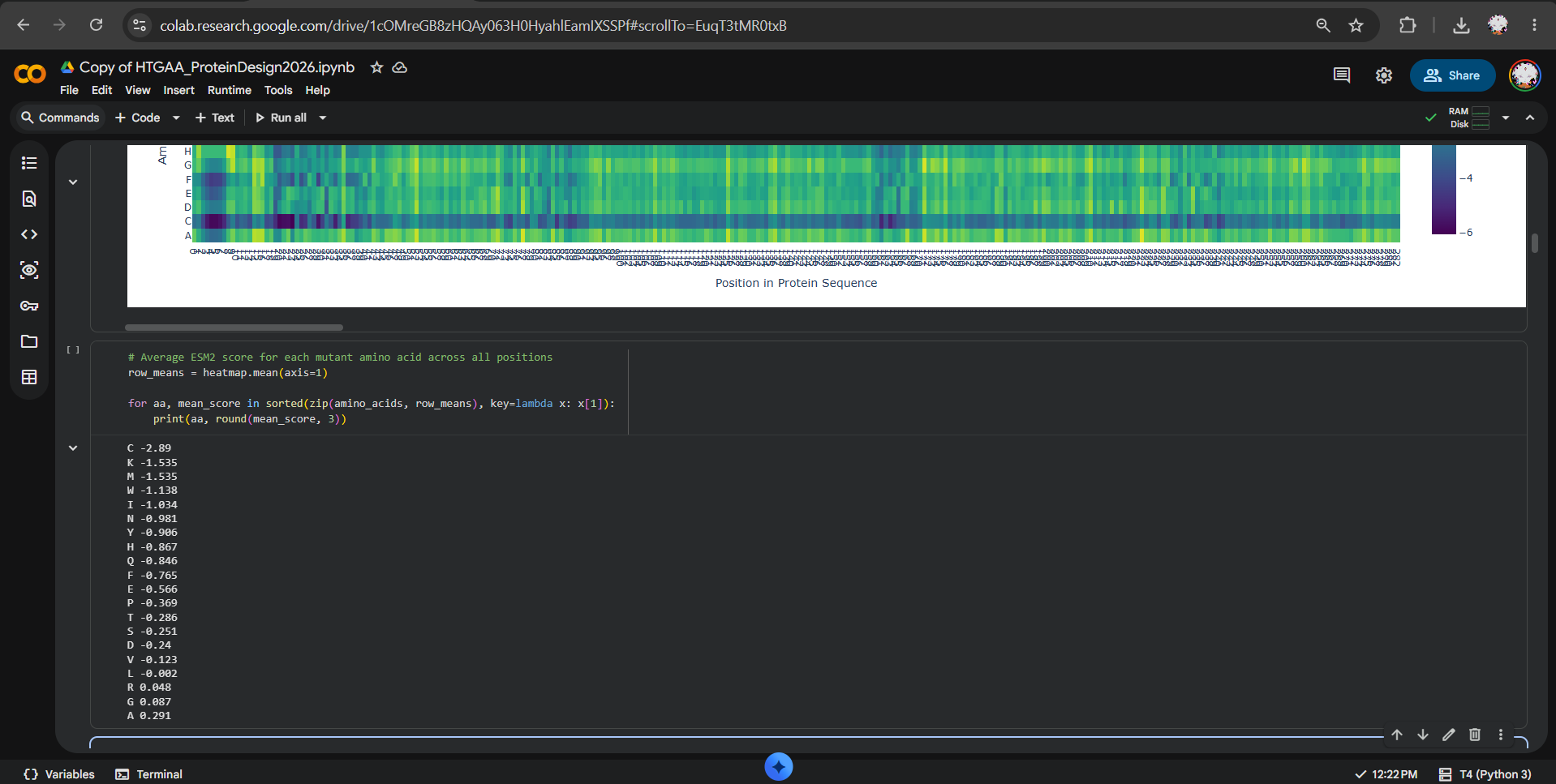
In fact, in the heatmap, the cysteine row apparently shows many strongly negative scores, suggesting that the model predicts cysteine mutations to be poorly compatible with this protein. This makes biological sense because cysteine can introduce reactive thiol chemistry, unwanted disulfide-like interactions, or local structural constraints that may disrupt folding or stability, especially in a soluble bacterial enzyme where cysteine is not broadly used as a tolerated replacement.
Question 2
During the latent space analysis, I tried to use the provided SCOPe/Astral sequence dataset from the notebook, but I could not load it correctly in Colab. When I attempted to display sequences, I got an IndexError: list index out of range, which indicated that no sequence records had been parsed.
At first, I tested whether the issue was caused by comment lines before the first FASTA entry and tried using the fasta-pearson parser. After further debugging with Gemini/AI assistance in Colab, the issue appeared to be that the dataset URL was not returning the expected FASTA file, but HTML content instead.
I also tried opening the SCOPe/Astral page manually in the browser, but the site displayed an anti-bot verification page and did not provide access to the dataset.
Because of this, Biopython could not parse the dataset, so I was not able to generate the reduced-dimensionality map or place my protein in it.
If the dataset had loaded correctly, my workflow would have been:
- Parse the SCOPe/Astral FASTA dataset.
- Add the MelC2 Chain B sequence to the dataset.
- Generate ESM2 embeddings for all sequences.
- Reduce the embeddings using t-SNE.
- Highlight MelC2 in the resulting map.
- Compare MelC2 to its nearest neighbors.
a) Use the provided sequence dataset to embed proteins in reduced dimensionality I attempted to use the provided SCOPe/Astral sequence dataset, but the file could not be accessed correctly. The downloaded content was HTML rather than a valid FASTA file, so I could not generate ESM2 embeddings from the provided dataset.
b) Analyze the different formed neighborhoods: do they approximate similar proteins? Since the dataset could not be parsed, I could not generate or analyze the embedding neighborhoods directly. Conceptually, I would expect ESM2 embeddings to place proteins with related sequence-level features, domains, motifs, or families closer together, but I could not verify this with the provided dataset.
c) Place your protein in the resulting map and explain its position and similarity to its neighbors My plan was to add MelC2 tyrosinase from PDB 6J2U Chain B to the dataset before embedding, then inspect whether it clustered near related proteins such as oxidoreductases, tyrosinases, or metal-binding enzymes. Since the dataset could not be accessed correctly, I could not place MelC2 in the final map, so this remains a planned analysis rather than a completed result.
C2. Protein Folding
Question 1
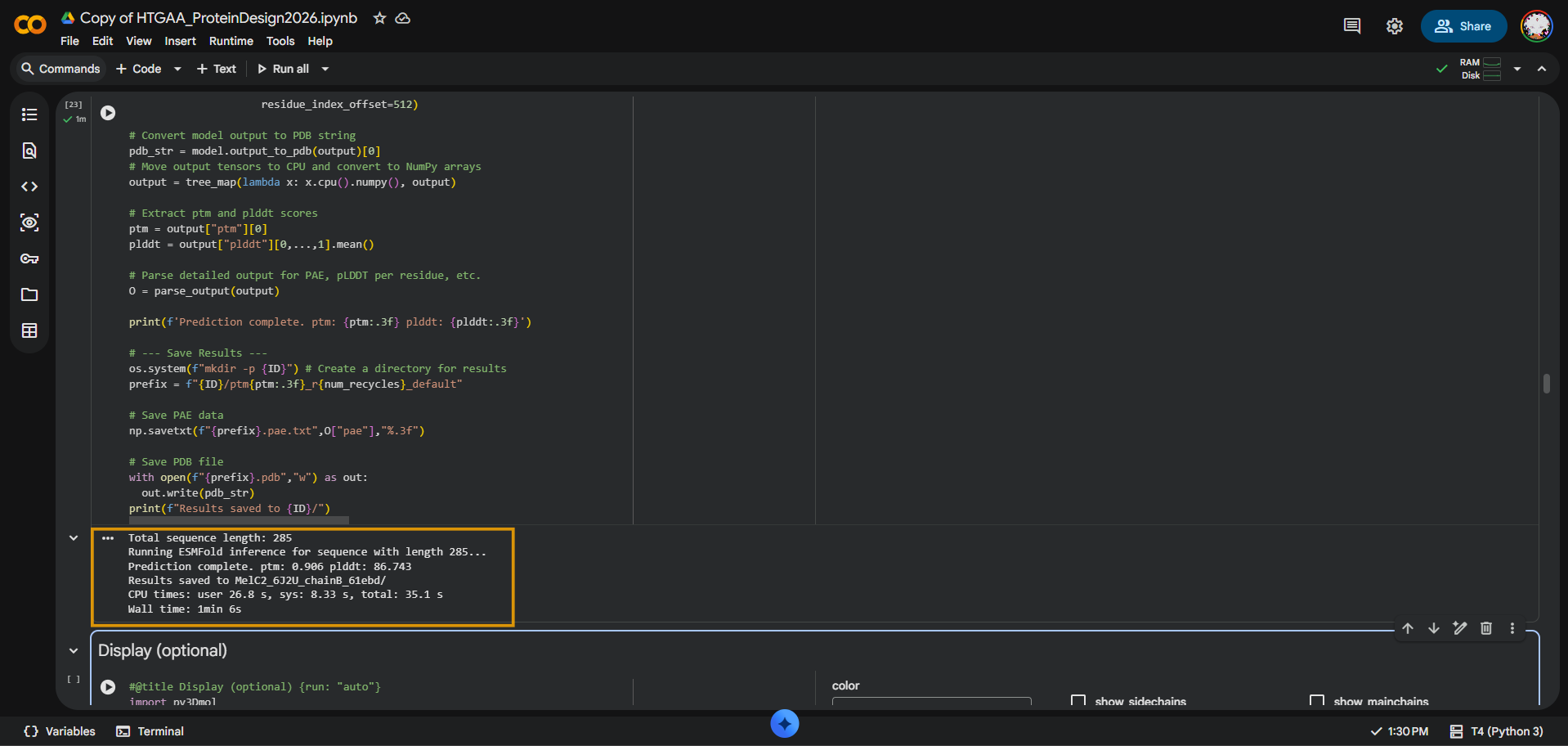
I folded the MelC2 tyrosinase Chain B sequence from PDB 6J2U using ESMFold. The input sequence was 285 amino acids long. The prediction completed successfully with pTM = 0.906 and average pLDDT = 86.743, suggesting that ESMFold produced a high-confidence global fold for MelC2.
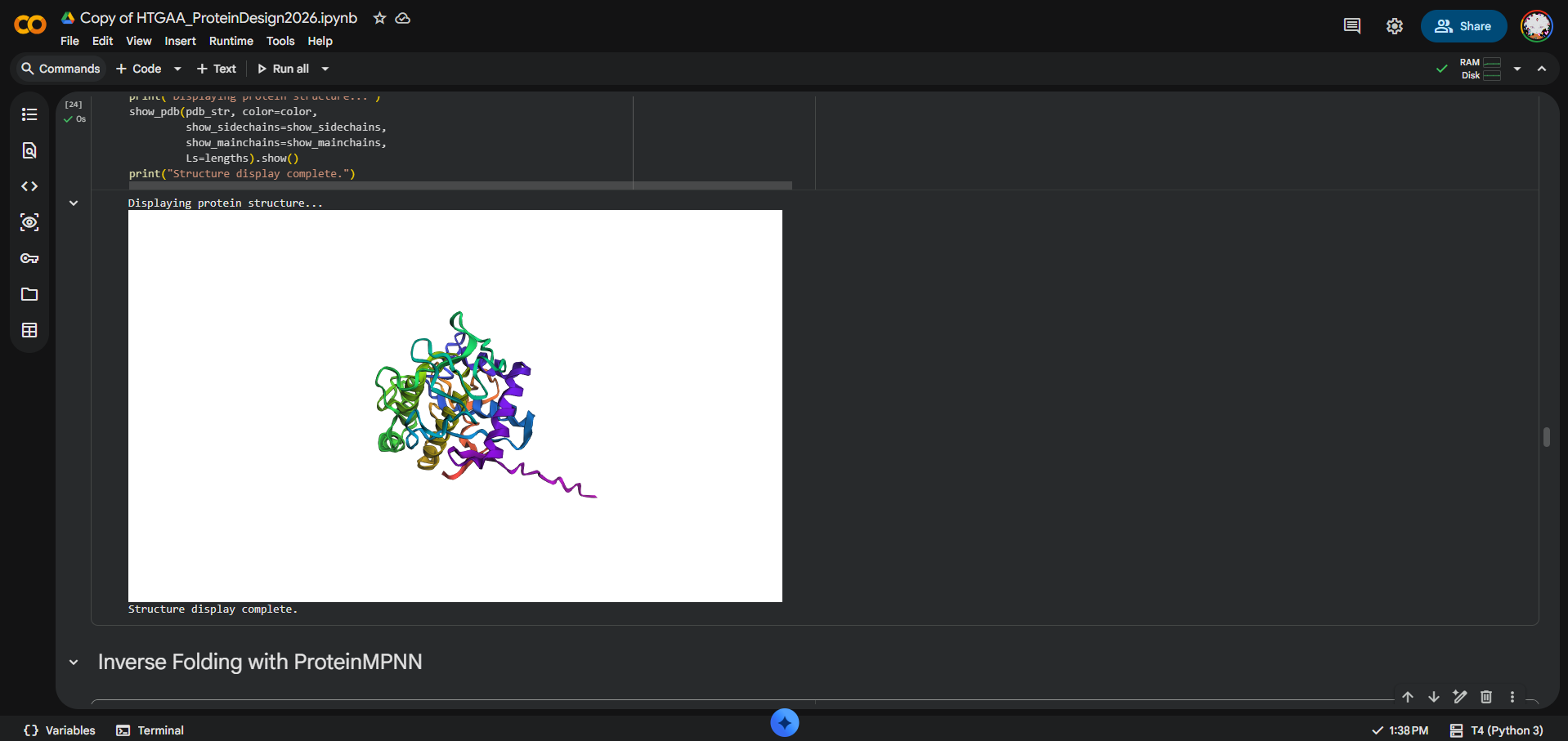
At the fold level, yes: the ESMFold prediction appears broadly consistent with the original MelC2 structure. However, I did not calculate RMSD, and the original PDB structure includes MelC2 in complex with MelC1 and metal ions, while ESMFold predicts from sequence alone. Therefore, I interpret the result as a strong qualitative fold-level match, not a precise coordinate-level comparison.
Question 2
Native MelC2
MGSHHHHHHSERTVRKNQATLTADEKRRFVDALVALKRSGRYDEFVTTHNAFIMGDTDSGERTGHRSPSFLPWHRRFLIEFEQALQAVDPSVALPYWDWSTDRTARASLWAPDFLGGSGRSLDGRVMDGPFAASTGNWPVNVRVDSRTYLRRTLGGGGRELPTRAEVDSVLAMSTYDMAPWNSASDGFRNHLEGWRGVNLHNRVHVWVGGQMATGVSPNDPVFWLHHAYIDRLWAQWQSRHPGSGYVPTGGTPNVVDLNETMKPWNDVRPADLLDHTAHYTFDTV
Length: 285 aa pTM = 0.906 pLDDT = 86.743
Mutant 1
To test whether the MelC2 predicted structure is resilient to a small sequence change, I first introduced a single point mutation into the Chain B sequence.
I used the following simple Python function to generate the mutant sequence here.
I selected position 100, where the native residue is serine (S), and mutated it to cysteine (C):
S100C MelC2 mutant
MGSHHHHHHSERTVRKNQATLTADEKRRFVDALVALKRSGRYDEFVTTHNAFIMGDTDSGERTGHRSPSFLPWHRRFLIEFEQALQAVDPSVALPYWDWCTDRTARASLWAPDFLGGSGRSLDGRVMDGPFAASTGNWPVNVRVDSRTYLRRTLGGGGRELPTRAEVDSVLAMSTYDMAPWNSASDGFRNHLEGWRGVNLHNRVHVWVGGQMATGVSPNDPVFWLHHAYIDRLWAQWQSRHPGSGYVPTGGTPNVVDLNETMKPWNDVRPADLLDHTAHYTFDTV
I then used this S100C MelC2 mutant sequence as the input for a new ESMFold prediction, so I could compare its predicted fold and confidence scores with the native MelC2 prediction.
The S100C mutant produced almost the same ESMFold confidence scores as the native sequence.
Length: 285 ptm: 0.906 plddt: 86.874
After introducing the S100C mutation, the predicted structure still appeared compact and globular, with no obvious large-scale disruption compared to the native model. This suggests that MelC2 is structurally resilient to this single substitution at the overall fold level.
Mutant 2
I generated this 16-amino-acid segment-level mutant using a short Python script suggested by ChatGPT here. The script replaced residues 120-135 of the native MelC2 sequence with a glycine-rich segment while preserving the original protein length. I used this to test whether the predicted MelC2 fold is resilient to larger local sequence disruption.
MelC2_segment_mutation_120_135_Gly
Original segment 120-135: RSLDGRVMDGPFAAST Replacement segment: GGGGGGGGGGGGGGGG Native length: 285 Segment mutant length: 285 MGSHHHHHHSERTVRKNQATLTADEKRRFVDALVALKRSGRYDEFVTTHNAFIMGDTDSGERTGHRSPSFLPWHRRFLIEFEQALQAVDPSVALPYWDWSTDRTARASLWAPDFLGGSGGGGGGGGGGGGGGGGGGNWPVNVRVDSRTYLRRTLGGGGRELPTRAEVDSVLAMSTYDMAPWNSASDGFRNHLEGWRGVNLHNRVHVWVGGQMATGVSPNDPVFWLHHAYIDRLWAQWQSRHPGSGYVPTGGTPNVVDLNETMKPWNDVRPADLLDHTAHYTFDTV
The segment mutant produced a lower-confidence ESMFold prediction than the native and S100C sequences. The native MelC2 model had pTM = 0.906 and pLDDT = 86.743, while the segment mutant dropped to pTM = 0.865 and pLDDT = 81.386.
Visually, the predicted structure still formed a compact globular fold, so the protein did not appear completely disrupted. However, the decrease in both pTM and pLDDT suggests that replacing residues 120-135 with glycines weakened the model’s confidence in the fold.
This makes sense because a glycine-rich replacement can increase flexibility and remove side-chain interactions that may help stabilize the local structure. Still, these are structure predictions only, experimental testing would be needed to know whether catalytic activity or copper/metal-related function is preserved.
| Sequence | Change | pTM | pLDDT | Interpretation |
|---|---|---|---|---|
| Native MelC2 | None | 0.906 | 86.743 | High-confidence compact fold |
| S100C | Single point mutation | 0.906 | 86.874 | Fold appears globally preserved; confidence essentially unchanged |
| Segment mutant | Residues 120-135 replaced with glycines | 0.865 | 81.386 | Fold still predicted, but confidence decreased, suggesting the perturbation affected structural stability more than the point mutation |
C3. Protein Generation
Question 1
I used ProteinMPNN to redesign the MelC2 chain from PDB 6J2U. I set Chain B as the designed chain and kept Chain A fixed, since Chain B is MelC2 tyrosinase and Chain A is the MelC1 caddy/cofactor protein.
ProteinMPNN used 273 resolved residues from Chain B and generated a redesigned sequence with:
Native score: 1.2305 Designed score: 0.7427 Sequence recovery: 0.5751
The sequence recovery means that about 57.5% of the redesigned residues matched the native MelC2 sequence. This suggests that ProteinMPNN found a sequence predicted to fit the same backbone while changing a substantial part of the original sequence.
However, this only suggests structural compatibility. It does not prove that the redesigned protein would preserve tyrosinase activity, metal binding, or melanin production.
Question 2
I folded the ProteinMPNN-designed MelC2 sequence with ESMFold to test whether the redesigned sequence still predicts a MelC2-like structure.
| Sequence | Length | pTM | pLDDT | Interpretation |
|---|---|---|---|---|
| Native MelC2 | 285 aa | 0.906 | 86.743 | High-confidence native fold prediction |
| ProteinMPNN design | 273 aa | 0.878 | 80.444 | Still folds with good confidence, but lower than native |
The ProteinMPNN-designed sequence produced pTM = 0.878 and pLDDT = 80.444. These scores are lower than the native MelC2 prediction, but still reasonably high, suggesting that the redesigned sequence remains structurally compatible with the MelC2 backbone.
Because the designed sequence had only 57.5% sequence recovery, it is substantially different from native MelC2. However, ESMFold still predicted a compact fold with good confidence. This suggests that ProteinMPNN generated a sequence that may preserve the overall structure, although this does not prove preservation of tyrosinase activity, metal binding, or melanin production.
Final Conclusions
| Sequence / model | Type of test | Change introduced | Length | pTM | avg pLDDT | Result / interpretation |
|---|---|---|---|---|---|---|
| Native MelC2 | Baseline ESMFold prediction | Original MelC2 Chain B sequence from PDB 6J2U | 285 aa | 0.906 | 86.743 | High-confidence compact fold. Used as the reference for comparison. |
| S100C mutant | Point mutation | Serine at position 100 replaced by cysteine | 285 aa | 0.906 | 86.874 | Scores were essentially unchanged compared with native MelC2. The global fold appears resilient to this single point mutation. |
| Segment mutant 120-135 Gly | Large local perturbation | Residues 120-135, RSLDGRVMDGPFAAST, replaced with 16 glycines | 285 aa | 0.865 | 81.386 | Still predicted to fold, but with reduced confidence. This suggests the global fold is not destroyed, but the perturbation affects structural confidence/stability more than the point mutation. |
| ProteinMPNN-designed MelC2 | Inverse-folding design + ESMFold validation | ProteinMPNN redesigned Chain B using the 6J2U backbone; sequence recovery = 0.5751 | 273 aa | 0.878 | 80.444 | Still predicted to fold with reasonably good confidence, despite only ~57.5% sequence recovery. Suggests the backbone can support substantial sequence variation, but function is not guaranteed. |
Overall, MelC2 appears structurally robust at the global fold level. However, all of these conclusions are structural predictions. A preserved fold does not prove preserved tyrosinase activity, copper/metal binding, or melanin production. Functional validation would still require experimental testing.
Part D. Group Brainstorm on Bacteriophage Engineering
Group Members: Diogo Custodio; Flo Razoux; Katharine Kolin; Mariana Kanbe; Marisa Satsia.
PROJECT MAIN GOAL in discussion:
- Increased stability (easiest)
- Higher titers (medium)
- Higher toxicity of lysis protein (hard)
My group and I are conducting research for the group phage project. We have set up a shared Google Docs (screenshot below).

Phage reading material
We reviewed the Week 4 phage reading material and used it to focus the proposal on the MS2 L protein, especially its stability, DnaJ dependence, membrane insertion, and lysis function.
From the proposed bacteriophage engineering goals, our group focused on: Increased stability of the L protein
Our short group plan was to use computational protein design tools to identify mutations that could improve the stability of the MS2 L protein. One possible direction was to make the L protein less dependent on the bacterial chaperone DnaJ by identifying mutations that could improve folding, membrane insertion, or oligomerization.
We proposed using:
- Protein language model mutational scoring
- In silico mutagenesis
- Experimental L-protein mutant data
- Biological reasoning based on known L-protein functional regions
These tools can help prioritize mutations before experimental testing. Protein language model scores can identify substitutions that are sequence-compatible, while experimental mutant data and biological reasoning can help filter candidates based on possible effects on DnaJ dependence, membrane behavior, and lysis function.
Potential pitfalls: One pitfall is that positive LLR scores may reflect sequence plausibility, but not necessarily improved lysis function. A second pitfall is that increasing protein stability may not always improve function, because L-protein activity may require flexibility, membrane disruption, or host-factor interaction.
Pipeline schematic
MS2 L-protein sequence: mutational scoring notebook → shortlist positive-scoring substitutions → compare with experimental L-protein mutant data → map candidates to functional regions → select mutations for future experimental testing
Individual plan / contribution
My individual contribution was to select candidate MS2 L-protein mutations by combining LLR scores, experimental mutant data, and biological reasoning.
I selected two soluble-region mutants, S9Q and C29R, to probe folding and possible DnaJ dependence. I also selected three transmembrane-region mutants, A45L, T52L, and N53L, to probe membrane insertion and oligomerization.
| Mutant | Region | LLR | Rationale |
|---|---|---|---|
| S9Q | Soluble / N-terminal | 2.014 | May affect folding or DnaJ-related surface chemistry |
| C29R | Soluble / N-terminal | 2.395 | Strong positive score; may alter chaperone-recognition surfaces |
| A45L | Transmembrane | 1.539 | May increase hydrophobic packing and membrane stability |
| T52L | Transmembrane | 1.814 | Polar-to-hydrophobic change that may improve membrane compatibility |
| N53L | Transmembrane | 1.865 | Additional transmembrane-stabilizing candidate |
Use of AI assistance
I used ChatGPT as a writing and organization assistant to help structure this section and make sure the required items were clearly addressed. I reviewed, edited, and finalized the scientific content myself.