Week 2 HW: DNA Read, Write, & Edit
Benchling & In-silico Gel Art
The lambda bacteriophage DNA sequence (Lambda_NEB) was imported into Benchling. Using the virtual restriction tool, simulated DNA digestion was performed with seven restriction enzymes: EcoRI, SacI, SalI, HindIII, BamHI, KpnI, and EcoRV. The results were visualized by agarose gel electrophoresis.

Simulated gel electrophoresis of lambda DNA after restriction digestion with EcoRI, SacI, SalI, HindIII, BamHI, KpnI, and EcoRV
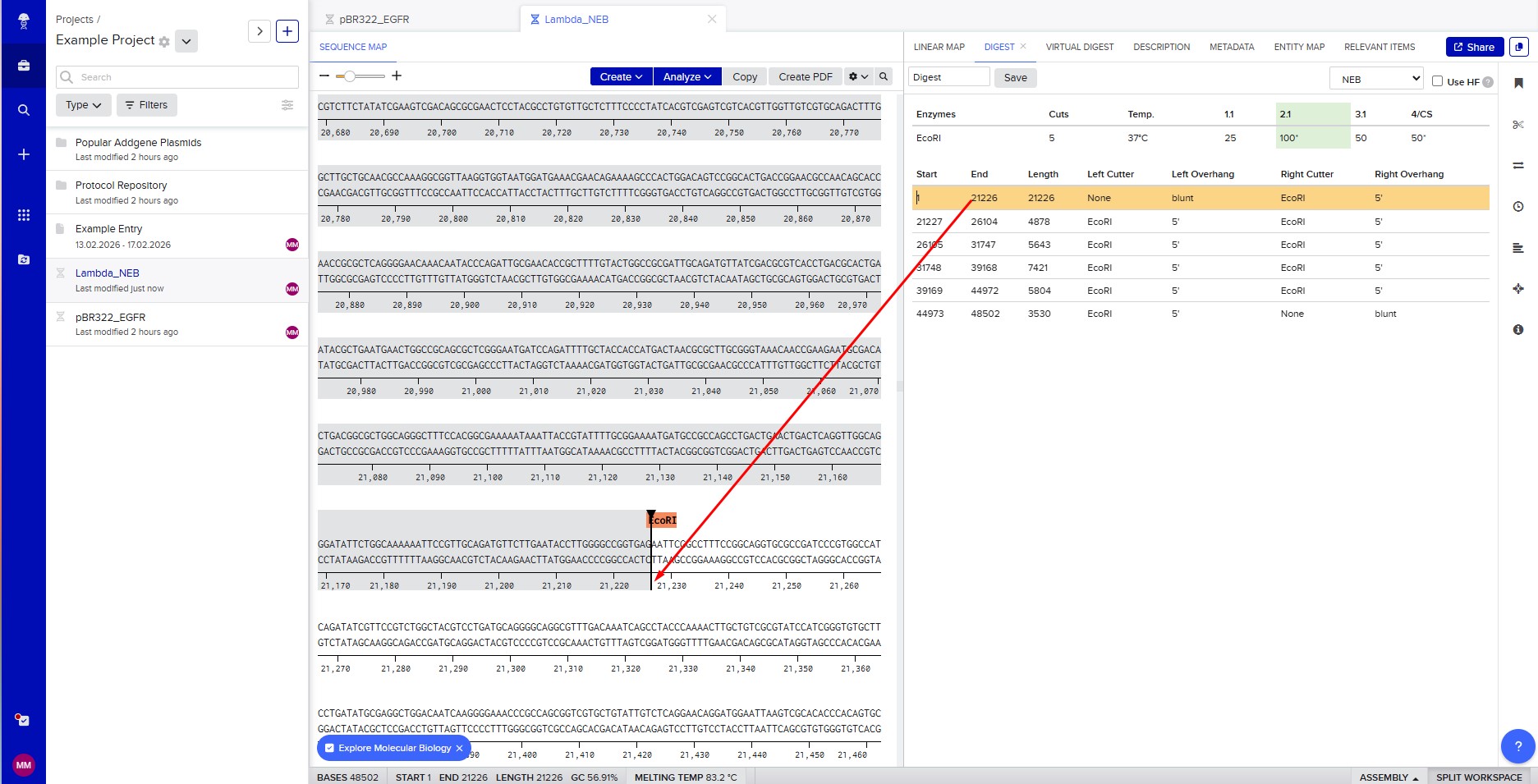
Restriction enzyme digestion with EcoRI
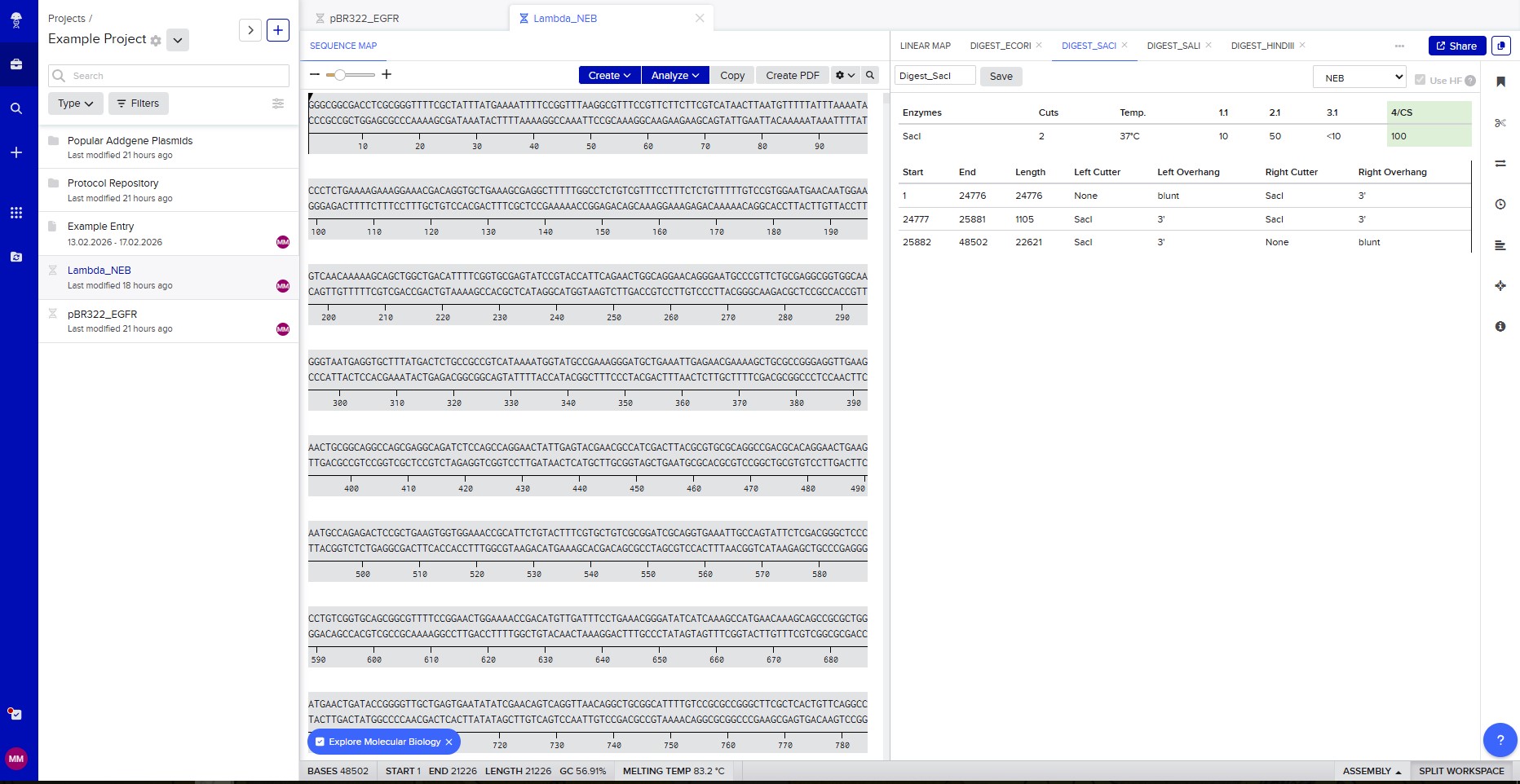 Restriction enzyme digestion with SacI
Restriction enzyme digestion with SacI
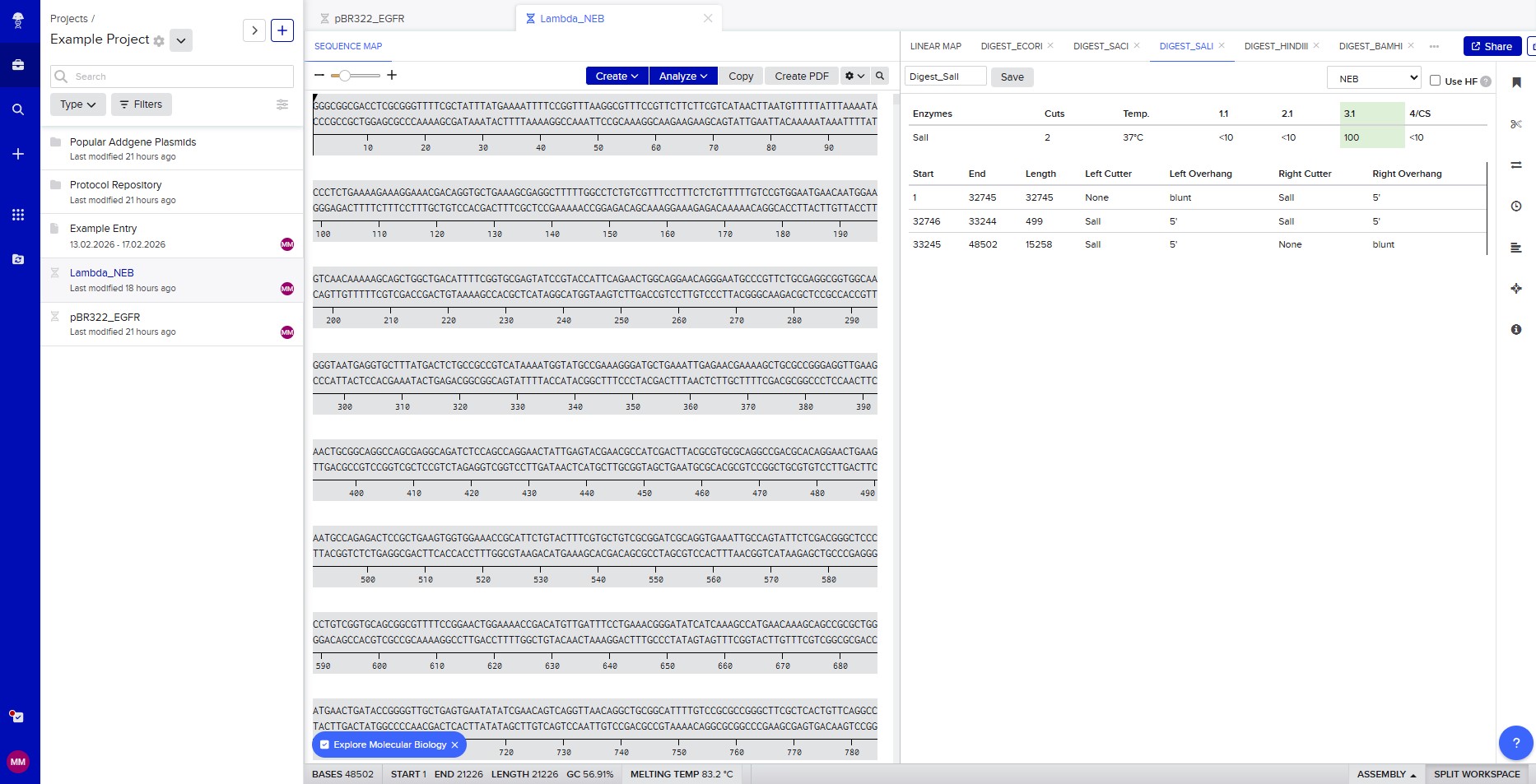 Restriction enzyme digestion with SalI
Restriction enzyme digestion with SalI
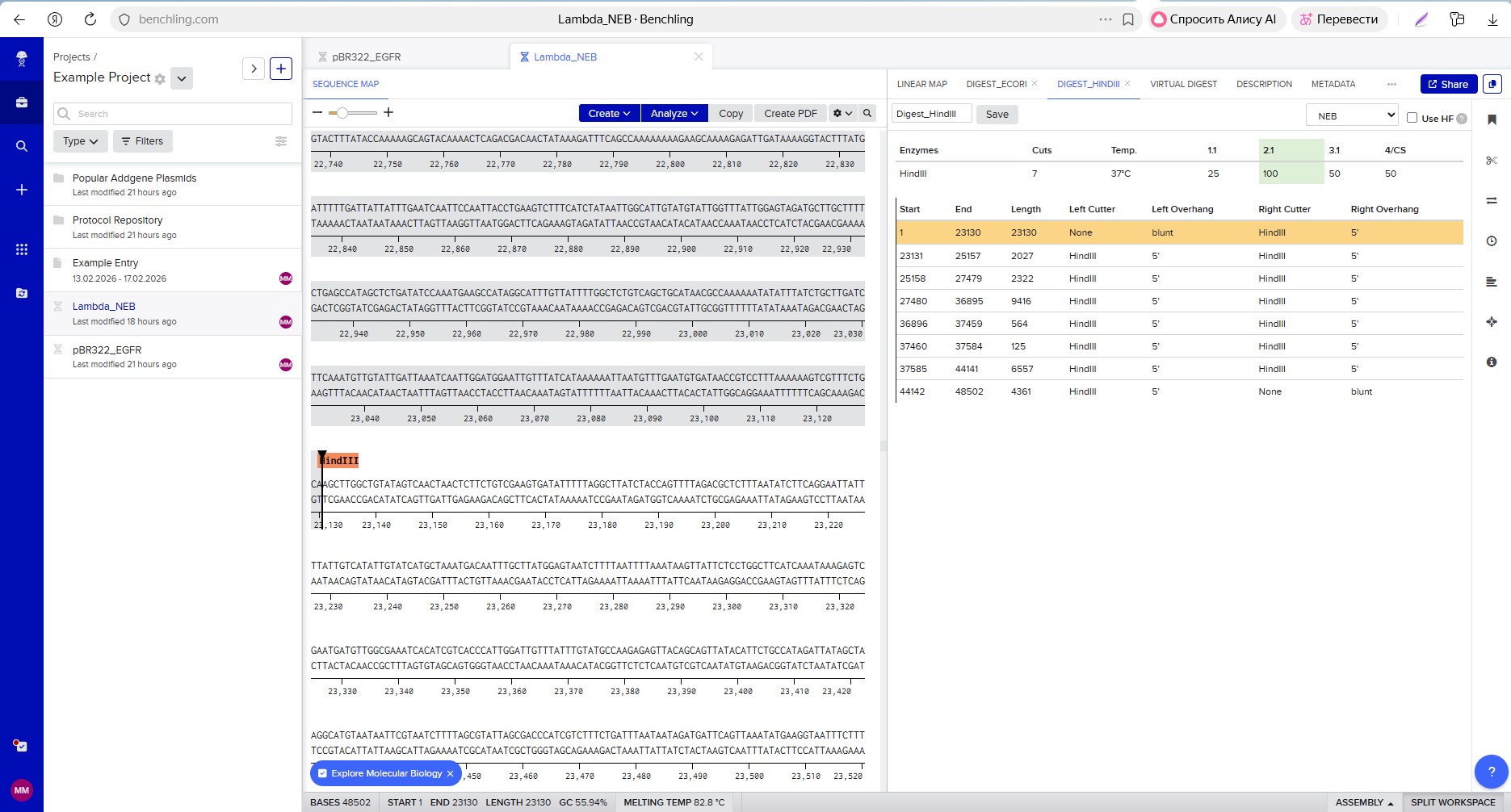 Restriction enzyme digestion with HindIII
Restriction enzyme digestion with HindIII
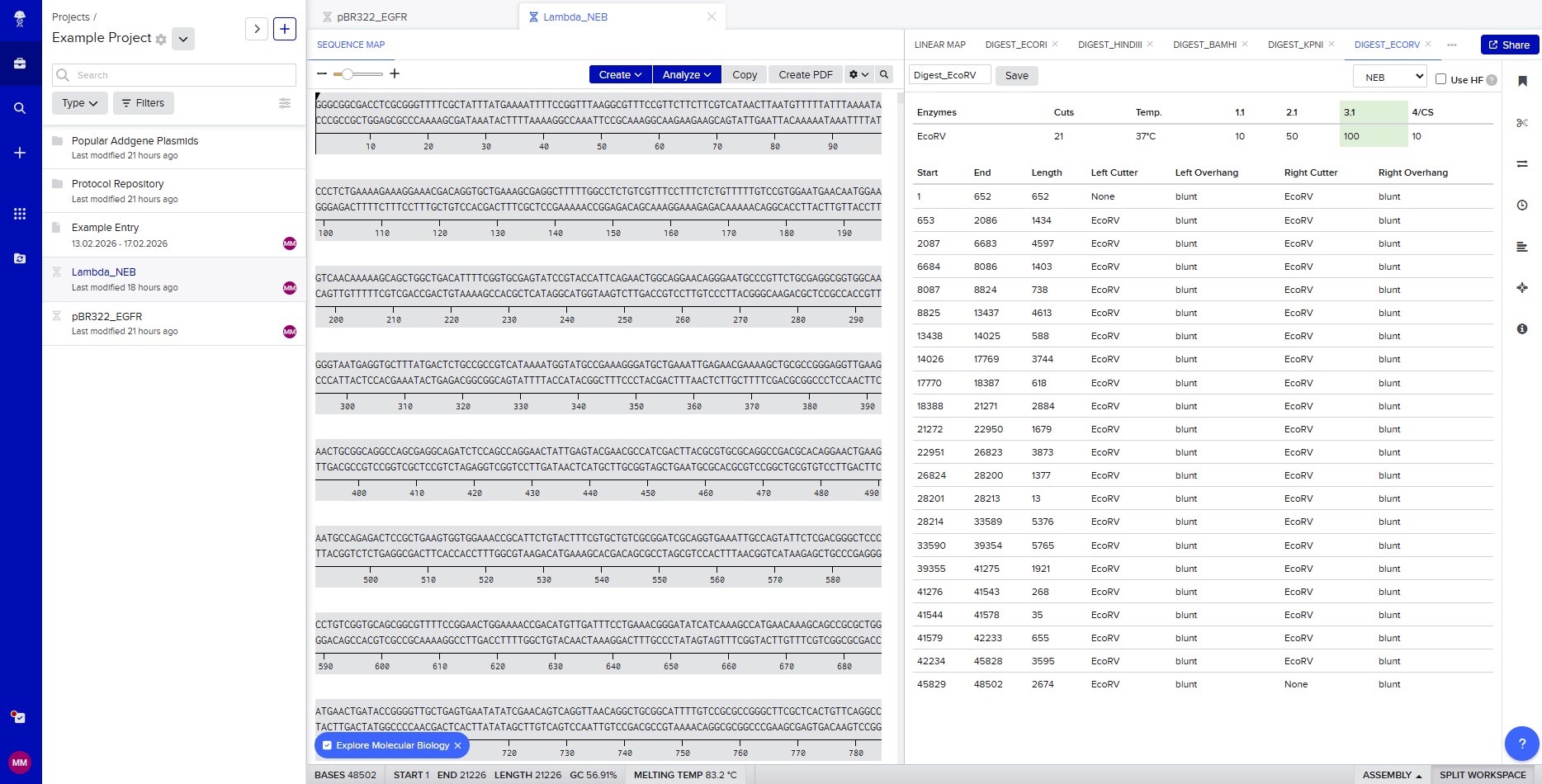 Restriction enzyme digestion with EcoRV
Restriction enzyme digestion with EcoRV
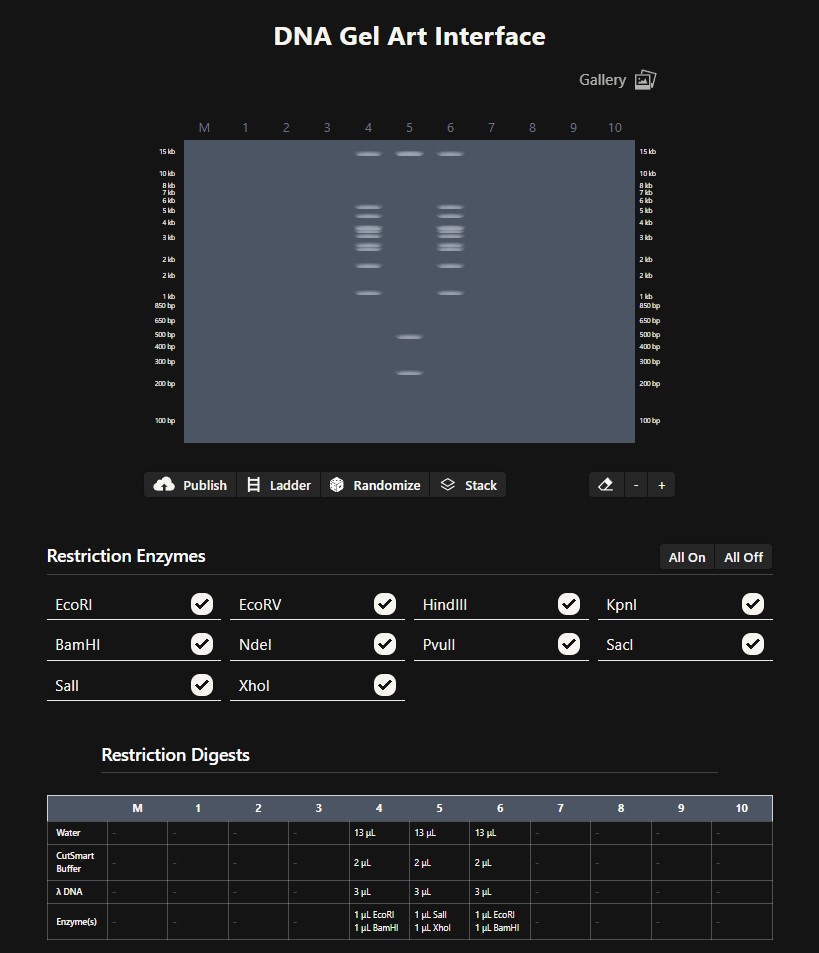 Gel art pattern resembling a face created using the Ronan Donovan interface
Gel art pattern resembling a face created using the Ronan Donovan interface
DNA Design Challenge
Choose your protein
I have chosen the protein mScarlet-I. This is a bright red fluorescent protein derived from mCherry and optimized for fast maturation and high photostability. It is widely used as a reporter in bacterial systems because its fluorescence can be easily detected and quantified. In my construct, mScarlet-I is expressed under the control of the RcsB‑dependent promoter P_rprA, allowing me to monitor the activation of the Rcs phosphorelay pathway in Escherichia coli by detecting red fluorescence.
The protein sequence of mScarlet-I was obtained from the FPbase database (FPbase ID: 6VVTK), which specializes in fluorescent proteins. The sequence is as follows:
MVSKGEAVIK EFMRFKVHME GSMNGHEFEI EGEGEGRPYE GTQTAKLKVT KGGPLPFSWD ILSPQFMYGS RAFIKHPADI PDYYKQSFPE GFKWERVMNF EDGGAVTVTQ DTSLEDGTLI YKVKLRGTNF PPDGPVMQKK TMGWEASTER LYPEDGVLKG DIKMALRLKD GGRYLADFKT TYKAKKPVQM PGAYNVDRKL DITSHNEDYT VVEQYERSEG RHSTGGMDEL YK
Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence
Using the NCBI Nucleotide database, I searched for the mScarlet-I gene and found entry KY021424.1. This entry contains the coding DNA sequence (CDS) for the mScarlet-I protein. The nucleotide sequence is: ATGGTGAGCAAGGGCGAGGCAGTGATCAAGGAGTTCATGCGGTTCAAGGTGCACATGGAGGGCTCCATGAACGGCCACGAGTTCGAGATCGAGGGCGAGGGCGAGGGCCGCCCCTACGAGGGCACCCAGACCGCCAAGCTGAAGGTGACCAAGGGTGGCCCCCTGCCCTTCTCCTGGGACATCCTGTCCCCTCAGTTCATGTACGGCTCCAGGGCCTTCATCAAGCACCCCGCCGACATCCCCGACTACTATAAGCAGTCCTTCCCCGAGGGCTTCAAGTGGGAGCGCGTGATGAACTTCGAGGACGGCGGCGCCGTGACCGTGACCCAGGACACCTCCCTGGAGGACGGCACCCTGATCTACAAGGTGAAGCTCCGCGGCACCAACTTCCCTCCTGACGGCCCCGTAATGCAGAAGAAGACAATGGGCTGGGAAGCGTCCACCGAGCGGTTGTACCCCGAGGACGGCGTGCTGAAGGGCGACATTAAGATGGCCCTGCGCCTGAAGGACGGCGGCCGCTACCTGGCGGACTTCAAGACCACCTACAAGGCCAAGAAGCCCGTGCAGATGCCCGGCGCCTACAACGTCGACCGCAAGTTGGACATCACCTCCCACAACGAGGACTACACCGTGGTGGAACAGTACGAACGCTCCGAGGGCCGCCACTCCACCGGCGGCATGGACGAGCTGTACAAG
Codon optimization
I optimized the mScarlet-I gene for expression in E. coli using the E. coli Codon Optimizer tool (efbpublic.org). Codon optimization is necessary because E. coli prefers specific codons over others; using rare codons slows down translation and reduces protein yield. I chose E. coli as the host because it is the standard organism for protein expression in my project. The optimized sequence has a GC content of 42.4% (down from 63.1%), which is ideal for E. coli.
Optimized sequence: ATGGTTAGCAAAGGTGAAGCAGTTATTAAAGAATTTATGCGTTTTAAAGTTCATATGGAAGGTAGCATGAATGGTCATGAATTTGAAATTGAAGGTGAAGGTGAAGGTCGTCCGTATGAAGGTACCCAGACCGCAAAACTGAAAGTTACCAAAGGTGGTCCGCTGCCGTTTAGCTGGGATATTCTGAGCCCGCAGTTTATGTATGGTAGCCGTGCATTTATTAAACATCCGGCAGATATTCCGGATTATTATAAACAGAGCTTTCCGGAAGGTTTTAAATGGGAACGTGTTATGAATTTTGAAGATGGTGGTGCAGTTACCGTTACCCAGGATACCAGCCTGGAAGATGGTACCCTGATTTATAAAGTTAAACTGCGTGGTACCAATTTTCCGCCGGATGGTCCGGTTATGCAGAAAAAAACCATGGGTTGGGAAGCAAGCACCGAACGTCTGTATCCGGAAGATGGTGTTCTGAAAGGTGATATTAAAATGGCACTGCGTCTGAAAGATGGTGGTCGTTATCTGGCAGATTTTAAAACCACCTATAAAGCAAAAAAACCGGTTCAGATGCCGGGTGCATATAATGTTGATCGTAAACTGGATATTACCAGCCATAATGAAGATTATACCGTTGTTGAACAGTATGAACGTAGCGAAGGTCGTCATAGCACCGGTGGTATGGATGAACTGTATAAA
You have a sequence! Now what?
To produce the mScarlet-I protein from my DNA sequence, I will use a cell-dependent method: expression in Escherichia coli.
My DNA construct contains the following elements:
- P_rprA: an inducible promoter that is activated by the RcsB protein. It initiates transcription of the downstream gene.
- B0034: a strong ribosome binding site (RBS) that recruits ribosomes to start translation.
- mScarlet-I: the coding sequence (CDS) for the red fluorescent protein.
- B0015: a strong terminator that stops transcription.
How the DNA sequence is transcribed and translated into protein:
First, the DNA is transcribed into mRNA. RNA polymerase binds to the P_rprA promoter and synthesizes a complementary RNA copy of the mScarlet-I coding sequence. Transcription continues until the terminator B0015 is reached, which causes RNA polymerase to detach from the DNA.
Second, the mRNA is translated into protein. The ribosome binds to the B0034 RBS on the mRNA and begins scanning for the start codon (ATG). Once it finds the start codon, it reads the mRNA in groups of three nucleotides (codons). Each codon specifies one amino acid. The ribosome links these amino acids together to form the mScarlet-I polypeptide chain, which then folds into a functional red fluorescent protein.
Prepare a Twist DNA Synthesis Order
Linear map DNA design in Benchling
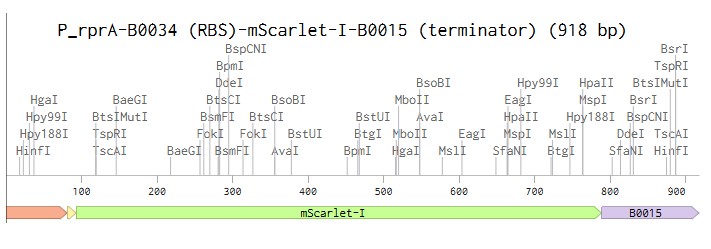
Linear map of the P_rprA-B0034-mScarlet-I-B0015 expression cassette (918 bp)
Gene design, vector selection and plasmid assembly
I selected the high-copy vector pTwist Amp High Copy with ampicillin resistance. After importing its GenBank file into Benchling, I assembled the final plasmid by inserting my construct P_rprA-B0034-mScarlet-I-B0015 into the vector using Gibson Assembly. The resulting circular plasmid pTwist_Amp_P_rprA-mScarlet-I_final (3139 bp) is ready for transformation into E. coli and expression of the red fluorescent protein under the control of the RcsB-dependent promoter.
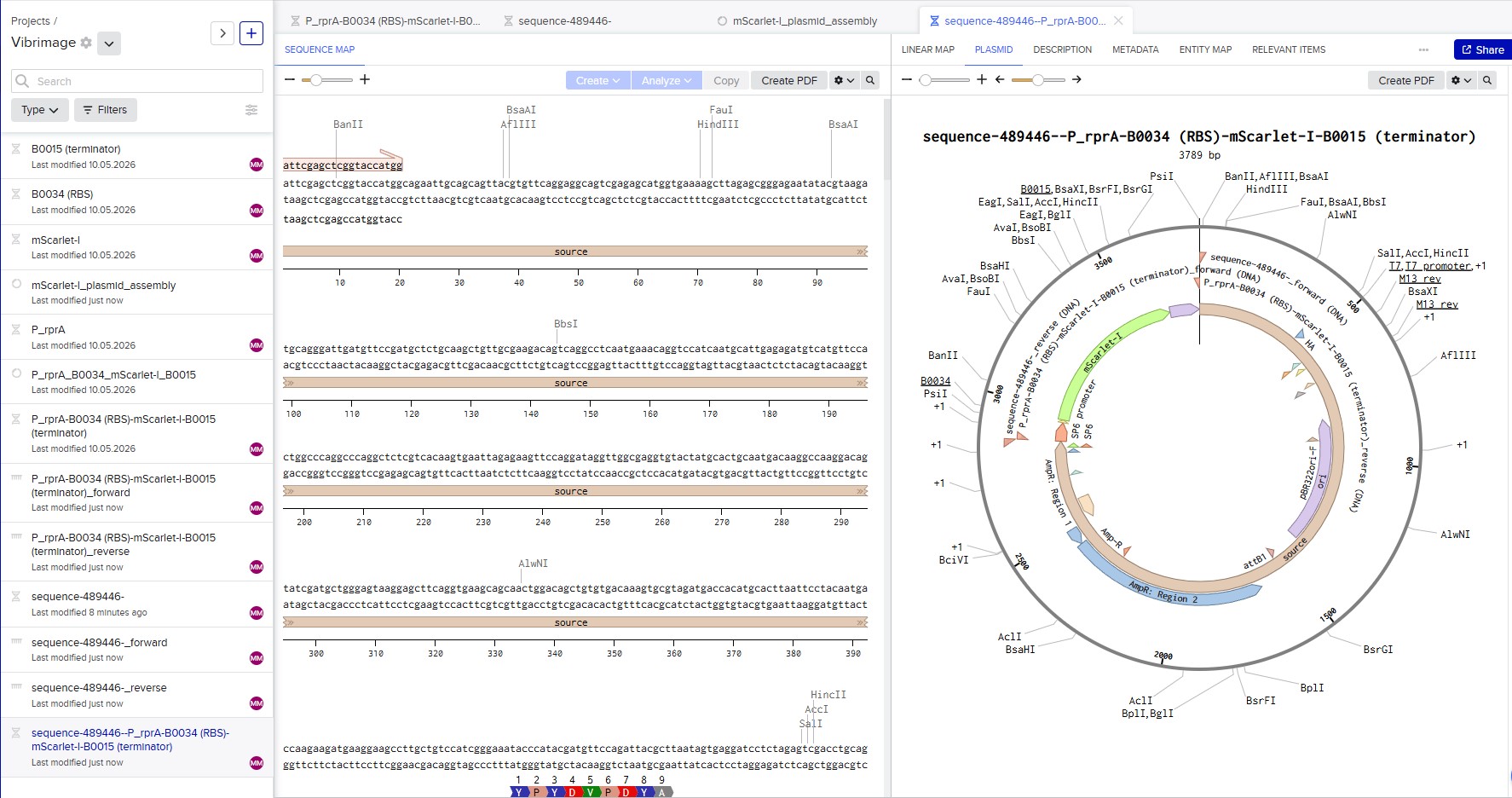
Circular map of the assembled expression plasmid pTwist_Amp_P_rprA-mScarlet-I_final (3139 bp)
DNA Read/Write/Edit
(i) What DNA would you want to sequence (e.g., read) and why?
I would like to sequence DNA reverse‑transcribed from mRNA (cDNA) of E. coli bacteria carrying my reporter construct P_rprA — B0034 — mScarlet‑I — B0015, before and after exposure to sound vibration. This would help me understand how mechanical oscillations affect gene expression, including activation of the RcsB pathway, and use sound as a non‑invasive tool to control recombinant protein expression in biotechnology, for example by optimizing bacterial growth conditions based on the fluorescence signal.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Technology: I would use Sanger sequencing to verify my assembled plasmid.
- Is your method first-, second- or third-generation or other? How so?
Sanger sequencing is a first‑generation method. It is defined by sequencing one DNA fragment at a time using chain termination (dideoxynucleotides), unlike massively parallel sequencing used in second‑ and third‑generation methods.
- What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
The input is purified plasmid DNA and a specific primer that binds to the vector next to my insert. Preparation consists of mixing the DNA, primer, DNA polymerase, regular nucleotides (dNTPs) and fluorescently labelled terminators (ddNTPs).
- What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
- Denature double‑stranded DNA by heating.
- Anneal the primer to the single‑stranded template.
- Chain elongation: polymerase adds nucleotides. When a terminator (ddNTP) is incorporated, synthesis stops on that strand.
- This produces a set of fragments of different lengths, each ending with a known fluorescent terminator.
- Fragments are separated by size using capillary electrophoresis.
- A laser excites fluorescence and a detector records the colour of each fragment.
- A computer converts the colour into a nucleotide (A, T, G or C). This is called base calling.
- What is the output of your chosen sequencing technology?
The output is a chromatogram (a graph with peaks for each nucleotide) and a text file containing the DNA sequence (.ab1 or .fasta). This sequence can be aligned to the expected sequence in Benchling to confirm that the construct has no mutations.
DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
I would like to synthesise my reporter construct P_rprA — B0034 — mScarlet-I — B0015 (918 bp). It contains the RcsB‑dependent promoter P_rprA, the strong ribosome binding site B0034, the mScarlet‑I red fluorescent protein gene and the B0015 terminator. This construct will make E. coli bacteria sensitive to sound vibration – when the RcsB pathway is activated, the bacteria will glow red. In the future, such a biosensor could be used for non‑invasive monitoring of bioreactors or for studying bacterial mechanosensitivity.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
To synthesise my construct, I would use a combination of solid‑phase chemical synthesis (phosphoramidite method) to make short oligonucleotides and Gibson assembly to build the full gene.
- What are the essential steps of your chosen sequencing methods?
- Sequence design and codon optimization
- Split the gene into short oligonucleotides (40–60 nt)
- Chemical oligonucleotide synthesis (cycles: deprotection, coupling, capping, oxidation)
- Assemble oligonucleotides into double‑stranded DNA by PCR
- Assemble fragments into a plasmid using Gibson assembly (exonuclease, polymerase, ligase)
- Clone into a vector and verify by sequencing
- What are the limitations of your synthesis method in terms of speed, accuracy, scalability?
- Accuracy: Error rate increases with gene length; GC‑rich and repetitive sequences are difficult to synthesise
- Speed: A full synthesis and assembly cycle takes 2–4 weeks
- Scalability: Thousands of genes can be ordered at once, but synthesising a whole genome (millions of bp) is very expensive and requires a library approach
DNA Edit
(i) What DNA would you want to edit and why?
Within my project, I would like to edit my own construct P_rprA — B0034 — mScarlet-I — B0015 in two ways:
- Replace the P_rprA promoter with a stronger or differently inducible promoter (e.g., a promoter that responds to a different signal instead of only the RcsB pathway). This would allow me to use the same reporter system to study different stress pathways in bacteria.
- Edit the mScarlet-I gene to obtain variants with different fluorescence colours (e.g., change a few key amino acids to shift the spectrum from red to yellow or blue). This would enable the creation of multicolour reporters for simultaneously monitoring several promoters. Why this is useful:
- Improve biosensor sensitivity (stronger promoter → brighter signal).
- Create a toolkit for multiplex analysis (different colours for different signals).
- Adapt the construct for other organisms or conditions.
(ii) What technology or technologies would you use to perform these DNA edits and why?
I would use PCR‑based site‑directed mutagenesis because it is suitable for introducing point mutations or small changes into an already assembled plasmid.
- How does your technology of choice edit DNA? What are the essential steps?
- Design two primers containing the desired mutation
- PCR that amplifies the entire plasmid
- DpnI treatment to digest the original template plasmid
- Transform bacteria and isolate the edited plasmid
- What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
- Plasmid DNA (template)
- Two mutagenic primers
- High‑fidelity DNA polymerase
- DpnI enzyme
- Competent E. coli cells
- What are the limitations of your editing methods (if any) in terms of efficiency or precision?
- Efficiency is 50–80%, depending on primer quality
- Works only for short changes (up to 20–30 bp)
- GC‑rich regions and long repeats are difficult to amplify