Week 4 HW:Protein Design Part I
Protein Design I
Part A. Conceptual Questions
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
The average molecular weight of an amino acid is ~100 g/mol. In 500 g of meat (if we consider all meat as amino acids) there are 5 moles, which gives 5 × 6 × 10²³ = 3 × 10²⁴ molecules. However, meat is not entirely protein: protein makes up only about 20% of the mass, i.e., 100 g of protein per 500 g of meat. After protein breakdown, this corresponds to 1 mole of amino acids, that is 6 × 10²³ molecules.
- Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Because meat proteins are broken down into individual amino acids in the stomach and intestines. These amino acids are absorbed into the bloodstream and used by the body to synthesize human proteins according to the instructions encoded in human DNA. Foreign DNA is not integrated into our genome during digestion.
- Why are there only 20 natural amino acids?
20 amino acids are an evolutionarily fixed optimum between chemical diversity, translational fidelity and mutational robustness, realised within the 64 codons of the genetic code.
- Where did amino acids come from before enzymes that make them, and before life started?
Amino acids were synthesised abiotically from simple molecules under the action of energy (electrical discharges, UV radiation, heat from hydrothermal vents), and were also delivered to Earth by meteorites — long before enzymes and life appeared.
- If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
An α-helix made of D-amino acids would be left-handed. Natural proteins made of L-amino acids form right-handed α-helices. D-amino acids are the mirror image of L-forms, so the helix will also be the mirror image and therefore left-handed.
- Can you discover additional helices in proteins?
Yes, proteins contain other types of helices besides the α-helix.
- Why are most molecular helices right-handed?
Most molecular helices are right-handed because they are built from L-amino acids. The chirality of L-amino acids makes the right-handed helix energetically more favourable.
- Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheets tend to aggregate because their edges have unpaired hydrogen bonds that seek to form bonds with other β-sheets. The main driving force is the hydrophobic effect: the side chains of amino acids (especially hydrophobic ones such as valine, leucine, isoleucine, phenylalanine) are exposed on the sheet surface and, to avoid water, tend to stick to the hydrophobic surfaces of other sheets. Inter‑sheet backbone hydrogen bonds also contribute.
- Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Misfolded proteins easily form β-sheets, which stick together into strong fibres (fibrils). These fibres are not broken down in the body and accumulate in tissues, causing disease. Amyloid β‑sheets can be used as materials due to their strength, stability, and biocompatibility.
Part B: Protein Analysis and Visualization
- Briefly describe the protein you selected and why you selected it.
I chose the mScarlet protein (a red fluorescent protein), because its mutant form mScarlet-I is directly used in my project as a reporter in the construct P_rprA — B0034 — mScarlet-I — B0015. This protein matures quickly, has high brightness and stability, making it convenient for imaging. For 3D structure analysis, I will use the mScarlet structure (PDB ID 5LK4), since mScarlet-I differs from it by only one amino acid (T74I) and has an almost identical fold.
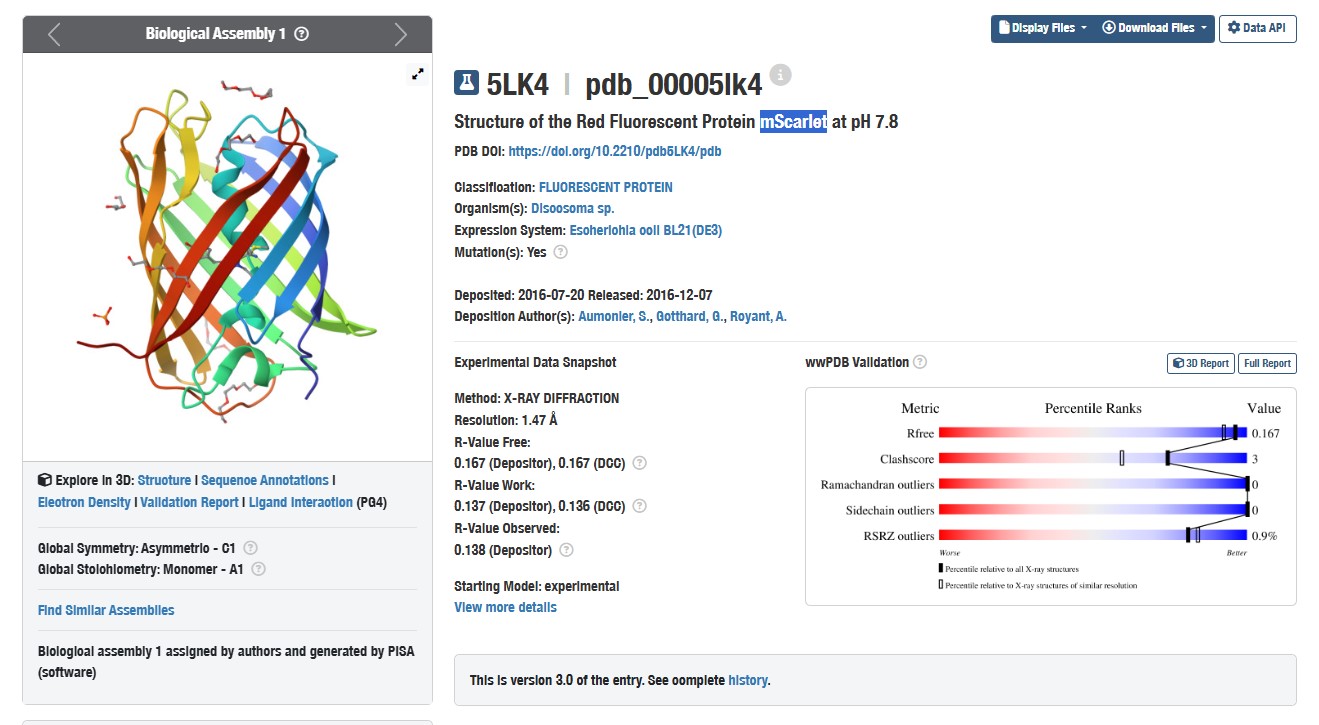
Structure of mScarlet (PDB 5LK4)
- Identify the amino acid sequence of your protein.
- How long is it? What is the most frequent amino acid?
The mScarlet protein has a chain length of 219 amino acids (in the structural model PDB 5LK4). The most frequent amino acid is leucine (Leu, L).
- How many protein sequence homologs are there for your protein?
According to the BLAST search against the UniProtKB database, the mScarlet protein has 225 homologs. This includes natural fluorescent proteins as well as engineered variants.
- Does your protein belong to any protein family?
mScarlet belongs to the GFP‑like fluorescent protein family (Pfam: PF01353). This family includes proteins that share a similar β‑barrel fold and chromophore formation mechanism. Within this family, it is classified as a red fluorescent protein (RFP) subfamily member.
- Identify the structure page of your protein in RCSB
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
The structure was determined in 2016. The resolution of 1.47 Å is good quality
Are there any other molecules in the solved structure apart from protein?
Yes, the structure contains PEG molecules and phosphate ions, which were used during crystallisation.
Does your protein belong to any structure classification family?
Yes. Structurally, mScarlet belongs to the GFP‑like fluorescent protein family (characteristic β‑barrel fold).
- Open the structure of your protein in any 3D molecule visualization software
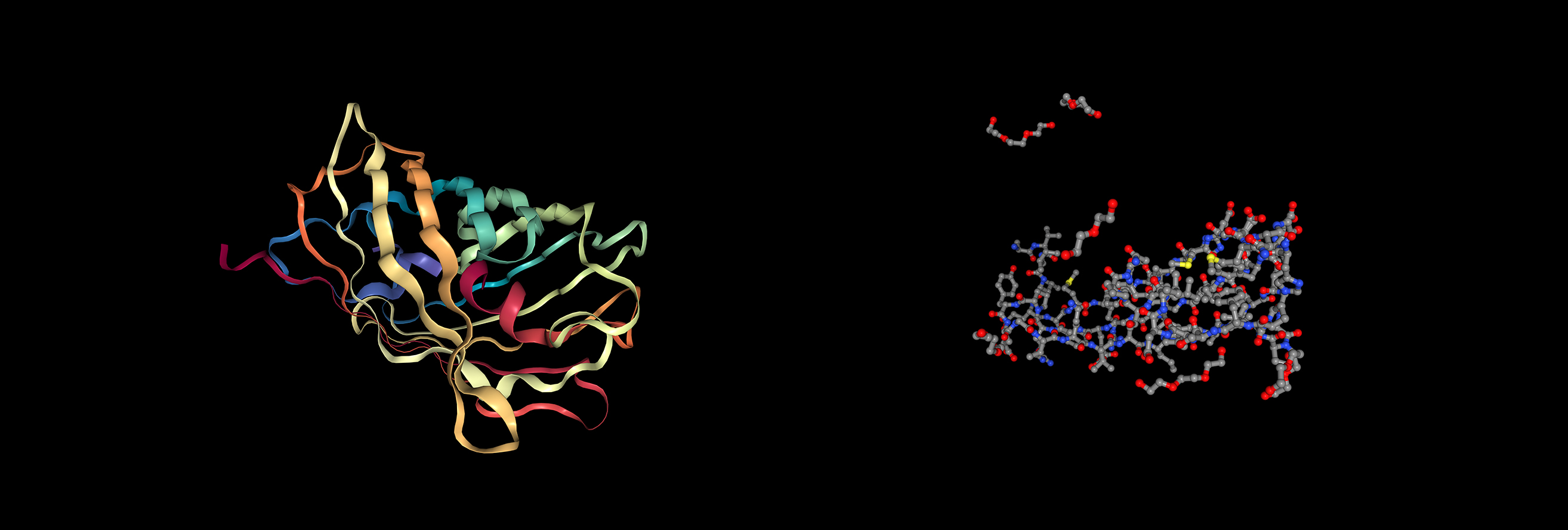
Visualization of the protein as “cartoon”, “ribbon” and “ball and stick”
The protein has more β-sheets (β-sheets dominate over α-helices), which is typical for the GFP‑like family with a β‑barrel fold.
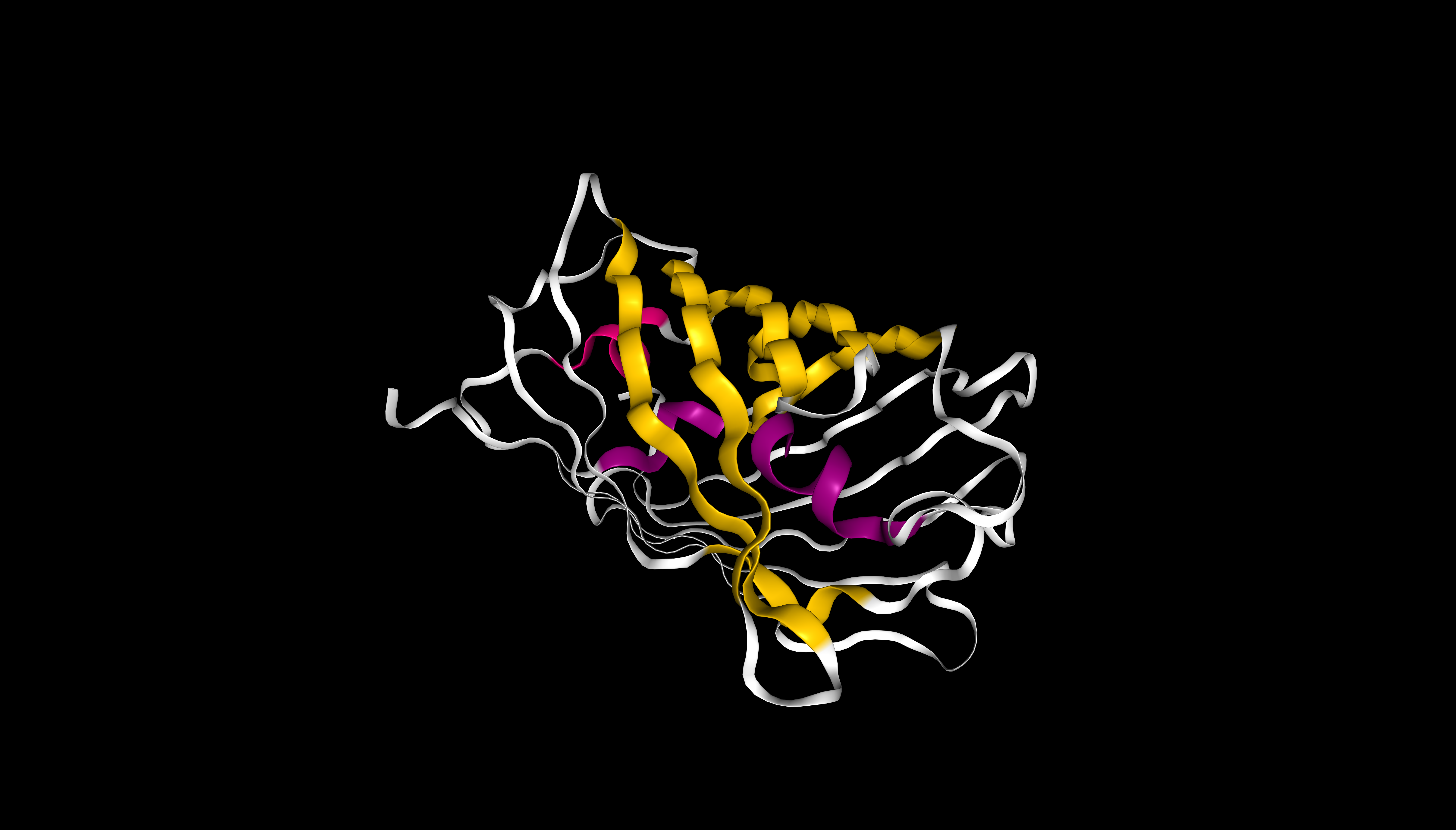
mScarlet coloured by secondary structure (sstruc). β-sheets are shown in yellow, α-helices in pink. β-sheets visually dominate, consistent with the β-barrel fold of this protein
The protein contains predominantly hydrophobic residues (beige/orange), which form the hydrophobic core. Hydrophilic residues (purple, magenta, turquoise) are scarce and are likely located on the surface, ensuring the protein’s solubility.
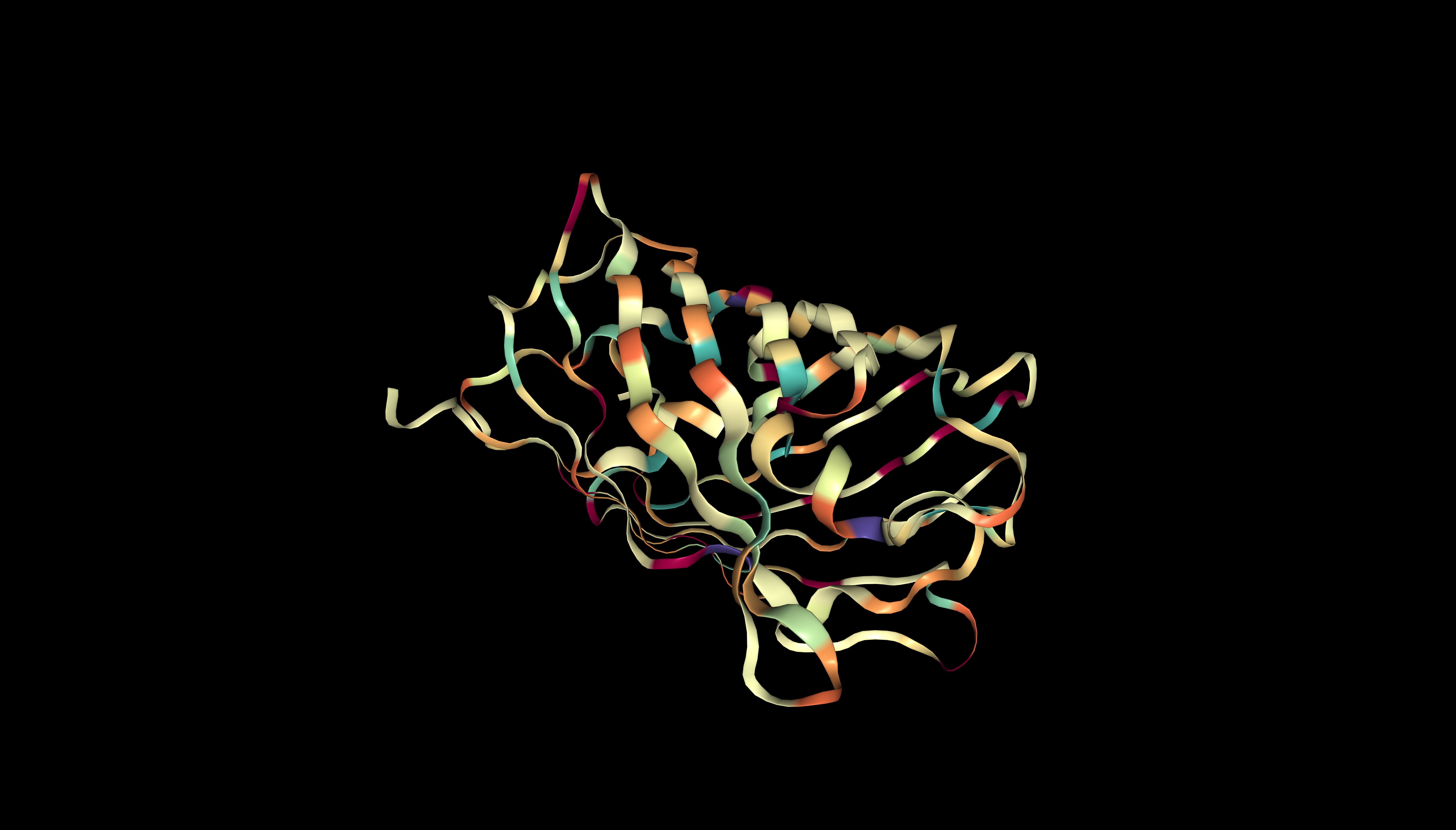
mScarlet coloured by hydrophobicity
The surface of mScarlet does not contain deep holes (binding pockets). Only minor irregularities are visible.
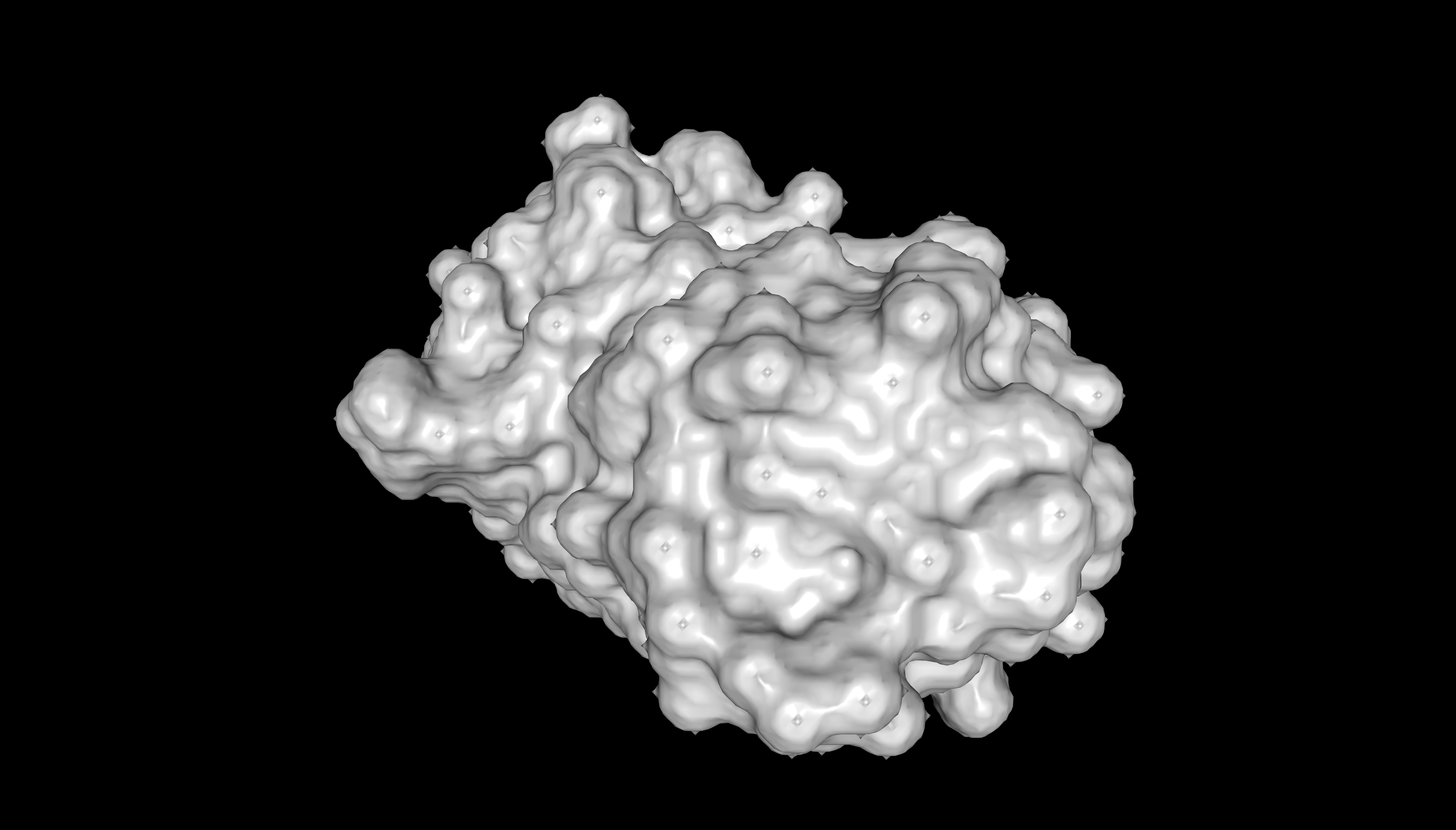
Surface representation of mScarlet (PDB 5LK4)
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
Deep Mutational Scans
a) Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods. Using ESM2, I generated a mutation tolerance heatmap for mScarlet.
protein_sequence: MVSKGEAVIKEFMRFKVHMEGSVNGHEFEIEGEGEGRPYEGTQTAKLKVTKGGPLPFAWDILSPQFMYGSKAYVKHPADIPDFLKLSFPEGFTWERVTRYEDGGVLTATQDTSLQDGCLIYNVKIRGVNFPSNGPVMQKKTLGWEAFTETLYPADGGLEGRNDMALKLVGGSHLIANIKTTYRSKKPAKNLKMPGFYFVDRRLERIKEADKETYVEQHEMAVARYCDLPSKLGHKLN
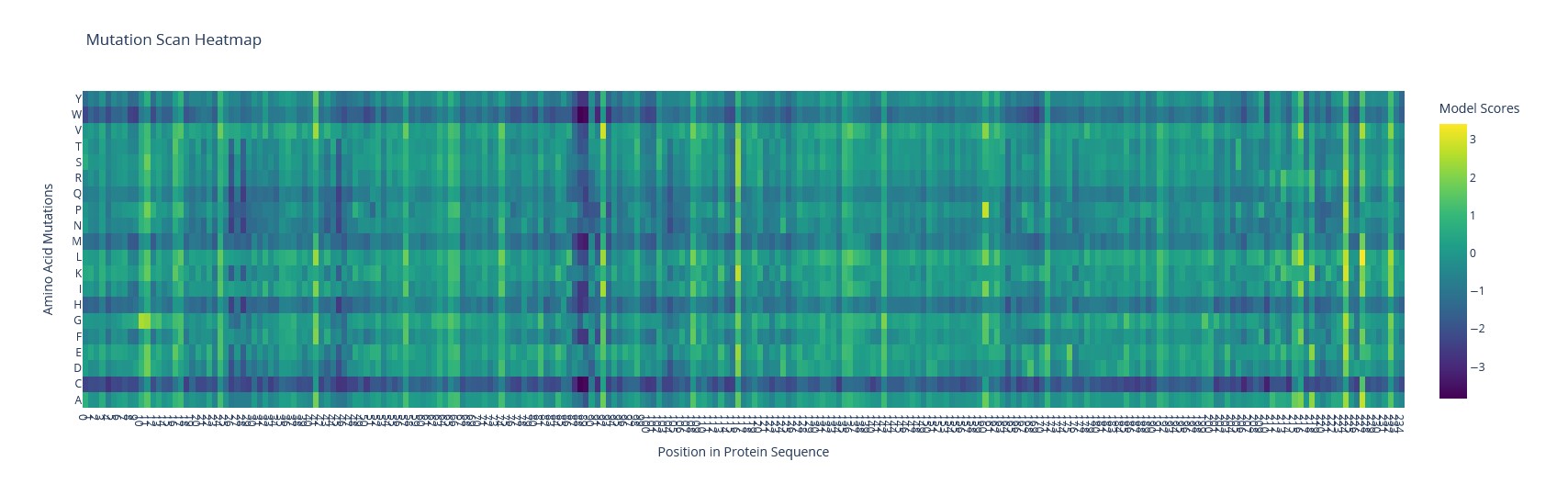 Mutation scanning heatmap of mScarlet
Mutation scanning heatmap of mScarlet
b) Can you explain any particular pattern?
The brightest spot is the T74I substitution (threonine → isoleucine) – a known mutation that accelerates maturation of mScarlet-I.
Latent Space Analysis
a) I built a 3D protein map using t‑SNE.
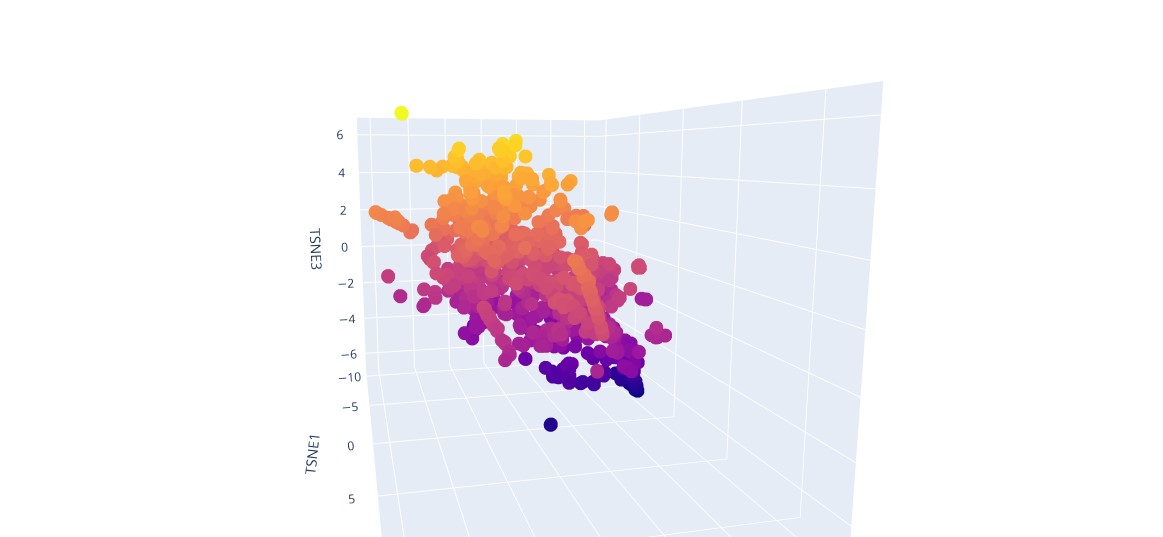
t‑SNE visualisation of proteins
b) Analyze the different formed neighborhoods: do they approximate similar proteins?
In latent space, proteins with similar sequences and structures form clusters.
c) Place your protein in the resulting map and explain its position and similarity to its neighbors.
The protein is located in the region corresponding to its structural family and clusters with homologous proteins that share similar sequence and three‑dimensional fold.
C2. Protein Folding
Folding a protein
- Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Using ESMFold, a structure of a 237 amino acid protein was predicted. The quality metrics are: plddt = 41.5, ptm = 0.376. These values indicate low confidence in the predicted folding: for plddt values below 70 the structure is considered unreliable. The color scheme of the structure is as follows: blue and green regions correspond to high confidence, while yellow and red regions indicate low confidence. The image shows a predominance of warm colors (orange, red), confirming low model confidence in these protein regions. The predicted coordinates are therefore unlikely to match the original (experimental) structure.
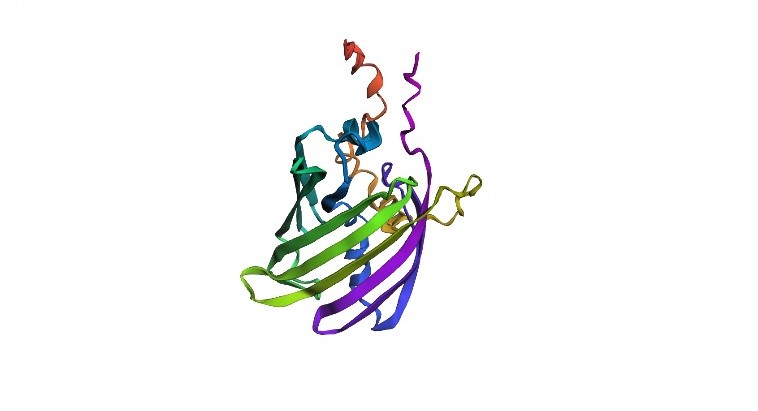
ESMFold‑predicted structure of mScarlet protein
- Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
To test the stability of the protein structure to mutations, a segment was deleted from positions 80 to 100 in the middle of the sequence, which is likely functionally important. The pLDDT and ptm values further decreased (pLDDT = 40.9, ptm = 0.326), indicating an additional decline in model confidence and reduced structural stability. After this large mutation, the model predicted significant structural changes: there was an increase in regions with low confidence (yellow and red), and the overall structure became less stable. This indicates that the protein structure is not stable to major mutations—removing a central segment seriously affects proper folding.
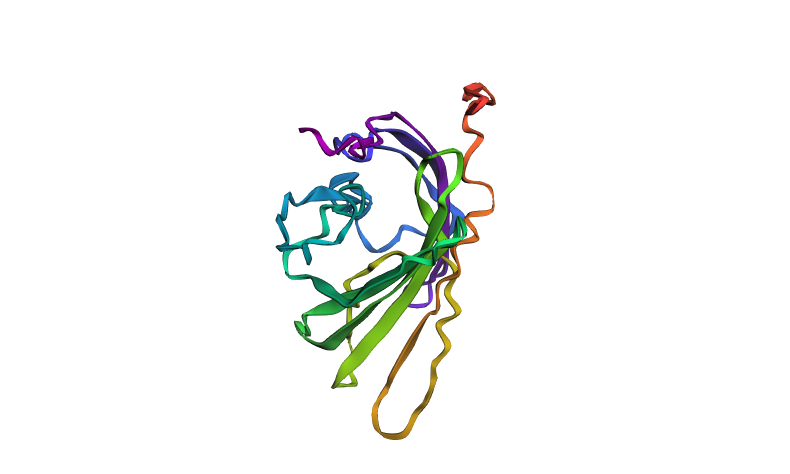
Predicted structure of the mutant mScarlet protein after central segment deletion (positions 80–100)
C3. Protein Generation
- Analysis of the predicted sequence probabilities shows that, for most positions, ProteinMPNN displays a clear preference for a single amino acid (high probability), indicating structural constraints. At some positions, the probabilities are more evenly distributed among different amino acids, reflecting variability. The predicted sequence generally matches the original in high-confidence regions, but there are differences at positions with lower confidence.
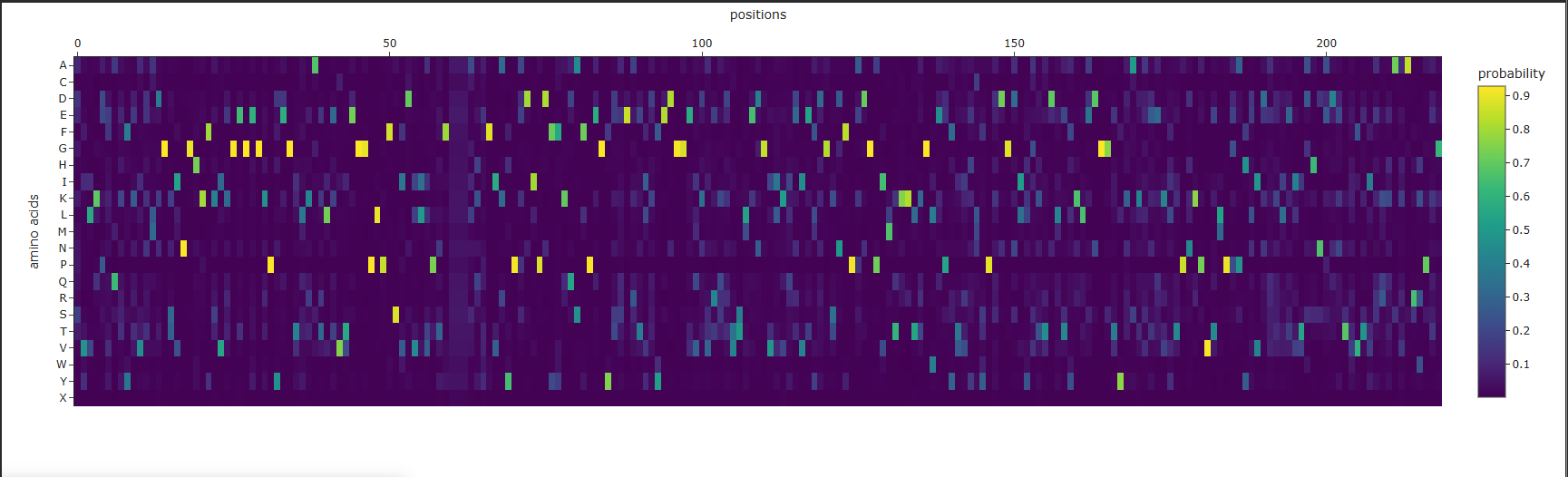
Amino acid probability map for mScarlet
- As a result of inputting the ProteinMPNN-designed sequence into ESMFold, a structure of 219 amino acids was predicted with pLDDT = 71.5 and ptm = 0.734, which is significantly higher than the original (pLDDT ~41). However, the predicted structure appears visually quite different from the original: the arrangement of secondary structure elements and the overall spatial organization have changed noticeably. This suggests that changes in the sequence led to a new fold, despite the model’s high confidence in the predicted structure.
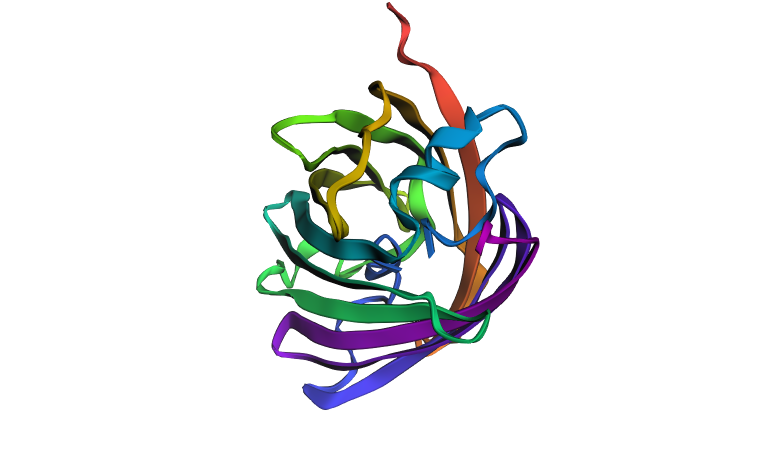
Predicted structure of the ProteinMPNN-designed sequence using ESMFold
Part D. Group Brainstorm on Bacteriophage Engineering
Goal
Improve the thermal stability and proper folding of the MS2 phage lysis protein L, with the aim of increasing phage burst size and overall production titer. A more stable lysis protein will function more efficiently, leading to synchronized and effective bacterial lysis, and thus higher phage yield.
Tools
- Protein Language Models (pLMs) for in silico mutagenesis, specifically ESM2 (as used in the previous assignment) to perform deep mutational scanning of the L protein.
- AlphaFold‑Multimer to validate that the selected mutations do not disrupt the L‑DnaJ chaperone complex, which is essential for the lysis function.
- Yeast surface display for high‑throughput experimental screening of the most promising variants from the computational library.
Why these tools can help solve the chosen task
- ESM2 learns evolutionary constraints from millions of protein sequences. It can predict which single‑amino‑acid substitutions are likely tolerated or even beneficial for stability without any experimental data. This allows us to rapidly filter thousands of mutations in silico.
- AlphaFold‑Multimer can model the 3D structure of the L‑DnaJ complex. Even if direct interaction evidence is not fully established, the model can flag mutations that would disrupt binding, saving us from testing non‑functional variants experimentally.
- Yeast display is a powerful, established method to screen libraries of protein variants (100 to 10⁶ members) for increased stability. It directly couples protein stability to a fluorescence readout, enabling selection of the best designs.
Potential challenges
- Lack of training data for phage‑bacteria interactions. AlphaFold‑Multimer may give unreliable predictions for the L‑DnaJ complex if no similar complexes are present in its training set. The direct interaction between L and DnaJ has not been strictly proven, adding uncertainty.
- Unpredicted in vivo behaviour. Mutations that stabilise the protein in a test tube (e.g., on the yeast surface) might not translate to the complex intracellular environment of E. coli. For example, the protein might become too stable and resist timely degradation, disrupting the delicate timing of lysis.
 Workflow of L‑protein design
Workflow of L‑protein design