Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
- The amino acid sequence of the human SOD1 protein was taken from the UniProt database (ID: P00441), and the A4V mutation was introduced.
Amino acid sequence of the human SOD1 protein from UniProt (ID: P00441):
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
The A4V mutation was introduced:
MVTKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Four peptides of 12 amino acids in length were generated based on the mutant SOD1 gene sequence.
The SOD1‑binding peptide FLYRWLPSRRGG was added to the generated list. PepMLM successfully generated four candidate peptides for binding to the SOD1 A4V mutant.
| index | Binder | Pseudo Perplexity |
|---|---|---|
| 1 | HLYYPAALRHKX | 13.094736350750331 |
| 2 | HRYVAAXLRWKE | 21.045882766316033 |
| 3 | HLYPAAAIELKX | 11.060795760122568 |
| 4 | WLYPVVXVEWKX | 17.854789099765938 |
| 5 | FLYWRLPSRRGG | 19.011551773463413 |
The pseudo‑perplexity values (from 11.06 to 21.05) are comparable to the known peptide FLYWRLPSRRGG (19.01), and the best candidate, HLYPAAAIELKX, shows the highest predicted affinity (11.06).
Part 2: Evaluate Binders with AlphaFold3
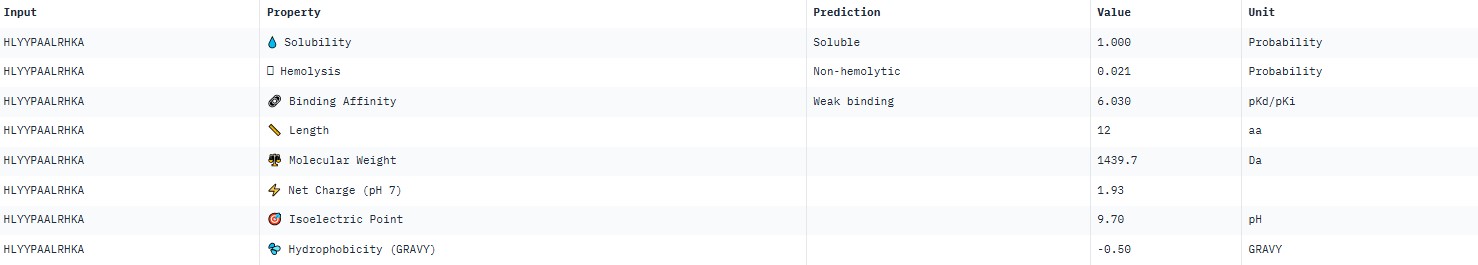
For peptide HLYYPAALRHKA (where the undefined position X was replaced by alanine), AlphaFold predicted a complex with SOD1 A4V giving ipTM = 0.4, which indicates low prediction confidence. The peptide likely does not bind specifically; visually it appears to be located on the protein surface, without interacting with the N‑terminal region containing the A4V mutation, the dimer interface or the β‑barrel region. This candidate underperforms expectations and does not surpass the known peptide (modelling of the latter is needed for direct comparison).
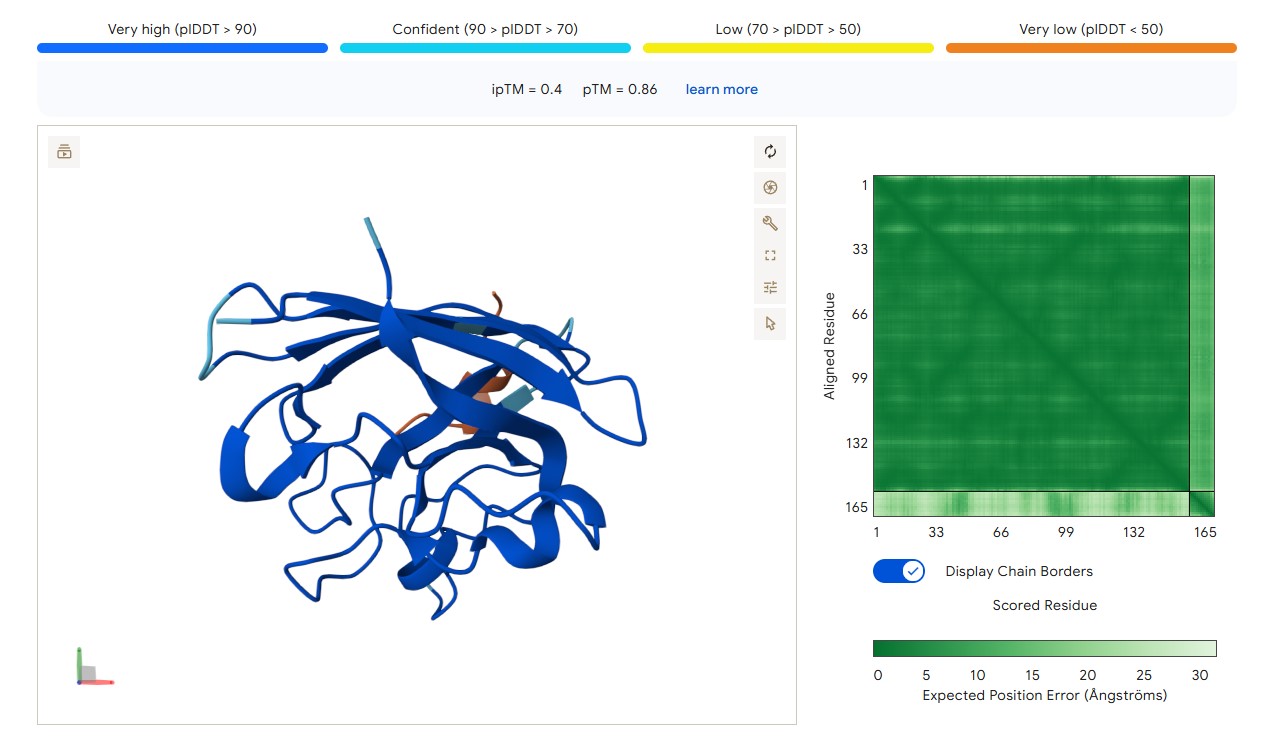
protein-peptide complex SOD1 A4V + HLYYPAALRHKA
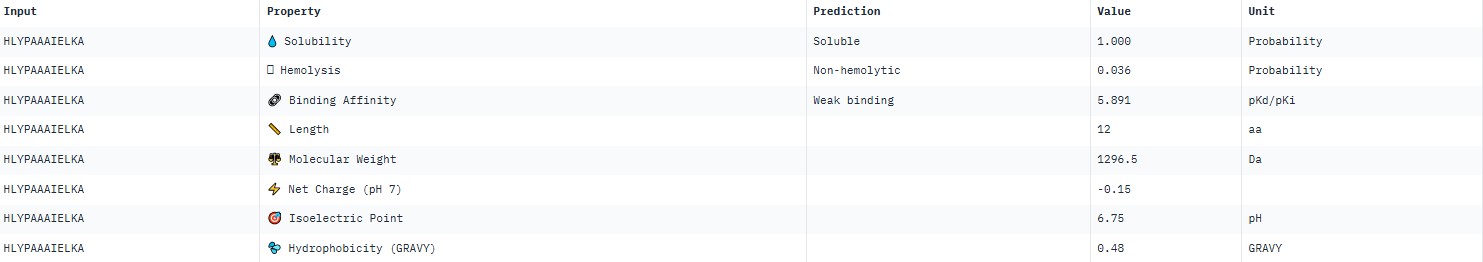
For peptide HLYPAAAIELKA (where the undefined position X was replaced by alanine), AlphaFold predicted a complex with SOD1 A4V giving ipTM = 0.31, which indicates very low prediction confidence (well below the 0.7 threshold). The peptide likely does not bind specifically; visually it appears to be located on the protein surface, without interacting with the N‑terminal region (where the A4V mutation resides), the dimer interface or the β‑barrel region.
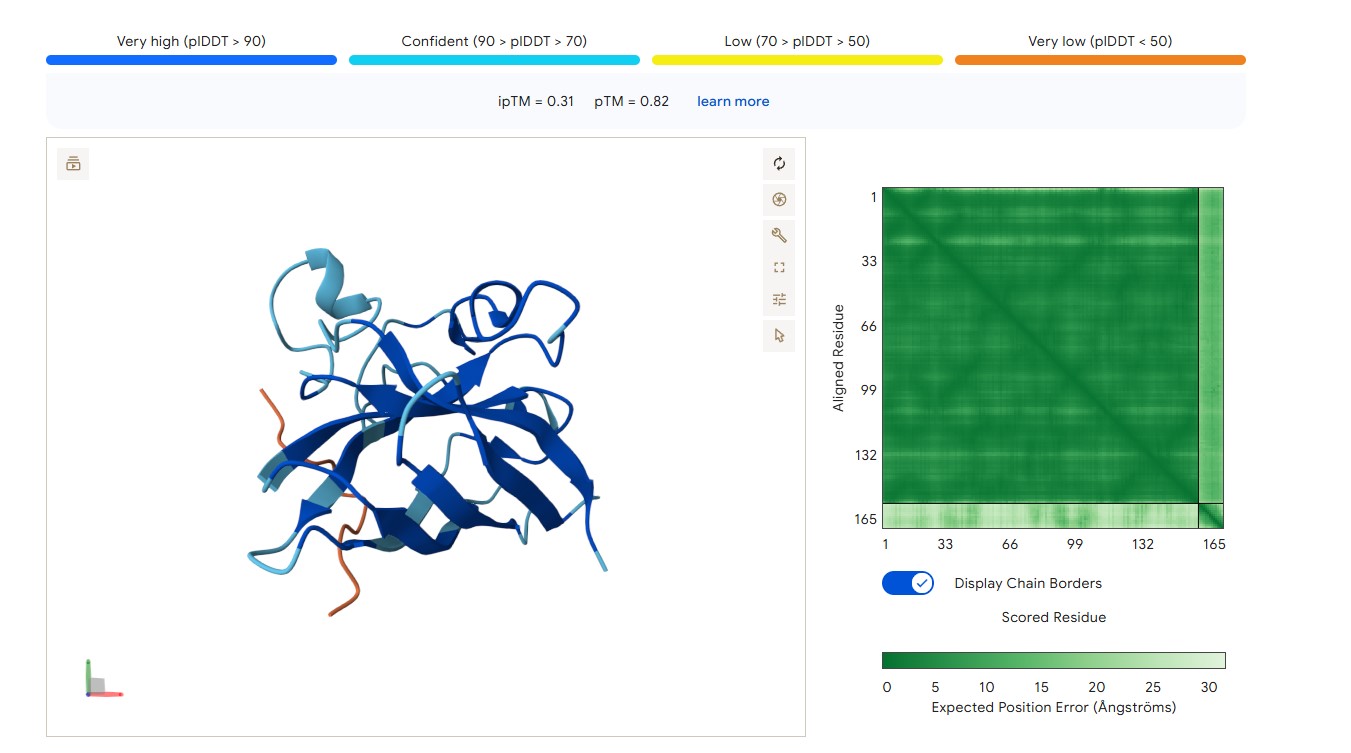
protein-peptide complex SOD1 A4V + HLYPAAAIELKA
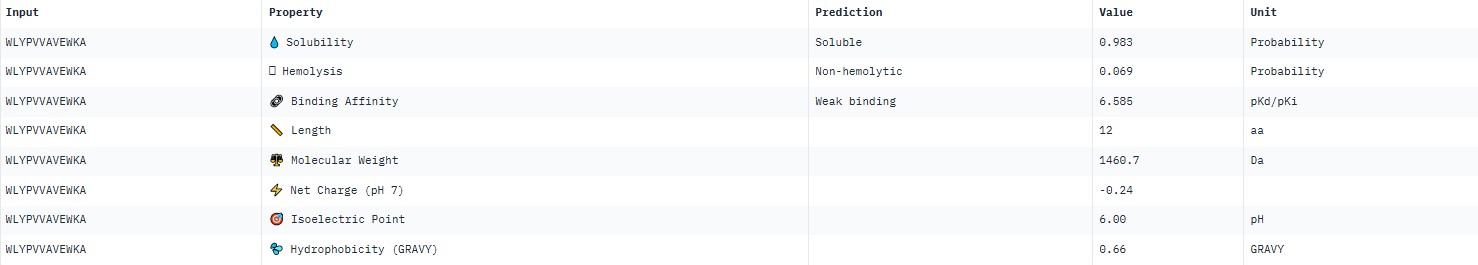
For peptide WLYPVVAVEWKA (where the undefined position X was replaced by alanine), AlphaFold predicted a complex with SOD1 A4V giving ipTM = 0.34, which indicates very low prediction confidence (well below the 0.7 threshold). The peptide likely does not bind specifically; visually it appears to be located on the protein surface, without interacting with the N‑terminal region (where the A4V mutation resides), the dimer interface or the β‑barrel region.
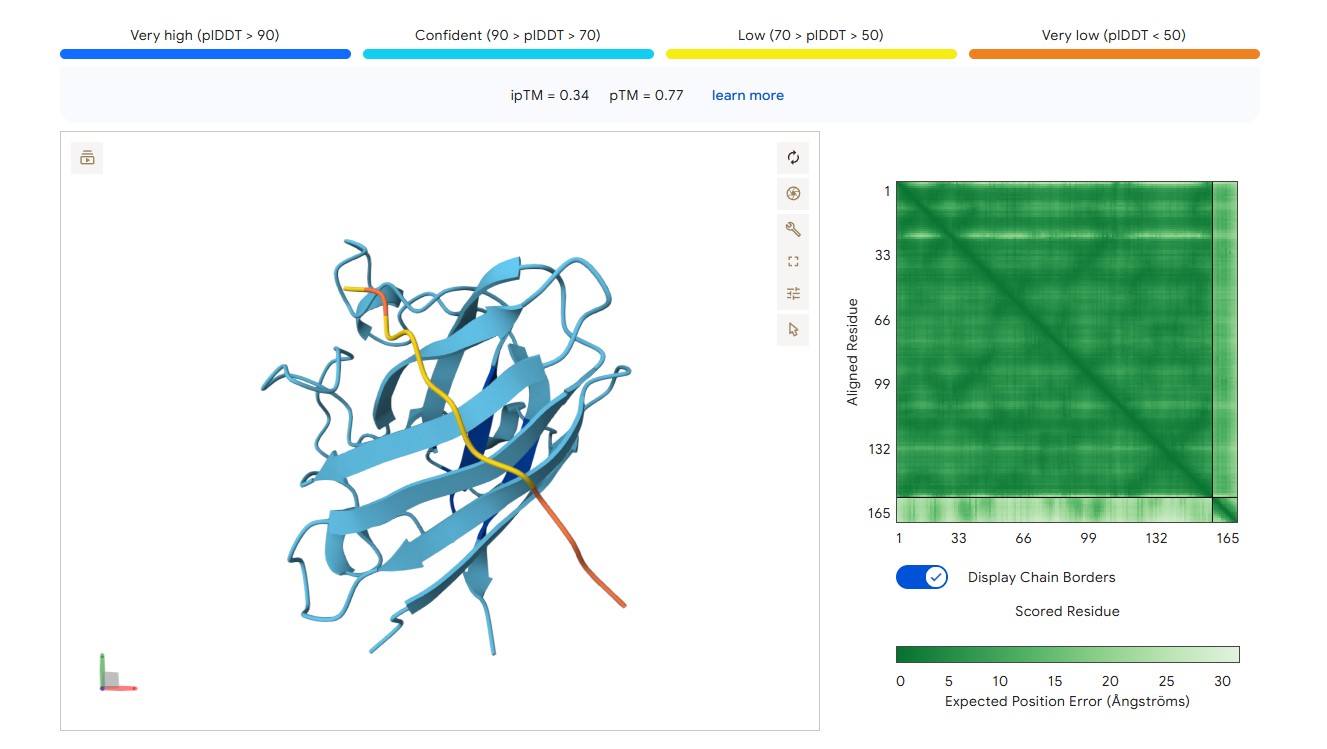
protein-peptide complex SOD1 A4V + WLYPVVAVEWKA
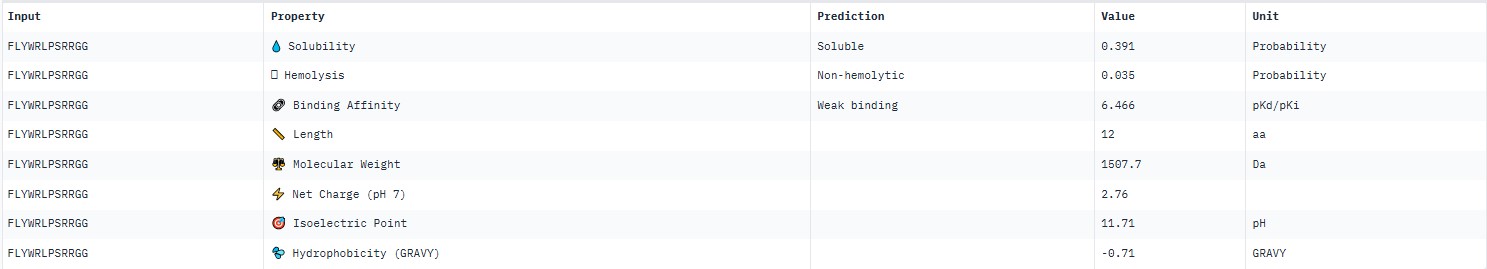
For the known peptide FLYWRLPSRRGG, AlphaFold predicted a complex with the SOD1 A4V mutant giving ipTM = 0.36, which indicates very low prediction confidence (well below the 0.7 threshold). The peptide likely does not bind specifically; visually it appears to be located on the protein surface, without interacting with the N‑terminal region (where the A4V mutation resides), the dimer interface or the β‑barrel region.

protein-peptide complex SOD1 A4V + FLYWRLPSRRGG
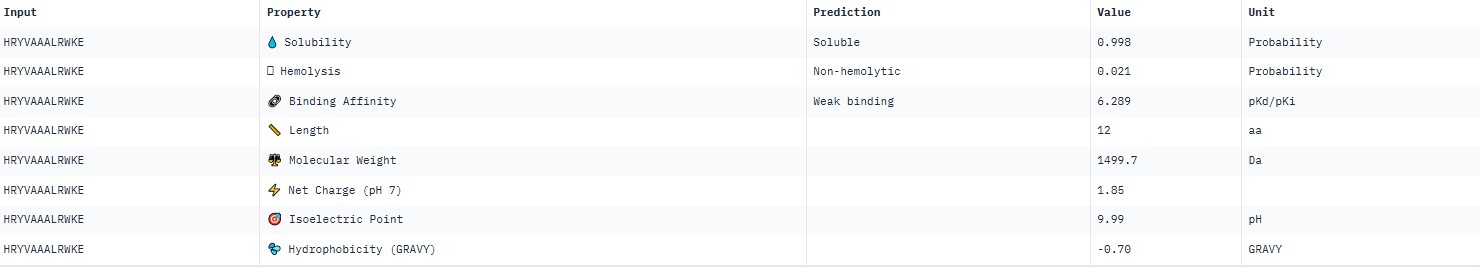
For peptide HRYVAAALRWKE (where the undefined position X was replaced by alanine), AlphaFold predicted a complex with SOD1 A4V giving ipTM = 0.23, which indicates very low prediction confidence (well below the 0.7 threshold). The peptide likely does not bind specifically; visually it appears to be located on the protein surface, without interacting with the N‑terminal region (where the A4V mutation resides), the dimer interface or the β‑barrel region.
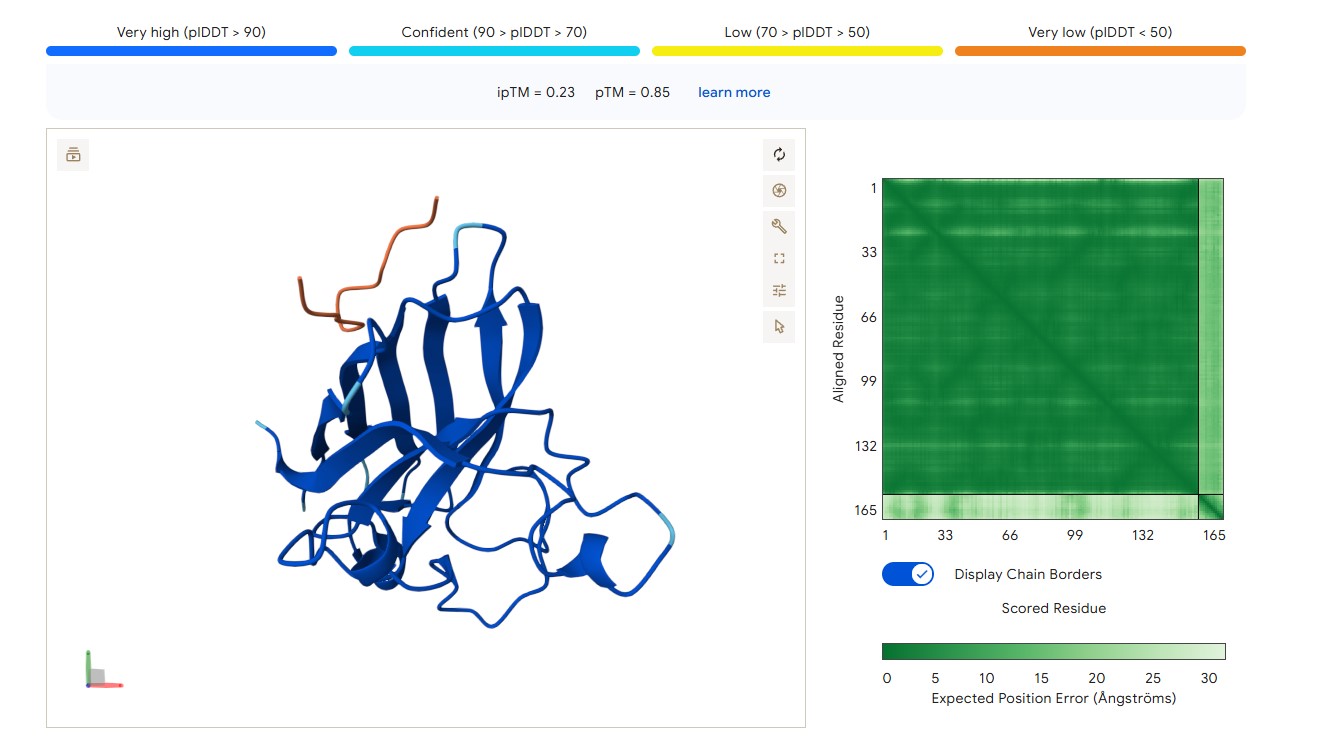
protein-peptide complex SOD1 A4V + HRYVAAALRWKE
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
AlphaFold3 gave very low ipTM values (<0.7) for all peptides, indicating a lack of confidence in specific binding. The highest ipTM (0.40) was for peptide HLYYPAALRHKA, and the lowest (0.23) for HRYVAAALRWKE.
PeptiVerse predicted “weak binding” for all peptides, with pKd values in a narrow range of 5.89–6.59. The highest affinity (6.585) was for WLYPVVAVEWKA, and the lowest (5.891) for HLYPAAAIELKA.
Hemolysis and solubility: all peptides were predicted to be non-hemolytic (probability <0.07) and soluble (probability >0.39). Even the lowest solubility value for FLYWRLPSRRGG (0.391) still falls into the “soluble” category. Thus, among these weak binders, there are neither hemolytic nor poorly soluble peptides – all of them have good therapeutic profiles in these parameters.
Although all peptides bind weakly, WLYPVVAVEWKA shows the highest predicted affinity (6.585) together with excellent solubility (0.993) and low hemolysis (0.069). According to the PeptiVerse data, it is the best candidate. However, AlphaFold3 does not confirm specific binding (ipTM=0.34). If relying on PeptiVerse, this peptide is the best.
Part 4: Generate Optimized Peptides with moPPIt
In moPPIt, the mutant SOD1 A4V sequence was pasted, the motif (residues 1–10, the N‑terminus containing the A4V mutation) was selected, peptide length was set to 12 amino acids, and guidance for motif, affinity, solubility and hemolysis was enabled. Five candidate peptides were generated.
| № | Peptide | Hemolysis | Solubility | Affinity | Motif |
|---|---|---|---|---|---|
| 1 | CTGGLPVGVGAA | 0.0445 | 0.9808 | 6.2601 | 0.5489 |
| 2 | ADPEFAAPSCTH | 0.0279 | 1.0000 | 5.9598 | 0.5326 |
| 3 | ESEKQCVKTHFT | 0.0483 | 1.0000 | 6.0218 | 0.5422 |
| 4 | MAAGIFKKQKQK | 0.0145 | 1.0000 | 5.5228 | 0.6357 |
| 5 | QEPCEELQFNHF | 0.0245 | 1.0000 | 6.2746 | 0.5575 |
All candidates show low hemolysis (<0.05) and high solubility (>0.98). The best affinity is observed for QEPCEELQFNHF (6.27) and CTGGLPVGVGAA (6.26). Compared to PepMLM, moPPIt peptides are preferable for further development due to their targeted design and consideration of safety.
Part C: Final Project: L-Protein Mutants
Mu2 (F60W, L64I)
The mutation was selected to accelerate lysis: replacing phenylalanine with tryptophan (F60W) increases the hydrophobicity of the transmembrane helix, while replacing leucine with isoleucine (L64I) increases helix rigidity, promoting faster oligomerization and pore formation. This shortens the window for bacteria to develop resistance.
Rationale: Tryptophan increases hydrophobicity and membrane stability; replacing leucine with isoleucine makes the alpha helix more rigid, accelerating oligomerization and pore formation. Faster lysis leaves less time for resistance to emerge.
Full sequence: METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKWNQILILLSLLEAVIRTVTLLQLLT
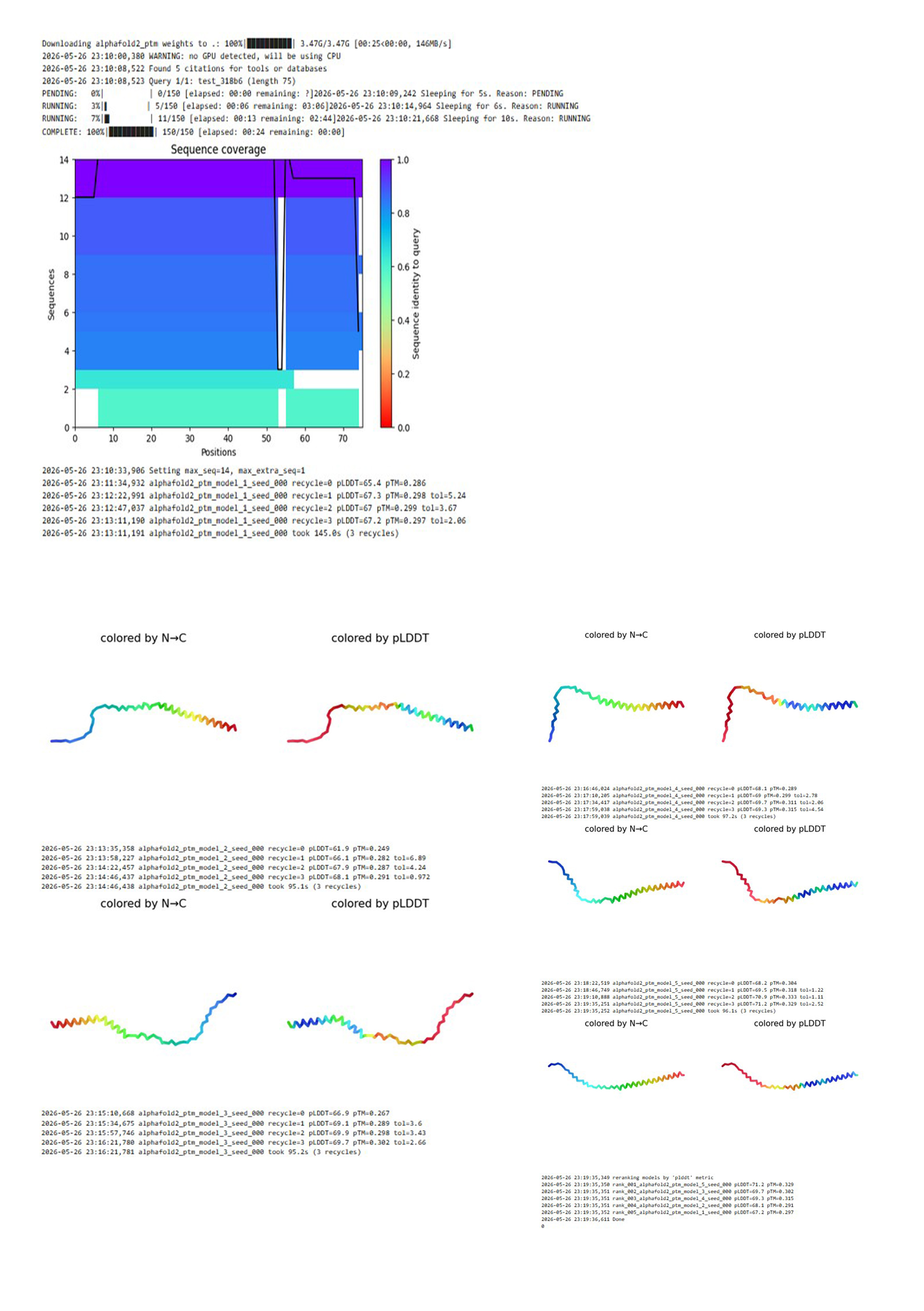
The mutant Mu2 sequence was submitted to ColabFold (AlphaFold2). The program performed homology search (MMseqs2), predicted 5 structural models, and ranked them by pLDDT.
ColabFold results for the Mu2 mutant (F60W, L64I)
AlphaFold2 predicted 5 independent models of the protein structure. After ranking by confidence (pLDDT), the best model, model_5, showed pLDDT = 71.2 and pTM = 0.329.
A low pTM (<0.5) indicates poor global reliability of the prediction. This is typical for small membrane proteins. Nevertheless, the pLDDT value of 71.2 suggests acceptable local confidence (e.g., for the transmembrane helix). The model does not show complete disruption of the structure.
Predicted structure of the Mu2 mutant (F60W, L64I) of MS2 lysis protein
Display 3D structure
The model has acceptable local confidence for the central part of the protein, but the global fold (pTM = 0.329) is unreliable. This is typical for membrane lytic proteins. The F60W and L64I mutations did not lead to complete structural degradation.
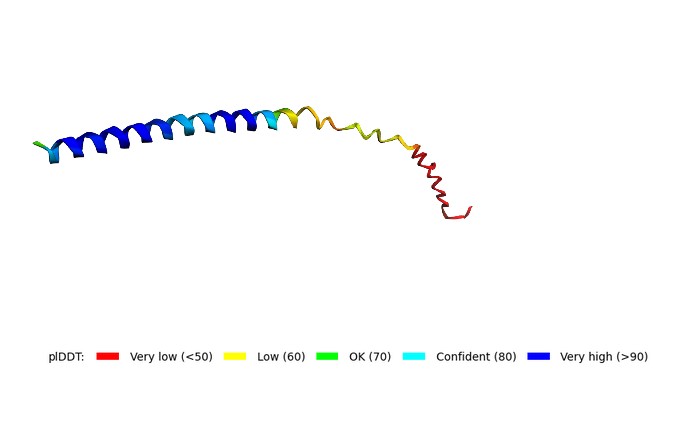
3D structure of the Mu2 mutant (F60W, L64I) of MS2 lysis protein predicted by ColabFold