My visit at one of the 2 fungi farms in Cyprus in 2023
First weeks assignment Describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
Geeking out over protein structures and data banks, DNA storage in plants, clouds and decoding DNA into sound I love that artist Antoine Bertin has decoded the RNA of SARS COV 2 into this track! check it out.
Antoine Bertin · Meditation on SARS-CoV-2 This is the RNA of the Coronavirus translated into sound (viruses are made of RNA, not exactly DNA). Each nucleotide of the RNA (A,U,G or C) is transformed into a note so the virus sequence can be heard. The tempo of the track follows the rhythm at which the epidemic is growing (exponential curve) and how this curve flattens if we all stay home :) I wanted to create a track that can help with relaxation in times of isolation, and meditate on the fact all life on earth, including viruses, are made of the same material. We (humans, animals, trees, bacteria, viruses) are the continuation of a same common ancestor. Anyway; I hope this will helps everyone explore in their own sonic way what we are going through! Here is an extract of the RNA sequence :)
[E-INK] MICROFLUIDICS <3 I have actually been interested in microfluidics in a while because I am into inflatables and soft robotics since 2020. I started working with bodily fluids and liquids in 2023. I love this little sweat collection and analysis wearable microfluidic system device.
You can find another example here and the paper.
What is protein design? Objective:
Learn basic concepts: +amino acid structure +3D protein visualization +the variety of ML-based design tools Brainstorm as a group how to apply these tools to engineer a better bacteriophage (setting the stage for the final project). Part A. INTUITIVE PART OF THE HOMEWORK! Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
Part A: SOD1 Binder Peptide Design (From Pranam) Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Acropora Millepora, Photo from Reefbuilders
This week we learn core molecular biology tools and techniques for processing and assembling DNA, including PCR and Gibson Assembly. Here is the updated HTGAA2026 Gibson assembly lab protocol document.
Homework PART A: PCR and DNA Assembly 1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? Phusion High-Fidelity PCR Master Mix is a 2X, ready-to-use mixture where the exact formulation is partly proprietary, but the functional components are documented in the manufacturer’s manual:
From the lecture with Ron Weiss "…central dogma, if you will, in synthetic biology, is the notion that almost everything that we build is based on sensing, processing, and actuation. So we want to be able to sense everything that's going on inside and outside the cell, have that information fed into some kind of controller, and have that regulate things that are going on in the cell". Week 7 Lab - Neuromorphic Circuits - Intracellular Artificial Neural Networks (IANNs) Download Neuromorphic Wizard.
This week introduces synthesis of proteins using cellular machinery outside of a cell. I LOVED THE LECTURE and I loved Kate Adamalas work.
We have to solve terrestrial problems before extraterrestrial problems…we need to shift away from a petroleum based bioeconomy…we need a paradigm shift. Kate Adamala <3 General homework questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell free expression is more beneficial than cell production. A cell-free system allows biological reactions to occur outside of living cells. By extracting and using cellular components like ribosomes, RNA polymerase, amino acids, and ATP, this method enables reactions in a controlled, simplified environment. Cell-free systems allow for the engineering, expression, and analysis of genetic constructs without the complexity of living cells.
WEEK 10 HW Homework is based on data that will be generated in the Waters Immerse Lab in Cambridge, MA. Students will be characterizing green fluorescent protein (eGFP, a recombinant protein standard) structure (primary, secondary/tertiary) in the lab using liquid chromatography and mass spectrometry. Data generated in the lab will be available on-line for Committed Listeners.
WEEK 11 HW This week examines how modern bioproduction pipelines, from strain engineering to fermentation and downstream processing, are increasingly designed, executed, and optimized through cloud lab platforms and automation — enabling remote, high-throughput, and reproducible synthetic biology at industrial scale.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork How it started….
Week 12 HW This week focuses on designing, synthesizing, and editing whole genomes, from minimal cells to refactored microbes and synthetic chromosomes.
WEEK 13 HW This week covers designing, programming, and fabricating engineered living materials — such as self-healing concretes, adaptive biofilms, and responsive biomaterials — by integrating genetic circuit design, materials science, and bioprocess engineering.
My visit at one of the 2 fungi farms in Cyprus in 2023
First weeks assignment
Describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals. Below is one example framework (developed in the context of synthetic genomics) you can choose to use or adapt, or you can develop your own. The example was developed to consider policy goals of ensuring safety and security, alongside other goals, like promoting constructive uses, but you could propose other goals for example, those relating to equity or autonomy.
Describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”). Try to outline a mix of actions (e.g. a new requirement/rule, incentive, or technical strategy) pursued by different “actors” (e.g. academic researchers, companies, federal regulators, law enforcement, etc). Draw upon your existing knowledge and a little additional digging, and feel free to use analogies to other domains (e.g. 3D printing, drones, financial systems, etc.).
Purpose: What is done now and what changes are you proposing?
Design: What is needed to make it “work”? (including the actor(s) involved - who must opt-in, fund, approve, or implement, etc)
Assumptions: What could you have wrong (incorrect assumptions, uncertainties)?
Risks of Failure & “Success”: How might this fail, including any unintended consequences of the “success” of your proposed actions?
Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
• By helping respond
Foster Lab Safety
• By preventing incident
• By helping respond
Protect the environment
• By preventing incidents
• By helping respond
Other considerations
• Minimizing costs and burdens to stakeholders
• Feasibility?
• Not impede research
• Promote constructive applications
Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties. For this, you can choose one or more relevant audiences for your recommendation, which could range from the very local (e.g. to MIT leadership or Cambridge Mayoral Office) to the national (e.g. to President Biden or the head of a Federal Agency) to the international (e.g. to the United Nations Office of the Secretary-General, or the leadership of a multinational firm or industry consortia). These could also be one of the “actor” groups in your matrix.
Reflecting on what you learned and did in class this week, outline any ethical concerns that arose, especially any that were new to you. Then propose any governance actions you think might be appropriate to address those issues. This should be included on your class page for this week.
An exploration into abandoned copper mines in Cyprus and the non existent restoration of toxic environments
I have been extremely interested in mycelium, plants and lichen. Lichen is a super queer and hybrid species that is a cross between fungi and algae. In recent years. Lichen are biosensors and bioindicators and some local plants are hyperaccumulators of heavy metals and they aid in bioremediation (through phytoremediation) and enviromental restoration of abandoned mines in Cyprus and other waste lands. Here you can find a conversation between the artist Helene Black and myself documenting my project We forgot how to forage as part of an artist residency. Helene Black is an artist, educator and cofounder of the interdisciplinary NGO, NeMe in Limassol, Cyprus. She has been researching abandoned copper mines and extractivism in Cyprus.
Here is a 3D scan of a cultivated petri dish. We tested the bodies of water of the river of abandoned Lefke mine.
As part of the Re(Grounding) program, myself and Ukrainian biohacker Dariia Dantseva of Yane lab completed a DIY biology workshop with a variety of groups of local citizens focusing on enviromental justice and restoration of abandoned copper mines through testing waters from Lefke mine river and Skouriotissa mine. We used readily available water test strips and then we proceeded with taking a water sample with a swab and trasfered it into the LB agar nutrient medium petri dishes. The participants used copper coins (british pennies and european cents) to test how resistant the existing micoorganisms in our water samples were to the copper inside the coin as as well as mixing samples from their microbiome (saliva, breath) with the contaminated water samples from the mines. The participants learned how to label their petri dishes, complete water pH tests with readily available test strips, and learned how to test fluids and swab them on petri dishes.
Lichen, plants and fungi for bioremediation, plastic degradation down plastics and monitoring enviromental changes and bioremediation
Alternatively, lichen and other endemic local plants are also being researched as biosensors or indicators of enviromental pollution and bioremediation of heavily pollutted environments. I live in Cyprus, where the british colonised us and started a bunch of copper mines that exported resources to nazi germany. After the 1974 war a lot of the mines were abandoned and some of them have been there since roman times. The mines have been abandoned and have never been rehabilitated and as a result the pollution still leaks through into our vegetables, fruit, drinking water and various bodies of water. I have completed a bunch of site specific visits to collect samples of water and to observe the flora and fauna of the abandoned mines. In the interview with Helene I talk about the local reseach around bioremediation of abandoned mines from Cypriot scientists.
1. A biological engineering application or tool you want to develop and why
MYCOREMEDIATION and SCAFFOLDS
Apart from my interest in bioremediation and phytoremediation, I am also extremely eager to explore a form of mycoremediation such as plastic or organic waste degrading mycelium for plastic pollution and waste management (mycoremediation). In the last 3 years I have been making a lot of biocomposite materials and working with crystallisation as well. The common root of crystallisation and mycelium cultivation is that both use scaffolds. Mycelium degrades organic or other material as a nutrient scaffold and as a helping hand in its cultivation journey and crystallisation can be combined with a scaffold that guides, support and induces the purification and formation of crystalline structure on pretty much made out of anything, organic or inorganic materials. Check my fabricademy page for more crystallisation scaffold techniques and tips.
I am quite curious as to which fungi can already break down and digest petroleum derived plastics and as to which fungi can be trained or modified. We have all heard about the fungi munching on radioactivity in Chernobyl and how mycelium is being utilised in bioremediation too! In addition, from my own research on abandoned mines and the flora and possible bioremediation of these sites I have discovered that some plants and organisms and microbes in soil and water have evolved to digest and breakdown different types of material waste and have evolved to accumulate heavy metals as well as bacteria in the polluted bodies of water have been evolved too.
In 1991, a species of fungi (Cladosporium sphaerospermum) was found growing inside the highly radioactive Chernobyl Exclusion Zone – an area deadly to most life. Fungi are already known for their extreme tolerance, often thriving in harsh environments, but this one does something scientifically compelling: it uses a process called radiosynthesis to absorb radiation (a form of energy, like sunlight) and uses it to fuel its cellular processes.found in petriandpen substack
“Fungi such as Pestalotiopsis microspora, Pleurotus ostreatus (oyster mushrooms), and Parengyodontium album, use enzymes to break down plastics, converting them into organic nutrients or harmless byproduct, Coastal pollution toolbox.org”.
In order to be able to understand how mycoremediation works we need to study how fungi degrade plastic and other organic or agricultural materials, their enzyme and metabolic actions and the mechanisms in which they grow, adhere and break down the plastic. In plastic degradation the mycelium adheres to the plastic using is also as a scaffold. Then we can study different types of plastics and if the plastics need pre-treatments for the fungi to be able to degrade the surface.
Here is a list of papers on fungi that degrade plastic!
Colonization: Fungi, such as Aspergillus terreus, Engyodontium album, and Pleurotus ostreatus (oyster mushroom), grow onto the plastic surface, often aided by pre-treatment (heat, UV light) to increase efficiency.
Enzymatic Degradation: Fungal mycelium secretes enzymes that hydrolyze and oxidize complex polymer chains (LDPE, polyurethane, PET) into simpler compounds.
Bioassimilation: The fungi consume these smaller molecules for energy and carbon, converting the plastic into organic biomass
2. Describe one or more governance/policy goals related to ethical futures
What are our governance or policy goals and our audience and what is the application of this idea?
Break big goals down into two or more specific sub-goals.
Governance or policy goals for our idea
Household everyday DIY small scale level application- relating to equity, autonomy and empowerment of citizens to manage their own family waste in the comfort of their own homes or offices or businesses.
Reproductibility of homeowners or business owners- the process, tools and resources need to be accessible using simple diy tech and in different environments.
Protect the environment- Environmental Application- Researchers are exploring these fungi for use in landfills and specialized recycling, with studies showing significant degradation within weeks. While promising, scaling this technology for industrial use is a major focus for future research, with potential for implementation in 3–5 years. Helping respond to the management of tonnes of single plastic produced, not reused or upcycled and discarded every year.
Promoting constructive uses.
3. Potential governance “actions”
By considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”) we can discuss potential governance actions for our idea :)
Action 1
Create and instruct workshops and create citizen science groups around mycoremediation and empower citizens to learn to degrade their own plastic waste anywhere. Another branch for a variety of actors in other sectors is to again create specially designed workshops and training sessions for companies, offices and other corporate actors.
Purpose
Mycoremediation and alternative waste management processes are still quite unknown and are still being researched.
Design
NGOs and environmental non profits as well citizens initiatives.
Assumptions
That people are mentally ready to deal with their own plastic waste especially in an age where everything is bought ready made from food to clothes to anything.
Risks of failure
People do not want to take responsibility and it might overwhelm them since it is a newish field.
SUCCESS!!!
Increasing interest and autonomy from individual citizens to offices and businesses to manage their waste and become more sustainable and equitable.
Action 2
Enviromental and larger scale actions such as mycoremediating plastic waste landfills or other types of material waste on the stop in the affected sites.
Purpose
Locally nothing is being done as far as mycoremediation or regenerative waste management.
Design
Local governments, corporations, ngos, academic bodies for research and development.
Assumptions
That people will be willing to try it.
Risks of failure
Might be too costly and time consuming to get it right and need 3-5 years to scale up.
SUCCESS!!!
Citizens, home and officer owners degrade and manage their own waste, easily, diy etc.
Action 3
Create a citizen science group that tackles environmental purposes and goals and disseminate knowledge, resources, diy tools to become stewards of environmental justice and more autonomous.
Purpose
Not much is being done and locally there are not that many citizen science groups that are autonomous.
Design: What is needed to make it “work”? (including the actor(s) involved - who must opt-in, fund, approve, or implement, etc)
NGOS, non profits, research groups.
Assumptions: What could you have wrong (incorrect assumptions, uncertainties)?
Cannot really think right now!
Risks of Failure & “Success”: How might this fail, including any unintended consequences of the “success” of your
SUCCESS!!!
People become more autonomous.
4. Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
• By helping respond
Foster Lab Safety
• By preventing incident
• By helping respond
Protect the environment
• By preventing incidents
*
• By helping respond
*
Other considerations
• Minimizing costs and burdens to stakeholders
*
• Feasibility?
• Not impede research
• Promote constructive applications
*
• Promote autonomy, equity
*
5. Drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why.
For example: Outline any trade-offs you considered as well as assumptions and uncertainties. For this, you can choose one or more relevant audiences for your recommendation, which could range from the very local (e.g. to MIT leadership or Cambridge Mayoral Office) to the national (e.g. to President Biden or the head of a Federal Agency) to the international (e.g. to the United Nations Office of the Secretary-General, or the leadership of a multinational firm or industry consortia). These could also be one of the “actor” groups in your matrix.
In our ideas case, I prioritize the household owners as main actors in my matrix; those who want to degrade and manage their personal plastic waste in the comfort of their own home and become more autonomous in managing their own waste without feeling like they are doing all the recycling and the waste management companies just burn them in a landfil. In the case of Cyprus this is what happens. People gather and recycle their waste but the companies are just burning them while charging people and the government.
Another idea is to create a mycoremediation start up that works similarly as a bio waste management company that goes around collecting the waste from users, businesses, offices etc and carry out the whole process in a “factory” but then the goal of equity, empowerment and autonomy in every household would not be valid, the goals will change once the main actors change.
New information
I took so many notes during the read, write and edit DNA lecture last night. Most of these concepts are new to me but I think I learned a bunch of new things that intrigued me and activated my curiosity. Below I will try to answer the homework questions with just going over the slides and doing some searching online if I must. Below you can find HW 2 PREP.
Homework Questions from Professor Jacobson
Q1. Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
A1. Polymerase has a high error rate and in biological synthesis of DNA, DNA polymerase is used and the Error Rate is 1:10 ^6 and throughput 10 mS per Base Addition [Beese et al. (1993), Science, 260, 352-355. .] The human genome consists of about 3 billion base pairs.
Biology has a way of dealing with discrepancies and errors through highly sophisticated processes of sensing, detecting, reporting, and repairing.
Q2. How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
A2. There are multiple codons to express the same aminoacid which gives us abundant possibilities for coding for an average human protein. An average protein consists of 400-500 aminoacids and most aminoacids have similar codons among them so the possibilities of coding are extremely high with many combinations being created.
Homework questions from Dr. Natalie LeProust
Q1. What’s the most commonly used method for oligo synthesis currently?
A1. The method that is still being used since the 80’s- phosphoramidite DNA synthesis cycle. It is a 4 step cycle and it is based on light based deprotection.
Q2. Why is it difficult to make oligos longer than 200nt via direct synthesis?
A2. If you look at the yield of the oligo sythesis on the graph on the image above you will notice that it is decreasing over time according to the number of coupling. More coupling more time passes the yield decreases and more errors are starting to accumulate. The longer the length of the oligonucleotide the more errors and discrepancies it will carry. Cumulative inneficiency, yield loss over time and increased errors.
Q3. Why can’t you make a 2000bp gene via direct oligo synthesis?
There are length constains in direct oligo synthesis, especially for one continuous strand and through this method we cannot create 2000bp genes. With direct oligosynthesis you can make up to 150 bases. As I mentioned above long chains have low yield, increased errors and will be incredibly hard to purify. Longer sequences >200 bp require different methods such as the Gibson assembly 2009.
Homework questions from Dr. George Church
Choose ONE of the following three questions to answer; and please cite AI prompts or paper citations used, if any. I chose question number 1 and used multiple sources from the internet and Prof. Church’s slide #4.
Q1. Using Google & Prof. Church’s slide #4, What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
A1. The 10 essential amino acids in all animals are:
In addition, the Lysine Contintency Jurassic-pedia, is a foolproof fall-back plan in Jurassic Park in order to ensure that the animals never left the island. It is about Henry Wu in Jurassic park had to come up with a contingency plan in case the dinosaus decided to escape the island and made a genetic alteration in the dinosaur genome and switched off their ability to produce the aminoacid Lysine. As a result they could not produce their own Lysine inside their bodies and had to depend on a constant external supply of Lysine by humans and therefore to become dependent on humans, veterinarians etc. It is quite inhumane in my opinion.
The 10 essential aminoacids as named above affect my view of the Lysine contingency and makes me think what would happen to all animals including humans that depend on these essential amino acids to survive as the essential aminoacids have to be consumed through food intake and cannot produced by our own bodies. What if someone played with our food and gatekept these essential to life aminoacids to create a contingency plan? How would we as humans react? Our food is already genetically modified and empty in nutrients in some cases. Makes me wonder…
Week 2 HW: DNA, READ, WRITE AND EDIT!
Geeking out over protein structures and data banks, DNA storage in plants, clouds and decoding DNA into sound
I love that artist Antoine Bertin has decoded the RNA of SARS COV 2 into this track! check it out.
This is the RNA of the Coronavirus translated into sound (viruses are made of RNA, not exactly DNA). Each nucleotide of the RNA (A,U,G or C) is transformed into a note so the virus sequence can be heard. The tempo of the track follows the rhythm at which the epidemic is growing (exponential curve) and how this curve flattens if we all stay home :) I wanted to create a track that can help with relaxation in times of isolation, and meditate on the fact all life on earth, including viruses, are made of the same material. We (humans, animals, trees, bacteria, viruses) are the continuation of a same common ancestor. Anyway; I hope this will helps everyone explore in their own sonic way what we are going through! Here is an extract of the RNA sequence :)
I had twisted sister in my mind while I was saying this, particularly I WANNA ROCK.
The 2nd week has been again packed with new information but I cannot wait to read, write and edit DNA as this it totally new information.
Week 2- DNA Read, Write, & Edit HW
This week explores the read–write–edit toolkit: sequencing and synthesis workflows, restriction digests and gel electrophoresis, and early genome-editing frameworks.
Make sure to document every step of the in-silico and lab experiments. Make sketches, screenshots, notes, drawings… anything that helps you - and others - understand the experiment.
Part 0: Basics of Gel Electrophoresis
Gel electrophoresis separates DNA fragments based on size using:
Negatively charged DNA backbone
Electric field
Agarose matrix
Size-dependent migration
I attended and watched all lecture and recitation videos apart from the one last week on Thursday, the first meetup with Tokyo Bioclub node because I was setting up an exhibition and because with the time difference I did not see the email on time but I watched the recording :)
How does gel electrophoresis work?!
…and what does it look like?
I have known for a while how it looks like but I never really looked properly into it. I have been working with agar for a while now due to making biomaterials for textiles and edible materials too. In addition, I have also worked with other polymers too such as different kinds of alginate, gelatin and different kinds of starch.
Part 1: Benchling & In-silico Gel Art
See the Gel Art: Restriction Digests and Gel Electrophoresis protocol for details.
This is what the DNA sequence looks like in FASTA SEQUENCE FORMAT! I saved the file in a file document because it was the only available option on the the neb.com website. I did right click and saved in file format. Let’s see if we can import it like this in benchling!
Importing the lambda DNA sequence in benchling
First I created a new project on benchling named ‘htgaa week 2 - MARISA SATSIA’.
Then i imported the DNA!
Then I clicked on open sequence and VOILA!
Simulate Restriction Enzyme
You might wonder what a restriction enzyme is right?!
Simulate Restriction Enzyme Digestion with the following Enzymes:
EcoRI
HindIII
BamHI
KpnI
EcoRV
SacI
SalI
Here is the enzyme digest simulation with all the enzymes!
I made this using Ronan’s website. I think it is pretty cool to simulate this whole process and have a visual because I do not know when I am actually gonna do the lab!
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
Perform the lab experiment you designed in Part 1 and outlined in the Gel Art: Restriction Digests and Gel Electrophoresis protocol.
Unfortunately I cannot do that here in Cyprus, but I am actively looking for a lab to let me practice a bit.
Part 3: DNA Design
Part 3.1. Choose your protein
In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose.
[Example from our group homework, you may notice the particular format — The example below came from UniProt]
I choose the HPV genome proteins L1 (HPV16-L1) and L2(HPV16-L2). The HPV genome is surrounded by an icosahedral capsid consisting of two structural proteins: the major capsid protein L1 (HPV16-L1) and the minor capsid protein L2 (HPV16-L2). The L1 proteins are highly conserved and aggregate to form 72 fivefold capsomers. The L2 protein binds viral DNA. There are multiple types of HPV unfortunately and each affects us differently. Some types cause cervical cancer and some warts. There is an mRNA vaccine which I got when it first came out in 2007 or 2008 or 2009, when I was 18 or 19, I do not exactly remember.
L1 Protein Lengths by HPV Type
The L1 gene encodes the major capsid protein of the Human Papillomavirus (HPV), which spontaneously self-assembles into virus-like particles (VLPs)).
Because HPV has over 100 different genotypes, the exact sequence length varies slightly:
Below is the FASTA sequence for the L1 Major Capsid Protein of HPV Type 16, the strain responsible for approximately 50% of all cervical cancer cases worldwide. HPV 16 L1 Protein Sequence (UniProt P03101). This protein is 505 amino acids long and is the primary antigen used in HPV vaccines like Gardasil.
L1 SEQUENCE
sp|P03101|VL1_HPV16 Major capsid protein L1 OS=Human papillomavirus type 16 OX=333760 GN=L1 PE=1 SV=1
I also got this Pentamer Structure of Major Capsid protein L1 of Human Papilloma Virus type 11 from the 3d viewer from the RCSB PDB I love the 3d visualisation tool and the fact that you can isolate things and make animations and download 3d models.
Part 3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence
The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
This is the FASTA sequence of phage MS2 DNA genome on the website:
For the HPV 16 L1 Protein Sequence (UniProt P03101) the reverse translation or reverse engineering sequence iiiiisssss:
NC_001526.4:5560-7077 Human papillomavirus type 16 (HPV16), L1 major capsid protein
ATGAGCCTGTGGCTGCCCAGCGAGGCCACCGTGTACCTGCCTCCCGTGCCCGTGTCCAAG
GTGGTGAGCACCGACGAGTACGTGGCCCGGACCAACATCTACTACCACGCCGGCACCAGC
CGCCTGCTGGCCGTGGGCCACCCCTACTTCCCCATCAAGAAGCCCAACAACAACAAGATC
CTGGTGCCCAAGGTGAGCGGCCTGCAGTACCGGGTGTTCCGGATCCACCTGCCCGACCCC
AACAAGTTCGGCTTCCCCGACACCAGCTTCTACAACCCCGACACCCAGCGGCTGGTGTGG
GCCTGCGTGGGCGTGGAGGTGGGCCGGGGCCAGCCCCTGGGCGTGGGCATCAGCGGCCAC
CCCCTGCTGAACAAGCTGGACGACACCGAGAACGCCAGCGCCTACGCCGCCAACGCCGGC
GTGGACAACCGGGAGTGCATCAGCATGGACTACAAGCAGACCCAGCTGTGCCTGATCGGC
TGCAAGCCCCCCATCGGCGAGCACTGGGGCAAGGGCAGCCCCTGCACCAACGTGGCCGTG
AACCCCGGCGACTGCCCCCCACTGGAGCTGATCAACACCGTGATCCAGGACGGCGACATG
GTGCACACCGGCTTCGGCGCCATGGACTTCACCACCCTGCAGGCCAACAAGAGCGAGGTG
CCCCTGGACATCTGCACCAGCATCTGCAAGTACCCCGACTACATCAAGATGGTGAGCGAG
CCCTACGGCGACAGCCTGTTCTTCTACCTGCGGCGGGAGCAGATGTTCGTGCGGCACCTG
TTCAACCGGGCCGGCGCCGTGGGCGAGAACGTGCCCGACGACCTGTACATCAAGGGCAGC
GGCAGCACCGCCACCCTGGCCAACAACTACTACCCCACCCCCAGCGGCAGCATGGTGACC
AGCGACGCCCAGATCTTCAACAAGCCCTACTGGCTGCAGCGGGCCCAGGGCCACAACAAC
GGCATCTGCTGGGGCAACCAGCTGTTCGTGACCGTGGTGGACACCACCCGGAGCACCAAC
ATGAGCCTGTGCGCCGCCATCAGCACCAGCGAGACCACCTACAAGAACACCAACTTCAAG
GAGTACCTGCGGCACGGCGAGGAGTACGACCTGCAGTTCATCTTCCAGCTGTGCAAGATC
ACCCTGACCGCCGACGTGATGACCTACATCCACAGCATGAACAGCACCATCCTGGAGGAC
TGGAACTTCGGCCTGCAGCCCCCCCCCGGCGGCACCCTGGAGGACACCTACCGGTTCGTG
ACCAGCCAGGCCATCGCCTGCCAGAAGCACACCCCCCCCGCCCCCAAGGAGGACGACCCC
CTGAAGAAGTACACCTTCTGGGAGGTGAACCTGAAGGAGAAGTTCAGCGCCGACCTGGAC
CAGTTCCCCCTGGGCCGGAAGTTCCTGCTGCAGGCCGGCCTGAAGGCCAAGCCCAAGTTC
ACCCTGGGCAAGCGGAAGGCCACCCCCACCACCAGCAGCACCAGCACCACCGCCAAGCGG
AAGAAGCGGAAGCTGTAA
Official Reference Information
Database: NCBI GenBank / RefSeq
Accession Number: NC_001526.4
Locus Tag: HPV16gp6 (L1)
Coordinates: 5560 to 7077 (1518 base pairs)
Function: Major capsid protein; self-assembles into virus-like particles (VLPs) used in vaccines.
Part 3.3. Codon optimization
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
For the HPV 16 L1 protein DNA sequence with codon-optimization
According to AI the preferred codon optimization tool for HPV16 and HPV18, particularly for designing vaccines, is the Java Codon Adaptation Tool (JCat). JCat is used to adapt the codon usage of the HPV genes to the host organism (e.g., E. coli or humans) to improve protein expression.
In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
For humans and for vaccine development.
Part 3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
AI Overview
The protein used to make HPV vaccines (specifically the L1 major capsid protein) is produced using recombinant DNA technology to create Virus-Like Particles (VLPs). These particles mimic the structure of the actual HPV virus but contain no genetic material, making them non-infectious and incapable of causing disease.
Here is the breakdown of the technology and production systems used:
1. Recombinant Expression Systems
The L1 genes are inserted into host cells that act as "factories" to produce large quantities of the protein. The two main systems are:
+Yeast Cells (Saccharomyces cerevisiae): Used in the production of Gardasil and Gardasil 9.
+Insect Cells (Baculovirus Expression Vector System - BEVS): Trichoplusia ni (Hi-5) cells infected with recombinant baculovirus are used for Cervarix.
2. VLP Self-Assembly and Purification
+Self-Assembly: Once the L1 protein is produced, it spontaneously assembles into VLPs within the host cells.
+Purification: The cells are broken open, and the VLPs are purified through complex physical and chemical processes.
+Adsorption: The purified VLPs are adsorbed onto an aluminum-based adjuvant to improve immune response.
3. Alternative & Emerging Technologies
Research is ongoing to reduce costs and increase production efficiency:
Bacteria-based systems: Escherichia coli (E. coli) is being tested for production of L1 proteins and L1 capsomeres (a cheaper alternative to full VLPs).
Transgenic Plants: Tobacco plants are being researched for plant-based VLP production.
Part 3.5. How does it work in nature/biological systems?
+Describe how a single gene codes for multiple proteins at the transcriptional level.
A single gene can code for multiple proteins known as isoforms at the transcriptional level primarily through a mechanism called alternative splicing, along with alternative promoter usage and alternative polyadenylation. This process allows the 20,000-25,000 human genes to generate over 90,000 different proteins, greatly expanding the coding capacity of the [genome](https://pmc.ncbi.nlm.nih.gov/articles/PMC4360811/).
Here is a detailed description of how a single gene codes for multiple proteins at the transcriptional level:
Alternative Splicing (The Primary Mechanism). Alternative splicing occurs when the pre-mRNA (primary transcript) is processed, and different combinations of exons are joined together while introns are removed.
+Transcriptional Processing: The entire gene, including exons (coding regions) and introns (non-coding regions), is transcribed into pre-mRNA.
+Splicing Variations: During maturation, splicing machinery (spliceosomes) can skip certain exons or retain specific introns, resulting in different mature mRNA molecules.
+Protein Diversity: These varied mRNA molecules are translated into proteins with different, sometimes opposing, functions.
+Key Types of Alternative Splicing:
-Cassette Exon (Exon Skipping): An exon may be included or excluded from the final mRNA (e.g., Exon 1-2-3 or 1-3-4).
-Alternative 5’ or 3’ Splice Sites: Changes the length of the exon, affecting the coding sequence.
-Intron Retention: An intron is kept in the final mRNA, usually leading to non-functional protein or degradation, but sometimes contributing to diversity.
-Mutually Exclusive Exons: Only one of two adjacent exons is retained.
Alternative Promoter Usage
+A single gene may have multiple promoters (transcription start sites).
+Different Transcription Starts: The cell can choose to start transcription at a different location (e.g., a “fast” or “slow” promoter), resulting in different 5’ exons (first exons).
+Functional Impact: This can produce N-terminal variants of a protein, which may have different localization or enzymatic properties.
Alternative Polyadenylation Alternative Termination. Genes can have multiple polyadenylation sites at the 3’ end.
+Different 3’ Ends: Transcription can end at different points, producing mRNAs with varying 3’ untranslated regions (UTRs) or different final exons.
+Impact on Stability: This affects the length of the transcript and its stability, often altering the binding sites for miRNAs, thus regulating translation.
Trans-splicing
+A more unusual, yet significant, mechanism where exons from two completely different primary transcripts are spliced together to form a new, chimeric, or “fusion” mRNA molecule.
+Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein!!! See example below.
[Example shows the biomolecular flow in central dogma from DNA to RNA to Protein] Special note that all “T” were transcribed into “U” and that the 3-nt codon represents].
I will see if I have time later to do this!
Part 4: Prepare a Twist DNA Synthesis Order
Part 4.1. Create a Twist account and a Benchling account
Part 4.2. Build Your DNA Insert Sequence
For example, let’s make a sequence that will make E. coli glow fluorescent green under UV light by constitutively (always) expressing sfGFP (a green fluorescent protein).
In Benchling, select New DNA/RNA sequence
I added my protein dna sequence of hpv16 l1 in jcat to get it optimised. Then I got the optimised codon result and added it here on the bases section!
Give your insert sequence a name and select DNA with a Linear topology (this is a linear sequence that will be inserted into a circular backbone vector of our choosing).
Here you can find my benchling page with the dna insert seq.
Go through each piece of the given DNA sequences highlighted below (Promoter, RBS, Start Codon, Coding Sequence, His Tag, Stop Codon, Terminator) and paste the sequences into the Benchling file one after the other (replacing the coding sequence with your codon optimized DNA sequence of interest!). Each time you add a new piece of the sequence, make sure to annotate by right clicking over the sequence and creating an annotation that describes what each piece (e.g., Promoter, RBS, etc.) is (see image below).
I have not managed to find a way to make annotations even from the previous section so I will do some more troubleshooting!
Once you’ve completed this, click on Linear Map to preview the entire sequence. If you intend to have a TA review a sequence in the future, this is a good way to verify that all sections are annotated!
Still having issues since number 3 is still confusing me! I need someone to explain it to me so I can finish this part -.-
This insert sequence you built is commonly referred to as an expression cassette in molecular biology (a sequence you can drop into any vector and it’ll perform its function). Go ahead and download the FASTA file for the sequence you made.
It’s helpful to visualize DNA designs using SBOL Canvas and Synthetic Biology Open Language to convey your designs. Here’s an example of what you just annotated in Benchling:
Part 4.3. On Twist, Select The “Genes” Option
Part 4.4. Select clonal genes option
Part 4.5. Import your sequence
Annnd I run into an issue!
I clicked on codon optimization option on the top right! I think something went wrong from the time I got the nucleotide sequence of the hpv16 l1 protein so I am having a hard time to optimize in jcat and subsequenstly to continue this section on benchling and then twist. It is taking a while so I am gonna document how it went and continue some other time.
I optimised again in Twist and I proceeded.
Part 4.6. Choose Your Vector
I chose the cloning vector.
I had a look at the construct viever after I added the cloning vectors like pTwist Amp High Copy into my hpv l1 sequence.
I clicked onto the cloning sequence and select download construct (GenBank) to get the full plasmid sequence.
AAAAAaaannnd back to my Benchling account. Inside of a folder, I clicked the import DNA/RNA sequence button and upload the GenBank file I just downloaded.
Part 5: Read, write, edit!🔮
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank)?
I would like to read HPV16 AND/or HPV18. It is important to me because of personal reasons.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Several advanced technologies are used to analyze HPV DNA and RNA sequences, ranging from established clinical screening methods to cutting-edge research tools for detecting viral integration. The primary techniques include Next-Generation Sequencing (NGS), PCR-based methods, and molecular hybridization.
Here is a breakdown of the technologies used for HPV DNA/RNA sequence analysis
Next-Generation Sequencing (NGS), NGS is used for high-throughput, comprehensive genomic analysis, including identifying multiple HPV subtypes, mutations, and integration sites.
+Nanopore Sequencing (Third-Generation): This technology is used for long-read sequencing, allowing for the characterization of complete HPV genomes and the identification of HPV integration into the host genome. It is particularly useful for identifying chimeric cellular–viral reads.
+Illumina Sequencing: Often combined with hybrid capture for high-accuracy sequencing of full HPV genomes.
+HPV-KITE: A specialized algorithm that uses k-mer data analysis for rapid HPV detection from NGS data.
Nucleic Acid Amplification & Detection (DNA/RNA)
+Real-Time PCR (qPCR): The most common method, using primers (e.g., L1, E6/E7) to amplify and quantify HPV DNA. Examples include Cobas HPV and BD Onclarity.
+RT-PCR (Reverse Transcription PCR): Used specifically for detecting mRNA expression of E6 and E7 oncoproteins.
+Transcription-Mediated Amplification (TMA): Used in the Aptima HPV Assay to detect E6/E7 mRNA for high-risk HPV.
+Isothermal Amplification (IATs): Methods like Loop-Mediated Isothermal Amplification (LAMP) and Nucleic Acid Sequence-Based Amplification (NASBA) are used for rapid, isothermal detection without a thermocycler.
+Droplet Digital PCR (ddPCR): Used for absolute quantification of HPV DNA/RNA with high sensitivity.
Signal Amplification & Hybridization
+Hybrid Capture (HC2): A signal amplification method that uses RNA probes to hybridize with HPV DNA, which is then captured and detected via chemiluminescence.
+Invader Technology: A signal amplification method (used in Cervista tests) that uses special enzymes to cleave DNA, creating a fluorescent signal.
+DNA Microarray/Chips: Technologies like Linear Array or PapilloCheck detect multiple HPV types by hybridizing amplified DNA to specific probes.
+Is your method first-, second- or third-generation or other? How so?
Let’s say we chose the APTIMA test to see if a patient has an active infection of HPV. The Aptima HPV Assay is an advanced molecular test that represents the latest generation of HPV screening. Unlike older tests that look for viral DNA, Aptima is an mRNA-based test.
+What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
The input is oncogenic E6/E7 viral messenger RNA (mRNA) from high-risk human papillomavirus (HPV) strains, cervical cells or vaginal swabs suspended in Aptima Specimen Transport Media. You do not need to prepare your input using DNA fragmentation, adapter ligation, or standard PCR. The Aptima assay is an isothermal molecular test. Sample preparation is entirely automated by the Panther System using target capture and Transcription-Mediated Amplification (TMA).
+What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
The Aptima HPV assay does not actually sequence DNA or perform traditional base calling. Instead, it is an RNA-based nucleic acid amplification test (NAAT) designed to detect active viral E6/E7 messenger RNA (mRNA) from 14 high-risk HPV types). It determines the presence of HPV by detecting specific genetic sequences rather than decoding individual bases.
Essential steps of APTIMA HPV ASSAY with AI overview
The entire testing process is performed in a single tube and consists of three main automated steps:
Target Capture: The sample (collected in a liquid cytology vial) is lysed to release its contents. Magnetic microparticles attached to sequence-specific "capture oligomers" are added. These oligomers bind only to the specific mRNA sequences of the targeted high-risk HPV types. A magnet is then used to pull these beads out of the solution, washing away cellular debris and potential contaminants.
Target Amplification (TMA): Rather than using PCR, the assay uses Transcription-Mediated Amplification (TMA), which rapidly creates billions of RNA copies of the target HPV E6/E7 mRNA at a single, constant temperature. This allows for highly sensitive detection.
Detection (HPA): The amplification products are detected using the Hybridization Protection Assay (HPA). Chemiluminescent (light-emitting) DNA probes are introduced that bind exclusively to the amplified HPV RNA. The assay uses a chemical wash to destroy the labels on any unbound probes, leaving only the bound probes to emit a light signal.
+What is the output of your chosen sequencing technology?
The output can be negative, meaning no active high risk RNA of the virus has been detected and positive for high risk HPV E6/E7 mRNA is present and active.
5.2 - DNA WRITE
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
Let’s say we want to make a vaccine for HPV so we would need to synthesize the L1 capsid protein [you can find the DNA sequence above].
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Twist bioscience to synthesize the isolated L1 capsid surface protein of the virus that will be inserted and expressed in yeast later on.
Also answer the following questions:
What are the essential steps of your chosen sequencing methods?
We will need to do codon optimization on twist bioscience for saccharomyces cereviseae (yeast) expression. The L1 protein is then incorporated into a plasmid and inserted into the yeast cells. Then the genetically modified yeast is placed into large fermentation tanks and the expression of the protein begins. The L1 proteins will begin to stick together and self assemble into virus-like particles. Then we purify the particles by breaking open the yeast cells to isolat the particles and then the particles go through purification.
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
Recombinant yeast expression has its limitations in terms of speed because it takes time to generate a stable genetically engineered strain of yeast. In terms of accuracy the size of VLP’s (virus-like particles) might vary and we can also see protein folding errors. In terms of scalability, we need to break open the yeast cells and the VLP’s also require extensive purification processes that drive up the cost of production significantly.
5.3 - DNA EDIT
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
Thinking around my final project, I would like to edit the DNA of pleurotus ostreatus (Oyster mushroom) in order to enhance its ability to decompose different types of plastics faster. There is a variety of ways of doing this. I could design a variety of peptides to optimize the decomposing process through directed signal peptide secretion or just create a whole plasmid synthesis that includes fast-petase, a gpdA constitutive promoter, a glucoamylase signal peptide for directed secretion of fast-petase, a trcp terminator, cloned into a pan7-1 backbone.
(ii) What technology or technologies would you use to perform these DNA edits and why?
I would do some manual bioinformatics using benchling to synthesise a whole plasmid and directly order the whole plasmid from twist bioscience. If I want to create a library of signal peptides I could use aplafold to generate new mutants and peptides that bind to specific plastic degrading enzymes.
Also answer the following questions:
How does your technology of choice edit DNA? What are the essential steps?
I need to do a lot of manual bioinformatics and the preparation is outlined below.
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
I need to introduce the mutations in the fast-petase dna manually on benchling, backtranslate the sequence and optimize it for expression in Aspergillus Niger, optimize the glucoamylase signal peptide as well and assemble the whole plasmid in twist bioscience.
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
In terms of precision we do not know if the glucoamylase signal peptide for directed secretion will succeed in producing fast pet-ase and might need to create new signal peptides and enhance existing pathways.
Apart from the cool robots for liquid handling in the lab I also really enjoyed the presentation on toehold switches/biosensors. I read some papers about the detection of HPV. I read some papers, have a look below!
Another paper was about High-sensitivity electrochemical detection of HPV DNA via enzyme-amplified target-induced hairpin opening on a thermally controlled paper-based digital microfluidic platform. You can find it here. I have been interested in bioelectronics for a while. Furthermore, the developed platform was successfully evaluated for HPV16 DNA detection from clinical cervical swab samples without requiring direct target amplification.
An electrochemical sensor integrated with a thermally controllable paper-based DMF (e-pDMF) device for target-induced hairpin opening with an enzyme-assisted signal amplification strategy (Scheme 1). This sensing platform is applied to detect HPV type 16 DNA (HPV16 DNA), a high-risk strain known to be a significant cause of cervical cancer. The e-pDMF device is designed to operate both transport and thermal features for precise droplet delivery and temperature control. The delivery mode enables efficient droplet manipulation and mixing, while the thermal zone on the device generates precise temperatures for optimal enzyme activity. Combining these functionalities allows for seamless operation, covering all steps from sample loading and mixing to signal amplification and electrochemical measurement. The HPV16 DNA opens the stem-loop structure of hairpin DNA (HP DNA) to form a duplex. Then, Exo III catalyzes the degradation of the duplex, releasing cleaved DNA and target DNA parts. The released target DNA part continues in cyclic enzymatic amplification, producing a large amount of cleaved DNA. This cleaved DNA is captured by a probe immobilized on the electrode surface. The decrease in current caused by electron transfer at the interface is measured using differential pulse voltammetry (DPV).
Week 03 HW
Assignment: Python Script for Opentrons Artwork — DUE BY YOUR LAB TIME!
Your task this week is to Create a Python file to run on an Opentrons liquid handling robot. Review this week’s recitation and this week’s lab for details on the Opentrons and programming it.
Generate an artistic design using the GUI at opentrons-art.rcdonovan.com.
Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script which draws your design using the Opentrons.
You may use AI assistance for this coding — Google Gemini is integrated into Colab (see the stylized star bottom center); it will do a good job writing functional Python, while you probably need to take charge of the art concept.
If you’re a proficient programmer and you’d rather code something mathematical or algorithmic instead of using your GUI coordinates, you may do that instead. If you use AI to help complete this homework or lab, document how you used AI and which models made contributions.
I also downloaded the 96 well plate python code from the website and here is a screenshot.
If you want copy and paste my code!
>from opentrons import types
import string
metadata = {
'protocolName': '{YOUR NAME} - Opentrons Art - HTGAA',
'author': 'HTGAA',
'source': 'HTGAA 2026',
'apiLevel': '2.20'
}
Z_VALUE_AGAR = 2.0
POINT_SIZE = 0.5
mclover3_points = [(27,23), (25,21), (29,21), (-29,19), (-25,19), (23,19), (-27,17), (-19,17), (19,17), (23,17), (-27,15), (-23,15), (-17,15), (-15,15), (15,15), (19,15), (21,15), (23,15), (-27,13), (-23,13), (-21,13), (-19,13), (15,13), (17,13), (19,13), (21,13), (23,13), (25,13), (-23,11), (-21,11), (-19,11), (-17,11), (-15,11), (-13,11), (-9,11), (11,11), (13,11), (15,11), (17,11), (19,11), (21,11), (23,11), (-27,9), (-23,9), (-21,9), (-19,9), (-17,9), (-15,9), (-13,9), (-11,9), (11,9), (13,9), (15,9), (17,9), (19,9), (21,9), (23,9), (-27,7), (-23,7), (-21,7), (-19,7), (-17,7), (-15,7), (-13,7), (-5,7), (-3,7), (-1,7), (1,7), (3,7), (5,7), (13,7), (15,7), (17,7), (19,7), (21,7), (23,7), (-25,5), (-23,5), (-21,5), (-19,5), (-17,5), (-9,5), (-7,5), (-5,5), (-3,5), (-1,5), (1,5), (3,5), (5,5), (7,5), (9,5), (17,5), (19,5), (21,5), (-25,3), (-23,3), (-21,3), (-19,3), (-13,3), (-11,3), (-9,3), (-7,3), (-5,3), (-3,3), (-1,3), (1,3), (3,3), (5,3), (7,3), (9,3), (11,3), (13,3), (19,3), (21,3), (23,3), (-21,1), (-15,1), (-13,1), (-11,1), (-9,1), (-7,1), (-5,1), (5,1), (7,1), (9,1), (11,1), (13,1), (15,1), (21,1), (-21,-1), (-17,-1), (-15,-1), (-13,-1), (-11,-1), (-9,-1), (-7,-1), (7,-1), (9,-1), (11,-1), (13,-1), (15,-1), (17,-1), (-17,-3), (-15,-3), (-13,-3), (-11,-3), (-9,-3), (-7,-3), (7,-3), (9,-3), (11,-3), (13,-3), (15,-3), (17,-3), (-19,-5), (-17,-5), (-15,-5), (-13,-5), (-11,-5), (-9,-5), (-7,-5), (7,-5), (9,-5), (11,-5), (13,-5), (15,-5), (17,-5), (19,-5), (-19,-7), (-17,-7), (-15,-7), (-13,-7), (-11,-7), (-7,-7), (-5,-7), (5,-7), (7,-7), (11,-7), (13,-7), (15,-7), (17,-7), (19,-7), (-21,-9), (-19,-9), (-17,-9), (-15,-9), (-13,-9), (1,-9), (11,-9), (15,-9), (17,-9), (19,-9), (-21,-11), (-19,-11), (-17,-11), (13,-11), (17,-11), (19,-11), (-19,-13), (-17,-13), (-15,-13), (-9,-13), (9,-13), (15,-13), (17,-13), (19,-13), (-29,-15), (-19,-15), (-17,-15), (11,-15), (19,-15), (-15,-17), (11,-17), (-17,-19), (9,-19), (17,-19), (-11,-21), (-1,-21), (1,-21), (11,-21), (-17,-23), (-13,-23), (13,-23), (17,-23), (19,-23)]
mrfp1_points = [(-29,21), (-27,21), (27,21), (-27,19), (25,19), (27,19), (-25,17), (-23,17), (21,17), (25,17), (27,17), (-25,15), (-21,15), (-19,15), (17,15), (25,15), (27,15), (-25,13), (-17,13), (-15,13), (-13,13), (11,13), (13,13), (-27,11), (-25,11), (-11,11), (9,11), (-25,9), (-5,9), (-3,9), (-1,9), (1,9), (3,9), (5,9), (9,9), (25,9), (-25,7), (-11,7), (-9,7), (-7,7), (7,7), (9,7), (11,7), (25,7), (-15,5), (-13,5), (-11,5), (11,5), (13,5), (15,5), (23,5), (25,5), (-17,3), (-15,3), (15,3), (17,3), (25,3), (-25,1), (-23,1), (-19,1), (-17,1), (17,1), (19,1), (23,1), (25,1), (-25,-1), (-23,-1), (-19,-1), (19,-1), (21,-1), (-21,-3), (-19,-3), (19,-3), (21,-3), (-21,-5), (21,-5), (-21,-7), (-9,-7), (9,-7), (21,-7), (-11,-9), (-5,-9), (-3,-9), (-1,-9), (3,-9), (5,-9), (13,-9), (21,-9), (-15,-11), (-13,-11), (15,-11), (21,-11), (-21,-13), (-11,-13), (11,-13), (21,-13), (-21,-15), (-15,-15), (-11,-15), (-9,-15), (9,-15), (15,-15), (17,-15), (21,-15), (27,-15), (-21,-17), (-19,-17), (-17,-17), (-11,-17), (-9,-17), (9,-17), (15,-17), (17,-17), (19,-17), (-19,-19), (1,-19), (19,-19), (-19,-21), (19,-21)]
mscarlet_i_points = [(-3,1), (-1,1), (1,1), (3,1), (-5,-1), (5,-1), (-5,-3), (-3,-3), (3,-3), (5,-3), (-5,-5), (-3,-5), (3,-5), (5,-5), (-3,-7), (1,-7), (3,-7)]
azurite_points = [(29,23), (-31,21), (-23,19), (21,19), (29,19), (-21,17), (17,17), (13,15), (-11,13), (9,13), (27,13), (25,11), (-9,9), (23,-1), (25,-1), (-23,-3), (-9,-9), (1,-11), (13,-13), (-25,-15), (-23,-15), (-7,-15), (7,-15), (23,-15), (29,-15), (-31,-17), (-29,-17), (-27,-17), (-25,-17), (-23,-17), (-13,-17), (7,-17), (13,-17), (21,-17), (23,-17), (25,-17), (27,-17), (29,-17), (31,-17), (-29,-19), (-27,-19), (-25,-19), (-23,-19), (-21,-19), (-15,-19), (-13,-19), (-9,-19), (11,-19), (13,-19), (15,-19), (21,-19), (23,-19), (25,-19), (27,-19), (29,-19), (-25,-21), (-21,-21), (-9,-21), (9,-21), (13,-21), (21,-21), (25,-21), (-19,-23), (15,-23)]
mwasabi_points = [(-27,-15), (25,-15)]
point_name_pairing = [("mclover3", mclover3_points),("mrfp1", mrfp1_points),("mscarlet_i", mscarlet_i_points),("azurite", azurite_points),("mwasabi", mwasabi_points)]
# Robot deck setup constants
TIP_RACK_DECK_SLOT = 9
COLORS_DECK_SLOT = 6
AGAR_DECK_SLOT = 5
PIPETTE_STARTING_TIP_WELL = 'A1'
# Place the PCR tubes in this order
well_colors = {
'A1': 'sfGFP',
'A2': 'mRFP1',
'A3': 'mKO2',
'A4': 'Venus',
'A5': 'mKate2_TF',
'A6': 'Azurite',
'A7': 'mCerulean3',
'A8': 'mClover3',
'A9': 'mJuniper',
'A10': 'mTurquoise2',
'A11': 'mBanana',
'A12': 'mPlum',
'B1': 'Electra2',
'B2': 'mWasabi',
'B3': 'mScarlet_I',
'B4': 'mPapaya',
'B5': 'eqFP578',
'B6': 'tdTomato',
'B7': 'DsRed',
'B8': 'mKate2',
'B9': 'EGFP',
'B10': 'mRuby2',
'B11': 'TagBFP',
'B12': 'mChartreuse_TF',
'C1': 'mLychee_TF',
'C2': 'mTagBFP2',
'C3': 'mEGFP',
'C4': 'mNeonGreen',
'C5': 'mAzamiGreen',
'C6': 'mWatermelon',
'C7': 'avGFP',
'C8': 'mCitrine',
'C9': 'mVenus',
'C10': 'mCherry',
'C11': 'mHoneydew',
'C12': 'TagRFP',
'D1': 'mTFP1',
'D2': 'Ultramarine',
'D3': 'ZsGreen1',
'D4': 'mMiCy',
'D5': 'mStayGold2',
'D6': 'PA_GFP'
}
volume_used = {
'mclover3': 0,
'mrfp1': 0,
'mscarlet_i': 0,
'azurite': 0,
'mwasabi': 0
}
def update_volume_remaining(current_color, quantity_to_aspirate):
rows = string.ascii_uppercase
for well, color in list(well_colors.items()):
if color == current_color:
if (volume_used[current_color] + quantity_to_aspirate) > 250:
# Move to next well horizontally by advancing row letter, keeping column number
row = well[0]
col = well[1:]
# Find next row letter
next_row = rows[rows.index(row) + 1]
next_well = f"{next_row}{col}"
del well_colors[well]
well_colors[next_well] = current_color
volume_used[current_color] = quantity_to_aspirate
else:
volume_used[current_color] += quantity_to_aspirate
break
def run(protocol):
# Load labware, modules and pipettes
protocol.home()
# Tips
tips_20ul = protocol.load_labware('opentrons_96_tiprack_20ul', TIP_RACK_DECK_SLOT, 'Opentrons 20uL Tips')
# Pipettes
pipette_20ul = protocol.load_instrument("p20_single_gen2", "right", [tips_20ul])
# Deep Well Plate
temperature_plate = protocol.load_labware('nest_96_wellplate_2ml_deep', 6)
# Agar Plate
agar_plate = protocol.load_labware('htgaa_agar_plate', AGAR_DECK_SLOT, 'Agar Plate')
agar_plate.set_offset(x=0.00, y=0.00, z=Z_VALUE_AGAR)
# Get the top-center of the plate, make sure the plate was calibrated before running this
center_location = agar_plate['A1'].top()
pipette_20ul.starting_tip = tips_20ul.well(PIPETTE_STARTING_TIP_WELL)
# Helper function (dispensing)
def dispense_and_jog(pipette, volume, location):
assert(isinstance(volume, (int, float)))
# Go above the location
above_location = location.move(types.Point(z=location.point.z + 2))
pipette.move_to(above_location)
# Go downwards and dispense
pipette.dispense(volume, location)
# Go upwards to avoid smearing
pipette.move_to(above_location)
# Helper function (color location)
def location_of_color(color_string):
for well,color in well_colors.items():
if color.lower() == color_string.lower():
return temperature_plate[well]
raise ValueError(f"No well found with color {color_string}")
# Print pattern by iterating over lists
for i, (current_color, point_list) in enumerate(point_name_pairing):
# Skip the rest of the loop if the list is empty
if not point_list:
continue
# Get the tip for this run, set the bacteria color, and the aspirate bacteria of choice
pipette_20ul.pick_up_tip()
max_aspirate = int(18 // POINT_SIZE) * POINT_SIZE
quantity_to_aspirate = min(len(point_list)*POINT_SIZE, max_aspirate)
update_volume_remaining(current_color, quantity_to_aspirate)
pipette_20ul.aspirate(quantity_to_aspirate, location_of_color(current_color))
# Iterate over the current points list and dispense them, refilling along the way
for i in range(len(point_list)):
x, y = point_list[i]
adjusted_location = center_location.move(types.Point(x, y))
dispense_and_jog(pipette_20ul, POINT_SIZE, adjusted_location)
if pipette_20ul.current_volume == 0 and len(point_list[i+1:]) > 0:
quantity_to_aspirate = min(len(point_list[i:])*POINT_SIZE, max_aspirate)
update_volume_remaining(current_color, quantity_to_aspirate)
pipette_20ul.aspirate(quantity_to_aspirate, location_of_color(current_color))
# Drop tip between each color
pipette_20ul.drop_tip()
Unfortunately I am having issues on google colab and cannot run the simulation. Find my colab here. I will try again. I keep following the errors but i am a bit lost.
More designs I made!
our grid plate looks amazing on ronans website!
I made this for a dear friend <3 You can find it here.
The grid looks amazing!!!! I also imported a png picture of a cute rainbow i found online and then added more points or edited out existing ones on ronans website!
This design had waaaay to many points and the code was 30 pages long. It has 7 colours or something like this!
Post-Lab Questions — DUE BY START OF FEB 24 LECTURE
One of the great parts about having an automated robot is being able to precisely mix, deposit, and run reactions without much intervention, and design and deploy experiments remotely.
For this week, we’d like for you to do the following:
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
I found 2 papers but none of them uses opentrons particularly. I am interested in bacterial and textile dyes.
is this amazing paper on DIY liquid handling robots for integrated STEM education and life science research.
Here you can find the second paper on Automated phenotyping of microalgae: scalable solution for high-throughput analysis.
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
While your description/project idea doesn’t need to be set in stone, we would like to see core details of what you would automate. This is due at the start of lecture and does not need to be tested on the Opentrons yet.
Example 1: You are creating a custom fabric, and want to deposit art onto specific parts that need to be intertwined in odd ways. You can design a 3D printed holder to attach this fabric to it, and be able to deposit bio art on top. Check out the Opentrons 3D Printing Directory.
Example 2: You are using the cloud laboratory to screen an array of biosensor constructs that you design, synthesize, and express using cell-free protein synthesis.
Echo transfer biosensor constructs and any required cofactors into specified wells.
Bravo stamp in CPFS reagent master mix into all wells of a 96-well / 384-well plate.
Multiflo dispense the CFPS lysate to all wells to start protein expression.
PlateLoc seal the plate.
Inheco incubate the plate at 37°C while the biosensor proteins are synthesized.
XPeel remove the seal.
PHERAstar measure fluorescence to compare biosensor responses.
For my final project I would be interested in making a printer or a diy dye handling machine that works with natural or bacterial dyes and prints directly on fabric or maybe make an open source one. I think I need a bit more time to find more opentrons examples in textile dyeing!
Final Project Ideas — DUE BY START OF FEB 24 LECTURE
To be completely honest I am more interested in natural and bacterial dyes, food and crystallisation more than ever. My personal work reflects this. I have a lot of ideas but I will try stick to these even though I want to focus more on edible delights, food, bacterial and natural dyes and pigments.
My final project idea number one explores the mining of colour inducing bacteria from the human microbiome for food, cosmetics, the production of sustainable textile dyes and other natural fibres such as human hair, natural weaving material such as straw, palm tree leaves etc.
here is me using my DIY inoculation loop that I designed and printed to inoculate some Janthinobacterium lividum bacteria that produces the
Violacein pigment and give off a Purple colour
According to this paper these are the pigment producing bacteria.
Color-producing bacteria exist in various environments, including on human skin, in water, and in soil, where they produce pigments as a survival mechanism against UV radiation, oxidative stress, or to compete with other microbes. These bacteria, often found in the human skin microbiome, produce natural, biodegradable, and often non-toxic pigments such as carotenoids (yellow/red/orange), violacein (purple), prodigiosin (red), and melanin (black/brown).
Key Color-Producing Bacteria on Human Skin
Staphylococcus aureus (Golden Yellow): Produces staphyloxanthin, a carotenoid pigment that gives it a golden color. This pigment acts as an antioxidant, helping the bacterium withstand oxidative bursts from human immune cells.
Micrococcus luteus (Yellow): Frequently found on human skin, this bacterium produces yellow carotenoid pigments that can absorb UV radiation.
Pseudomonas aeruginosa (Blue-Green): Often found in infections (e.g., burns, wounds), it produces pyocyanin (blue-green) and pyoverdine (yellow-green).
Corynebacterium species (Various/Creamish): Some species in the skin microbiome produce pigments like indogoidine (blue).
Streptococcus agalactiae (Orange-Red): Known to produce a pigment called granadaene, which is linked to its virulence.
Common Pigments and Their Sources
Prodigiosin (Red): Produced by Serratia marcescens, a bacterium that can be found on skin or in the environment.
Violacein (Purple): Produced by Chromobacterium violaceum and Janthinobacterium lividum, these are found in water and soil, but sometimes on skin.
Melanin (Black/Brown): Produced by various bacteria, including Pseudomonas and Bacillus species, providing photoprotection.
Significance to Humans
Clinical Diagnosis: The distinct colors of these bacteria on agar plates are used in clinical labs for rapid identification (e.g., the “golden” S. aureus).
Skin Health/Pathogenesis: Pigments like staphyloxanthin help pathogens evade the immune system, acting as virulence factors.
Industrial/Medical Applications:
i. Textiles: Bacteria like Janthinobacterium lividum are used to dye fabrics (silk, cotton, wool) with natural purple colors.
ii. Cosmetics/Medicine: Bacterial pigments are being researched as natural, UV-protective ingredients for sunscreens and as anti-cancer agents.
iii. Food: Some, like prodigiosin, are explored for use as natural food colorants, though many are still under study for safety.
These pigments are not just for color; they are essential for bacterial survival under stress.
I have a few ideas on how I can work with this concept based on the significance to humans section above. Just like natural pigments I suspect that you can also create a fully circular system with utilising bacterial pigments too.
Here you can find my own open source resource that I created for my students for a zero waste circular journey in natural dyes! I am interested in this model for my project idea too.
Here is a screenshot of the circular system design. I did not design it. Its Cecilia Raspanti of textile lab in Waag academy that did for fabricademy.
My second idea that is again based on circularity is A domestic DIY mycelium lab for breaking down household single use plastics. How to train your mycelium…to eat plastic
I asked chatGPT to make me a picture but this is not my vision to be honest. I just appreciate the humour of chatGPT, haha. I did not imagine my domestic diy mycelium lab like this.
Inspired by this project called Fungi mutarium (2011) by Katharina Unger, recycles plastic while growing edible treats. It is a prototype system that uses fungi to grow edible biomass (mycelium) on plastic waste. The process involves placing plastic in agar cups (“FU”) filled with fungi. The aim of Livin Studio’s project is to use commonly uneaten parts of fungi to break down plastic while simultaneously producing a novelty food product.
She began working with two widely consumed types of fungus: Pleurotus Ostreatus, more commonly known as Oyster Mushroom and found on Western supermarket shelves, and Schizophyllum Commune, colloquially named Split Gill that is eaten in Asia, Africa and Mexico.
Producing edible treats from this process adds more dimensions to the project and creates a zero waste circular journey adding to the circularity of the system explored <3 I LOVE THIS PROJECT AND I HAD THIS IDEA OF MAKING EDIBLE FUNGI SCAFFOLDS MYSELF. MORE LINKS COMING SOON!
Single-use plastics are goods that are made primarily from fossil fuel–based chemicals (petrochemicals) and are meant to be disposed of right after use—often, in mere minutes. Single-use plastics are most commonly used for packaging and serviceware, such as bags, bottles, wrappers, and straws.
Single use plastics
PET (Polyethylene Terephthalate): Used for drink bottles, water bottles, and food containers.
HDPE (High-Density Polyethylene): Found in milk jugs, shampoo bottles, and sturdy, often reusable shopping bags.
LDPE (Low-Density Polyethylene): Used for flexible plastics like cling wrap, bread bags, and grocery bags.
PP (Polypropylene): Common in microwaveable food containers, yogurt tubs, potato chip bags, and bottle caps.
PS (Polystyrene) & EPS (Expanded Polystyrene): Used for disposable cutlery, plates, cups, and foam food packaging.
As well as recycling nutrients and helping plants and crop grow efficiently, fungi provide us with compounds that produce antibiotics, statins for treating cholesterol and immunosuppressants. Fungarium projects like at Kew Gardens, focused on breaking down plastic, often termed mycoremediation, involve using specialized fungi to degrade synthetic polymers into organic matter. Research from institutions like Kew Gardens and various university teams has identified fungi capable of breaking down plastics—specifically polyurethane and polypropylene—in a matter of weeks, rather than centuries.
Kew Gardens Research: Scientists are mapping the “terrestrial plastisphere” to identify how fungal enzymes can degrade common, hard-to-recycle plastics.
Dr Irina Druzhinina has been studying hundreds of fungal species, as well as bacteria, that make their home on the surface of plants like Welwitschia and certain palms. What makes these plants interesting is their thick, waxy leaf cuticles made of polymers with remarkably similar traits to plastic. To avoid being swept away from their leaf surface home by the elements, fungi secrete enzymes that digest waxy leaf polymers, allowing for better grip. If they can easily digest plant polymers, it stands to reason they may have some ability to digest plastic too. Already, Irina and her international collaborators have identified more than 180 species whose enzymes could digest basic plastics in a lab setting. Identifying the genes associated with this ability and making use of a huge new fungal DNA dataset, could accelerate the finding of other fungi with plastic-eating potential far more quickly than we can now. A fungus-based solution to the enormous issue of plastic pollution could be just years away.
here is Kew gardens fungarium collection <3
and a beautiful video by Katharina Unger on her process for Fungi mutarium
My third idea is a wearable microfluidic sweat collector and biosensor that enables the detection of hormones and endocrine-disrupting chemicals (EDC), including xenoestrogens.
Here is a video from Gao Research Group on their Wearable Estrogen Sensor
In addition, there is a portable sensor that could detect PFAS on site. Led by PhD student Henry Bellette and Dr Saimon Moraes Silva, Director of La Trobe’s Biomedical and Environmental Sensor Technology (BEST) Research Centre, the research has been published in the journal ACS Sensors.
“Most PFAS testing relies on expensive laboratory equipment and specialist analysis, which makes regular monitoring difficult,” he said.
“This biosensor could be used on site and provides a simple yes or no result, allowing water to be screened quickly and easily.”
As you can see on the top section of the page i added some more microfluidics research based on sweat collection <3
Paper microfluidics are also very fun to do at a DIY level. I read this paper!
Another amazing example on paper microfluidics is the 3D Paper Mmicrofluidic Device Fabricated by Embossing fabricated on two layers of omniphobic paper containing different microchannels. Liquids flowing on the upper layer get transfer to the layer underneath to avoid mixing.
Week 4 HW: Protein design- PART I
What is protein design?
Objective:
Learn basic concepts:
+amino acid structure
+3D protein visualization
+the variety of ML-based design tools
Brainstorm as a group how to apply these tools to engineer a better bacteriophage (setting the stage for the final project).
Part A. INTUITIVE PART OF THE HOMEWORK!
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Assuming that the average molecular weight of each amino acid (M) is 100 Daltons or g/mol and mass (m) is 1g, leads to n = m/M = 0.01 mol of amino acids. Taking Avogadro’s constant 1 mol = ~6.022𝐸23 mol(-1) then, X = 0.01 mol * 6.022𝐸23 mol(-1) = 6.022𝐸21 amino acids. Considering that 100 g serving of red meat provides around 28g of protein, this approximates to 3.011E26 amino acid molecules.
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
We have the ability to digest, break down and transform proteins from other species without becoming what we eat.
Why are there only 20 natural amino acids?
While only 20 are used in protein synthesis, others exist for specific metabolic roles. These 20 have proven sufficient to build all known life on Earth. The reason isn’t that 20 is a “magic number,” but rather a combination of biological efficiency, evolutionary history, and chemical necessity. One of the most prominent theories (proposed by Francis Crick) suggests that the genetic code became “frozen” early in evolution. Once early life forms settled on these 20 amino acids, any mutation that tried to add a 21st or swap one out would have been lethal. Because every protein in the organism would have changed simultaneously, the organism likely wouldn’t survive. Therefore, we are stuck with the “standard kit” that worked for our most ancient ancestors.
Can you make other non-natural amino acids? Design some new amino acids.
Ofcourse you can! You can make new building blocks and more synthetic aminoacids :) There are a few different methods to design and create new synthetic aminoacids!
According to this paper these are the methods for reprogramming natural proteins using unnatural amino acids-
Methods for Creating Non-Natural Amino Acids
Chemical Synthesis: This is the most common approach, utilizing organic chemistry reactions such as asymmetric synthesis and transition metal-catalyzed reactions to produce diverse amino acids.
Gold Catalysis: Recent breakthroughs include using gold catalysis to create amino acids ready for peptide assembly, which simplifies the process of making complex peptides.
Modifications: This includes modifying natural amino acids by adding, for example, fluorine atoms to enhance stability or altering the backbone through
methylation.
Biological Synthesis (Biosynthesis): Engineered microorganisms, such as E. coli, are designed to produce specialized amino acids through metabolic engineering.
Enzyme Catalysis: Enzymes can be used to convert substrates into amino acids with high specificity and, often, in an environmentally friendlier way.
Genetic Code Expansion: Scientists reprogram the cellular machinery to incorporate UAAs into proteins during translation (in vivo).
Applications of these synthetic amino acids are crucial in:
Drug Discovery: Developing more stable and effective peptide-based drugs.
Protein Engineering: Creating enzymes with enhanced activity and stability under harsh conditions.
Antibody-Drug Conjugates (ADCs): Creating unique linkages to bind drugs to antibodies for targeted cancer therapy.
Where did amino acids come from before enzymes that make them, and before life started?
I have a feeling I have to dive into astrobiology for this, haha!
Overall, the findings of the study indicate that slight differences in the conditions present during aqueous alteration on planetesimals can have big effects on the end abundances of amino acids. Some amino acids can be destroyed and others created and this in turn will affect the availability of amino acids at the origin of life on Earth.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
A a helix made of D-amino acids will form a left-handed helix. Because D-amino acids are mirror images of the naturally occurring L-amino acids (which form right-handed helices), their sterically favored conformation results in an enantiomeric, left-handed structure.
L-Amino Acids: Form Right-Handed helices.
D-Amino Acids: Form Left-Handed helices.
Mechanism: The reversal occurs because the steric hindrance caused by side chains is mirrored in D-amino acids, favoring the opposite twist.
Can you discover additional helices in proteins?
Yes, additional helices in proteins can be discovered and analyzed beyond the common helix, as modern structural biology, computational methods, and tools like AlphaFold continue to reveal more complex and rare structural motifs. While helices and sheets constitute the majority of secondary structures, other helices, such as a helices and b helices, are frequently identified in protein structures
Why are most molecular helices right-handed?
Most molecular helices, such as DNA and alpha-helices in proteins, are right-handed because this configuration is energetically more stable and allows for tighter packing without steric hindrance (clashing) between atoms. This structural preference is rooted in the intrinsic chirality of their building blocks—left-handed amino acids in proteins and right-handed sugars in DNA.
β-sheets have a natural, intrinsic tendency to aggregate due to their structural properties, which make them ideal building blocks for stable, intermolecular arrangements known as “cross-β” spines or amyloid fibrils. They are prone to aggregating when “edge strands” of a sheet—which contain open hydrogen bond donors/acceptors—encounter other strands, facilitating the growth of intermolecular hydrogen-bonded networks.
Protein β-sheet structures are formed by the lateral alignment of β-strands in parallel or antiparallel orientations and are stabilized by hydrogen bonding, hydrophobic interactions, and other forces Figure 1.
What is the driving force for β-sheet aggregation?
The driving force for sheet aggregation—commonly referred to as amyloid fibril formation is a complex, multi-faceted process primarily driven by the hydrophobic effect and the maximization of intermolecular hydrogen bonding. These forces allow, often unstructured or partially folded, peptide monomers to overcome the energetic penalty of unfolding and rearrange into highly stable, stacked sheet structures.
Why do many amyloid diseases form β-sheets?
Amyloid diseases form sheets because this structure represents a highly stable, low-energy, and thermodynamically favorable conformational state for misfolded proteins to aggregate. The sheets form “cross-” spines, where peptide strands align perpendicularly to the fiber axis, allowing for tight"dry" (water-free) interlocking of amino acid side chains (steric zippers) and maximum hydrogen bonding.
More specifically, here and here I found more answers-
Here is why amyloid diseases specifically form sheets:
Thermodynamic Stability:
The sheet conformation is extremely stable, allowing misfolded, often disordered proteins (e.g., Alzheimer’s A) to aggregate into insoluble amyloid fibrils.
Dry Steric Zippers: Amyloid fibrils are held together by a “dry steric zipper” where two-sheets interdigitate, leaving no room for water between them, creating a very strong, stable structure.
Maximum Hydrogen Bonding: The in-register sheets maximize inter-strand hydrogen bonding, which stabilizes the fibrous structure.
“Generic” Protein Property: Many proteins have the potential to form sheets if subjected to specific conditions (e.g., PH changes, denaturation), meaning it is a common, almost universal"default" misfolding state.
Templated Growth:
The sheet structure allows for an “indefinitely repeating” pattern, which enables the rapid recruitment of more monomers to the growing fiber.
Can you use amyloid β-sheets as materials?
Yes!! Amyloids are Building Blocks for Macroscopic Functional Materials. Amyloid sheets are used as highly stable, versatile building blocks for advanced functional materials, including hydrogels, nanofibrils, and conductive coatings. Due to their ordered cross-structure, they are engineered for applications like 3D cell culture, tissue engineering, water filtration, and drug delivery, exploiting their remarkable strength and resistance to environmental degradation.
Design a β-sheet motif that forms a well-ordered structure.
Secondary structural features- b pleated sheet. N-H groups in the backbone of one strand of aminoacids form HYDROGEN BONDS with C=O groups in the backbone of adjacent strands. Less stearic constraint compared to alpha helices. 2 orientations-> parallel and antiparallel.
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
NGLViewer: NGL Viewer is a collection of tools for web-based molecular graphics. WebGL is employed to display molecules like proteins and DNA/RNA with a variety of representations.
PyMOL: PyMOL is a user-sponsored molecular visualization system on an open-source foundation, maintained and distributed by Schrödinger.
Chimera: A highly extensible program for interactive visualization and analysis of molecular structures and related data, including density maps, supramolecular assemblies, sequence alignments, docking results, trajectories, and conformational ensembles.
Briefly describe the protein you selected and why you selected it. I chose hpv 16 L1 protein because I used it during the second week for reading, writing and editing DNA.
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
It is a 531 amino acid, highly conserved major capsid structural protein that self-assembles into virus-like particles (VLPs). The prototype sequence, often derived from HPV-16 reference genomes (e.g.UniProt P03101), features key neutralizing epitopes, particularly within surface-exposed loops. Common variants show minor mutations (e.g., H76Y, T176N, T266A) depending on geographical lineage. Serine (S) is generally identified as the most frequently occurring amino acid in the L1 major capsid.
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs. Here is my search for HPV 16 L1 PROTEIN HOMOLOGS. I have 181 holomogs. On the map red indicated close relation to other families
Does your protein belong to any protein family? It is a part of the papillomaviridae L1 protein family.
Identify the structure page of your protein in RCSB
Here is the DOI of the protein on RCSB. I went to the homepage and searched in the top right for my protein and this is what i got!
Then to explore the 3D structure I clicked under the image on the left.
I also made an animation of the unwind assembly of the HPV16 L1 PROTEIN.
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
L1 protein of human papillomavirus 16
PDB DOI: https://doi.org/10.2210/pdb1DZL/pdb
Classification: VIRUS
Organism(s): Human papillomavirus 16
Expression System: Escherichia coli
Mutation(s): Yes
Deposited: 2000-03-01 Released: 2000-03-09
Deposition Author(s): Chen, X.J., Garcea, B., Goldberge, I., Harrison, S.C.
Protein C, also known as autoprothrombin IIA and blood coagulation factor XIV is a zymogen, that is, an inactive enzyme. The activated form plays an important role in regulating anticoagulation, inflammation, and cell death and maintaining the permeability of blood vessel walls in humans and other animals. Activated protein C (APC) performs these operations primarily by proteolytically inactivating proteins Factor Va and Factor VIIIa. APC is classified as a serine protease since it contains a residue of serine in its active site. In humans, protein C is encoded by the PROC gene, which is found on chromosome 2.
The zymogenic form of protein C is a vitamin K-dependent glycoprotein that circulates in blood plasma. Its structure is that of a two-chain polypeptide consisting of a light chain and a heavy chain connected by a disulfide bond. The protein C zymogen is activated when it binds to thrombin, another protein heavily involved in coagulation, and protein C’s activation is greatly promoted by the presence of thrombomodulin and endothelial protein C receptors (EPCRs).
This is the structure with symmetry indices. I exported the animation on the rcsb website and rendered it too. Then i downloaded the mp4 and converted it into a gif.
I also made another animation by choosing the unwind assembly under the export animation section on the right hand side.
I will use both the sequence of HPV 16 L1 protein for this and the C PROTEIN (let’s see if i manage! 0.0). I am having a lot of issues with my internet in the last 10 days due to the situation in Iran, Lebanon and here in Cyprus because of drone attacks on the british military bases.
Picture Source: Bordin, Nicola et al (2023). Novel machine learning approaches revolutionize protein knowledge. Trends in Biochemical Sciences, Volume 48, Issue 4, 345 - 359
I am waiting for aaaaaages for the cells to excute -.-
I did run all and added the sequence in the sections like MUTATION SCANS and RUN ESM FOLD and then I got an error about hugging face. I did take a screnshot and forgot to save it (no comment!). It was something like get a token from hugging face to continue and something about a secret tab. No idea! I then signed up on hugging face because that was the response of the 1st or 2nd cell in the googlecolab file.
I have 2 modes to choose from for the deep mutational scan for my protein sequence- RELATIVE or ABSOLUTE. I chose RELATIVE.
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
I noticed there are different models I can choose from. After reading a lot about latent space and encoding and transformers I can tell you that ESM stands for Evolutionary Scale Modeling are protein language models designed to design and predict 3d structures based on evolutionary information! We are using ESM2 models that are transformer based models that learn representation from massive protein sequence databases, which can be used to predict structure (ESMFold).
I used the model esm2_t6_8M_UR50D.
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
(Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
I will see if I have extra time to answer this in the following day!
Latent Space Analysis
I did some ML and AI in 2020 in Berlin. I remember learning about latent space analysis and through HTGAA I am remembering things I learned in the past but connecting them to bio stuff, which is something that I honestly enjoy a lot but I feel I need a bit more time to experiment more and get used to these concepts again. I did some phython too as well as algorithmic botany. <3
I found this website that explains latent space in deep learning also.
At the end of cell 10 while its developing the 3d figure to show I got this message -> Shape of embeddings array before 3D t-SNE: (15177, 320)
/usr/local/lib/python3.12/dist-packages/sklearn/manifold/_t_sne.py:1164: FutureWarning:’n_iter’ was renamed to ‘max_iter’ in version 1.5 and will be removed in 1.7.
Shape of embeddings array after 3D t-SNE: (15177, 3)
Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins?
I explored the neighbooring organisms and added screenshots of similar protein structures to less similar. Found some interesting ones!
Here is an examples with less similar proteins!
Place your protein in the resulting map and explain its position and similarity to its neighbors.
Part C2. Protein Folding
Picture Source: Lin et al (2023). Evolutionary-scale prediction of atomic-level protein structure with a language model.
Folding a protein
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
I managed to figure it out, I did not have enough space and was using the wrong memory option.
Total sequence length: 505
Running ESMFold inference for sequence with length 505...
Prediction complete. ptm: 0.120 plddt: 28.209
Results saved to test_18a92/
CPU times: user 1min 24s, sys: 8.67 s, total: 1min 33s
Wall time: 2min 10s
Display (optional)
I made this gif by getting screenshots of my 3d model and then using photoshop I turned the pngs into a sequence and made an mp4. I then went to this website to turn the mp4 into a gif an optimize it!
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
Part C3. Protein Generation
Picture Source: 1. Post from Sergey Ovchinnikov 2. Roney, Ovchinnikov et al (2022). State-of-the-art estimation of protein model accuracy using AlphaFold. Phys. Rev. Lett. 129, 238101
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Aaaaand here is a better picture showing the position of the aminoacids and their probability :)
Input this sequence into ESMFold and compare the predicted structure to your original.
EXTRA PYMOL PLAY FOR C PROTEIN
Ribbon
Surface
AAAnnnnnd I made another animation in pymol that I then converted to an optimized gif!
Part D. Group Brainstorm on Bacteriophage Engineering
1. Find a group of ~3–4 students
I formed a group with the following people in our node :)
2. Read through the Phage Reading material listed under “Reading & Resources” below.
3. Review the Bacteriophage Final Project Goals for engineering the L Protein:
Increased stability (easiest)
Higher titers (medium)
Higher toxicity of lysis protein (hard)
4. Brainstorm Session
+ Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”).
+ Write a 1-page proposal (bullet points or short paragraphs) describing:
Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
+ Why do you think those tools might help solve your chosen sub-problem?
+ Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”).
+ Include a schematic of your pipeline.
+ Each individually put your plan on your HTGAA website.
+ Include your group’s short plan for engineering a bacteriophage
5. Each individually put your plan on your HTGAA website
+ Include your group’s short plan for engineering a bacteriophage
Week 5 HW: PROTEIN DESIGN PART II
Part A: SOD1 Binder Peptide Design (From Pranam)
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Your challenge:
Design short peptides that bind mutant SOD1.
Then decide which ones are worth advancing toward therapy.
You will use three models developed in our lab:
PepMLM: target sequence-conditioned peptide generation via masked language modeling.
So in the mutant, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
Binder
Pseudo Perplexity
SWYDVVAGVYKAAAK
8.742062391716917
WWYGELVAVVKARAX
14.3777905642455
WRWPVYAGVKAAARK
7.102400937372668
SWWPELAGRKKWRRK
21.326166626669973
Unfortunately my connection was timed out and I had to run the cells again and I generated another 4 binders so I am going to be using these to compare the custom binder below as control!
Binder
Pseudo Perplexity
WWYPVYAGVVALKKK
13.334819029505825
SWYDPYVAVVKAKAK
13.884064880953778
ARWDPYAARKKWARX
20.375727062709792
WWYPEYVVVVELKKK
26.13860234017482
To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
So I assume we are using FLYRWLPSRRGG as control of our experiment. So we need to find a way to add to the code and add our peptide to the list and to get the perplexity score.
I checked out Diogos documentation for and I noticed he asked Gemini to help him with this and i did the same!
Here is the code that gemini helped me write in order to introduce a custom binder into the mix! In all honesty, Diogos documentation was right haha.
And we have generated the custom binder and a table for comparison :)
Buuuuut I notice that the additional binder only has 12 peptides instead of 15 like the binders I chose to generate. So I will run this again so I can generate the same length peptides as the control/custom binder!
Here we go again -.-
Binder
Pseudo Perplexity
WRVPAVAVRHKK
12.133492
WHYYAAAVRLWE
20.173057
WRVYPVAVEWKK
18.690124
WRVGAAGVAWKX
8.172350
I run the last code cell I made again to get the value of the custom binder in relation to the binders generated previously.
Record the perplexity scores that indicate PepMLM’s confidence in the binders.
+Navigate to the AlphaFold Server: alphafoldserver.com
+For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
I added the mutant SOD1 sequence and now I am attempting to add the separate chains to model the protein-peptide complex!
I have no idea if I am doing this right and I already got an error as you can see in the image above regarding the peptide residue X of the peptide generated WRVGAAGVAWKX. I then scrolled below or above and read the FAQ’s.
What should I do if I have unknown residues or nucleotides in my protein, DNA or RNA sequence? AlphaFold Server was not designed to model unknown residues or nucleotides (e.g. X for the unknown residues and N for unknown nucleotides). Please substitute by one of the standard residues or nucleotides that is appropriate for your particular case. In general, consider following substitutions:
-Proteins: replace unknown protein residues with alanine (A)
-DNA: replace unknown nucleotides by poly-T (T), but other nucleotides are also suitable
-RNA: replace unknown nucleotides by poly-U (U), but other nucleotides are also suitable
Based on this information i substitute X with A -> WRVGAAGVAWKX to WRVGAAGVAWKA!
Next steps!
Then continue and preview job but this is what I got.
I then realised I have to run the mutant sod1 sequence and each peptide separately and run each job separately otherwise when i run all peptides together with the sequence this is what i get.
CONTROL PEPTIDE- FLYRWLPSRRGG
I continued by running 5 different jobs for all my different peptides and I started from the control peptide FLYRWLPSRRGG.
PEPTIDE 1-WRVPAVAVRHKK
PEPTIDE 2-WHYYAAAVRLWE
PEPTIDE 3-WRVYPVAVEWKK
PEPTIDE 4-WRVGAAGVAWKX to WRVGAAGVAWKA
+Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
About the ipTM score:
ipTM measures the accuracy of the predicted relative positions of the subunits forming the protein-protein complex. Values higher than 0.8 represent confident high-quality predictions, while values below 0.6 suggest likely a failed prediction. ipTM values between 0.6 and 0.8 are a grey zone where predictions could be correct or wrong. These values assume modelling with multiple recycling steps, so the process of prediction reaches a degree of convergence. In large-scale screenings for protein-protein interactions, often settings optimised for the speed of prediction are used, e.g. very few or no recycling steps. In such cases ipTM thresholds as low as 0.3 have been used for initial screening; importantly though, all pairs of proteins with ipTM scores higher than 0.3 were then subjected to additional examination (e.g. Weeratunga et al., 2023). Disordered regions and regions with low pLDDT score may negatively impact the ipTM score even if the structure of the complex is predicted correctly.
As you can see from the visuals generated above in alphafold and the iptM scores above of the control peptide and generated peptides have an iptM score under 0.6 that suggests failed prediction.
Peptide
Binding info
FLYRWLPSRRGG
Peptide does not bind-floats away from the protein and not engaging with the N-terminus, the beta barrel or the Dimer Interface
WRVPAVAVRHKK
Peptide does not bind-floats away from the protein and not engaging with the N-terminus, the beta barrel or the Dimer Interface
WHYYAAAVRLWE
Peptide does not bind-floats away from the protein and not engaging with the N-terminus, the beta barrel or the Dimer Interface
WRVYPVAVEWKK
Peptide does not bind-floats away from the protein and not engaging with the N-terminus, the beta barrel or the Dimer Interface
WRVGAAGVAWKA
Peptide does not bind-floats away from the protein and not engaging with the N-terminus, the beta barrel or the Dimer Interface
+In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
WRVGAAGVAWKA with an iptM score of 0.45 is the only peptide generated that exceeds the known binder FLYRWLPSRRGG.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
+Paste the peptide sequence.
+Paste the A4V mutant SOD1 sequence in the target field.
+Check the boxes
Predicted binding affinity
Solubility
Hemolysis probability
Net charge (pH 7)
Molecular weight
Peptide 1, WRVPAVAVRHKK
Peptide 2, WHYYAAAVRLWE
Peptide 3, WRVYPVAVEWKK
Peptide 4, WRVGAAGVAWKA
+Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Peptides with higher ipTMP do show stronger predicted affinities as we can see above. All of our binders have weak binding properties and none of them really best balances predicted binding and therapeutic properties.
+Choose one peptide you would advance and justify your decision briefly.
I would choose the peptide with the higher binding affinity and the ones that are permeable and in this case it is peptide WHYYAAAVRLWE.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
Open the moPPit Colab linked from the HuggingFace moPPIt model card
Make a copy and switch to a GPU runtime.
In the notebook:
-Paste your A4V mutant SOD1 sequence.
-Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
-Set peptide length to 12 amino acids.
-Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
I generated 3 peptides for the A4V mutated SOD1 sequence choosing the parameters I am more interested in in order to to optimize the the binding process such as affinity and motif.
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
PepMLM generated peptides for a4v sod1 mutant
The known binder is the first one on the table below.
Peptide
iptM
ptM
FLYRWLPSRRGG
0.32
0.80
WRVPAVAVRHKK
0.28
0.82
WHYYAAAVRLWE
0.39
0.81
WRVYPVAVEWKK
0.29
0.81
WRVGAAGVAWKA
0.45
0.88
moPPit generated peptides
After the command was executed (after a whiiiiile) I then found my moppit files on the sideback of the colab notebook and downloaded the csv file I saved through the code named-> a4vmutsodpept.csv
Binder
Affinity
Motif
KKCFLLAIIFER
7.779302597045898
0.7270036339759827
IFQKFFCVKKFH
8.377154350280762
0.7129143476486206
IVPCWVYYYDPL
8.104171752929688
0.695409893989563
I took the 3 moPPit peptides generated from the colab notebook above and analysed their properies in Peptiverse <3
Binder 1- KKCFLLAIIFER
Binder 2- IFQKFFCVKKFH
Binder 3- IVPCWVYYYDPL
I then generated another 3 moPPit peptides by checking more properties in the colab notebook such as solubility, hemolysis and specificity.
Here is the table:
Binder
Hemolysis
Solubility
Affinity
Motif
Specificity
KKSTYKSCLTKQ
0.9520842246711254
0.9166666865348816
6.306060791015625
0.3722824454307556
0.6474359035491943
KKKERCGGRSGK
0.967436034232378
1.0
7.427951812744141
0.018683239817619324
0.942307710647583
KNRSRKYCYHYR
0.9408624060451984
1.0
7.301994323730469
0.001087116077542305
0.9807692170143127
I then analysed these again on 1. peptiverse and finally I took the best binders and popped them up in alphafold. We focused on the binders that would bind to the N-terminus which was the motif we were interested in.
What is an N-terminus? The N-terminus (also amino-terminus) is the start of a protein or polypeptide chain, characterized by a free amino group on the first amino acid.
I then added KNRSRKYCYHYR and KKKERCGGRSGK on alphafold.
Moppit binder KKSTYKSCLTKQ on alphafold
Moppit binder KKKERCGGRSGK on alphafold
Moppit binder KNRSRKYCYHYR on alphafold
Part B: BRD4 Drug Discovery Platform Tutorial (Gabriele)- OPTIONAL
I will not be doing this part as I am really having a hard time with the protein weeks and need more time to digest this new knowledge.
Part C: Final project- L-Protein Mutants
High level summary: The objective of this assignment is to improve the stability and auto-folding of the lysis protein of a MS2-phage. This mechanism is key to the understanding of how phages can potentially solve antibiotic-resistance.
Note: Lysis protein contains a soluble N-terminal domain followed by a transmembrane protein (blue/last 35 residues). Transmembrane protein affects the lysis activity. The soluble domain (green) is the domain responsible for interaction with DnaJ.
After running the MS2 Lysis protein sequence on its own on Alphafold:
Redered interaction of MS2 lysis protein with DnaJ
1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion High-Fidelity PCR Master Mix is a 2X, ready-to-use mixture where the exact formulation is partly proprietary, but the functional components are documented in the manufacturer’s manual:
Component (Phusion 2X Master Mix)
Purpose
Phusion High-Fidelity DNA Polymerase
DNA synthesis with high fidelity + proofreading
dNTPs (dATP, dCTP, dGTP, dTTP)
Building blocks for new DNA strands
HF reaction buffer (salts + pH buffer)
Maintains optimal pH/ionic strength for enzyme function
Mg2+ (via buffer system; often MgCl2-derived)
Essential polymerase cofactor
Stabilizers / additives (partly proprietary)
Improve enzyme stability and consistency
Nuclease-free water
Solvent to reach correct 2X working concentrations
2. What are some factors that determine primer annealing temperature during PCR?
Key Factors Determining Phusion Annealing Temperature ( Tα )
Primer Melting Temperature (Tm): The primary determinant, usually calculated using the nearest-neighbor method.
Primer Length: For primers >20 nt, Ta = Tm + 3 degrees celcius. For primers < 20 nt, Ta equals the Tm of the lower primer.
Buffer Choice: Phusion HF Buffer generally provides higher fidelity, while GC Buffer is optimized for high-GC or complex templates, which may require higher.
Method of Calculation: results vary by method; Thermo Fisher Scientific recommends specific tools, as Phusion requires higher
than many other polymerases.
Template Complexity: High-GC or complex templates often require higher temperatures for specificity, whereas longer, lower-concentration targets may need optimization.
Gradient Optimization: For challenging PCRs, a temperature gradient is recommended to find the best
for both specificity and yield.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Incubation with restriction enzyme at constant temperature
AMOUNT OF DNA REQUIRED
Very small amount
Sufficient amount of PLASMID DNA
FLEXIBILIY
Can introduce mutations on new sequences
Limited to enzyme recognition sites
SPEED
1-2 hours
30-60 mins digestion
PRECISION
Depends on primer design
Cuts exactly at recognition sequence
When restriction digest is preferable
When PCR is preferable
Cloning using existing restriction sites
Introducing mutations
Cutting large plasmids
Creating new overlaps
Avoiding PCR errors
Amplifying small fragments
4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To ensure DNA sequences are appropriate for Gibson Assembly, you must:
Ensure of overlapping ends -> design primers that create specific overlaps
Precise preparation of fragmens usually via PCR
amplify with high-fidelity polymerase
purify the resulting DNA
ensure correct molar ratios of the fragments.
The success of Gibson Assembly relies on creating overlapping, complementary ends (15–40 bp) that allow the T5 exonuclease to chew back the 5′ ends and the Taq ligase to join them.
Here is the step-by-step guide to preparing your DNA
Primer Design and Amplification
Create Overlaps: Design primers for your insert (or backbone) to have a 5′ tail (15–40 bp) that is identical to the end of the adjacent fragment.
Melting Temperature (Tm): Ensure the Tm of the overlapping region is >50 degrees to facilitate stable annealing at 50 degrees celcius. .
High-Fidelity PCR: Use a high-fidelity DNA polymerase (e.g., Phusion, Q5) to minimize mutations during amplification.
Avoid Secondary Structures: Use software tools like SnapGene or NEBuilder to ensure the overlap regions do not form hairpins or dimers.
Post-PCR Processing and Purification
Gel Electrophoresis: Always verify the size and yield of your PCR product on an agarose gel.
DpnI Treatment: If your template was a circular plasmid, treat the PCR product with DpnI to degrade the template plasmid, which significantly reduces background colonies.
-> Purification:
Gel Extraction: Highly recommended if the PCR produces non-specific bands.
PCR Cleanup Column: If only one clean band is present, a standard column cleanup is fine. This is preferred over gel extraction to avoid contamination with salts that inhibit the reaction.
Elution Buffer: Elute the DNA in water or Tris-HCl, as EDTA in TE buffer can inhibit the enzyme mix.
Optimization and Quantification
Quantify DNA: Use a spectrophotometer (e.g., NanoDrop, Qbit) to determine the concentration of your purified fragments.
Molar Ratios: Use an equimolar ratio for optimal assembly. A common ratio is 2-3 fold molar excess of insert over a 50–100 ng vector. Use the NEBioCalculator to calculate the precise amounts based on length and nanograms.
Minimize Contaminants: Ensure that total unpurified PCR product in the assembly reaction does not exceed 20% of the total reaction volume.
Final Verification
Colony PCR/Sequencing: Screen colonies for the correct assembly using PCR or restriction digests, followed by sequencing to confirm the seams between fragments.
How does the plasmid DNA enter the E. coli cells during transformation?
During E. coli transformation , plasmid DNA (usually double-stranded and circular) enters competent cells via heat shock (42°C) or electroporation, which temporarily increase membrane permeability. The plasmid remains distinct from the chromosome, replicates independently, and expresses selective markers like antibiotic resistance.
E. coli transformation is the process where bacteria take up external plasmid DNA, usually facilitated by making cells “competent” via chemical (CaCl2 or electrical treatment.
The steps include
(1) Ice Incubation to mix plasmid with cells
(2) Heat Shock (42 degrees celcius) to create membrane pores for DNA entry
(3) Recovery in nutrient broth
(4) Selection on antibiotic plates.
Step-by-Step Mechanism of Plasmid Transformation
E. coli cells are treated with a chilled calcium chloride solution. This makes the cell membrane porous and permeable to DNA. The calcium ions neutralize the negative charges on both the phosphate backbone of the plasmid DNA and the phospholipids of the cell membrane, reducing repulsion.
Incubation with DNA: The plasmid DNA is added to the competent cells on ice. The mix is incubated on ice for 20-30 minutes, allowing the plasmid to adhere to the outside of the E. coli cells.
Heat Shock: The tube is quickly transferred to a water bath for 30–60 seconds. This sharp temperature change causes a convection current that pulls the DNA through the adhesion sites/pores created by the and heat treatment, allowing the plasmid to enter the cytoplasm.
Recovery Step: Immediately after heat shock, the cells are returned to ice for 2 minutes, then transferred to a nutrient-rich medium (like LB or SOC) and incubated at for 45–60 minutes. This allows the bacteria to recover and, importantly, begin expressing the antibiotic resistance gene (e.g. ) carried on the plasmid.
Selection (Plating): The cells are spread on an agar plate containing a selective antibiotic. Only those E. coli cells that successfully took up the plasmid (transformants) will survive and grow into colonies, while untransformed cells die.
Key Factors for Success
Competence: Using high-efficiency competent cells is crucial.
Heat Shock Time: Exactly 45 seconds at is often optimal.
Recovery time: Allowing enough time (typically 1 hour) for the resistance gene to express.
i. Describe another assembly method in detail (such as Golden Gate Assembly) 5 - 7 sentences w/ diagrams (either handmade or online). and ii. Model this assembly method with Benchling or Asimov Kernel!
Golden Gate Assembly (GGA) is an extremely powerful modular assembly technique in synthetic biology that allows for the efficient and precise assembly of multiple DNA fragments into a single construct. It can be used to create complex DNA constructs, such as expression plasmids, circuits or gene clusters.
The power of Golden Gate Assembly permits the creation of off the shelf libraries of DNA parts that can be used for single construct assembly or multiplexed assembly of many constructs. This technique also shines in its ease of multiplexing using automation.
Golden Gate Assembly comprises a number of constituent elements: Type IIS Restriction Enzymes, Parts and Plasmids, and Assembly Standards.
Golden Gate assembly utilizes a Type IIS restriction enzyme (REase), which cleaves outside of its non-palindromic recognition sequence and T4 DNA Ligase in a simultaneous, single-tube reaction. Inserts and vectors are designed to place the Type IIS recognition site distal to the cleavage site. Cut sites can be introduced by PCR primers, if needed. During the reaction, the Type IIS REase removes the recognition sequence from the assembly with each fragment bearing the designed 3- or 4-base complementary overhangs that direct the assembly. The fragments anneal, T4 DNA Ligase seals the nicks, and the final construct accumulates over time. Cycling between optimal restriction and ligation temperature further enhances the Golden Gate efficiency. Golden Gate Assembly can be used for ordered assembly of 2–50+ fragments simultaneously.
Multiple inserts could be assembled into a vector backbone using only the sequential (3) or simultaneous (4) activities of a single Type IIS restriction enzyme and T4 DNA ligase. Golden Gate Assembly and its derivative methods exploit the ability of Type IIS restriction endonucleases (REases) to cleave DNA outside of the recognition sequence. The inserts and cloning vectors are designed to place the Type IIS recognition site distal to the cleavage site, such that the Type IIS REase can remove the recognition sequence from the assembly. The advantages of such an arrangement are three-fold:
1. The overhang sequence created is not dictated by the REase, and therefore no scar sequence is introduced.
2. The fragment-specific sequence of the overhangs allows orderly assembly of multiple fragments simultaneously.
3. The restriction site is eliminated from the ligated product, so digestion and ligation can be carried out simultaneously. [NEb](https://www.neb.com/en/applications/cloning-and-synthetic-biology/dna-assembly-and-cloning/golden-gate-assembly)
Soooo I will attempt to do the lab on the top of the page (in silico) using benchling. I got totally confused whether this part of the assignment is asking me to do golden gate assembly or gibson and I noticed a lot of my classmates went with gibson so I will do that too. I followed what Nourelden did.
I then added added this gfp sequence on benchling manually!
I chose 2 restriciton enzymes, BamHI, and BstAPI inside the puc19 plasmid so I can linearize my vector and proceed with gibsol assembly.
And here is the virtual digest ladder!
Then I went to the assembly wizard on the bottom right of the page and i selected gibson!
And then I realised I made a mistake because I chose puc19 as both the backbone and the insert instead of selecting the gfp sequence for the insert so I corrected it.
I realised I was not selecting all of the gpf sequence so I did it again and again until it selected the whole gfp sequence as an insert. Then I hit assembly and benchling generates the final construct.
Unfortunately this does not seem right to me and it is a bit embarassing because I did it at least 10 times and I keep doing it wrong. There is a gap where the gfp was supposed to be.
Homework PART B: Asimov Kernel
1. Create a Repository for your work
2. Create a blank Notebook entry to document the homework and save it to that Repository
3. Explore the devices in the Bacterial Demos Repo to understand how the parts work together by running the Simulator on various examples, following the instructions for the simulator found in the “Info” panel (click the “i” icon on the right to open the Info panel)
4. Create a blank Construct and save it to your Repository
i. Recreate the Repressilator in that empty Construct by using parts from the Characterized Bacterial Parts repository
ii. Search the parts using the Search function in the right menu
iii. Drag and drop the parts into the Construct
iv. Confirm it works as expected by running the Simulator (“play” button) and compare your results with the Repressilator Construct found in the Bacterial Demos repository
v. Document all of this work in your Notebook entry - you can copy the glyph image and the simulator graphs, and paste them into your Notebook
5. Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo
i. Explain in the Notebook Entry how you think each of the Constructs should function
ii. Run the simulator and share your results in the Notebook Entry
iii. If the results don’t match your expectations, speculate on why and see if you can adjust the simulator settings to get the expected outcome
I finally got the chance to look into doing this and managed to navigate it. I made a blank notebook entry and made some constructs and simulated them!
I recreated the repressilator from the class by using the characterized bacterial parts repository and dragged and dropped the components to create this repressilator construct.
Construct 1
After dragging and dropping all the components I made this!
Week 7 HW: Genetic Circuits Part II: Neuromorphic Circuits
From the lecture with Ron Weiss
"...central dogma, if you will, in synthetic biology, is the notion that almost everything that we build is based on sensing, processing, and actuation. So
we want to be able to sense everything that's going on inside and outside the cell, have that information fed into some kind of controller, and have
that regulate things that are going on in the cell".
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
"you know, one day in particular, I looked at it and said, I actually want to flip that arrow. So rather than being inspired by biology and how to program computers, I want to flip that and say, what can we take about what we know about computing, and actually program biology?".
Ron Weiss
1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
From the lecture with Ron Weiss! Motivation-> need for non digital and non binary biological computing!
First of all let’s start from what are boolean functions or logic and traditional genetic circuits! The lecture was super interesting but I had to read a looooooot to understand the concept of the week and I am still digesting!
Traditional genetic circuits are the first and most fundamental synthetic circuits created and they carry out binary decision-making with YES/NOT gates.
examples- genetic toggle switch, repressilator (oscillator circuit).
While traditional circuits are better for precise, discrete switching, IANNs excel in complex sensing and continuous regulation.
Limitations of Boolean Logic : While useful for modeling, the Boolean model is an abstraction. The real biological system is continuous, where protein levels change smoothly over time, rather than switching instantly between 0 and 1.
To date, both the digital and analog computing paradigms have been implemented in living cells in an attempt to design and build genetic circuits efficiently. The digital paradigm, which abstractly computes with two discrete binary-coded levels, has inspired implementation of wide variety of genetic circuits, including logic gates, memory elements, a counter, state machines, a toggle switch, a digitizer, and highly complex logic functions. The analog paradigm, in contrast, computes on a continuous set of numbers and has been suggested as an alternative to the digital paradigm for tasks that don’t require decision-making. Efforts in synthetic biology have also focused on other aspects of circuit control, such as complex temporal dynamics, and integral feedback controllers for robust adaptation.
2. Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
I am still interested in HPV detection and therapy. There could be a useful Application for an Autonomous IANN for HPV therapy. The IANN acts as a synthetic “molecular sensor-actuator” within keratinocytes (the target cells of HPV). Its goal is to distinguish between healthy cells, transiently infected cells, and cells with high-risk oncogenic activity, and then trigger apoptosis (programmed cell death) only when the high-risk, cancer-prone state is detected.
Input 2 ( x2- Host Differentiation State): Specific markers of suprabasal cells where HPV replicates, such as high p16INK4a expression.
Input 3 ( x3 - Immune Evasion Status): Low interferon (IFN) response or downregulation of interferon-stimulated genes (ISGs), which is a common strategy of HPV to persist.
IANN Processing: The synthetic network acts as a threshold classifier. It weights the inputs—for instance, giving higher weight to the E6/E7-IFN downregulation combination, which strongly suggests imminent malignant progression.
Outputs (Actuator Response)
Low-Risk Scenario (Threshold not met): The IANN remains inactive, allowing natural immune clearance.
High-Risk Scenario (Threshold met): Activation of a synthetic genetic switch that expresses a pro-apoptotic protein (e.g., Caspase-3 or Bcl-2-associated X protein (BAX)), inducing apoptosis in that specific HPV-infected cell.
Limitations and Challenges
Implementing an IANN to achieve this goal faces significant biological and technological challenges:
Sensitivity and False-Positive Rates: The system must not activate in healthy cells, as excessive, unintentional apoptosis could lead to tissue damage. + Training the network to be highly specific to only oncogenic HPV types (16, 18) is difficult.
Delivery Mechanism: Getting the engineered DNA or RNA circuits into the cytoplasm and nucleus of the targeted cervical basal cells in vivo is a major bottleneck.
Signal Noise and Interference: The intracellular environment is crowded and noisy. The IANN’s molecular components (e.g., siRNA) might be degraded by natural cellular processes or interact unintentionally with host pathways (off-target effects).
Adaptability: If the HPV mutates or the cell changes, the pre-engineered, static IANN cannot “re-learn” or update its weights like a software-based ANN, unlike the immune system.
Immune Evasion of the IANN itself: The cell’s own immune system might recognize the IANN’s synthetic machinery as foreign (if using protein-based nodes) and destroy it.
3. Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
x1: input- dna encoding for csy4 endoribonuclease
x2 input - dna encoding for fluorescent protein whose mrna is regulated by csy4
Tx: transcription
Tl: translation
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
1. What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Fungal materials are biofabricated biocomposite materials that have been colonised by mycelium growth, the roots of filamentous fungi. The raw material/waste is our nutrient substrate as well as a scaffold for the mycelium to grow on. The raw materials can come from agricultural or household waste like straw, coffee grounds, pellets, wood chunks, sawdust, even legumes (look at tempeh), or even plastic. The nutrient medium is not only food to the fungus but it is also a scaffold. Some food is better than others but I believe that you can train your mushrooms to eat anything. Reishi usually grows on rye, Oyster mushrooms on straw or coffeegrounds or paper. Do not forget there are different types of mushrooms and each grows/feeds/prefers different food.
For fungal materials the most commonly used types of fungi are the ones below!
Image from recitation with bioengineer Ren Ramlan on Fungal materials
CLASSIFICATION BASED ON TYPES OF FEEDING
SAPROPHYTIC (THE DECOMPOSERS)
ENDOPHYTIC (Internal associates)
MYCORRHIZAL (Symbiotic partnership)
PARASITIC (Host feeders)
PLEUROTUS OSTREATUS (Oyster)
Fusarium sp.
Truffles
Cordyceps
Shitake
Trichoderma sp.
Chanterelle
Chaga
Lions Maine
Phomopsis sp.
Porcini
Honey fungus (armillaria mellea)
TYPE
FEEDING BEHAVIOR
HABITAT
SAPROPHYTIC
Breaks down organic material
Dead wood, soil, leaves
ENDOPHYTIC
Internal associate [unclear to me how yet]*
Lives inside plant tissues
MYCORRHIZAL
Symbiosis with living roots
Underground
PARASITIC
Attacks living host organism
Inside living plants, trees, insects
*still figuring it out.
For my own final project and my research interest in mycoremediation and plastic degrading fungi I have focused more into SAPROPHYTC NUTRITION and specifically on saprobionts that digest their food externally and then absorb the products.
Fungi have the ability to transform organic materials into a rich and diverse set of useful products and provide distinct opportunities for tackling the urgent challenges before all humans. Fungal biotechnology can advance the transition from our petroleum-based economy into a bio-based circular economy and has the ability to sustainably produce resilient sources of food, feed, chemicals, fuels, textiles, and materials for construction, automotive and transportation industries, for furniture and beyond. Fungal biotechnology offers solutions for securing, stabilizing and enhancing the food supply for a growing human population, while simultaneously lowering greenhouse gas emissions. Fungal biotechnology has, thus, the potential to make a significant contribution to climate change mitigation and meeting the United Nation’s sustainable development goals through the rational improvement of new and established fungal cell factories.
Applications/Industry
Examples
Building and construction
Insulation panels, biobricks
Fashion and textiles
Mushroom leather
Art, design, stationery
Paper
Packaging
Biofoam alternatives
Decorative items and interior design
Lampshades, plant pots
Sports equipment
Styrofoam alternatives- Surf boards and canoes
Biomedical
Fungal textiles - rapid regenerative qualities
Food
Tempeh and so much more
Some screenshots from Ren Ramlans recitation presentation!
Another example of a self healing fungal material from Rens recitation presentation
Advantages and Disadvantages
Advantages
Disadvantages
Negative carbon emissions, cultivated with minimal energy
Contamination
Natural fire resistance, non toxic combustion
Need specialised knowledge to grow (but not a fancy lab or equipment)
Sustainable and circular process
Slow process*
Biodegradable
end results/products will not be identical to each other
Slow process-grown not manufactured
some shrinkage
Long life cycle
One of the most versatile biomaterials ever
renewable source
Lightweight
Slowness- for me its an advantage because I value slow making and cultivation but it can also seem like a disadvantage to someone who does not understand that biofabrication is a collaboration and co creation process and requires care and patience.
A lot of “disadvantages” on the list above can actually be seen as advantages.
3. What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
This question right here is THE MOST IMPORTANT ONE FOR ME AND MY FINAL PROJECT!!!! I will try to answer this questions in more depth and add any more research in my final project page during this week.
I am interested in plastic degrading fungi and mycoremediation. I am interested in researching the enzymes that break down plastics in saprophytic fungi and figure out how to train my mushrooms either naturally or with synthetic biology. Certain moulds and fungi have evolved to break down more than agricultural organic sources. Another interest is studying melanin producing fungi for protection from radiation and learning more about protein producing mechanisms by looking into their genome and editing it.
In addition, yeast (saccharomyces cerevisiae) is a type of unicellular, eukaryotic microorganism classified within the
Fungi kingdom. It has been often used as a biosensor in modern-day biotechnology. It was the first eukaryote to have its genome sequenced in its entirety in 1996. Along with that advance was the concerted effort to assign functions to all 6000 open reading frames.
The benefits of yeast being used as a biosensor have opened new avenues for drug discovery, understanding molecular pathways involved in disease pathogenesis, protein–protein interaction studies, understanding of the molecular architecture of complex protein assemblies, identifying mutations in proteins that have significance in determining the functional differences, and detecting pollutants from the environment. Yeast has already proved its benefits in studying protein–protein interactions, drug screening against several diseases, including cancer, Alzheimer’s disease, Parkinson’s disease, and others, detection of pollutants, and diagnosis of diseases.
Yeast-based biosensors are engineered microorganisms, modified to detect and quantify target compounds, toxins, or environmental pollutants. These biosensors use genetic modifications—such as reporter genes (fluorescence/luminescence) and modified receptors—to produce measurable signals (colorimetry, electrical) upon interaction with specific molecules. They are widely used in environmental monitoring, pathogen detection, and pharmaceutical development due to their ease of culturing and genetic tractability.
I also virtually attended a workshop by artist Mary Maggic who were also on htgaa 2015 on Becoming with fungi where they used remazol blue (endorcrine disrupting chemical) to test the ability of the Schizophyllum commune mushroom for bioremediation. Marry Maggic have also worked with yeast biosensors.
YES-HER YEAST BIOSENSORS-DOES IT SAY (YES) TO (hER)?
Because endocrine disrupting compounds are usually found in minimal amounts (ng/L-1) in the water, one of the most common techniques for their detection is liquid chromatography-tandem mass spectrometry (LC-MS). But this approach is very expensive to perform on a routine basis, requiring both skilled personnel and a robust quality assurance/control program. Maybe biology is the answer... The YES-YEAST (yeast estrogen sensor) are a genetically modified strain of Saccharomyces cerevisiae (W303) that contain Human Estrogen Receptor (HER). They act as a biosensor: the input is estrogen and the output is a yellow color change. More importantly, the YES yeast are extensions of our bodies: what binds to their receptors also bind to ours, demonstrating a DIRECT biological response to xenoestrogens on our bodies. The same process of estrogen binding and activation is reproduced in the yeast. This bioassay detection method is more sensitive than the chemical approach either detecting estrogenic target compounds at lower concentrations, other non-target compounds, and synergistic effects that chemical methods and machines fail to detect.
This is both a "part two" of the Open Source Estrogen project as well as the Final Project for HTGAA, which combines lectures (1) "Synthetic Minimal Cells" with Kate Adamala, (2) "Bio-production" with Patrick Boyle, (3) "Computational Protein Design" with Srivatsan Raman, and (4) "Tools, Automation, and Open Hardware.
Some yeasts can find potential application in the field of bioremediation. One such yeast, Yarrowia lipolytica, is known to degrade palm oil mill effluent, TNT (an explosive material), and other hydrocarbons, such as alkanes, fatty acids, fats and oils. It can also tolerate high concentrations of salt and heavy metals, and is being investigated for its potential as a heavy metal biosorbent. Saccharomyces cerevisiae has potential to bioremediate toxic pollutants like arsenic from industrial effluent.[84] Bronze statues are known to be degraded by certain species of yeast.
In Rens research she is working with Ascomycota and Basidiomycota are less conducive to existing engineering pipelines.
It seems that Agrobacterium is gonna be super important for my own research into how to incorporate synthetic biology in my final project.
You 11:52 PM (Edited)
I am interested in plastic degrading fungi. How can I apply synthetic bio to this concept? I have a few ideas but it will be nice to get expert advice.
TA, Val Thompson, ChiTownBio, Chicago 11:57 PM
You could look at the pathways responsible for making the enzymes in mushrooms like oysters, that break down plastic, like lactase, manganese peroxidase, and lignin peroxidase, and maybe boost one of those pathways in some manner? The specifics beyond that are above me at this point in time.
Advantages of doing synthetic biology in fungi as opposed to bacteria
While fungi offer superior PTMs and secretion, they often have longer cell cycles (12–24 hours) compared to bacteria (20–60 minutes), and their genetic toolkit is often considered less developed compared to Escherichia coli.
Doing synthetic biology in fungi, particularly filamentous fungi and yeasts, offers several advantages over bacteria (such as E. coli) due to their eukaryotic nature, metabolic complexity, and specialized secretion systems. Key advantages include superior protein folding and secretion, a vast repertoire of secondary metabolites, and higher environmental robustness.
In Prospects of Fungal Biotechnologies for Livestock Volume 2. Fungal Biology. Springer, Cham., it is being mentioned that “engineered fungi like Aspergillus, Trichoderma, and Saccharomyces are increasingly used to produce valuable biomolecules such as enzymes, insulin, and antimicrobial peptides. These organisms naturally secrete large quantities of proteins, making them particularly attractive for industrial-scale applications”.
Previous work with digital fabrication for fungal biocomposites
I have been a big fan of mushroom cloning since 2020. I was introduced to it by Rodolfo Acosta Castro in the Natural Machines program of the School of Machines, Making, and Make Believe in Berlin <3 I was also introduced to AI stuff, python, algorithmic botany and many other amazing stuff during my scholarship in the program.
I also worked a lot with 3d photogrammetry and simulating natural systems and I visited the natural history museum and made this model of a reef!
If you are into mushrooms you should read this book!
Fast forward to 2021 in fabricademy I made more can cultivated materials explorations. A few months ago I was invited to talk about my diy biolab and working with living systems in a New European Bauhaus presentation in a local university! Here is my presentation.
Mycelium as scaffold
I have done a lot of experiments with mycelium in the past 5 years! The craziest one was during Textile as scaffold week in Fabricademy in 2022 where I grew epsom crystals on a piece of mycelium biocomposite sample that I fabricated in a workshop earlier that year.
Mushroom cloning, fungal materials protocols and other resources <3
I visited a mushroom factory here in Cyprus a few years ago to get some inoculated substrate for my residency at the Cyprus University of Technology makerspace.
I am here to give you some inspiration before you get deep in mushroom cloning. I hope you enjoy my references.
I am interested in the illusion of technological mastery over nature. In response, I construct controlled environments that gradually yield to organic processes. When cultivating Lingzhi, I initiate growth by placing woodchips and spores into a mold and regulating humidity, temperature, and light. At first, the form appears engineered. Yet once the mycelium binds the substrate and the mold is removed, the sculpture continues to evolve according to its own biological logic. Fruiting bodies emerge, spores settle as fine dust across the surface, and authorship shifts from design to negotiation.
These works examine collaboration rather than control. Mycelium demonstrates adaptation, self-organization, regeneration, and repair.
Mushroom cloning and fungal materials protocol
Here you can go on my google drive and view or download the protocol Rodolfo shared with me in better quality!
You can clone any edible mushroom from the supermarket using agar nutrient medium in petri dishes OR you can even clone mushrooms from the supermaket without using agar nutrient medium or petri dishes. Another option for BEGINNERS is to buy something like a Grow-Your-Own kit and also have a look at the molds that come with the kit as it will help you understand how you can make a mold for your biocomposite fungal material <3
Images from Ren Ramlan recitation on Fungal materials
Other DIY RESOURCES
DIY STILL AIR BOX
DIY INCUBATOR
You can find so many out there it is confusing! But here I found an article that has some good ones. You can make an incubator only for petri dish cultivation.
Here is also a video by my friend Dariia at Yane Lab in Dnipro, Ukraine <3
LAMINAL FLOW HOOD
A step by step to create this beautiful portable laminar flow hood! Apart from the Still Air Box method you can also use a lamiral flow hood also inside your box with a HEPA filter.
And here is another step by step instruction manual.
Nutrient medium recipes and intructions
What you will need:
Mushroom from the supermarket
Scalpel
Parafilm to seal your petri dishes
Sterilised petri dishes
Pressure cooker to sterilise agar medium and petri dishes (if needed because you might choose to buy presterilised dishes to just pour in your medium into which could be better for a begginer imo)
Still air box or lateral flow hood
For cloning with agar agar nutrient medium in petri dishes
You can follow Rodolfos protocol above. I am just rewriting it here and adding another resource too. You cannot know if it works until you try it and figure it out but I think both are good.
Rodolfo’s recipe
To make 300 ml agar medium
- 7.2 gr of agar agar
- 6g malt extract
- 0.6 gr of nutritional yeast
For cloning without agar agar nutrient -> using cardboard or any other organic resources/waste
Cloning with Moist Cardboard is a useful technique for outdoor situations or when laboratory equipment is not available.
+ Cardboard Preparation
Soak the cardboard in boiling water to sterilize it, then let it cool.
+ Placement of Mushroom Tissue
Place a piece of mushroom tissue between two layers of damp cardboard, then roll it up and place it in a plastic bag.
+ Growth and Transfer
Let the mycelium develop inside the cardboard. Once it has fully colonized, you can transfer the mycelium to a more nutritious substrate.
How does mycelium grow? What is important for us to know about mycelial growth when designing or fabricating a mold? LIFE CYCLE AND MOLD FABRICATION
LIFE CYCLE
YOU NEED TO STOP THE GROWTH CYCLE OF THE MYCELIUM TO PREVENT FROM FRUITING TO GET A WHITE-ISH BIOCOMPOSITE (DEPENDING ON THE KIND OF STRAIN USED)
When the environmental conditions are right, mycelium will form mushrooms. Some fungi are very particular in what they need to switch over from mycelial growth to producing a mushroom. The most commonly cultivated mushrooms do not require much to induce fruiting. The mushroom’s main role in the life cycle is to produce spores. Spores are similar to seeds in that they are the reproductive elements of the organism. They are microscopic packets of genetic material that are distributed by insects, rain, and wind to hopefully find a new food source. Spores are produced by mushrooms in the tens of thousands. In fact every breath we take on this planet we inhale mushroom spores. In the wild, the mushroom life cycle rotates between these 3 phases -mushroom, spore, mycelium- in a constant evolution of change and growth. Amazingly mushroom tissue can revert to mycelial growth for many species of mushrooms. This is extremely useful for the cultivator as exact clones with the same DNA can be taken from mushroom tissue and further expanded. These clones from individual mushrooms are called strains.
YOUR MYCELIUM NEEDS TO BREATHE!!! FORGET ABOUT FANCY OVERLY DESIGNED DIGITALLY FABRICATED MOLDS AND LOOK FOR MATERIALS AND DESIGNS THAT ALLOW OUR MYCELIUM TO FORM ROOTS
If you ever wondered if there is a simulation of mycelium filamentous growth, there are a few! Also, if you are interested in generative design you can watch some tutorials of Grasshopper, an extension of the program Rhinoceros, which is all about generative design. You can for libraries and ready made definitions to experiment a bit and understand how Grasshopper works and its logic.
Apart from Grasshopper, I found a few cool websites that simulate mycelial growth.
Here is a really cool website that analyzes the algorithim for the growth rate of mycelium.
MOLD FABRICATION FOR FUNGAL MATERIALS/ MYCELIUM BIOCOMPOSITES
Mold examples screenshot from recitation with Ren Ramlan <3
Tips for your mould
No porous mould materials. Your mould needs to retain moisture, otherwise the mycelium composite will dry out before it has time to grow into the shape of your mould. In addition, porous materials that cannot be sanitised will harbour contaminants such as bacteria and mould spores which will compromise your project. Mycelium also has the ability to penetrate into moulds which are porous. That is why plastic, metal or glass works well, otherwise the mould should be covered with plastic cling film.
Your mycelium needs to breath since takes in oxygen and releases carbon dioxide. If your mould is completely sealed, the mycelium won’t be able to breathe and or grow. To provide controlled airflow, leave small holes or gaps in the mould and cover them with breathable filters, such as micropore tape, to keep contaminants out while still allowing gas exchange.
Start small. Make a small mould first and avoid making your mould very large. After the mycelium has colonised and taken shape, the object must be thoroughly dehydrated to stop growth and preserve it. Very large moulds are harder to dry evenly without specialised equipment, which increases the risk of spoilage or structural failure. If you have a large project in mind, consider creating several smaller, moulds that can be assembled into a larger final structure.
More mold examples like the ones above from Domingo club.
I use Rhinoceros to make a 2d technical drawing and I then extrude the surfaces to make a 3D printed mold but I would advise you to use any type of plastic vacuum formed molds (look for them in things you buy like biscuit packages or other stuff from the supermarket) or a silicone mold for candle making or resin art. Just avoid porous materials for your mold fabrication. You can use a 1 part mold or a mold with more parts. You can use freecad an open source parametric modeler or you can also download some open source designs from places like instructables. You can also use any plastic containers you have available, pack your material and then take another container as your negative part to create a positive negative space to make an object like a plant pot etc.
Another really good resource for making mycelium biocomposites is this infographic by my fabricademy peer Elsa Gil.
Submit this Google Form with your draft Aim 1, final project summary, HTGAA industry council selections, and shared folder for DNA designs. DUE MARCH 20 FOR MIT/HARVARD/WELLESLEY STUDENTS -> GOTTA CHECK WITH BIOCLUB
Review Part 3: DNA Design Challenge of the week 2 homework. Design at least 1 insert sequence and place it into the Benchling/Kernel/Other folder you shared in the Google Form above. Document the backbone vector it will be synthesized in on your website.
Week 9 HW — Cell-Free Systems
This week introduces synthesis of proteins using cellular machinery outside of a cell. I LOVED THE LECTURE and I loved Kate Adamalas work.
We have to solve terrestrial problems before extraterrestrial problems...we need to shift away from a petroleum based bioeconomy...we need a paradigm shift.
Kate Adamala <3
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell free expression is more beneficial than cell production.
A cell-free system allows biological reactions to occur outside of living cells. By extracting and using cellular components like ribosomes, RNA polymerase, amino acids, and ATP, this method enables reactions in a controlled, simplified environment. Cell-free systems allow for the engineering, expression, and analysis of genetic constructs without the complexity of living cells.
Applications of Cell-Free Systems include:
+ Synthetic Biology: Designing and testing biological circuits or pathways without cellular constraints.
+ Protein Engineering: Rapid protein production and screening, especially for proteins that are toxic or hard to express in cells.
+ Metabolic Engineering: Production of high-value chemicals, biofuels, and pharmaceuticals via synthetic pathways.
+ Biosensing: Creating diagnostic tools that are portable and easy to use, like paper-based biosensors.
Gene Editing Research: Testing CRISPR-based systems or genome editing tools in a controlled environment.
Cases where cell free expression is more benedicial than cell production?
Cell free expression is often more beneficial than conventional cell-based (in vivo) production in scenarios requiring speed, high-throughput screening, or when producing proteins that are toxic or difficult to fold. By bypassing the need for living cells, cell free protein expression removes constraints associated with cell viability, growth, and genetic stability, acting as an “open” system that allows higher flexibility, speed, control over the environment and direct manipulation.
Describe the main components of a cell-free expression system and explain the role of each component.
...when we say synthetic cell...we mean a liposome that encapsulates metabolism and genome inside.
If you use cell pre-expression or expression in synthetic cells, you can express your proteins from linear fragments, linear PCR fragments, so you no longer have to make plasmids.And you can express them in a survey system in the matter of hours. So basically synthesize your DNA, drop it in, and express it. With the cell free expression, you can basically do it all at once. You can get all of your proteins expressed in one pot by just adjusting the concentrations of the DNA.
Our chassis for engineering!
Explain the role of each component (the answers are from Kate Adamalas presentation):
1. Communication (Cell membrane/channels) -> to detect signals from the environment, we need to have membrane channels. And so that's what our cells do have, a way to communicate with the outside through membrane channels.
2. Lipid membrane (cholesterol and phospholipids) -> Cholesterol is an amazing molecule. It makes your membranes more fluid. It's cholesterol in the wrong place that's so deadly to us, multicellular organisms that happen to have veins. Usinegcholesterol in our synthetic cell membranes gives them fluidity, it gives them stability, it improves mechanical stability, shear stress stability.
3. Cytoplasm (small molecules) -> cytoplasm of our synthetic cell is really the cell-free protein expression system that's been purified, modified in some ways, and put inside a liposome. This is where a lot of flexibility comes in, because we're making those small molecules. Sometimes we have fatty acids, simple, non-modified fatty acids, and that's usually when we do experiments around the origin of life, like figuring out how life started.
4. Cell extract (ribosomes and other enzymes) -> to provide the essential machinery for transcribing DNA into RNA and translating RNA into proteins in an artificial, cell-like environment.
5. tRNAs -> for transcription to occur inside the synthetic cell we need the transcription proteins. This is where the genetic code gets decoded, and this is how you hack the genetic code. If you want to do recoding of a genetic code you need to have a tRNA.
6. Minimal set of genes (plasmids or linear fragments) -> minimal genome, so we can still understand it, but that genome has to be complex enough that it will be capable of expressing all the proteins that we need.
From the bioclub hangout-> slight confusion between cell free systems vs synthetic cells!
Cell-free systems use extracted, non-living cellular machinery (ribosomes, enzymes) in an open, in vitro environment to perform tasks like protein synthesis, offering high control and flexibility. Synthetic cells (or protocells) are bottom-up, engineered, microscopic compartments—often using lipid vesicles—that encapsulate biochemical reactions to mimic living cell structure and function, providing compartmentalization.
Cell-Free Systems (CFS)
Definition: Molecular machinery removed from cells (e.g., E. coli lysates) used in an open reaction mixture.
Key Advantage: Direct access to the machinery, allowing for easy monitoring, modulation of conditions, and elimination of cell-membrane transport issues.
Applications: Rapid prototyping of genetic circuits, producing toxic proteins, and building molecular sensors.
Limitations: Lack of compartmentalization leads to degradation over time and inability to maintain complex homeostasis.
Synthetic Cells (Protocells)
Definition: Artificial structures, such as liposomes, that package synthetic biology components (like a cell-free system) inside a membrane to simulate a functional cell.
Key Advantage: Compartmentalization allows for local concentration of components, mimicry of cell architecture, and protection of the internal environment.
Applications: Studying the origin of life, creating artificial organisms for drug delivery, and modeling cellular interactions.
Limitations: High complexity in building the membrane and transporting materials in/out of the structure.
Key Comparisons
Control: Cell-free systems allow direct, open control, while synthetic cells require managing artificial membrane boundaries.
Complexity: Synthetic cells are more complex because they aim to reconstruct a minimal, living-like unit.
Stability: Cell-free reactions can be rapid but fleeting, whereas synthetic cells can be designed for higher stability.
In summary, cell-free systems are a technology for utilizing machinery, while synthetic cells are a platform for engineering life-like structures.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Supplying energy for cell-free protein synthesis reactions is one of the biggest challenges to the success of these systems. Oftentimes, short reaction duration is attributed to an unstable energy source. Traditional cell-free reactions use a compound with a high-energy phosphate bond, such as phosphoenolpyruvate, to generate the ATP required to drive transcription and translation. However, recent work has led to better understanding and activation of the complex metabolism that can occur during cell-free reactions. We are now able to generate ATP using energy sources that are less expensive and more stable. These energy sources generally involve multistep enzymatic reactions or recreate entire energy-generating pathways, such as glycolysis and oxidative phosphorylation. We describe the various types of energy sources used in cell-free reactions, give examples of the major classes, and demonstrate protocols for successful use of three recently developed energy systems: PANOxSP, cytomim, and glucose.
Method for Continuous ATP Supply: Phosphoenolpyruvate (PEP) Regeneration System. A highly effective method to ensure continuous ATP supply is using the PEP/pyruvate kinase (PK) system.
Component: Add Phosphoenolpyruvate (PEP) (approx. 20–30 mM) and Pyruvate Kinase enzyme to the reaction mixture.
Mechanism: Pyruvate kinase catalyzes the transfer of a phosphate group from PEP to ADP, converting it back into ATP, thereby maintaining high ATP/ADP ratios.
Alternative Approach: For reduced cost and inorganic phosphate buildup, using [Glucose or Glucose-6-Phosphate in combination with NAD+]((https://pmc.ncbi.nlm.nih.gov/articles/PMC4651010/) can enable sustained ATP generation through glycolysis.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic cell-free protein synthesis (CFPS) systems, notably E. coli, offer high-yield, low-cost production for simple proteins, while eukaryotic systems (e.g., wheat germ, rabbit reticulocyte) excel in folding complex proteins requiring post-translational modifications. Prokaryotic systems are faster and produce higher protein concentrations (up to 2 mg/mL), whereas eukaryotic systems provide superior machinery for complex post-translational modifications
Prokaryotic vs. Eukaryotic Cell-Free Expression Systems
Prokaryotic (E. coli):
Advantages: Rapid, cost-effective, high yield of protein (>2μg/ml). Ideal for high-throughput screening.
Disadvantages: Lacks endoplasmic reticulum/Golgi, meaning it cannot perform complex PTMs (glycosylation) and often fails to fold complex eukaryotic proteins properly.
Eukaryotic (Wheat Germ, Rabbit Reticulocyte Lysate - RRL):
Advantages: Contains necessary chaperones, folding enzymes, and membranes to support disulfide bonds, glycosylation, and proper post-translational modifications.
Disadvantages: Generally lower yield, higher costs, and slower, more complex protocols compared to E. coli.
Systems and proteins chosen
Prokaryotic-> Green Fluorescent Protein (GFP) As a relatively small, fast-folding protein that does not require glycosylation, GFP is ideal for high-yield expression in E. coli extracts. The goal here is sheer quantity, speed, and cost-effectiveness for imaging or structural studies.
Eukaryotic-> Human Insulin Active insulin requires the formation of specific disulfide bonds to link the A and B chains. Eukaryotic systems (such as Wheat Germ or RRL) provide the necessary chaperone environment to fold this protein correctly, whereas E. coli would likely produce it as an inactive inclusion body.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Protein Aggregation & Misfolding: MPs are hydrophobic and aggregate without a lipid bilayer.
Solution: Introduce pre-formed liposomes or nanodiscs into the reaction, or use detergents to create artificial membrane environments for direct insertion during synthesis.
Low Expression Yields: High costs and time can be wasted if expression is low.
Solution: Use automated liquid handling for small-scale (50
L) screening and optimize the ATP regeneration system.
Template Degradation: Linear templates are vulnerable to endogenous nucleases.
Solution: Use Gam protein to inhibit exonucleases or switch to circular plasmid templates.
Incorrect Redox Potential: The cytoplasm of the lysate is reducing, while many membrane proteins require an oxidizing environment for disulfide bonds.
Solution: Add glutathione (GSH/GSSG) to balance the redox state and promote correct folding.
Degradation by Proteases: Cell lysates contain proteases.
Solution: Add protease inhibitor cocktails directly to the reaction mixture.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
I imagine this has to do with rapid energy depletion. A synthetic cell also has a limited lifespan and components and does not have the ability to regenarate.
Rapid Energy Depletion: Cell-free systems rely on high-energy molecules like ATP and phosphoenolpyruvate (PEP) to fuel transcription and translation. These energy sources are quickly used up in a batch reaction, stopping protein synthesis within 1-3 hours.
Inhibitory Byproduct Accumulation: The metabolic activities within the cell lysate produce byproducts, such as inorganic phosphate, that accumulate and inhibit further synthesis.
Limited Lifespan of Machinery: The transcriptional and translational machinery (ribosomes, enzymes) is not being regenerated as it would be in a living cell, leading to a loss of functionality over time.
- Continuous Exchange Cell-Free (CECF): Using dialysis systems to constantly add energy and remove byproducts can increase reaction times up to 24 hours and boost yields significantly.
- Optimized Energy Systems: Switching from PEP to more efficient systems like glucose or maltodextrin can prevent byproduct accumulation.
- GamS Supplementation: Adding GamS protein helps protect linear DNA from degradation, boosting yield.
Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
Artificial cells translate chemical signals for E. coli.
(a) In the absence of artificial cells (circles), E. coli (oblong) cannot sense theophylline.
(b) Artificial cells can be engineered to detect theophylline and in response release IPTG, a chemical signal that induces a response in E. coli.
Pick a function and describe it.
a. What would your synthetic cell do? What is the input and what is the output?
My synthetic cell will produce vitamin D (cholecalciferol) on demand. Input (Precursor): 7-Dehydrocholesterol (7-DHC). This molecule, present in cell membranes, serves as the provitamin D3. The secondI input (Energy): UVB light (typically 290–315 nm). Output (Product): Previtamin D3, which subsequently rearranges into Vitamin D3 (Cholecalciferol) (see ref below for sources 1 and 2 that helped me figure this out).
Based on synthetic biology principles and the cutaneous vitamin D synthesis pathway, a synthetic minimal cell engineered to produce Vitamin D functions as a specialized, self-contained factory, transforming a specific sterol precursor into the vitamin.
b. Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
Yes, this function can be realized by cell-free Transcription-Translation (Tx/Tl) systems without encapsulation, essentially treating the reaction as a “liquid catalyst” rather than a cell. In a cell-free setup, you would provide the DNA templates for the necessary enzymes (like the cytochrome P450s) directly to a lysate (such as E. coli or wheat germ).
c. Could this function be realized by genetically modified natural cell?
Yes, this function has already been successfully realized using genetically modified natural cells, primarily in yeast
(Saccharomyces cerevisiae) and plants like tomatoes. Unlike minimal synthetic cells, these natural hosts provide an existing metabolic framework (the mevalonate pathway) that can be redirected to accumulate Vitamin D precursors (3).
d. Describe the desired outcome of your synthetic cell operation.
The desired outcome of this synthetic cell operation is the autonomous, high-yield production of active Vitamin D in a controlled environment.
By stripping away the “noise” of a natural organism, the operation achieves three specific goals (1,2,3):
Continuous Synthesis: Unlike humans, who rely on intermittent sun exposure and complex organ signaling (skin to liver to kidney), the synthetic cell provides a one-stop shop. It takes in a simple precursor (7-DHC) and UVB light to output the final, bioactive form (Calcitriol) immediately.
Purity and Specificity: In natural systems, UVB exposure often creates inactive “side products” (like lumisterol) to prevent Vitamin D toxicity. The synthetic cell is engineered to bypass these safety brakes, ensuring that nearly 100% of the input is converted into the useful metabolite without “wasted” chemistry.
Deployment Flexibility: The ultimate goal is to create a “biomachine” that can function where natural cells cannot—such as in a wearable patch that synthesizes Vitamin D through the skin, or as a fortification factory in a bioreactor that produces vegan-sourced Vitamin D3 at an industrial scale.
Essentially, the outcome is a standardized, programmable source of Vitamin D that removes the unpredictability of diet, geography, and skin pigmentation.
Design all components that would need to be part of your synthetic cell. I used google search for this and AI.
To design a synthetic minimal cell for Vitamin D production, you must integrate four primary structural and functional “modules.” This bottom-up design ensures the cell can host the necessary chemistry while remaining lean and efficient.
1. Structural Chassis: The Boundary Layer
The cell requires a physical barrier to concentrate enzymes and protect the reaction.
Composition: A Phospholipid Bilayer made of a mix of zwitterionic lipids like Phosphatidylethanolamine (PE) and anionic lipids like Phosphatidylglycerol (PG).
Stabilizers: Adding Cholesterol or its analogs increases membrane toughness and prevents the leakage of small molecules.
Encapsulation: These lipids are assembled into Giant Unilamellar Vesicles (GUVs), which are large enough (10–100 µm) to be easily monitored and handled.
2. Metabolic Machinery: Membrane-Bound Enzymes
The conversion of precursors into active Vitamin D relies on specific proteins that must be embedded in the membrane.
Internal Organelle (Nanodiscs): Since Vitamin D enzymes are membrane-bound, the cell should contain internal lipid nanodiscs or micelles to provide docking sites for proper protein folding.
Key Catalysts:
25-hydroxylase (CYP2R1): Anchored to the internal membrane to convert cholecalciferol into calcidiol.
1-alpha-hydroxylase (CYP27B1): The “final step” enzyme that produces the active hormone, Calcitriol.
3. Genetic Information Unit: The Software
Instead of a full genome, this cell uses a minimal genetic circuit to maintain its production line.
Genome Chassis: A stripped-down DNA template based on the JCVI-syn3.0 model, which contains only the ~473 genes essential for basic survival.
Custom Operon: A specialized DNA “plug-in” containing the genes for the CYP enzymes mentioned above, controlled by a light-sensitive or chemical promoter to turn production “on” or “off.”
4. Energy and Support: The “Fuel” Module
A minimal cell cannot generate all its own energy, so these components must be supplied or engineered.
External Power: UVB Light (290–315 nm) serves as the primary energy input for the first non-enzymatic step of synthesis.
Cofactor Regeneration: A system to recycle NADPH and provide molecular oxygen, which are required for the hydroxylation steps to continue without stalling.
Transport Proteins: Embedded pore proteins (like alpha-hemolysin) to allow the 7-DHC precursor in and the finished Vitamin D out.
Component Category
Specific Requirement
Justification
Boundary
Phospholipid Bilayer
Maintains the internal environment and chemical gradients.
Genotype
Minimal DNA Circuit
Provides the blueprints for enzyme production with zero “waste”.
Enzymes
CYP2R1 & CYP27B1
Necessary for the multi-step hydroxylation of the sterol backbone.
Logistics
Membrane Pores
Facilitates the “Input-Output” flow of precursors and products.
a. What would be the membrane made of?
To ensure the membrane can support the specific “greasy” chemistry of Vitamin D synthesis, it would be composed of a hybrid phospholipid bilayer designed for stability and enzyme integration. The recipe for the membrane-
The Base: DOPC and DOPE
DOPC (Dioleoyl-phosphatidylcholine): This provides the flexible, fluid matrix. It is the “standard” lipid that forms the stable bilayer structure.
DOPE (Dioleoyl-phosphatidylserine): This adds “curvature” to the membrane, which is essential for helping the bulky CYP enzymes (like CYP2R1) wedge themselves into the surface without destabilizing the cell.
The Functional Anchor: DGS-NTA(Ni)
Since your enzymes are the stars of the show, you include a small percentage of Nickel-chelating lipids.
Why? These act like “Velcro” for your proteins. If your enzymes are engineered with a Histidine-tag (His-tag), they will snap onto these lipids, ensuring they stay anchored to the membrane where the reaction happens.
The Fluidity Regulator: Cholesterol
While the cell’s goal is to make a cholesterol-like product, you must include Cholesterol (around 10–20%) in the membrane itself.
Why? It acts as a “buffer” for membrane thickness and prevents the lipid wall from becoming too leaky or too stiff as temperature changes during UVB exposure.
The Protective Coating: PEG-Lipids
PEG-DSPE: A lipid with a “tail” of Polyethylene Glycol.
Why? This creates a “stealth” layer on the outside of the cell, preventing it from clumping together with other cells or sticking to the walls of your container (bioreactor or patch).
Transport Pores: Alpha-Hemolysin
Strictly speaking, this is a protein, but it functions as part of the membrane. These are “holes” that allow the 7-DHC input to enter the cell and the Vitamin D output to exit, as the lipid bilayer alone is too dense for them to pass through quickly.
Summary Profile:
Phase: Liquid-disordered (fluid)
Charge: Slightly negative (to mimic natural endoplasmic reticulum membranes)
Stability: High (enhanced by cholesterol)
b. What would you encapsulate inside? Enzymes, small molecules.
To make the synthetic cell functional, the interior (the lumen) must contain the “operating system” and the raw materials needed to build and power the Vitamin D synthesis machinery.
Since this is a minimal cell, you would encapsulate:
1. The Genetic Instructions (DNA/RNA)
DNA Plasmids: Coding for the three essential enzymes: 7-DHC reductase (to manage precursor flow), CYP2R1, and CYP27B1.
Alternative (mRNA): If you want a faster “start-up,” you can encapsulate the mRNA directly, bypassing the need for transcription.
2. Transcription/Translation Machinery (Tx/Tl)
Since the cell has no natural nucleus or ribosomes, you must “borrow” them:
Ribosomes: Sourced from E. coli or wheat germ lysates to build the enzymes from the DNA/RNA.
RNA Polymerase: To read the DNA templates.
tRNAs and Amino Acids: The 20 standard building blocks required to assemble the protein enzymes.
3. Energy and Redox Small Molecules
The Vitamin D “engine” (CYP enzymes) requires a constant flow of “fuel”:
ATP, GTP, UTP, CTP: The chemical energy needed to power protein synthesis.
NADPH: The critical electron donor for the hydroxylation steps.
Creatine Phosphate & Creatine Kinase: A “battery” system to regenerate ATP as it gets used up, extending the cell’s lifespan.
4. Supporting Small Molecules & Buffer
Magnesium and Potassium Salts: Essential ions that stabilize DNA and keep ribosomes functional.
Reducing Agents (DTT or TCEP): To prevent the enzymes from oxidizing and clumping in the watery interior.
Molecular Oxygen ($O_2$): While a gas, it must be dissolved in the internal fluid as a co-substrate for the hydroxylation reactions.
5. Specialized “Docking” Components
Nanodiscs or Small Unilamellar Vesicles (SUVs): Tiny internal lipid “rafts.” Since the CYP enzymes are hydrophobic, they need these internal membranes to fold correctly before they eventually migrate to the main outer membrane.
Summary of the “Inside”:
It would look like a concentrated molecular soup—highly viscous, packed with the machinery of life, and primed to start building enzymes the moment the temperature or light conditions are right.
c. Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
A bacterial Tx/Tl system (specifically an E. coli lysate like PURE) is actually the best fit for this minimal cell, provided we address one specific “mammalian” requirement.
d. How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
The synthetic cell will communicate with its environment through a hybrid permeability strategy. Since Vitamin D synthesis involves bulky, hydrophobic sterols, we cannot rely solely on the lipid bilayer’s natural diffusion rates. [1, 2]
1. Passive Diffusion (For Lipophilic Substrates)
The primary input, 7-Dehydrocholesterol (7-DHC), and the output, Vitamin D3 (Calcitriol), are highly lipophilic.
The Mechanism: They can naturally partition into and move through the lipid membrane.
The Limitation: This process is relatively slow and can cause the membrane to become “saturated” with grease, potentially destabilizing the cell or slowing down production as the product builds up inside. [3, 4]
2. Engineered Membrane Channels (For Speed and Small Molecules)
To make the cell a truly efficient “factory,” we must express or insert specific membrane pores and transporters:
Alpha-Hemolysin ($\alpha$HL): We will express this bacterial pore to create non-selective 1.4 nm “holes.” This allows for the rapid exchange of hydrophilic small molecules like ATP, NADPH, and metal ions (Mg²⁺/K⁺) if the cell is sitting in a nutrient-rich “feed” solution.
Cyclodextrin “Shuttles”: Since the sterols (7-DHC and Vitamin D) are so hydrophobic, we can add methyl-beta-cyclodextrin to the external environment. These act as molecular “taxis,” picking up the produced Vitamin D from the membrane surface and carrying it into the bulk liquid, maintaining the concentration gradient.
Stimuli-Responsive Gating: If we want to control when the cell releases its cargo, we could express a mechanosensitive channel like MscL. By engineering it to open under specific triggers (like pH changes or light), we can “flush” the cell’s contents on command. [5, 6, 7, 8, 9]
3. Light Communication
Because the first step of the pathway is photochemical, the environment “communicates” with the cell via UVB light (290–315 nm). This is a wireless input that doesn’t require a physical channel; it simply passes through the membrane to trigger the isomerisation of 7-DHC. [10]
Summary:
The cell uses passive diffusion for the “greasy” stuff (helped by external shuttles) and expressed $\alpha$HL pores for the “watery” energy inputs.
a. List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
To finalize the experimental blueprint for your Vitamin D-producing synthetic cell, here are the specific lipids and genes required for assembly.
a. Lipids (The Membrane Matrix)
The lipid composition is tailored to support CYP enzyme anchoring and maintain structural integrity during UVB exposure.
Lipid Component
Full Name
Molar Ratio
Role
DOPC
1,2-dioleoyl-sn-glycero-3-phosphocholine
50%
The primary fluid bilayer matrix.
DOPE
1,2-dioleoyl-sn-glycero-3-phosphoethanolamine
20%
Promotes membrane curvature for enzyme insertion.
Cholesterol
(Purified)
20%
Stabilizes the membrane against UVB-induced leakiness.
We will use a modular DNA plasmid approach. These genes are codon-optimized for the E. coli Tx/Tl system to ensure high expression.
Gene Symbol
Origin
Function
CYP2R1
Human/Mammalian
Vitamin D 25-hydroxylase: Converts cholecalciferol into calcidiol.
CYP27B1
Human/Mammalian
1$\alpha$-hydroxylase: Converts calcidiol into bioactive Calcitriol.
ADR
Bovine/Human
Adrenodoxin Reductase: Essential electron transfer partner for the CYP enzymes.
ADX
Bovine/Human
Adrenodoxin: Small iron-sulfur protein that shuttles electrons to the CYPs.
hlyA
S. aureus
Alpha-Hemolysin: Pore-forming protein for nutrient/product exchange.
tetR
E. coli
Tet Repressor: Provides the “On/Off” logic gate for the system.
The “Logic” Layout:
Pores: hlyA is expressed under a constitutive (always-on) promoter to ensure the cell can “breathe” immediately.
Synthesis: CYP2R1, CYP27B1, and the electron partners (ADR/ADX) are placed under a Tet-regulated promoter.
Activation: The system remains dormant until anhydrotetracycline (aTC) is added to the environment, triggering the “Start” of Vitamin D production.
b. How will you measure the function of your system?
To confirm the cell is actually “breathing” and producing Vitamin D, we need to track both the disappearance of the precursor and the appearance of the active hormone. We will use a three-tier approach to measure function:
1. The Gold Standard: LC-MS/MS
Since Vitamin D metabolites (7-DHC, D3, 25(OH)D, and Calcitriol) look very similar, Liquid Chromatography-Tandem Mass Spectrometry is the only way to be 100% sure of the chemical identity.
The Process: Take samples of the external “bath” liquid at specific time intervals.
What we look for: A clear shift in the mass-to-charge ratio (m/z) that matches the addition of hydroxyl groups (-OH) at the 25 and 1$\alpha$ positions.
2. Real-Time Tracking: Fluorescent Sterols
We can use a 7-DHC analog tagged with a fluorescent marker (like BODIPY).
The Process: Use a Confocal Microscope to watch the cell.
What we look for: If the fluorescent sterols move from the outside of the cell, concentrate in the membrane, and then change their emission wavelength (if using a “smart” probe sensitive to hydroxylation), we know the transport and metabolic machinery are working in real-time.
3. Biological Activity Assay (The “Calcitirol” Test)
To prove the output is biologically active and not just a “dead” chemical isomer, we can perform a VDR (Vitamin D Receptor) Reporter Assay.
The Process: Take the output from the synthetic cell and add it to a separate culture of natural cells (like HEK293) that have been engineered with a VDR-linked Luciferase gene.
What we look for: If the natural cells “glow,” it confirms that the synthetic cell successfully produced 1,25-dihydroxyvitamin D3 in a form that the body can actually use.
4. Metabolic Health: Oxygen Consumption
Since the CYP enzymes consume molecular oxygen ($O_2$) to function, we can use a Clark-type electrode or oxygen-sensitive fluorescent patches in the reaction vessel.
What we look for: A steady drop in $O_2$ levels immediately following the addition of the “On” switch (aTC), which acts as a proxy for enzymatic activity.
Homework question from Peter
Synthetic biology without living cells! Screenshots from the presentations.
Freeze-drying technology lyophilization, originated during World War II to transport blood plasma and penicillin, later evolving into a critical method for preserving food and pharmaceuticals. It works by freezing materials and reducing pressure, allowing water to transition from ice to vapor (sublimation).
Freeze-dried cell free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field - Architecture, Textiles/Fashion, or Robotics, and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept.
I chose to explore textiles/fashion applications of cell-free systems. Can I use this freeze fried cell free system to make a wearable that warns us against radiation exposure? I was really inspired by this project but it uses freeze fried engineered yeast (a wearable microbrewery) to save hospital lab workers from daily radiation exposure. Similarly I want to make a wearable biosensor that produces melanin when radiation is detected.
How will the idea work, in more detail? Write 3-4 sentences or more.
The core mechanism involves using tyrosinase enzymes to convert precursor molecules (like L-DOPA or L-Tyrosine) into melanin, which is dark brown/black, creating a visible, light-absorbing color change.
The Freeze-Dried Cell-Free System
Source Fungi: Use genes from radiotrophic fungi such as Cryptococcus neoformans or Cladosporium sphaerospermum.
Key Enzyme: The gene for tyrosinase (TYR) is essential, as it catalyzes the production of melanin, a pigment that changes color from colorless or light-colored to black/brown.
System Formulation: Use Escherichia coli lysate-based cell-free systems, which can be lyophilized (freeze-dried) and stored without refrigeration, designed to rehydrate instantly when exposed to moisture (like sweat).
Lyophilization: The reaction mixture—containing the enzyme-producing machinery, DNA blueprints (plasmid), and precursors (e.g., L-tyrosine)—is freeze-dried into a “paper-based” or powder format for integration into fabric.
Making the Wearable Device
Sensor Design: The cell-free reaction is embedded into a flexible substrate, such as a thin silicone film (PDMS) or directly into fabrics like cotton, to create a wearable “patch” or textile sensor.
Activation Mechanism: To make it change color upon encountering a specific condition (e.g., radiation, or simply a passive environmental check), the lyophilized capsule/paper is placed on the fabric. When the user sweats (or a small amount of water is added), the system rehydrates, initiates protein expression, and produces melanin.
Fabric Integration: The melanin-producing cell-free reaction, when dried, can be integrated into fabric microfluidic channels or porous materials that allow the reaction to proceed and display the color change on the surface.
Achieving the Color Change
Light/Dark Shift: Because melanin is highly black, the fabric will turn dark in the areas where the freeze-dried system is activated.
Enhanced Functionality: The produced melanin acts as a photo-protective layer, enhancing the wearable’s ability to absorb UV or ionizing radiation.
Conceptual Steps for Assembly
Engineered Cells: Create a cell-free reaction designed to express tyrosinase from C. neoformans.
Lyophilize: Freeze-dry the lysate in a protective sugar matrix (e.g., trehalose) on a small membrane or paper patch.
Encapsulate: Place this paper patch inside a flexible, sweat-permeable, transparent, or breathable membrane patch.
Wear: Apply to the skin (e.g., as a "smart tattoo", smart clothing layer or spray a coat on a wearable 3d printed piece).
Activate: Upon moisture/sweat, the melanin produces color, turning the patch dark brown or black.
What societal challenge or market need will this address?
In a world where we are encountering so much terrestrial pollution from nearby wars and destruction of the environment, radiation pollution and damage is becoming a real every day problem. In addition, this idea will be useful for astronauts for space exploration, nuclear plant workers and hospital radiology workers. Unfortunately, citizens encounter problems from radiation exposure on a daily basis too.
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
Challenge/limitations
Solution
High temperature denaturation
Use lyoprotectants (trehalose, maltodextrin)
Sensitivity issues
Use CRISPR-based tools & toehold switches
Rehydration failure
Integrate into hydrogels or porous substrates
Evaporation/Stability
Lamination and sealed reaction modules
Homework question from Ally
Georg asked me to use my critical side to the homework question, haha. My comment was that I do not believe in space colonisation by humans.
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
My challenge is to find ways to warn for radiation exposure as well as ways of protection from radiation which is a terrestrial as well an extraterrestrial issue for humans and more than human ecosystems. Some ecosystems and kingdoms have developed mechanisms for protection against radiation and oxidative stress, like fungi! Mushrooms secrete melanin as a protective mechanism agains oxidative stress and radiation exposure. We can extract melanin from fungi and freeze dry it.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
Cell-free, freeze-dried (lyophilized) melanin-producing systems involve extracting melanin precursors, such as purified mushroom tyrosinase, and using them in a dehydrated reaction format. Once freeze-dried, these preparations can be stored at room temperature without cold-chain requirements and rehydrated at the point-of-use for applications in biosensors, UV-resistant shielding, and medical diagnostics.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
Cell-free, freeze-dried melanin from fungi is a cutting-edge biomedical and bio-material technology being explored for deep-space missions. By utilizing extracted fungal enzymes (like tyrosinase) via reconstituted cell-free systems, melanin can be synthesized efficiently for radiation-shielding topical creams or structural composites to protect astronauts from hazardous cosmic rays.
Clearly state your hypothesis or research goal and explain the reasoning behind it (Maximum 150 words)
My hyporthesis is that by utilizing extracted fungal enzymes like tyrosinase through cell free systems we can efficiently synthesize radiation shielding materials, structural composites and even topical creams to protect astronauts from radioactivity.
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
We can use the biobits to express tyrosinase enzymes that are responsible for fungal melanin production in space.
AI overview
Experimental Samples & Plasmid Design
Experimental Sample (Melanin Production): A FD-CF reaction pellet rehydrated with water and a DNA plasmid containing the gene for the key melanin-producing enzyme (e.g., tyrosinase) sourced from a melanin-producing mushroom such as Cryptococcus neoformans or Coprinopsis cinerea.
Precursor Molecules: To facilitate melanin synthesis in the cell-free environment, the reaction will be supplemented with pathway substrates like L-DOPA or L-Tyrosine.
Experimental Controls
Positive Control 1 (Protein Synthesis): FD-CF pellets rehydrated with DNA encoding a stable, easily detectable reporter protein, such as Superfolder Green Fluorescent Protein (sfGFP) or mCherry. This ensures the cell-free machinery is active and viable.
Positive Control 2 (Enzyme Baseline): A cell-free reaction expressing the tyrosinase enzyme but lacking the L-DOPA/L-Tyrosine substrate to confirm the enzyme’s transcription and translation without the pigment end-product.
Negative Control (No DNA): A FD-CF reaction rehydrated with water only (no template DNA) to establish the baseline of autofluorescence and spontaneous, non-enzymatic melanin browning.
Colorimetric/Pigmentation Analysis: Visually document and photograph the samples over the 24-hour incubation period. Pigment buildup can be tracked by measuring the absorbance of the solution at (400 \text{ nm} - 475 \text{ nm}) using a microplate reader or a spectrophotometer.
Fluorescence Verification: Monitor the controls by measuring fluorescence emissions (e.g., excitation (\sim 485 \text{ nm}), emission (\sim 510 \text{ nm}) for GFP) using a portable tool like the P51 Molecular Fluorescence Viewer..
Radiation-Shielding Assay (Optional): Once melanin has been produced, test the cell-free samples for UV-blocking efficacy by subjecting the samples to UV irradiation and assessing enzymatic survival compared to non-melanized controls.
Homework Part B - Individual Final Project Report
Mandatory for Committed Listeners and MIT/Harvard Students. We’d like students to start exploring their final project in depth this week! The minimum requirement is filling out Aim 1.
See this link if you want examples of what you can include in your answers for each question. Note: the question numbers do not line up, and you can ignore any word requirements in this linked document.
Homework is based on data that will be generated in the Waters Immerse Lab in Cambridge, MA. Students will be characterizing green fluorescent protein (eGFP, a recombinant protein standard) structure (primary, secondary/tertiary) in the lab using liquid chromatography and mass spectrometry. Data generated in the lab will be available on-line for Committed Listeners.
Homework: Final Project
Before I decide what to measure I need to:
Decide which types of substrates or food or environmental contaminants I want to use for training my mycelium with. We will train it naturally to degrade a variety of different types of petroleum derived polymers and environmental contaminants but also synthetically through DNA sequencing and optimising the enzymatic degradation process through gene editing technologies and methods.
Decide which types of mycelium I am going to use for my experiment based on the type of contaminants or petroleum derived polymers I want my mycelium to remediate. I need to pick at least a strain that already has the capability of degrading petroleum derived polymers, one strain that is not particularly able to break down contaminants (for engingeering and optimising the enzymatic degradation pathways) and one that can break down other types of enviromental pollutants or contaminants like heavy metals.
The hypothesis is that we can measure the bioremediation process through looking into the enzymatic degradation process and the action potential (electrical activity) of mycelial growth/networks using different types of stimulation (aka food or substrates, contaminants etc). Enhancing or introducing plastic-degrading enzyme pathways—through selective exposure or through genetic modification will alter metabolic activity and produce distinct electrophysiological patterns that can be used as control inputs for actuating a soft robotic system.
For my final project in this weeks homework
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
+action potential (electrical charge)
+extracellular enzymes (mass spectrometry)
+DNA sequencing, amplification and editing to optimise extracellular pathways during mycoremediation of synthetic petroleum derived polymers (LDPE)
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
I need to learn how to DNA sequence specific fungal strains and look into how mass spectrometry can help me understand how mycelium interact and talk. For the extracellular enzymes mass spectrometry is needed.
I need to measure fungi action potential-> μV -> electrical spikes from stimulation and without stimulation.
Action potentials are measured in neuroscience to understand how neurons communicate, as they are the fundamental “language” of the brain. They are brief electrical impulses that travel along neurons, firing in an all-or-none manner to transmit signals, with the frequency of these spikes representing the intensity of stimuli or information. Similarly, Mycelium networks generate action potential-like electrical spikes to communicate environmental changes, such as changes in moisture, nutrients, or physical injury. I found some interesting papers like this one that talks about “Action potentials” in Neurospora crassa, a mycelial fungus.
I want to measure the environmental response or Stimulation: Electrical activity increases in frequency when the mycelium is stimulated with attractive resources (wood) or stressors (salt, chemical agents).
Technologies used to carry out my final project (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
For cultivation: Cultivation of mycelia using a variety of scaffolds, substrates and contaminants.
For optimising enzymatic degradation pathways: Mass spectrometry, DNA sequencing, amplification and editing.
For the fabrication of the robot: digital fabrication (technical drawing and 3D modeling on Rhinoceros, 3D printing)
For the sensing part and actuation of the soft robot: capture activity via electrodes, to interface and translate these signals into actuation with microcontroller-driven soft robotic systems.
Homework: Waters Part 1 — Molecular Weight
*We will be analyzing an eGFP standard onto a BioAccord LC-MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the denatured (unfolded) state. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
Based only on the predicted amino acid sequence of eGFP (see below), what is the calculated molecular weight? You can use an online calculator like the one at expasy.org.
From the recitation-> notes to remember on how to calculate the theoritical molecular weight of eGFP.
eGFP Sequence:
VSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNYNSHNV YIMADKQKNG IKVNFKIRHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LSTQSALSKD PNEKRDHMVL LEFVTAAGIT LGMDELYKLE HHHHHH
Note: This contains a His-purification (HHHHHH) tag and a linker (the LE before it).
The molecular weight of eGFP with added linker and His-tag is 28006.60 Da.
Soooo there is a trick here!
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
i. Determine z for each adjacent pair of peaks (n, n+1) using:
iii. Calculate the accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1 using:
Figure 1. Mass Spectrum of intact eGFP protein from the Waters BioAccord LC-MS (a mass spectrometer with 10,000 resolution) with individual charge state peaks labeled with m/z values.
Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
No because the charge state cannot be determined for the zoomed-in peak from this figure alone. In order to determin the charge state we need at least two adjacent peaks, so their spacing can be used to calculate z. In the zoomed region, only a single isolated peak is shown and no neighboring charge-state peak is visible.
Homework: Waters Part 2 — Secondary/Tertiary structure
We will be analyzing eGFP in its native, folded state and comparing it to its denatured, unfolded state on a quadrupole time-of-flight MS. We will be doing MS only analysis (no liquid chromatography) on the Xevo G3-QToF MS.
Based on bonus learnings in the lab, please explain the difference between native and denatured protein conformations. For example, what happens when a protein unfolds? How is that determined with a mass spectrometer? What changes do you see in the mass spectrum between the native and denatured protein analyses (Figure 2)?
Figure 2. Comparison of the mass spectra between denatured (top) and native (bottom) eGFP standard on the Waters Xevo G3 QTof MS.
In the native state -> eGFP stays folded and compact, so it has fewer exposed protonation sites and usually carries fewer charges, which gives peaks at higher m/z.
In the denatured state -> the eGFP protein unfolds, exposes more basic sites, and picks up more charges, so the spectrum shifts to a broader charge-state distribution at lower m/z.
Zooming into the native mass spectrum of eGFP from the Waters Xevo G3 Q-Tof MS (see Figure 3), can you discern the charge state of the peak at ~2800 m/z? What is the charge state? How can you tell?
Figure 3. Native eGFP mass spectrum from the Waters Xevo G3 Q-Tof MS. The inset is a zoomed-in view of the charge state at ~2800
on a mass spectrometer with 30,000 resolution.
At 2800 m/z the charge state is 10.
Homework: Waters Part 3 — Peptide Map Work - primary structure
We will be digesting eGFP protein standard into peptides using Trypsin (an enzyme that selectively cleaves the peptide bond after Lysine (K) and Arginine (R) residues. These peptides, resulting from the digested eGFP will be analyzed by LC-MS to measure their molecular weight and to fragment them to confirm the amino acid sequence within each peptide – generating a Peptide Map. This process is used to confirm the primary structure of the protein.
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the sequence listed above. (note: Adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
I found 6 lysines(K) and 20 Arginines(R) in eGFP.
There are a variety of tools available online to calculate protein molecular weight and predict a list of peptides generated from a tryptic digest. We will be using tools within the online resource Expasy (bioinformatics resource portal of the SIB Swiss Institute of Bioinformatics) to predict a list of tryptic peptides from eGFP.
How many peptides will be generated from Tryptic digestion of eGFP?
i. Navigate to https://web.expasy.org/peptide_mass/
ii. Copy/paste the sequence above into the input box in the PeptideMass tool to generate expected list of peptides.
iii. Use Figure 2 below as a guide for the relevant parameters to predict peptides from eGFP.
I chose the relevant parameters to predict peptides from eGFP.
Figure 2. Example conditions for predicting the number of tryptic peptides from eGFP standard. Please replicate all parameters shown above.
iv. Click “Perform the Cleavage” button in the PeptideMass tool and report the number of peptides generated when using Trypsin.
Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 3a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
Figure 3a. Example LC-MS Chromatogram for eGFP Peptide Map. The peak at 2.78 minutes is circled, and its MS data is shown in the mass spectrum in Figure 3b, below.
I counted 19 peaks before counging all peaks that are >10% relative abundance. The highest peak is at 4.78 minutes in which in which the counts are 1.2e7.
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from Step 2.3 and 2.4? Are there more peaks in the chromatogram or fewer?
There are only 19 peaks or peptides generated from expasys above in steps 2.3 and 2.4. In the chromatogram we can see more peaks but we should only cound the >10% relative abundance. The little spikes have to do with impurities in the sample. Linsday mentioned even something from your hair or fingernails even when you are wearing gloves could add impurities to the mix.
Identify the mass-to-charge (m/z) of the peptide shown in Figure 3b. What is the charge (z) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide ([M+H]+) based on its m/z and z.
Figure 5b. Mass spectrum figure to show m/z for the chromatographic peak at 2.78 min from Figure 5a above. The inset is a zoom-in of the peak at m/z
525.76, to discern the isotope peaks.
The most prominent peak in Figure 3b is at m/z 525.76712. From the other zoomed-in inset in the image, the m/z values of two adjacent major isotopic peaks are approximately 525.76712 and 526.25918.
m/z= 525.76712
The difference between these values is 526.25918-525.76712 = 0.492
z= 0.492 -> 0.5
Singly Charged Mass (MH+): (525.767 * 2) - 1.0078 = 1050.53 Da.
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm.
Comparing the mass to the predicted list, the peptide is FEGDTLVNR (Theoretical MH+ = 1049.52 Da).
What is the percentage of the sequence that is confirmed by peptide mapping? (see Figure 6)
Figure 6. Amino Acid Coverage Map of eGFP based on BioAccord LC-MS peptide identification data.
It is 88%.
Homework: Waters Part IV — Oligomers
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS). CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
Using the given subunit masses below-
Polypeptide Subunit Name
Subunit Mass
7FU
340 kDa
8FU
400 kDa
Table 1: KLH Subunit Masses
Figure 7. Mass spectrum of Keyhole Limpet Hemocyanin (KLH) acquired on the CDMS.
Please fill out this table with the data you acquired from the lab work done at the Waters Immerse Lab in Cambridge, or else the data screenshots in this document if you were unable to have lab work done at Waters.
Theoretical
Observed/measured on the Intact LC-MS
PPM Mass Error
Molecular weight (kDa)
28.006
27.982
252.6 ppm
Week 11 — Bioproduction & Cloud Labs
WEEK 11 HW
This week examines how modern bioproduction pipelines, from strain engineering to fermentation and downstream processing, are increasingly designed, executed, and optimized through cloud lab platforms and automation — enabling remote, high-throughput, and reproducible synthetic biology at industrial scale.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
How it started….
How it evolved…
How it ended… Highjacked!
Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST.
+A personalized URL was sent to the email address associated with your Discourse account, and you can discuss the artwork on the Discourse.
+If you did not have a chance to contribute, it’s okay, just make sure you become a TA this fall! 😉
Make a note on your HTGAA webpages including:
+what you contributed to the community bioart project (e.g., “I made part of the DNA on the bottom right plate”)
+what you liked about the project, and
+what about this collaborative art experiment could be made better for next year.
I made the part of the DNA in the middle of the upper 2 plates but unfortunately it was highjacked as you can see on the top! I really enjoyed this collaborative part.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase) -> BL21 (DE3) Star strains feature a crucial mutation in the rne gene (encoding RNase E).
Salts/Buffer
Potassium Glutamate -> Supplies a high concentration of potassium ions to mimic the intracellular environment. It supports protein-nucleic acid interactions, ensures optimal charge balancing, and provides a mild counter-ion for metabolic efficiency
HEPES-KOH pH 7.5 -> A zwitterionic organic chemical buffering agent used to maintain the reaction at a physiological pH of 7.5, which is necessary to prevent enzymes from denaturing during transcription and translation.
Magnesium Glutamate -> Supplies magnesium ions ((Mg^{2+})) required by enzymes to synthesize proteins, specifically for proper ribosome function and stabilizing mRNA.
Potassium phosphate monobasic and Potassium phosphate dibasic -> These two salts are combined in a specific ratio to act as an inorganic pH buffer. They also supply essential inorganic phosphate ((P_{i})) which is required to continuously drive the energy regeneration (ATP/GTP) cycle during protein expression.
Ribose -> gives main energy source for ATP production.
Glucose -> energy source, provides a long lasting source of energy.
AMP
CMP
GMP
UMP
Guanine
All the above are the basic building blocks we need to make rna.
Translation Mix (Amino Acids)
These are all building blocks for making proteins.
17 Amino Acid Mix -> Serves as the primary raw material supply for the translation machinery to build nascent polypeptide chains.
Tyrosine -> A standard, aromatic amino acid used directly to build proteins with specific hydrophobic and hydrogen-bonding motifs. In certain specialized CFPS applications (like the in vitro maturation of hydrogenases), Tyrosine acts as a direct biochemical substrate for radical SAM enzymes to synthesize complex reaction intermediates and cofactors.
Cysteine -> Cysteine contains a highly reactive thiol group. It is essential for the proper formation of di-sulfide bridges, which dictate the tertiary structure, stability, and function of secreted or complex proteins.
Nicotinamide-> Nicotinamide fundamentally directs cell-free reactions. It acts as a biochemical building block for synthesizing (NAD^{+}) and (NADP^{+}) via salvage pathways, functions as an inhibitor of NAD-consuming enzymes like sirtuins to prevent consumption, and helps regulate metabolic networks by driving cofactor availability and recycling.
Backfill
Nuclease Free Water-> Prevents the degradation of DNA templates and RNA transcripts by DNases and RNases. It ensures maximum protein yield, reaction reproducibility, and prevents premature halting of the translation machinery.
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
Main differences:
Speed: PEP-NTP is designed for fast, high-intensity, short-term production (1-hour), whereas NMP-Ribose-Glucose is designed for slow, sustained, long-term production (20-hours).
Cost & Economics: The 1-hour PEP-NTP mix is more expensive due to substrates like phosphoenolpyruvate, while the 20-hour mix is more economical, using Glucose/Ribose.
Metabolic Byproducts: The 20-hour mix often has better control over metabolic byproducts (such as inorganic phosphate) due to its slower, more balanced energy consumption, leading to longer reaction stability.
Bonus question: How can transcription occur if GMP is not included but Guanine is?
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
Superfolder GFP is a basic (constitutively fluorescent) green fluorescent protein published in 2005, derived from Aequorea victoria. It is reported to be a very rapidly-maturing weak dimer. Engineered for robust folding, even under non-optimal conditions, making it highly reliable in cell-free systems.
It has slow maturation and lower photostability, which delays fluorescence signal and reduces effective brightness in short or energy-limited cell-free reactions. Slow maturation can delay readout despite successful expression.
It is commonly used as a fusion tag for tracking protein localization and dynamics, particularly in long-term imaging due to its high photostability. mKO2 is known for relatively rapid maturation compared to other fluorescent proteins, which is critical for studies requiring fast protein turnover tracking. It is generally stable within a wide pH range, though like most fluorescent proteins, its fluorescence decreases in highly acidic environments. It is often preferred over dsRed-type proteins for studies in slightly acidic compartments, although it is not specifically designed as a strict pH sensor. The protein is highly monomeric and folds well, making it suitable for fusions to proteins where dimerization might cause dysfunction or localization issues.
High quantum yield and fast maturation. Faster but limited still folding limited compared to GFP making its fluorescence more sensitive to translation kinetics and folding efficiency. mTurquoise2 is highly stable in acidic environments, exhibiting very low acid sensitivity.
It has moderate acid sensitivity, with a high tolerance to acidic environments. A faster-maturing variant than mScarlet (T74I mutation) matures in roughly 25–36 minutes at 37°C. mScarlet-I is generally preferred for fusions requiring fast-maturing, bright red tags.
Displays oxygen-dependent chromophore formation, so fluorescence requires sufficient oxygen availability, which can be limiting in dense or sealed reactions. Its performance is typically influenced by trade-offs between brightness, folding efficiency, and maturation kinetics, meaning signal output depends strongly on how well it folds and matures in the cell-free environment.
The amino acid sequences are shown in the HTGAA Cell-Free Benchling folder.
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
I will pick to maximize the fluoresence of Electra2 over a 36-hour icubation period. This protein displays oxygen-dependent chromophore formation, so fluorescence requires sufficient oxygen availability, which can be limiting in dense or sealed reactions. Its performance is typically influenced by trade-offs between brightness, folding efficiency, and maturation kinetics, meaning signal output depends strongly on how well it folds and matures in the cell-free environment. According to AI we have to prevent the rapid depletion of the energy regeneration system, avoiding nuclease-mediated mRNA degradation, and fine-tuning salt concentrations.
My suggestions are:
Optimize Magnesium and Potassium Ratios: Final protein yields depend heavily on the concentrations of both Magnesium and Potassium ions, which are vital for ribosome structural integrity over extended synthesis times. Perform a matrix screen to balance these ions against your PEG-8000 concentration for maximal yield.
Increase ribose to prevent the rapid depletion of energy since we want to maximize fluorescence over a 36-hour incubation period.
The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24). You can begin composing master mix compositions here.
Unfortunately all the wells I added have been highjacked so I did not know which wells to choose -.-
The final phase of this lab will be analyzing the fluorescence data we collect to determine whether we can draw any conclusions about favorable reagent compositions for our fluorescent proteins. This will be due a week after the data is returned (date TBD!). The reaction composition for each well will be as follows:
6 μL of Lysate
10 μL of 2X Optimized Master Mix from above
2 μL of assigned fluorescent protein DNA template
2 μL of your custom reagent supplements
Total: 20 μL reaction
PART D - BONUS- BUILD A CLOUD LAB
Ginkgo Nebula Cloud Laboratory Rendering, 2025
Use this simulation tool to create an interesting looking cloud lab out of the Ginkgo Reconfigurable Automation Carts. This is just a minimal implementation so far, but I would love to see some fun designs!
Week 12 HW - Building Genomes
Week 12 HW
This week focuses on designing, synthesizing, and editing whole genomes, from minimal cells to refactored microbes and synthetic chromosomes.
WEEK 13 HW: AI, SynBio, and Scaling Health Innovation
WEEK 13 HW
This week covers designing, programming, and fabricating engineered living materials — such as self-healing concretes, adaptive biofilms, and responsive biomaterials — by integrating genetic circuit design, materials science, and bioprocess engineering.

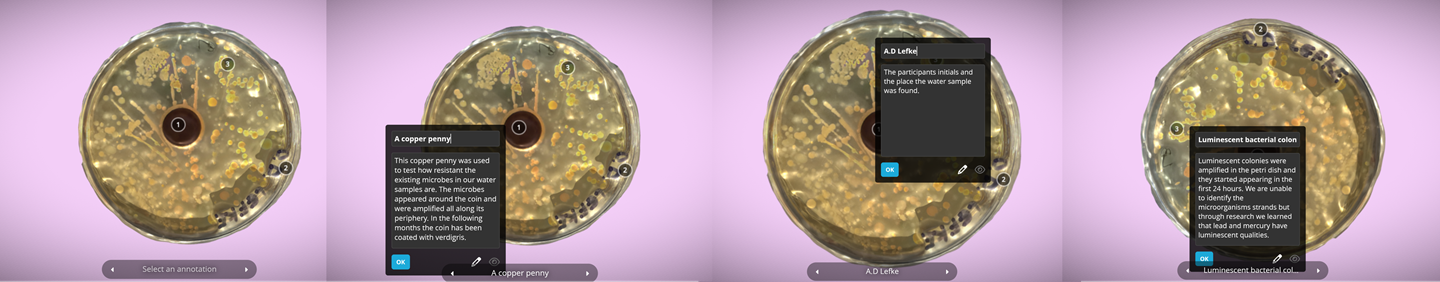

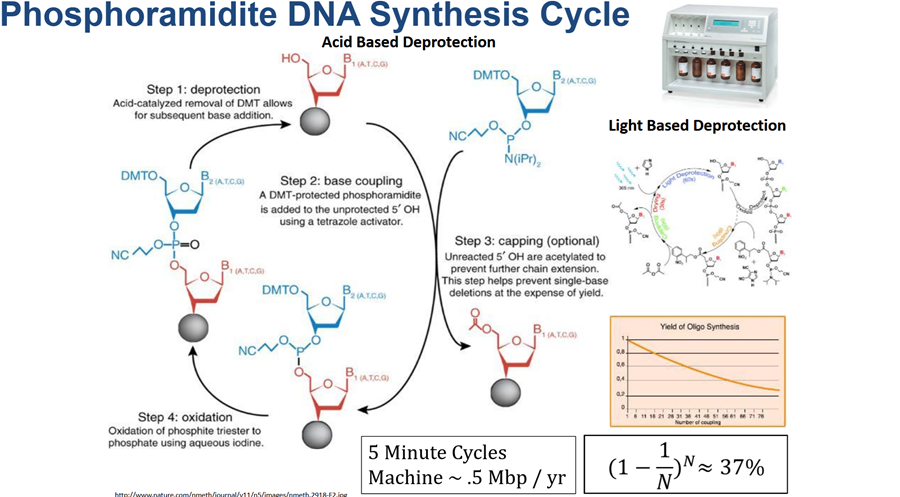
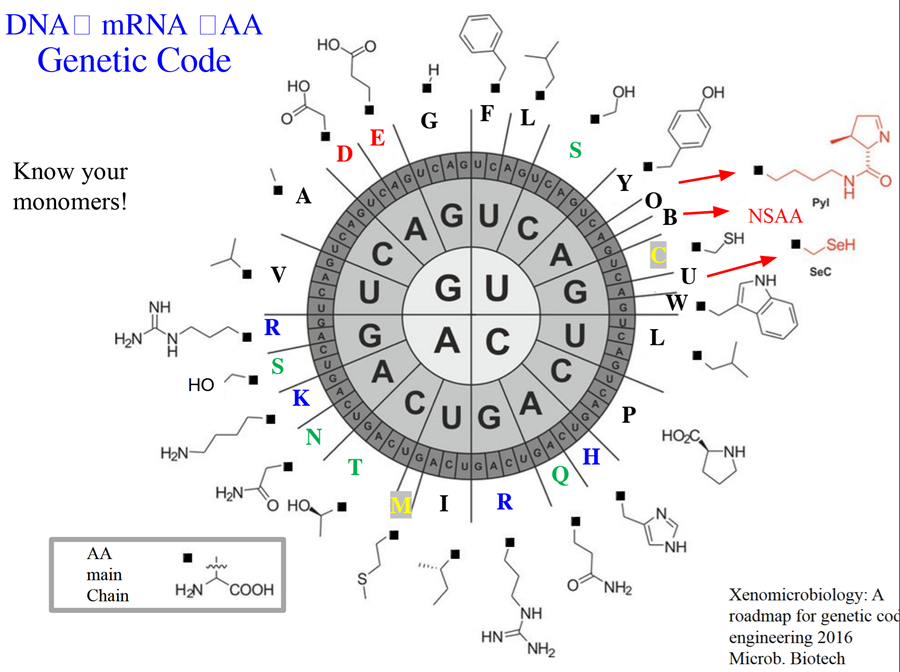


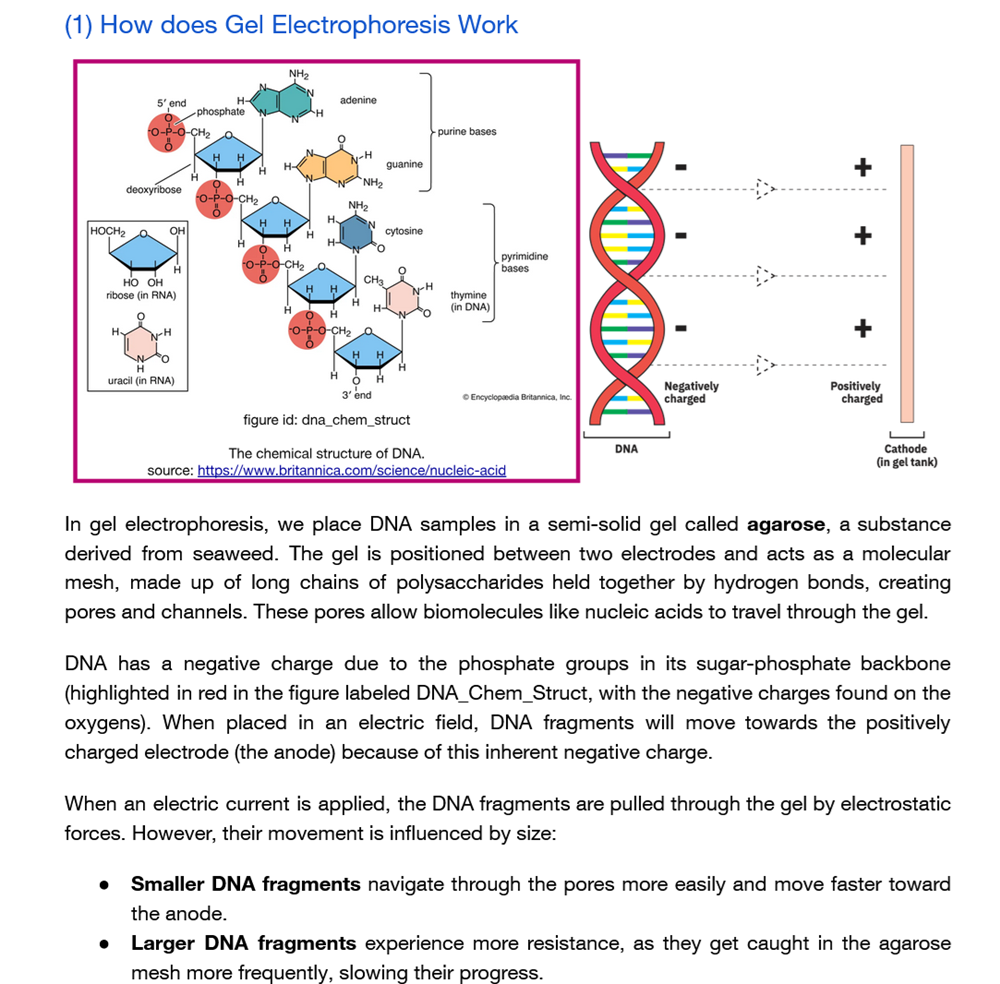
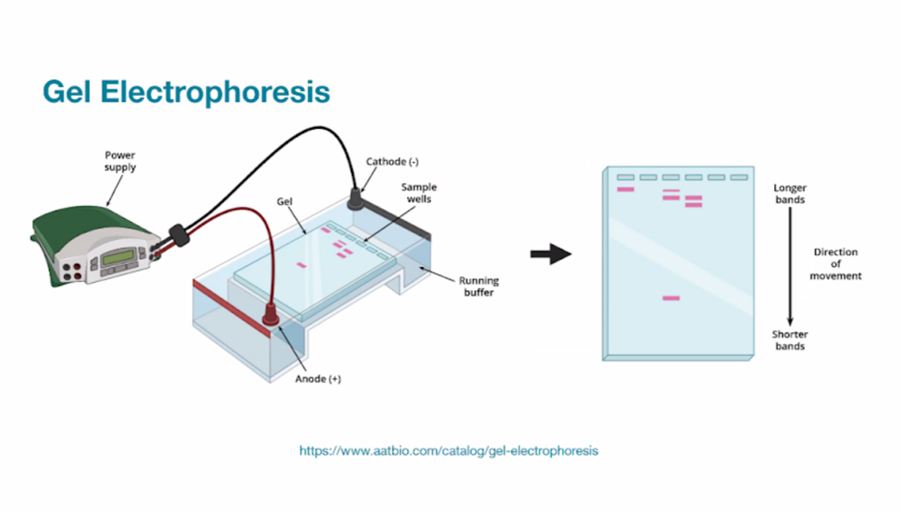
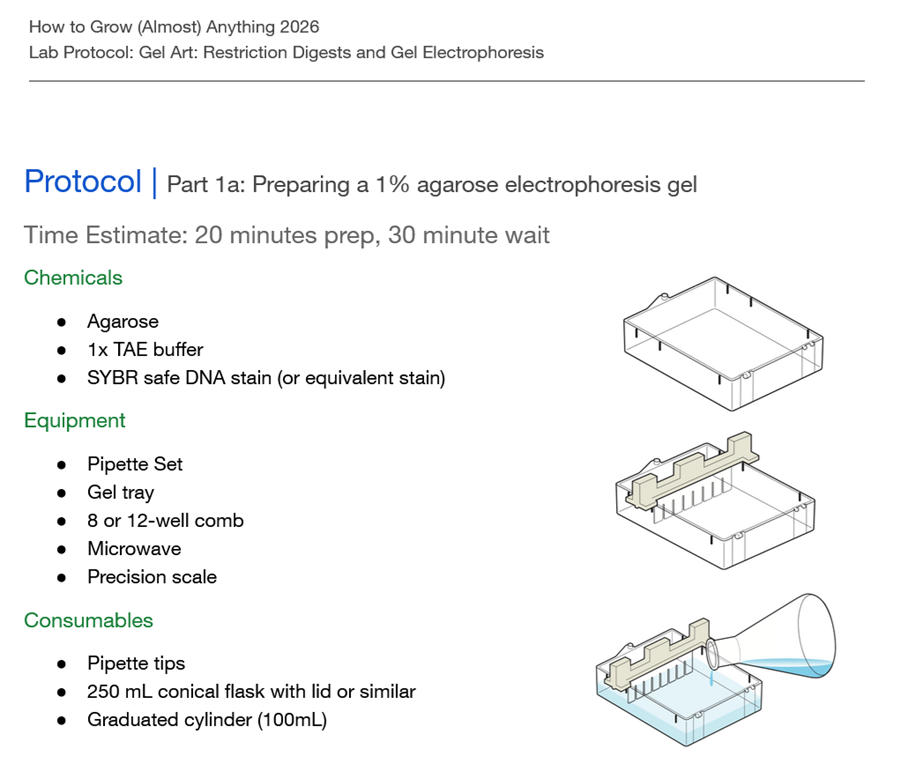
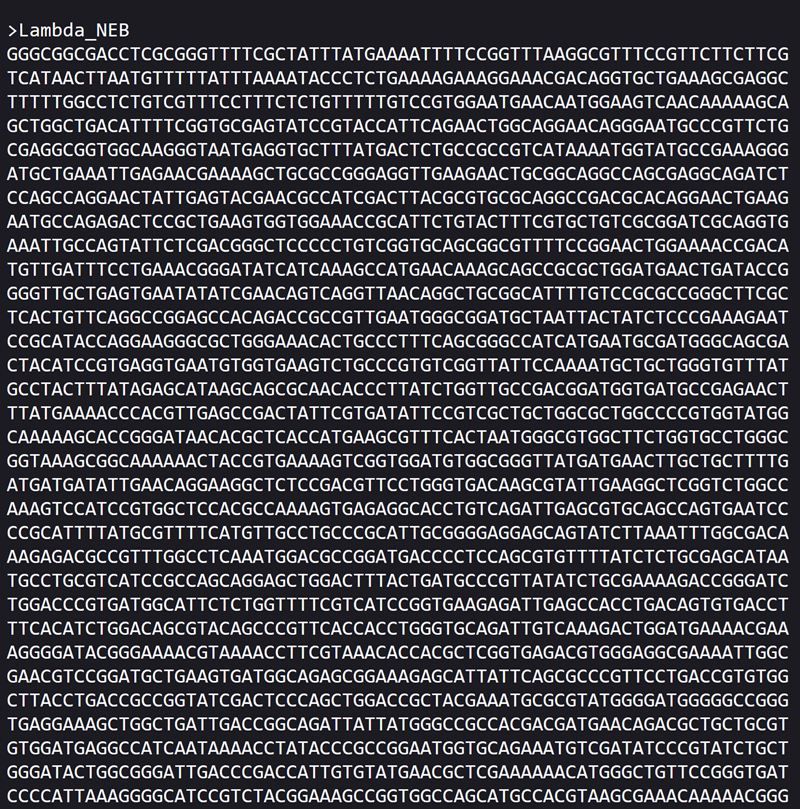
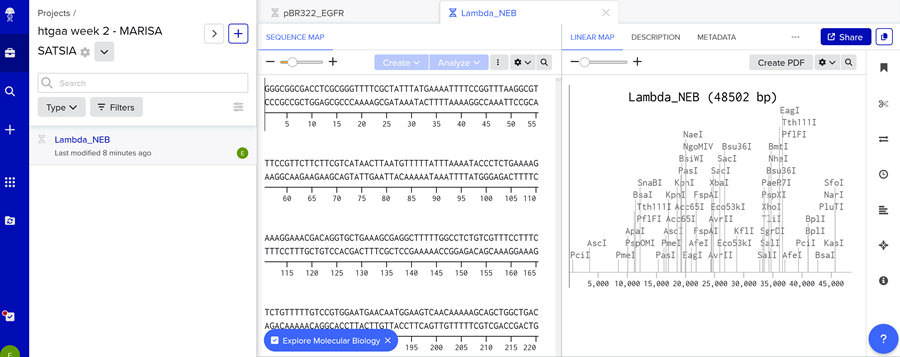

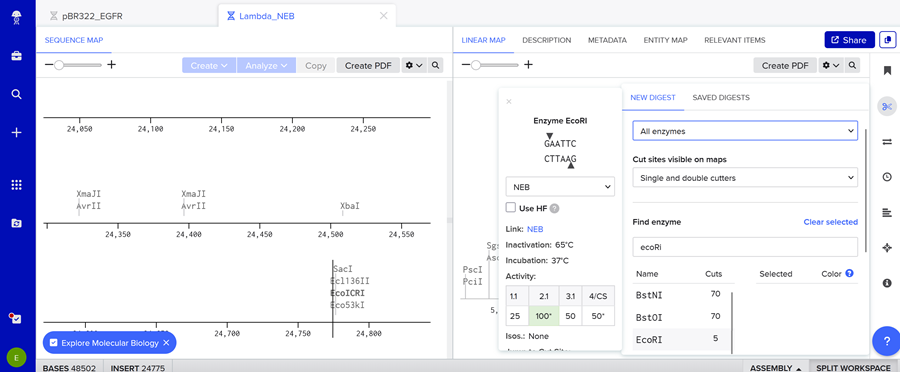
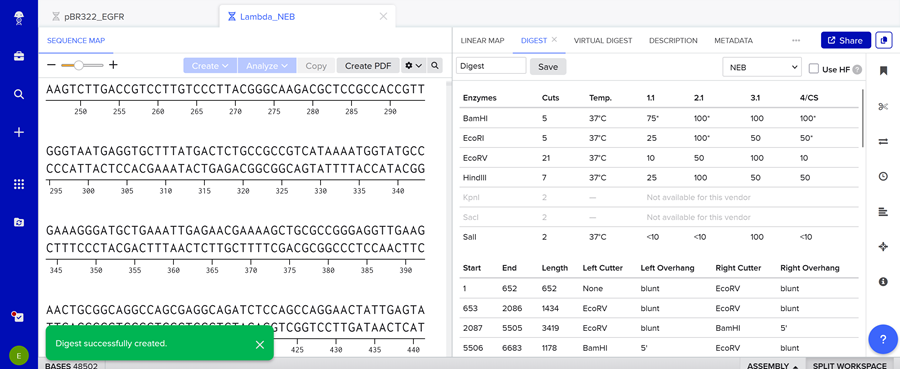
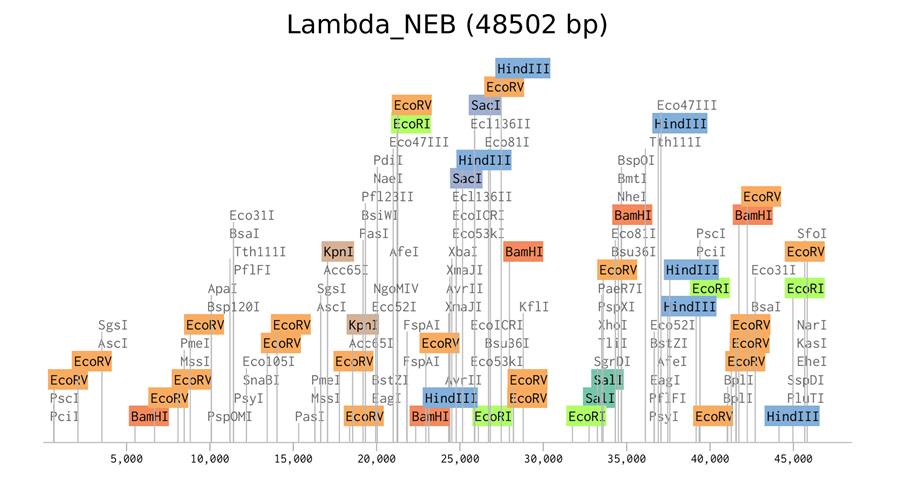
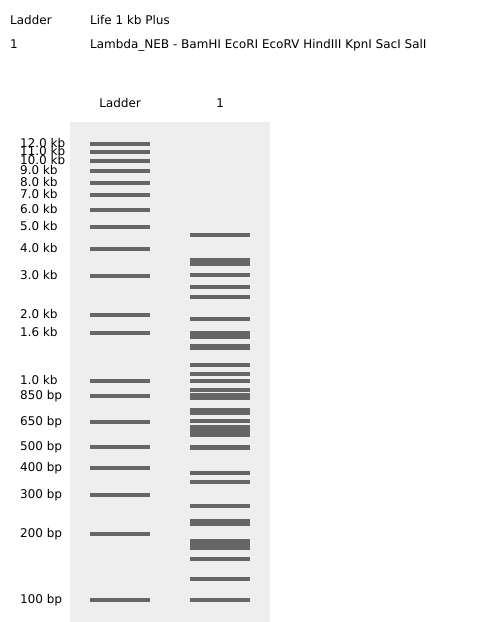



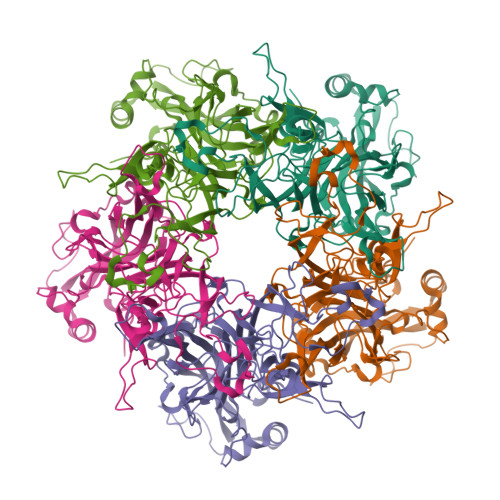

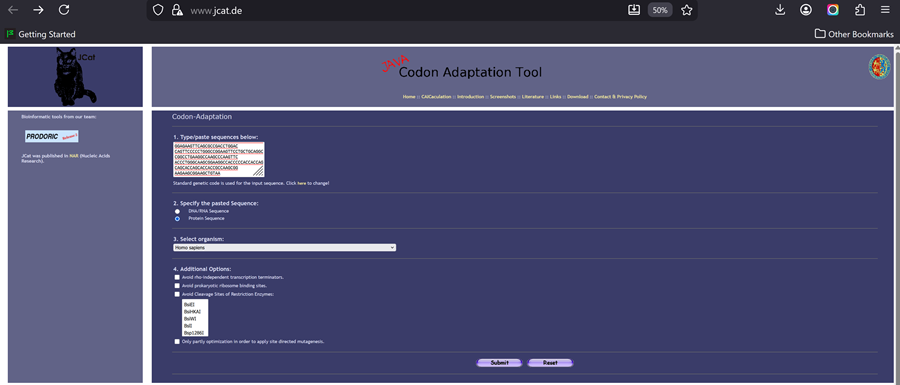
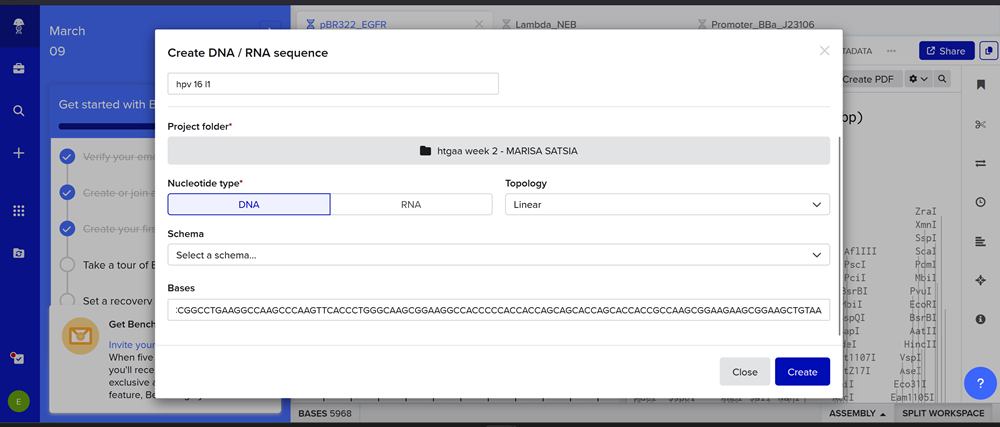
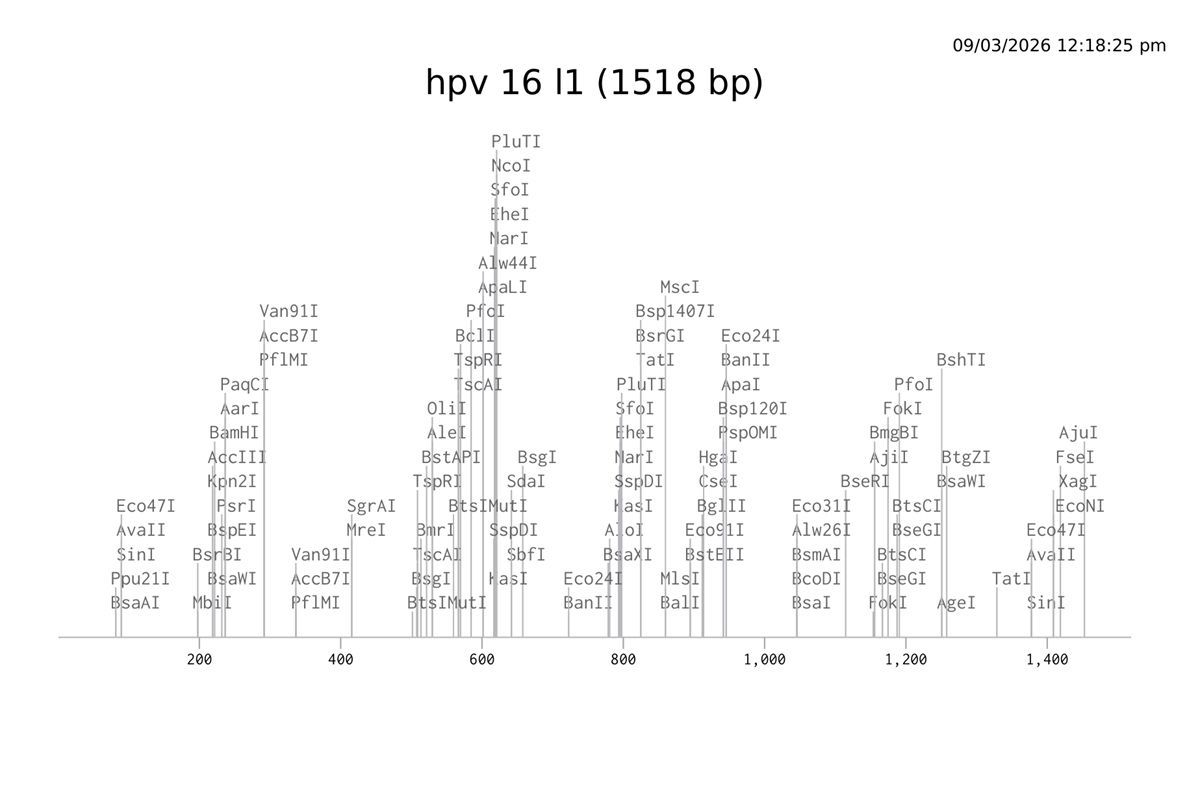

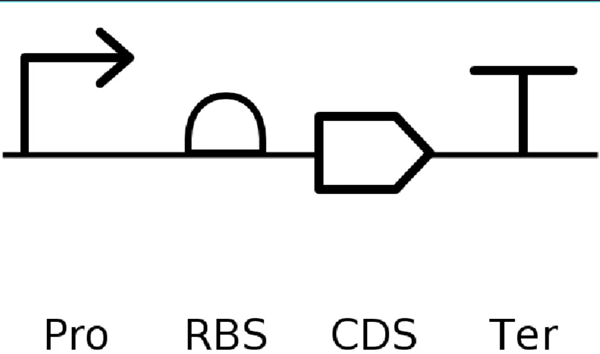
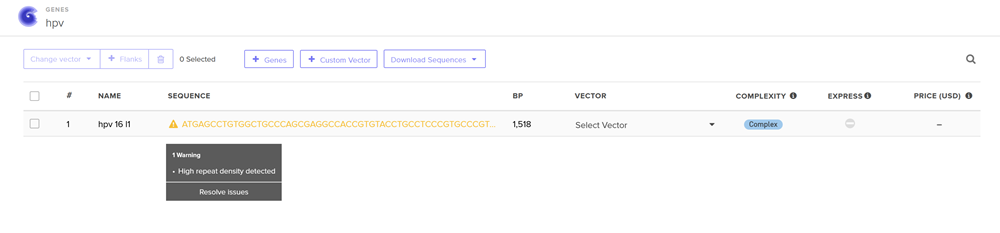
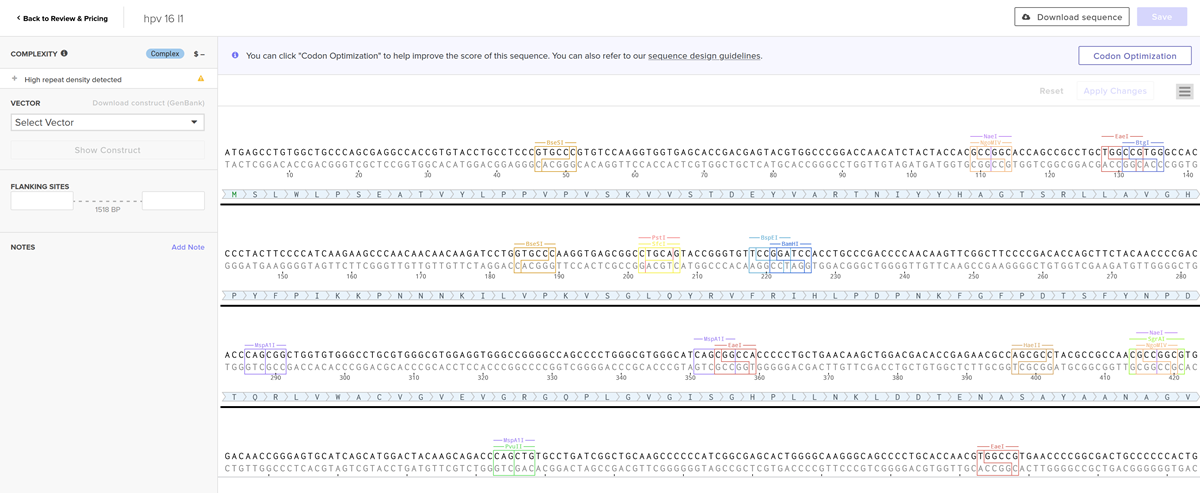
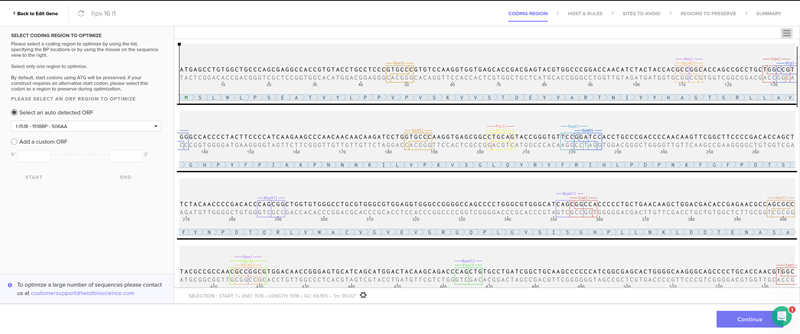


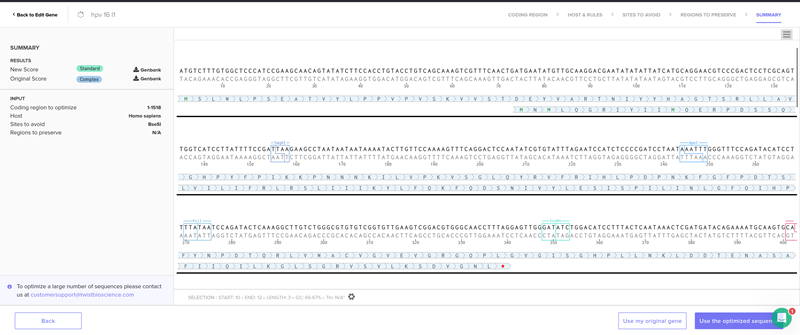
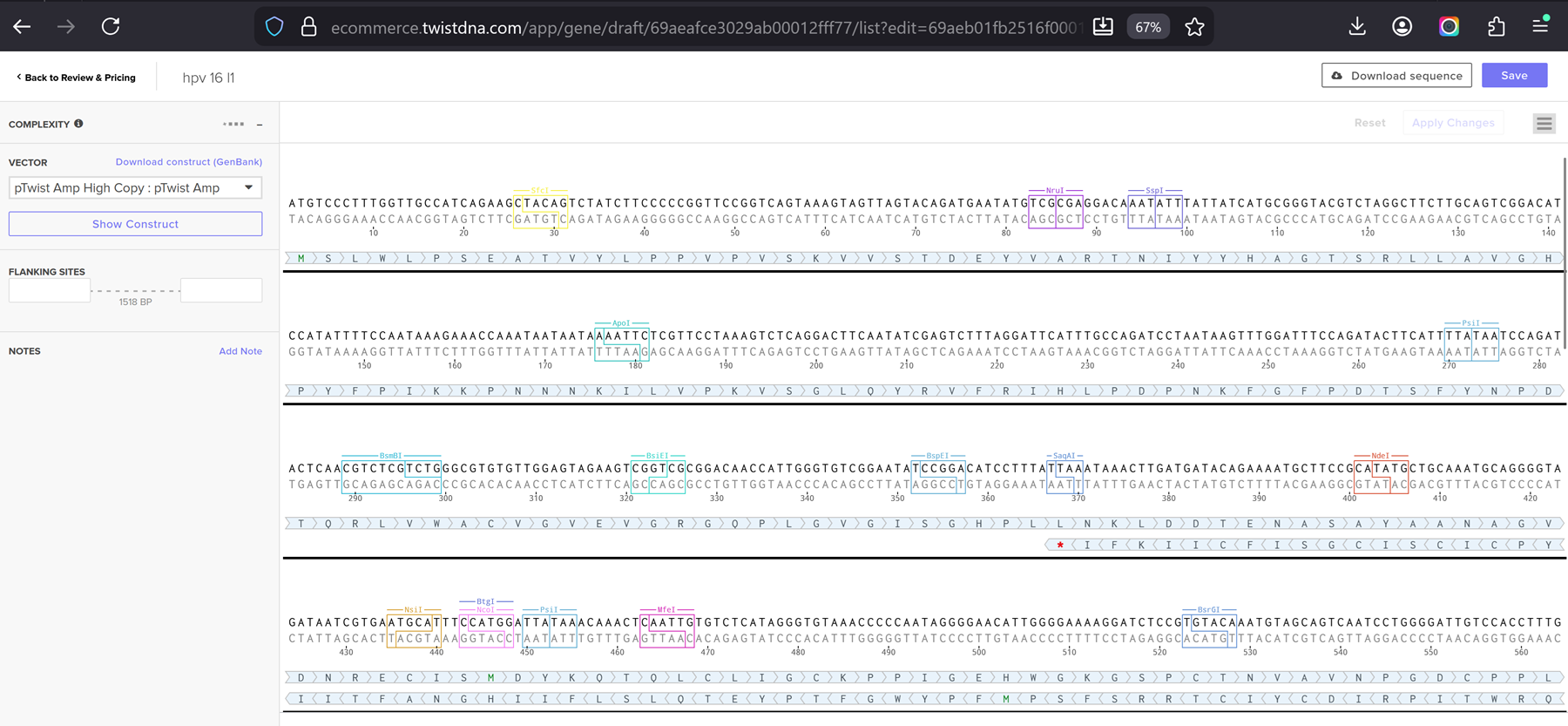
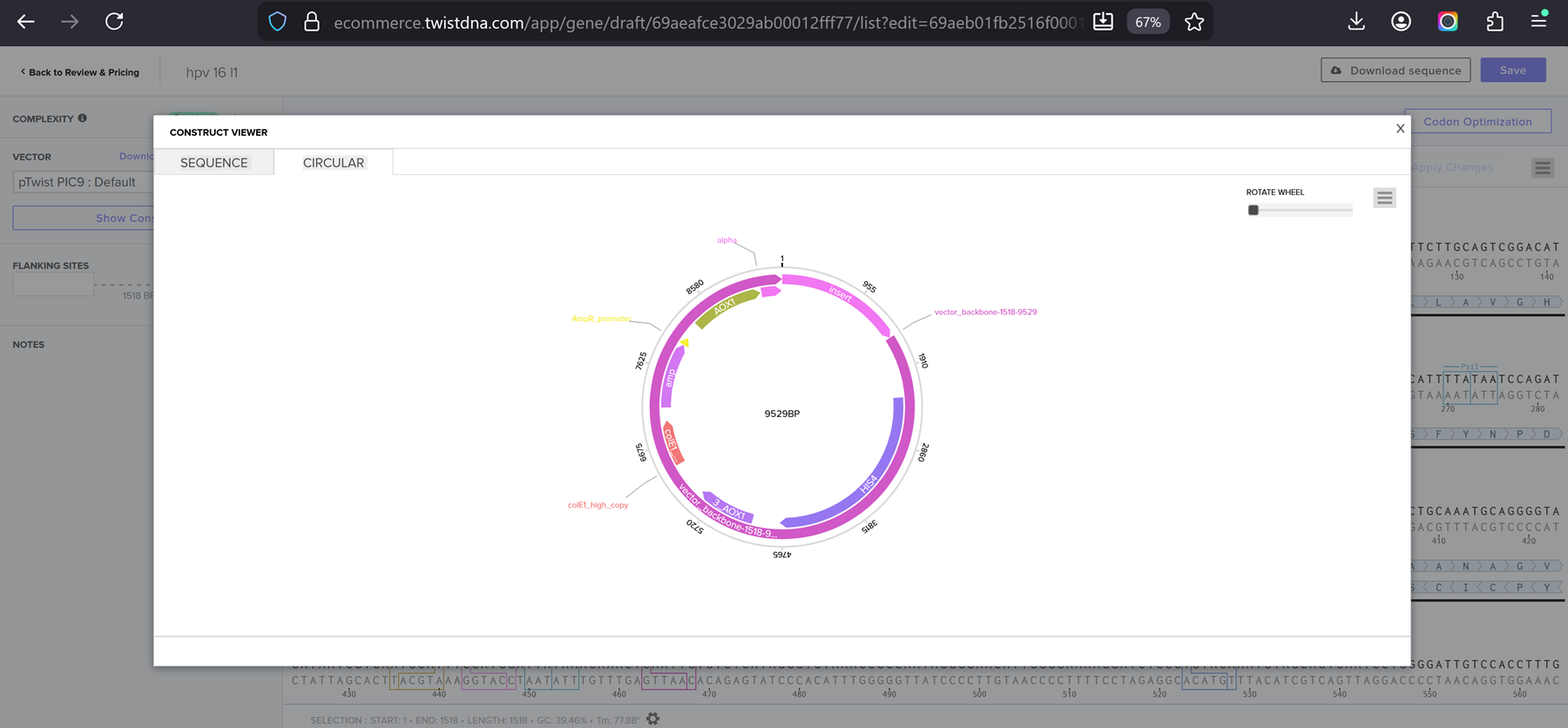
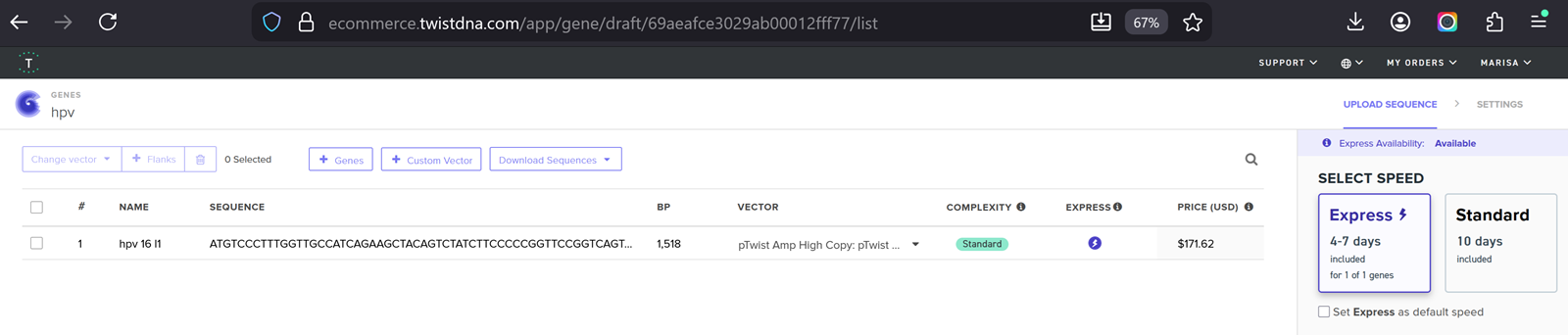
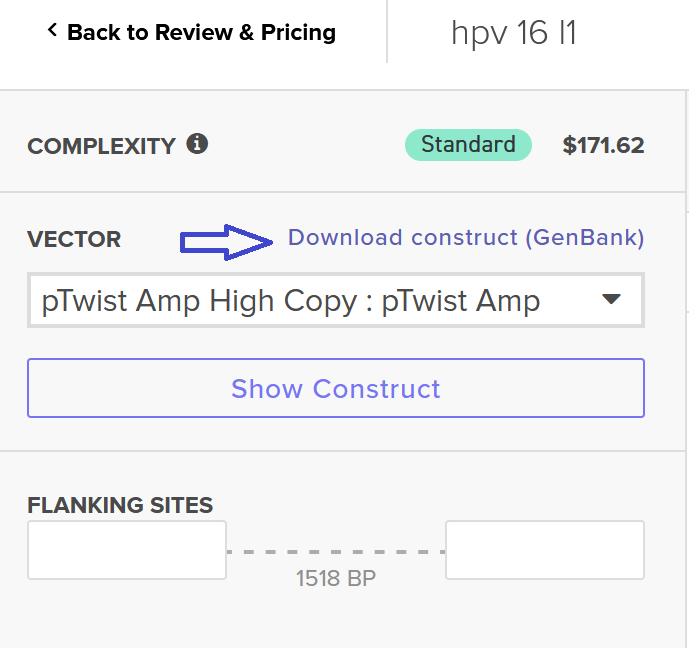
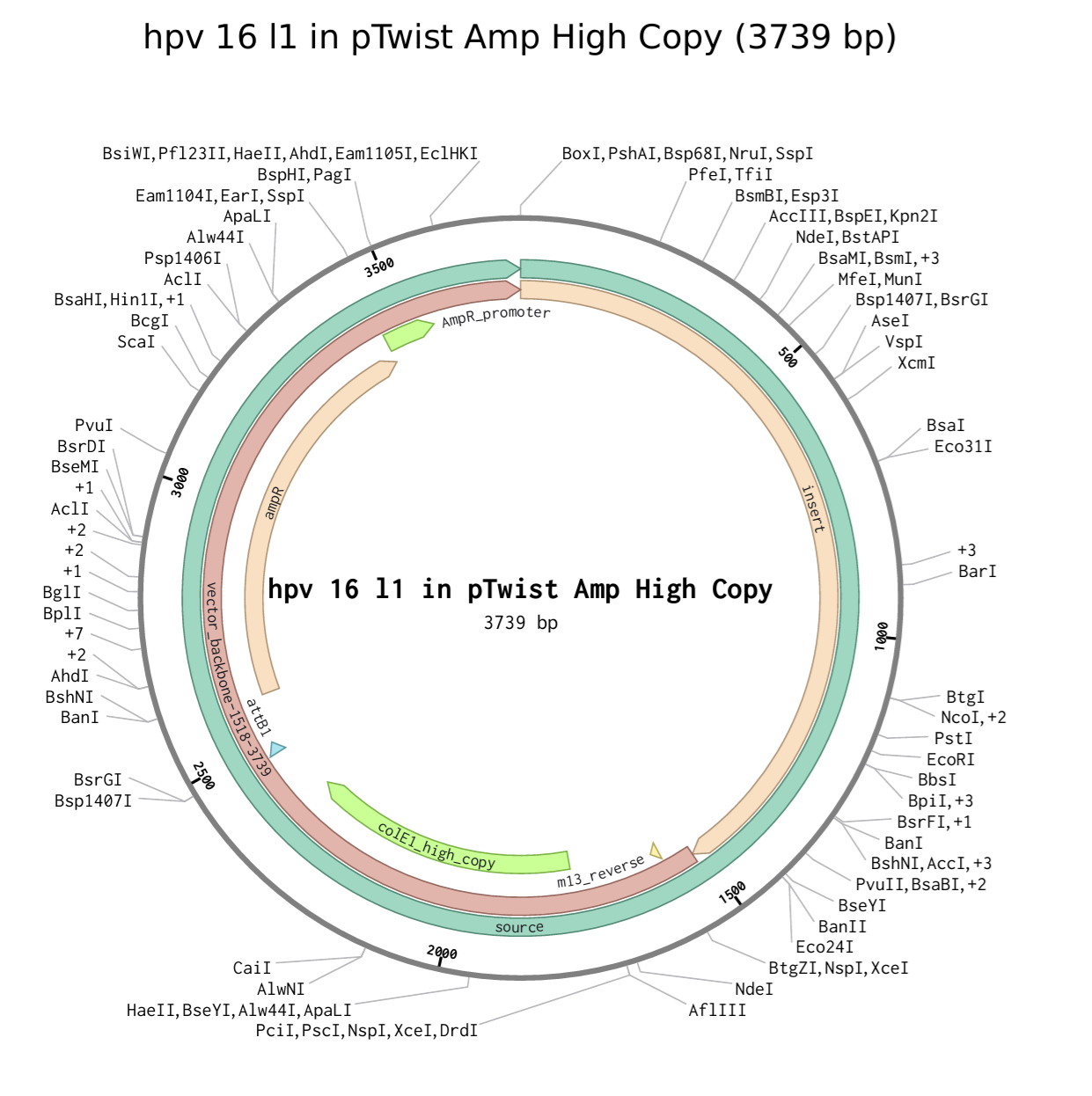
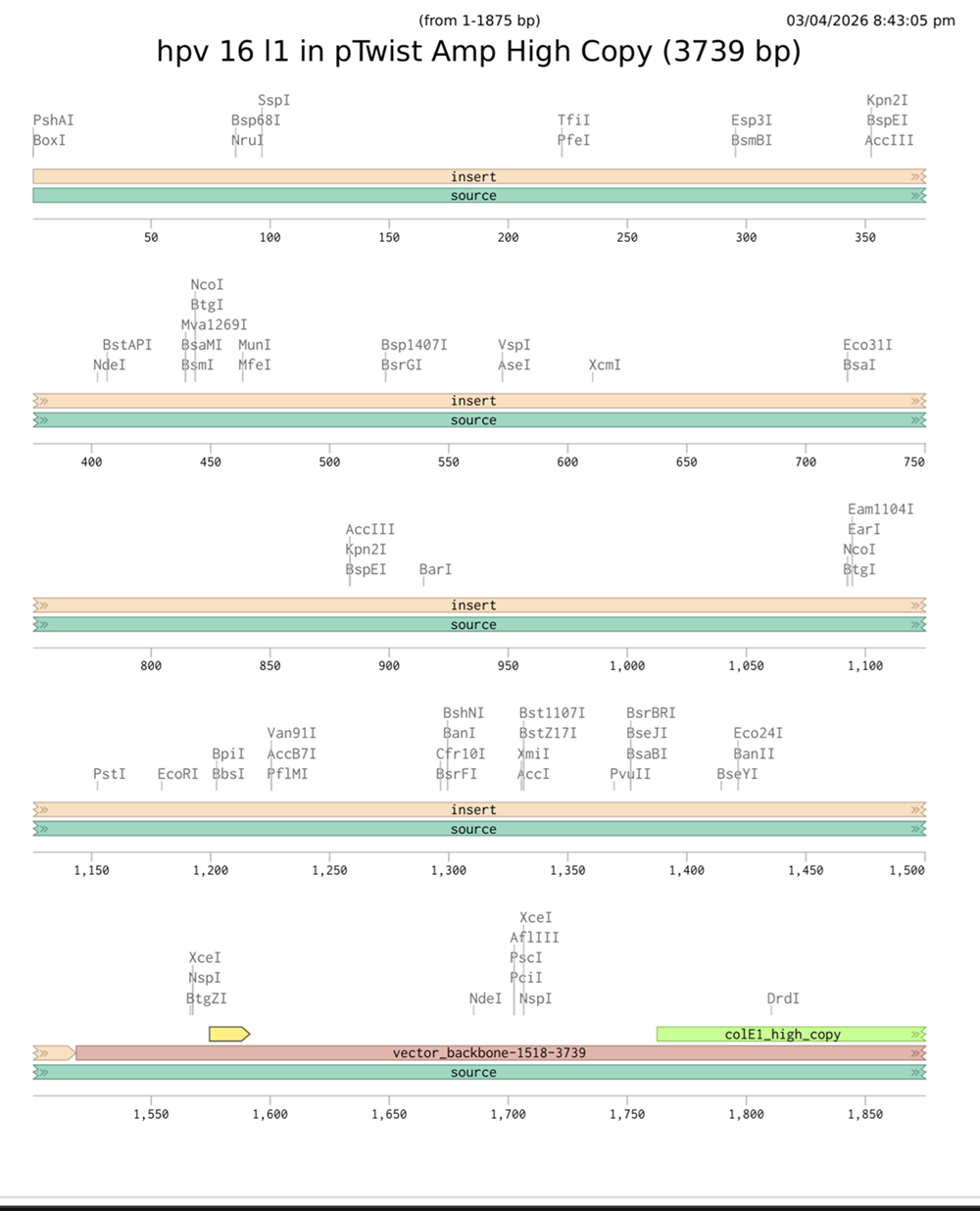
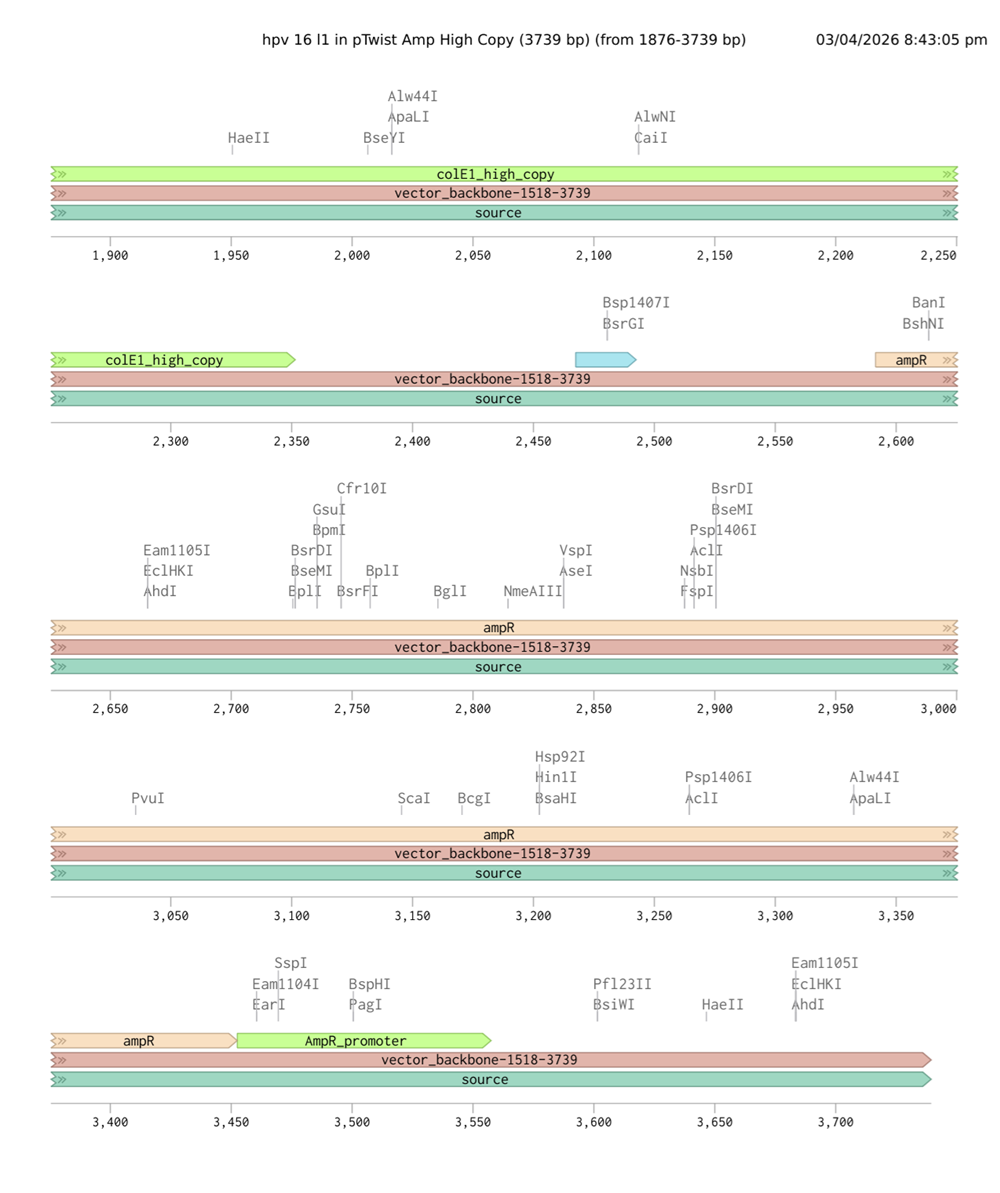


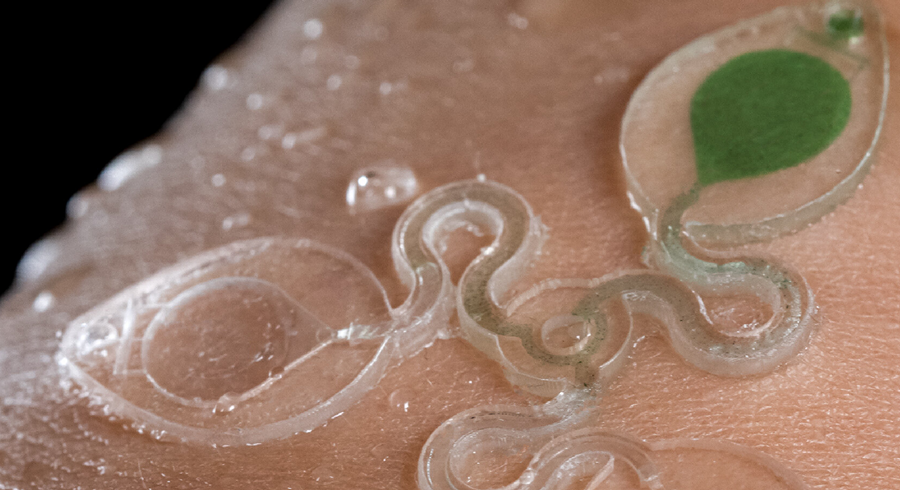
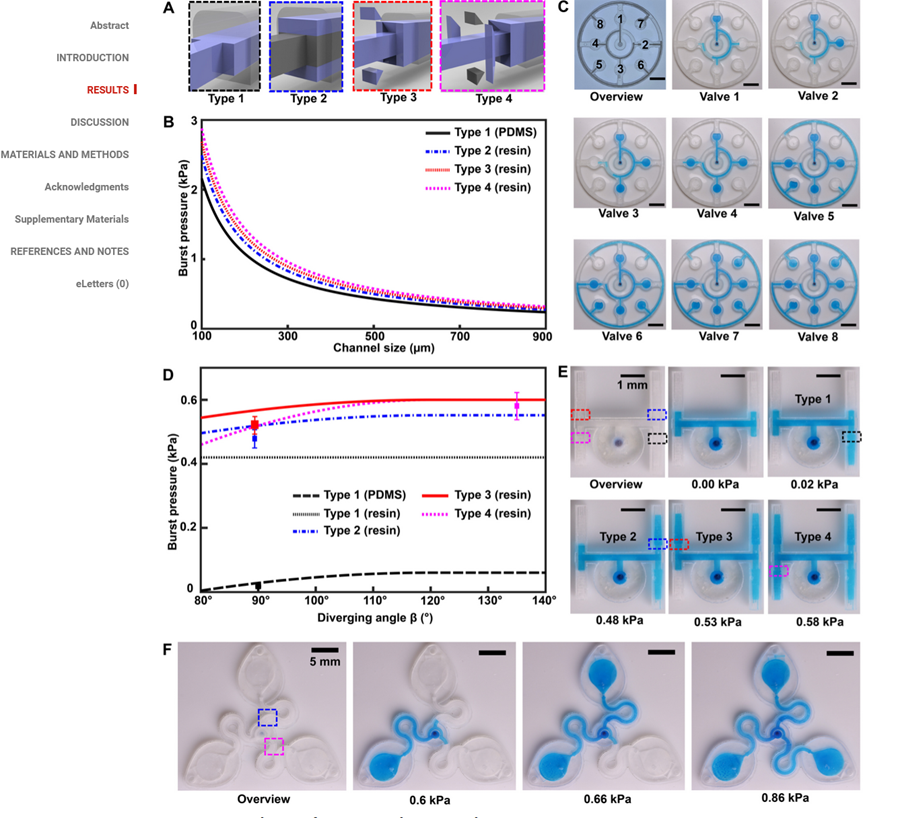
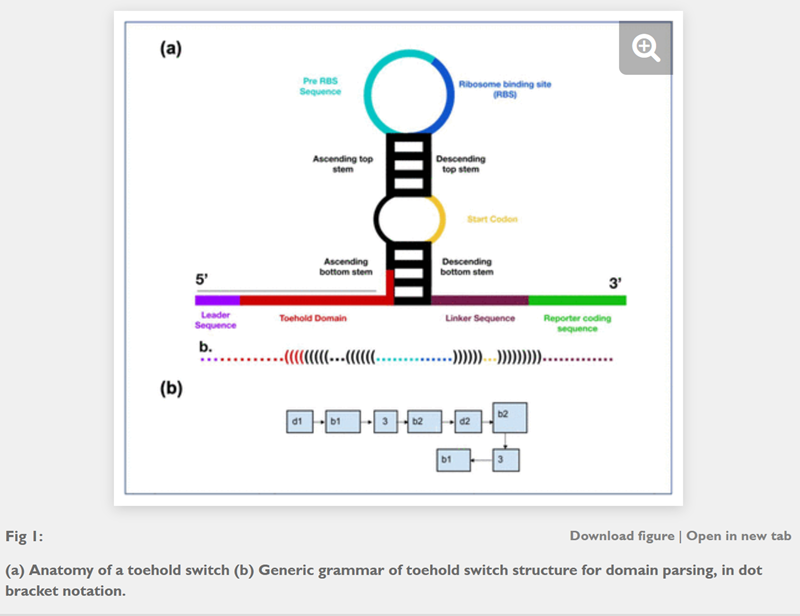

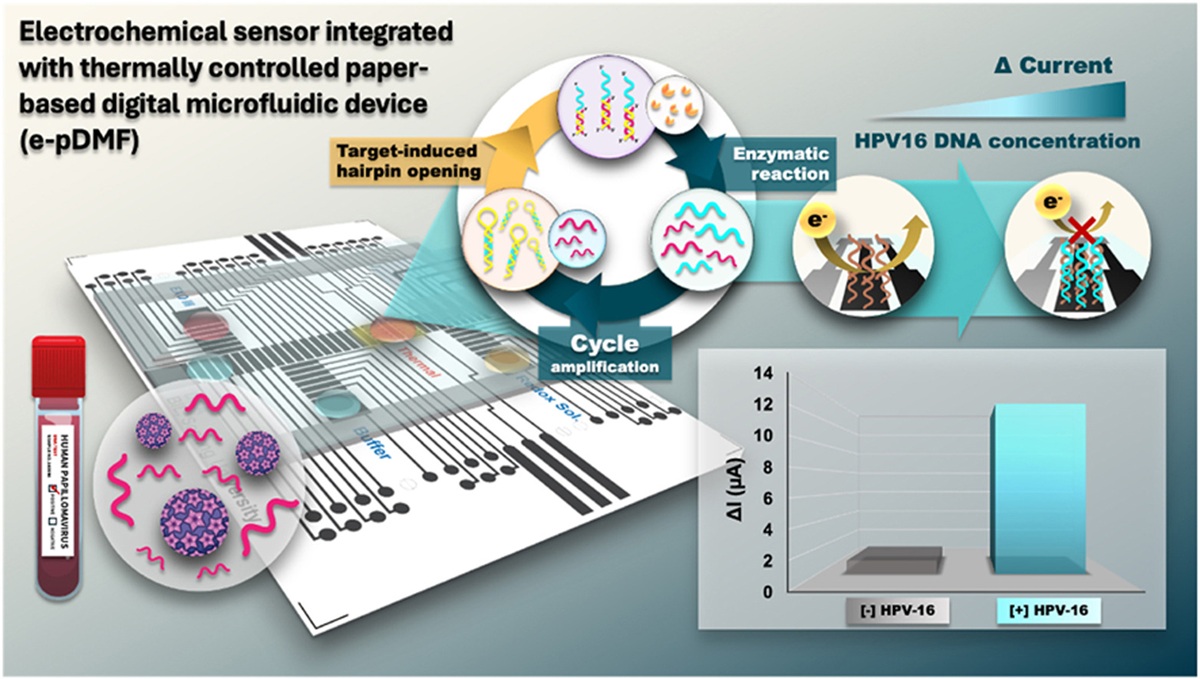
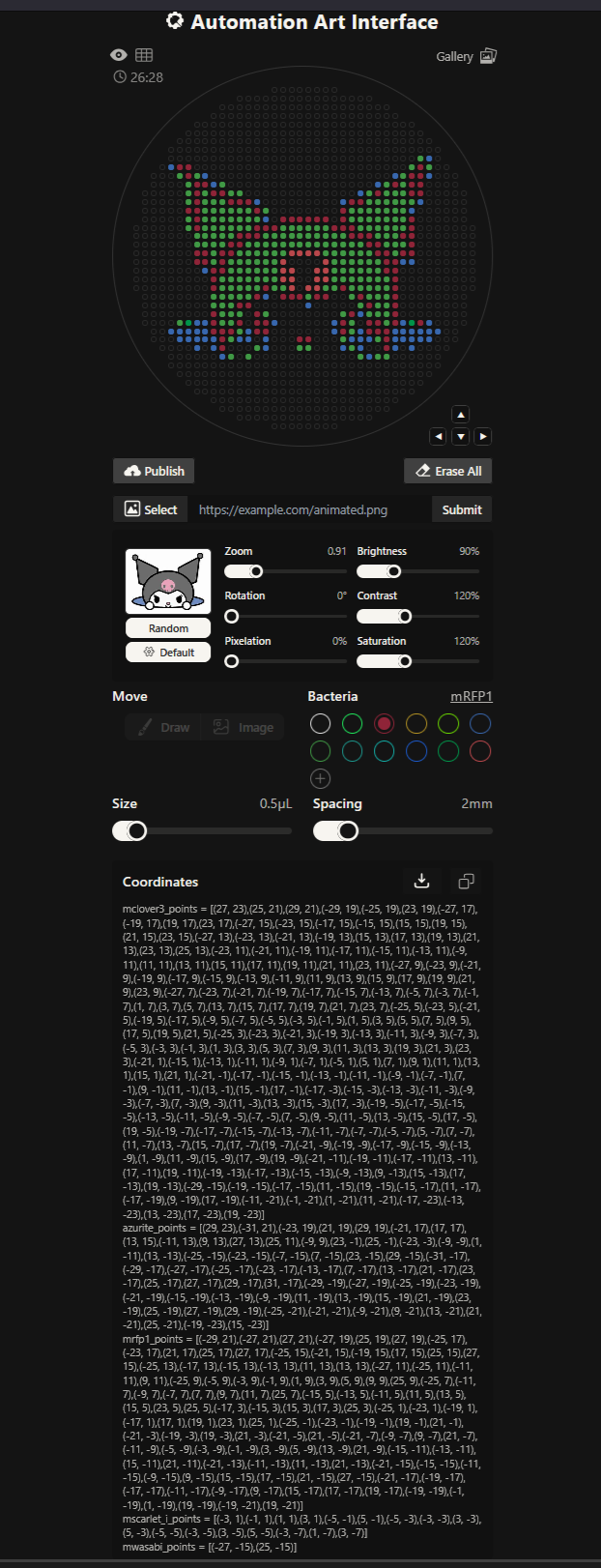
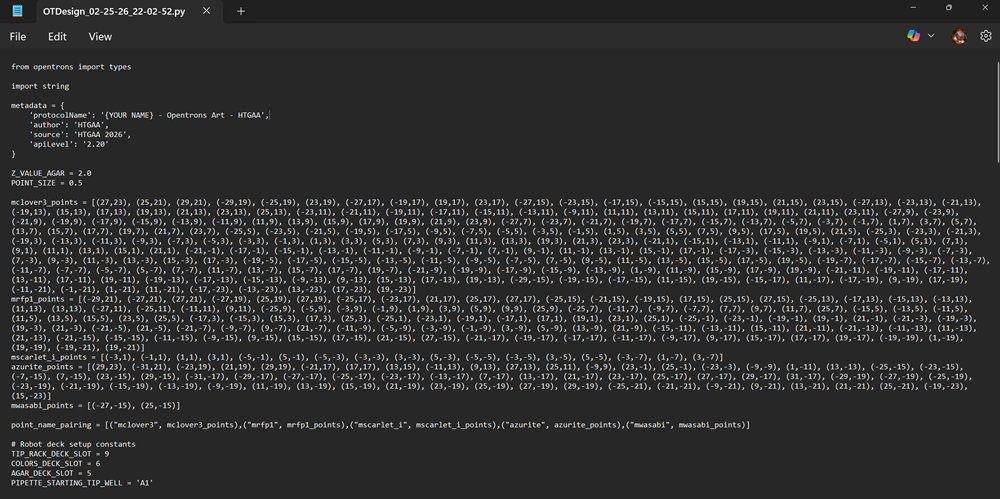

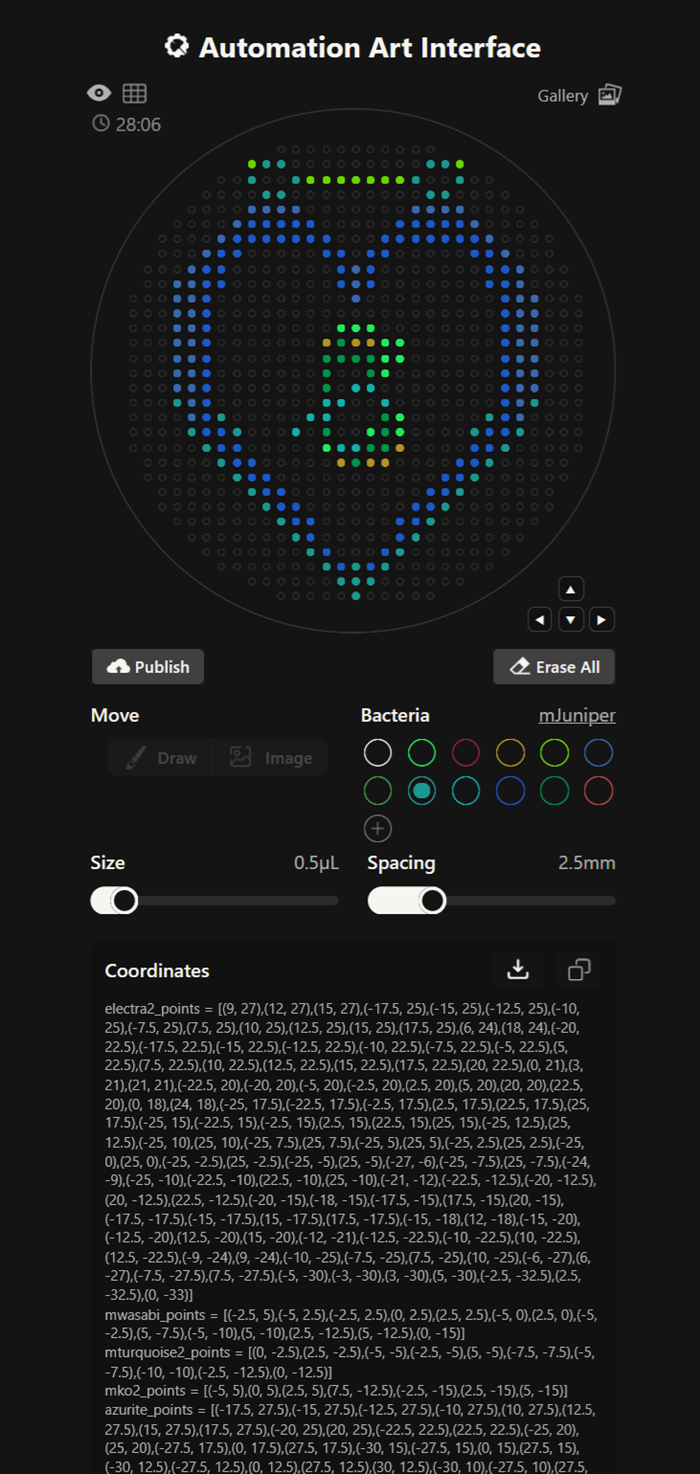

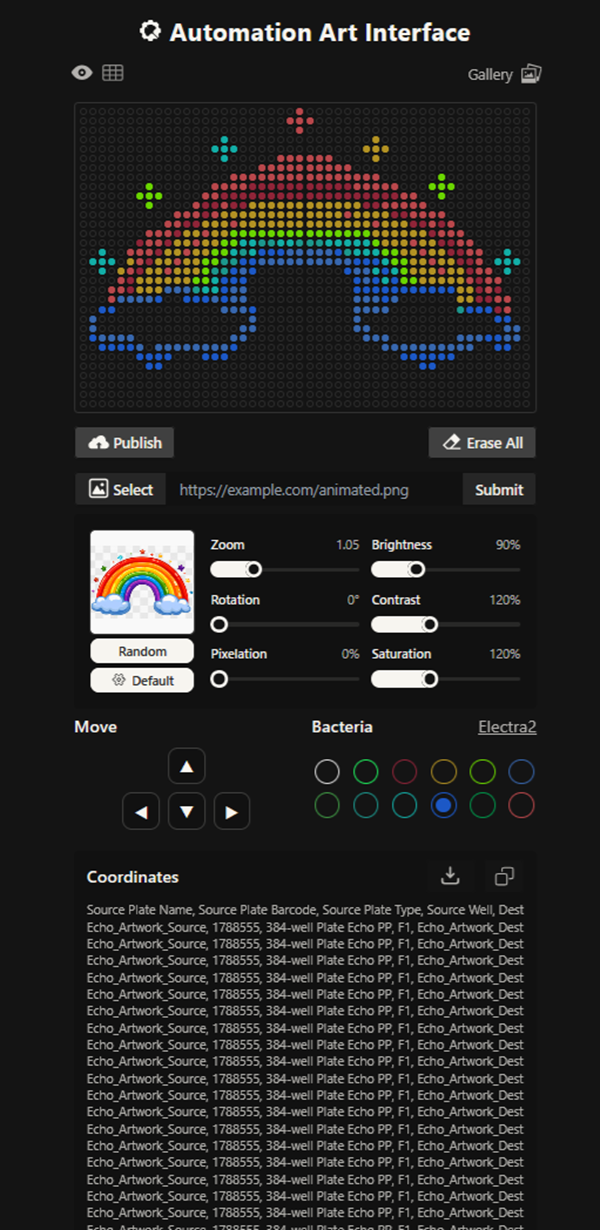


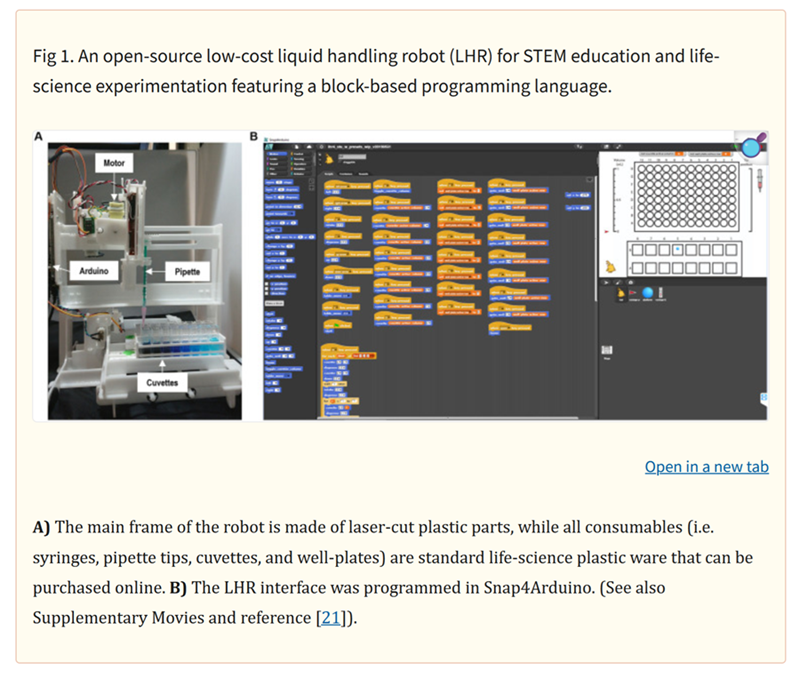

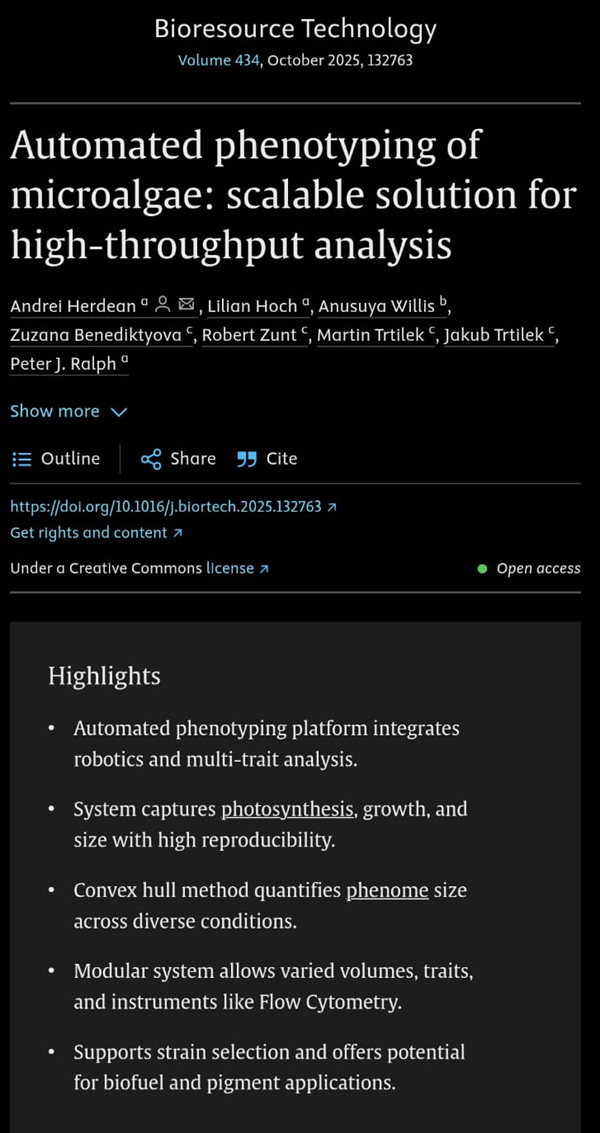



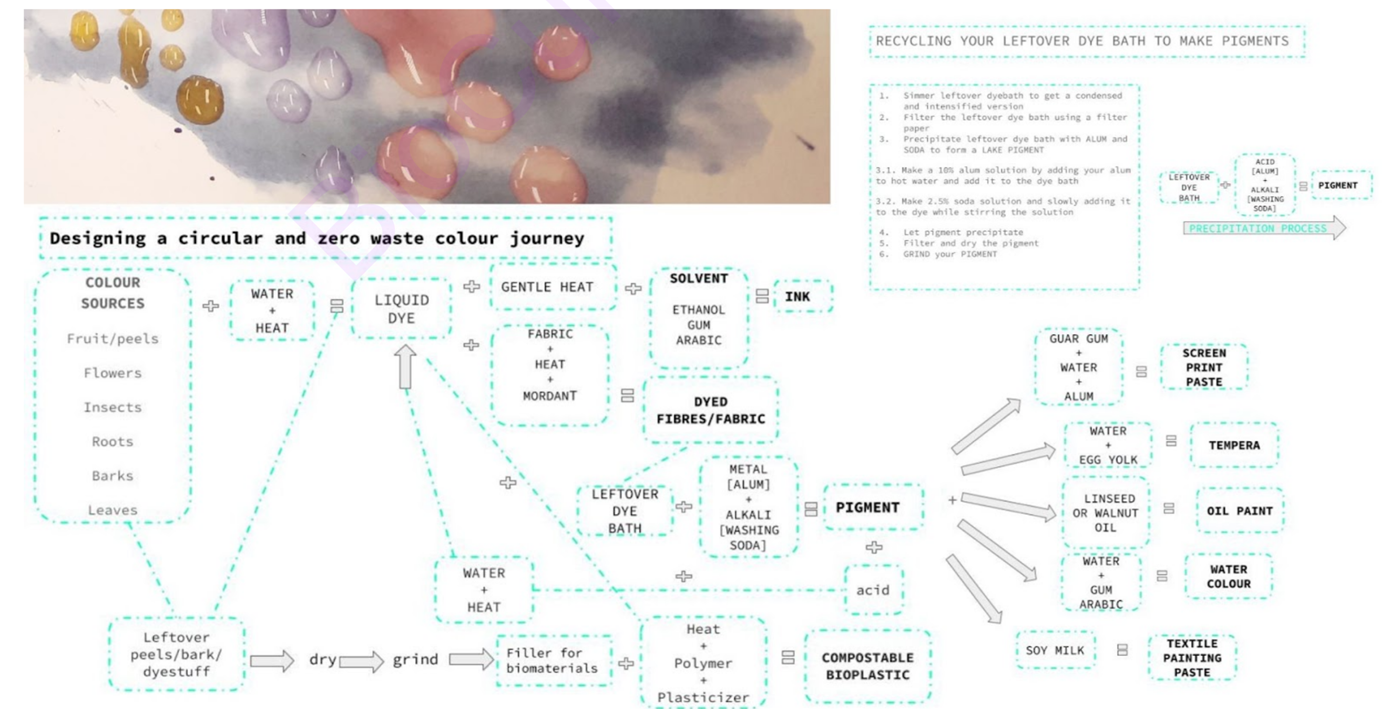








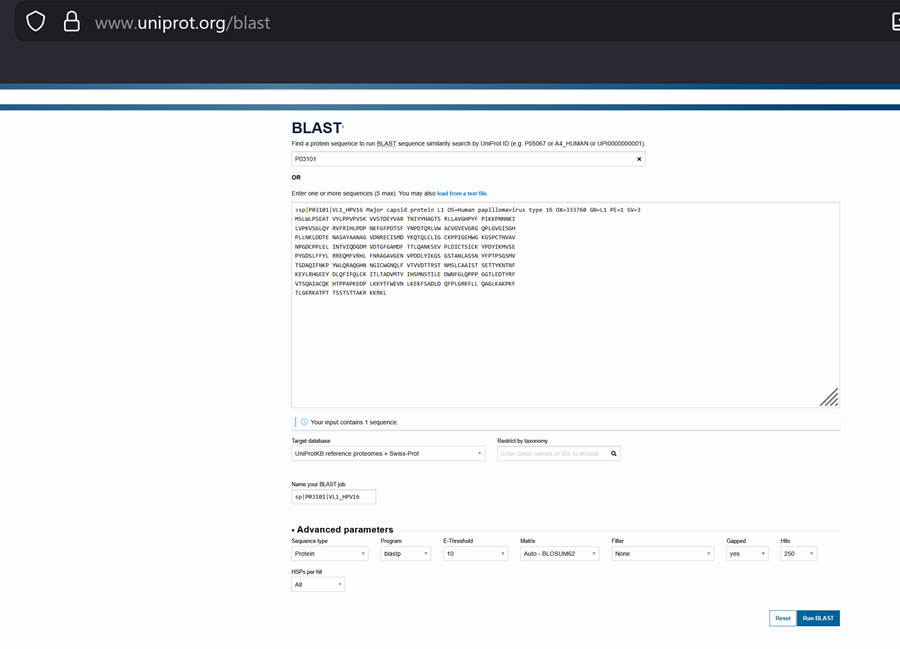
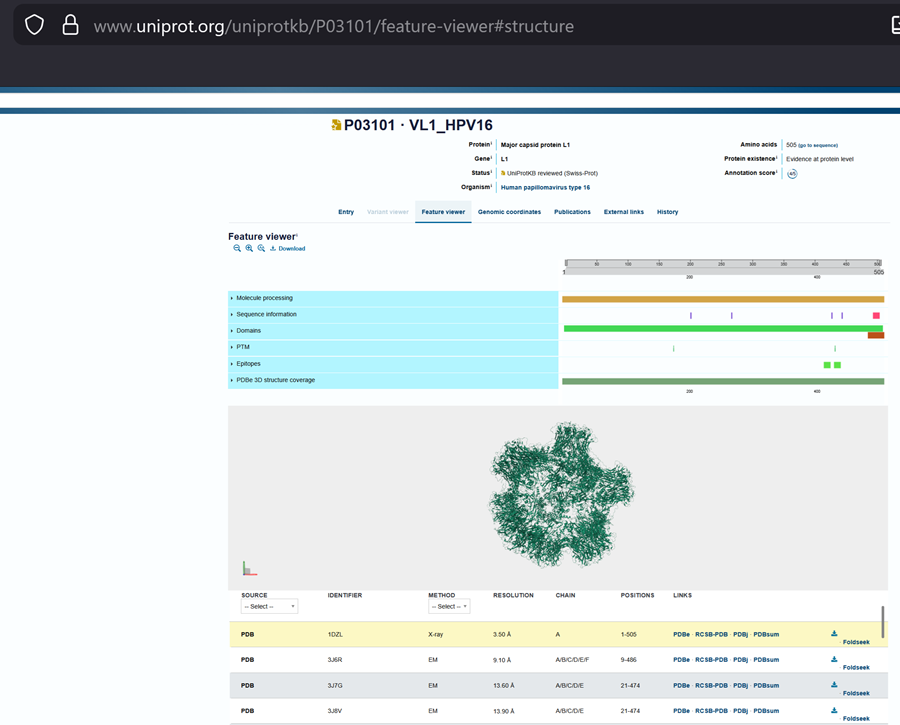
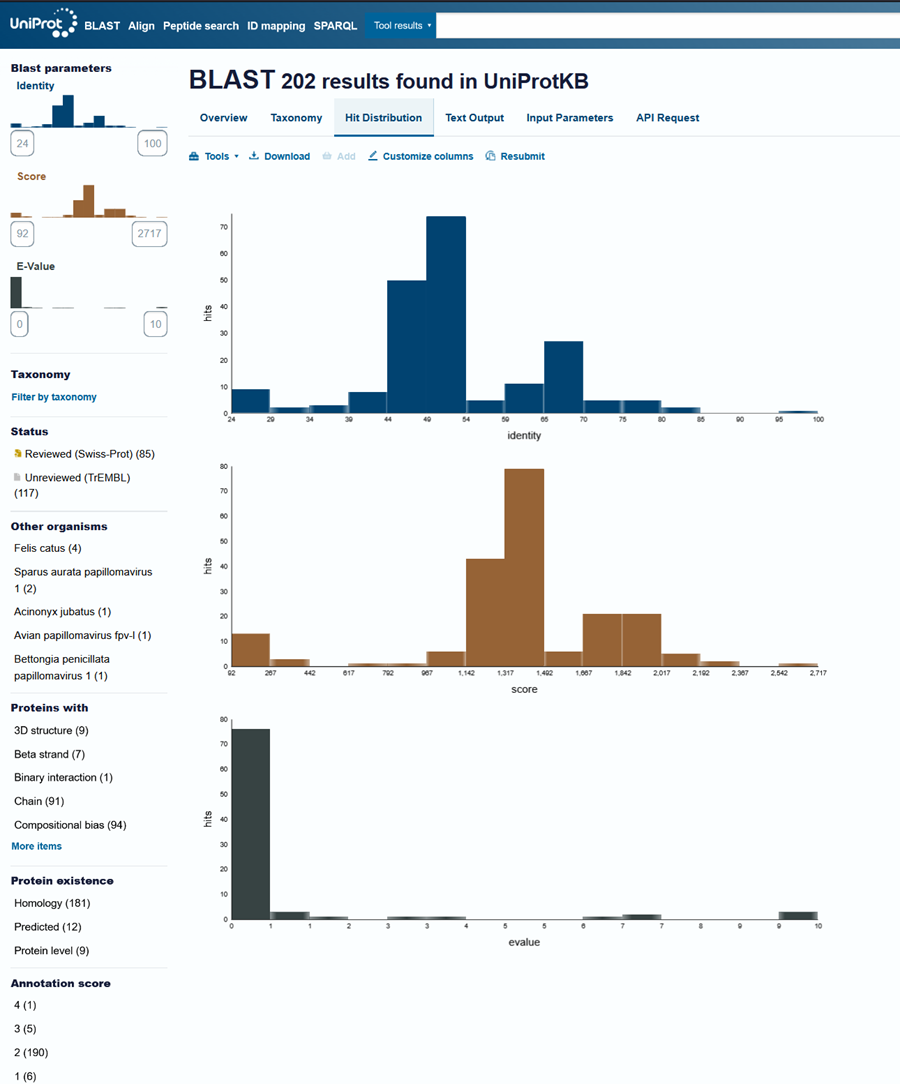
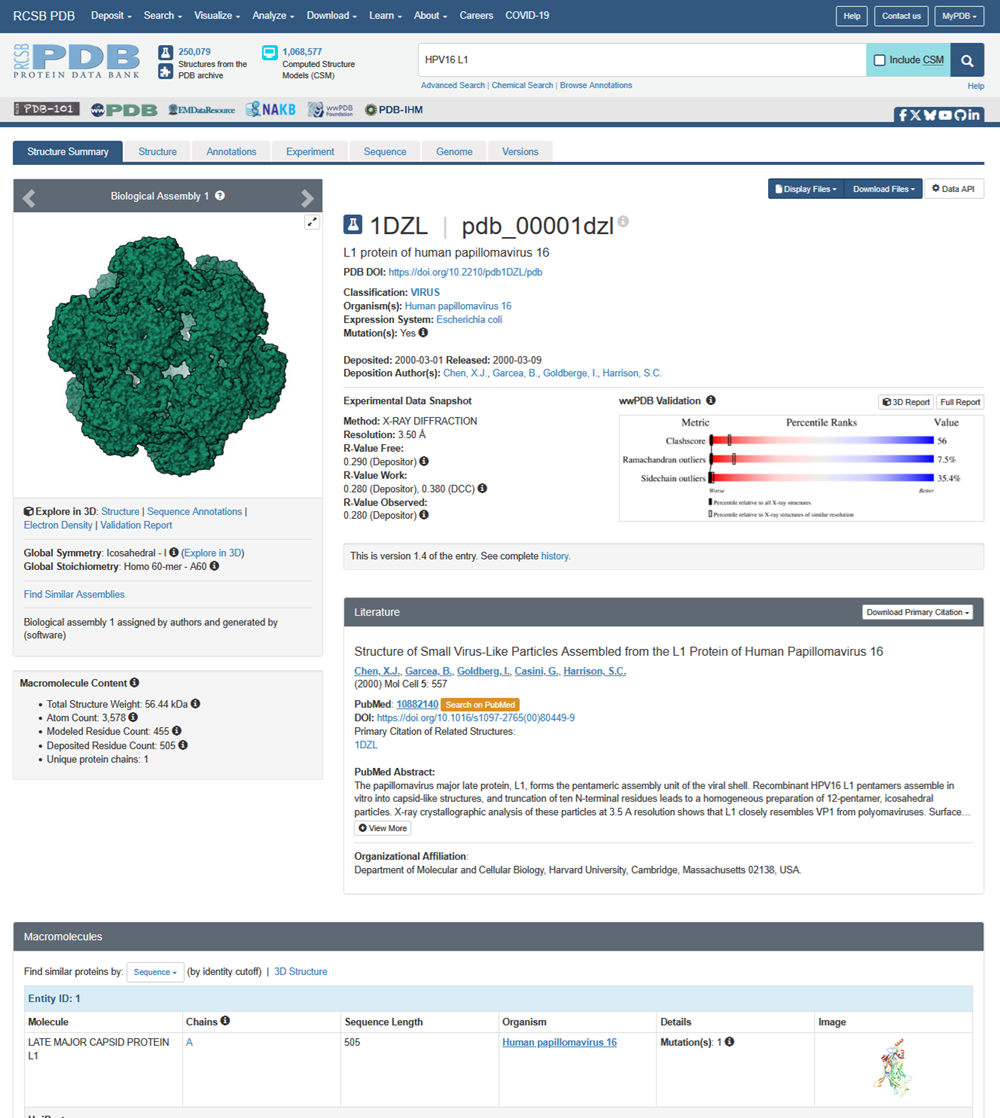
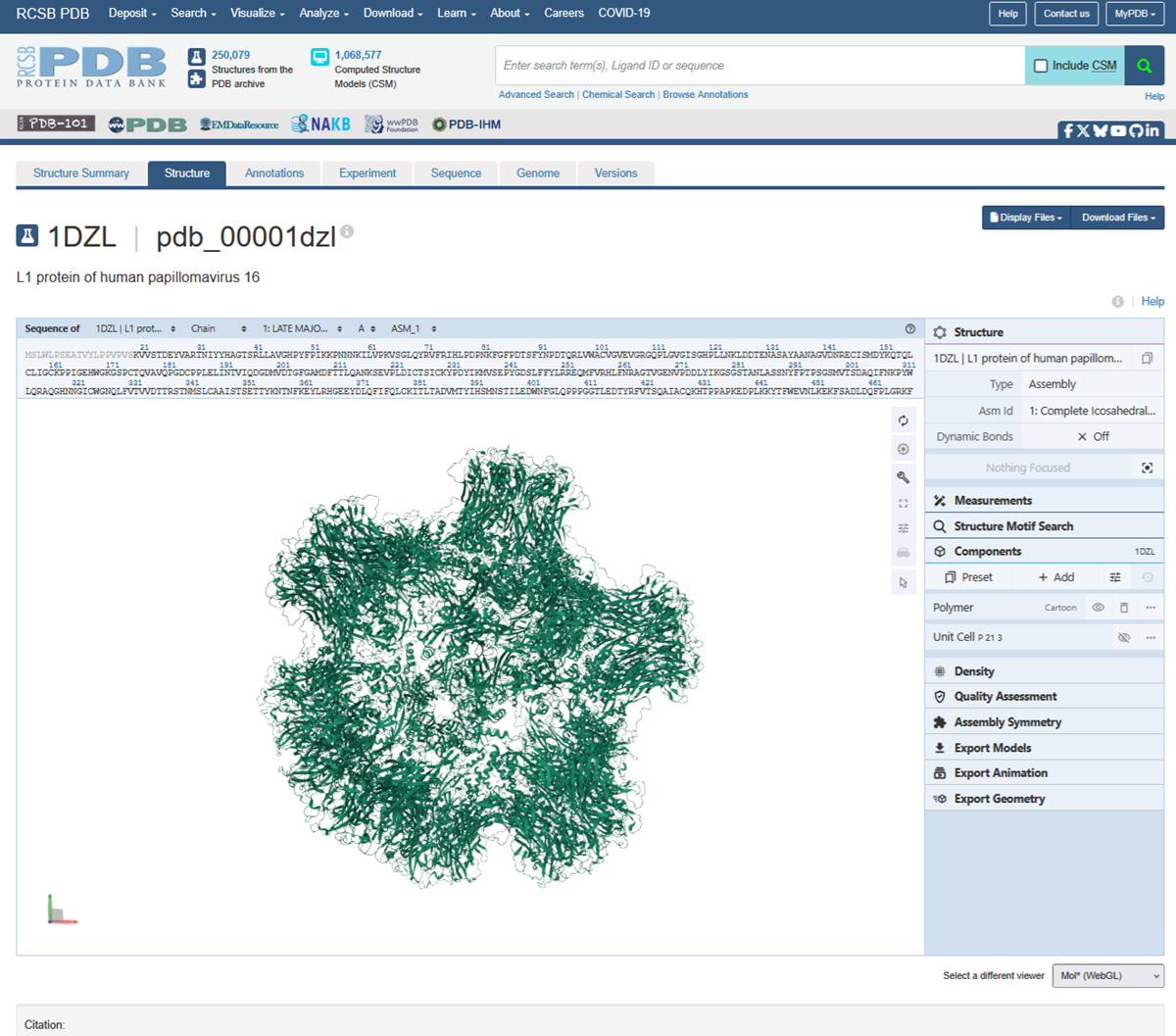
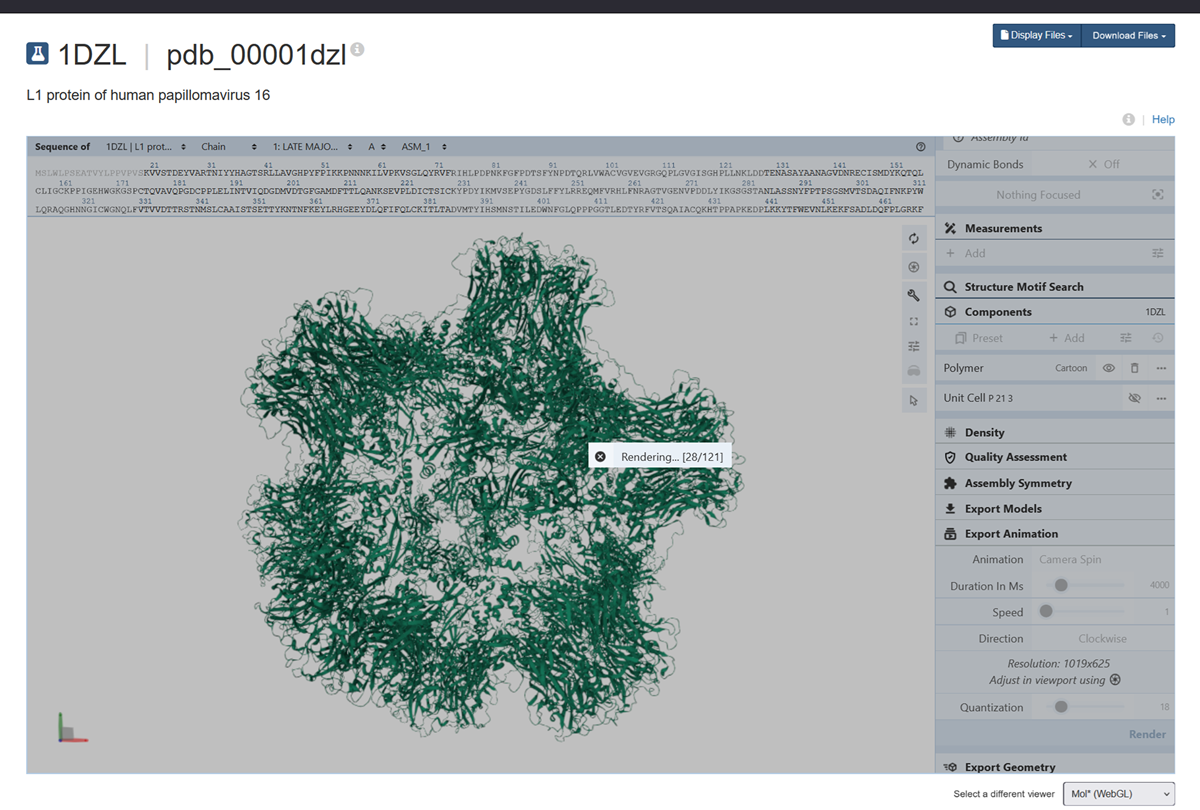

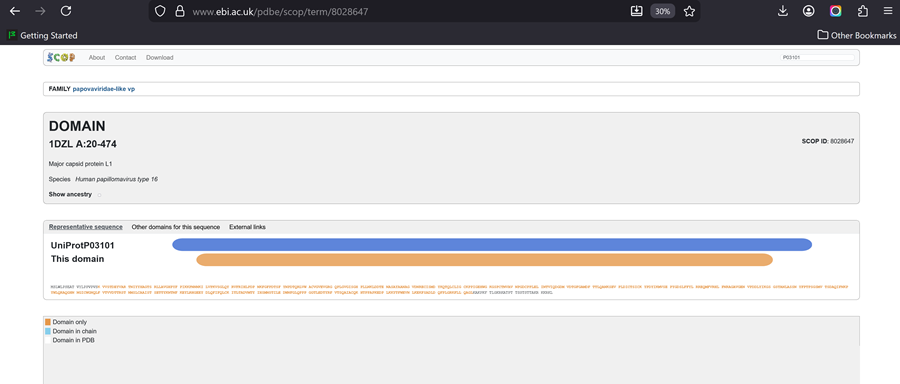
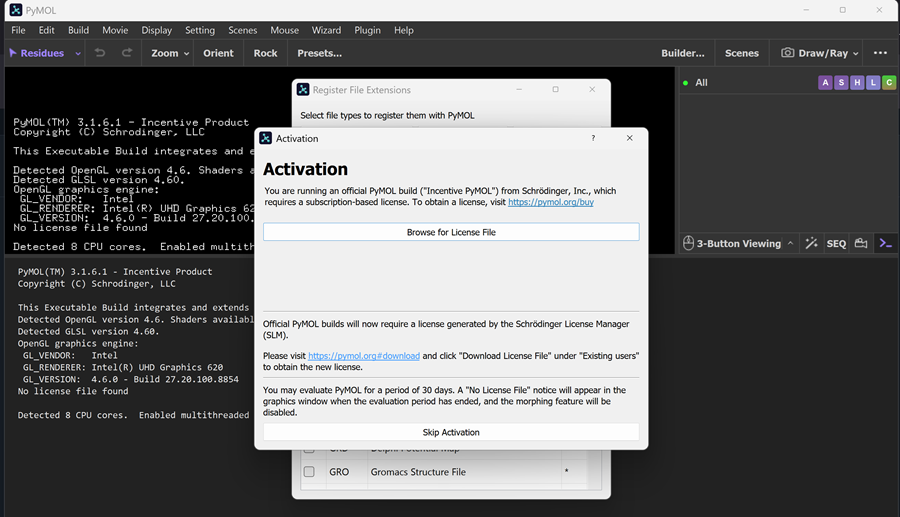
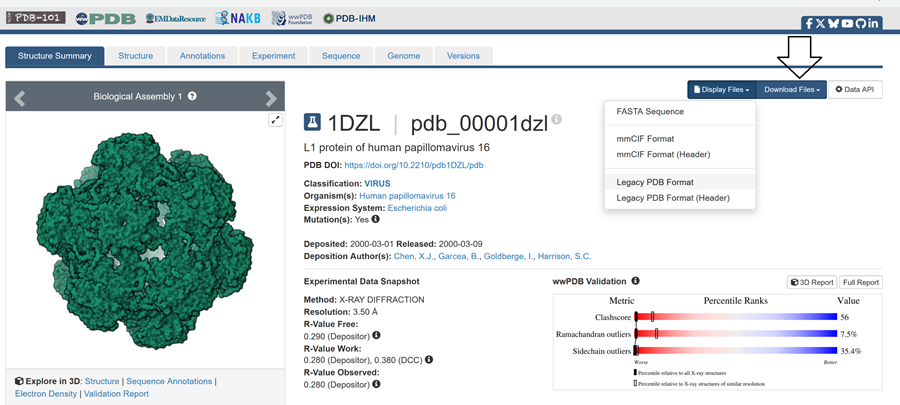
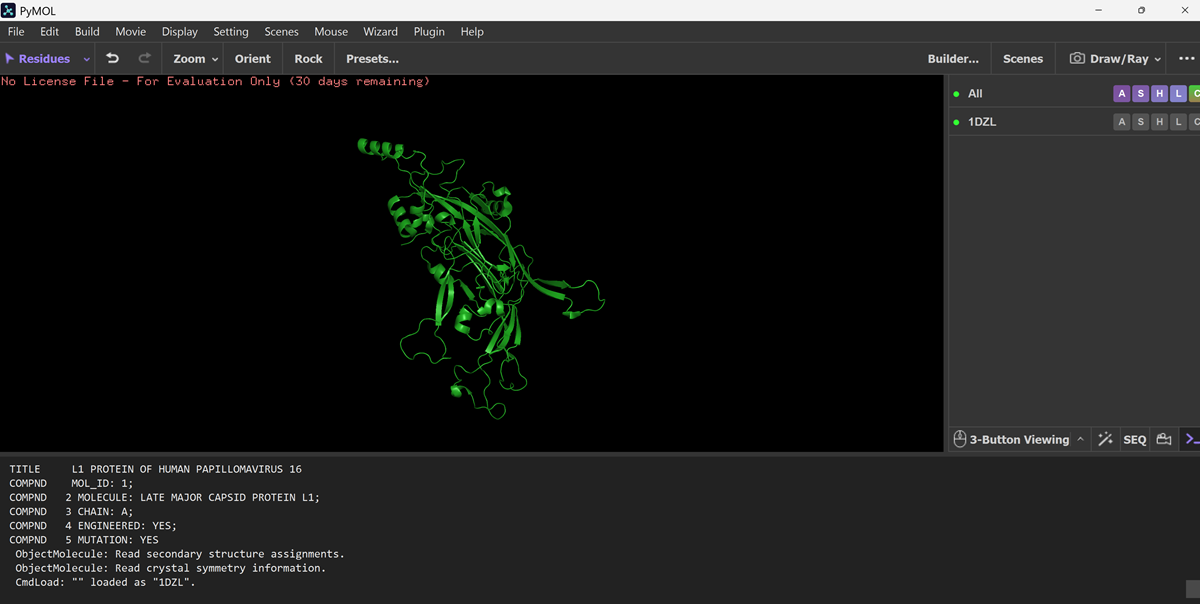
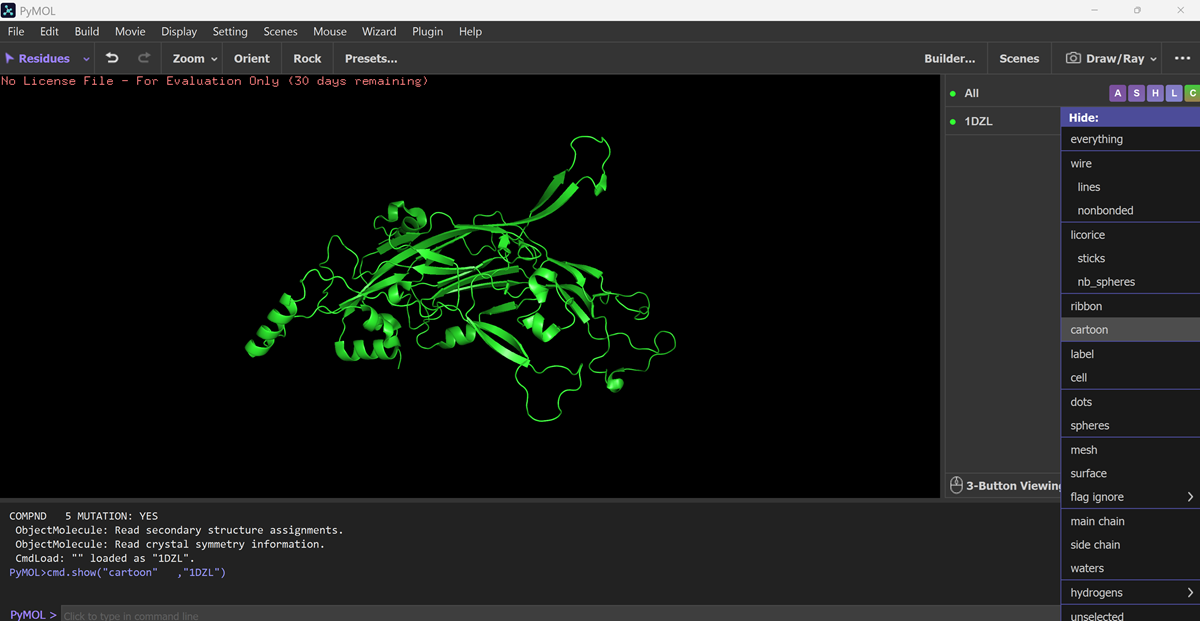
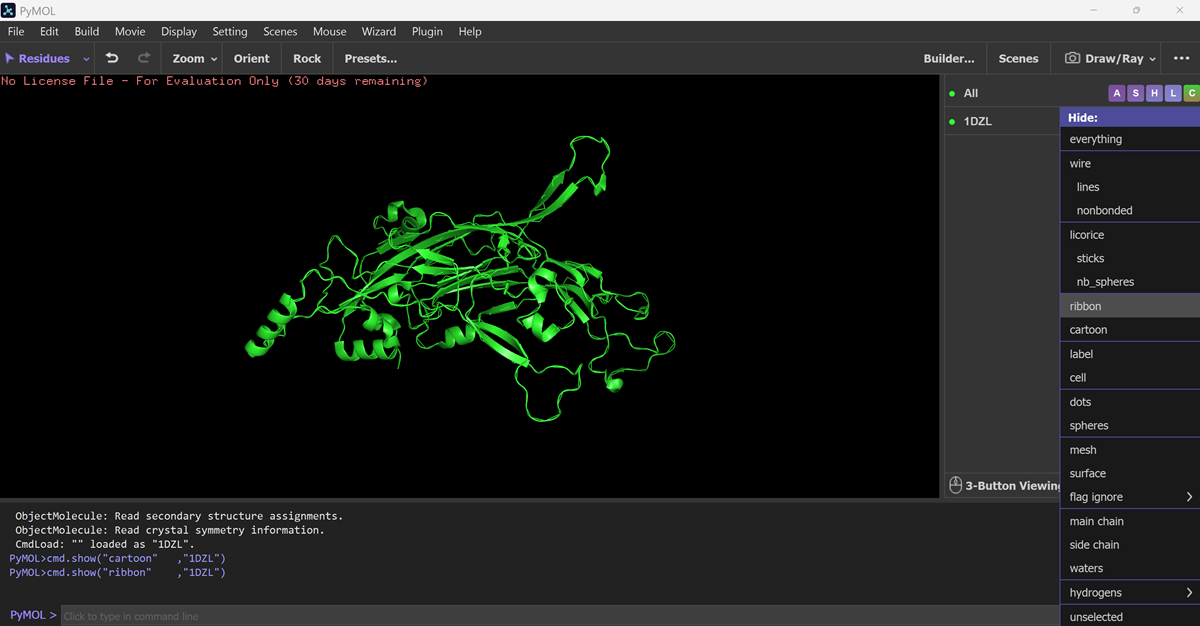
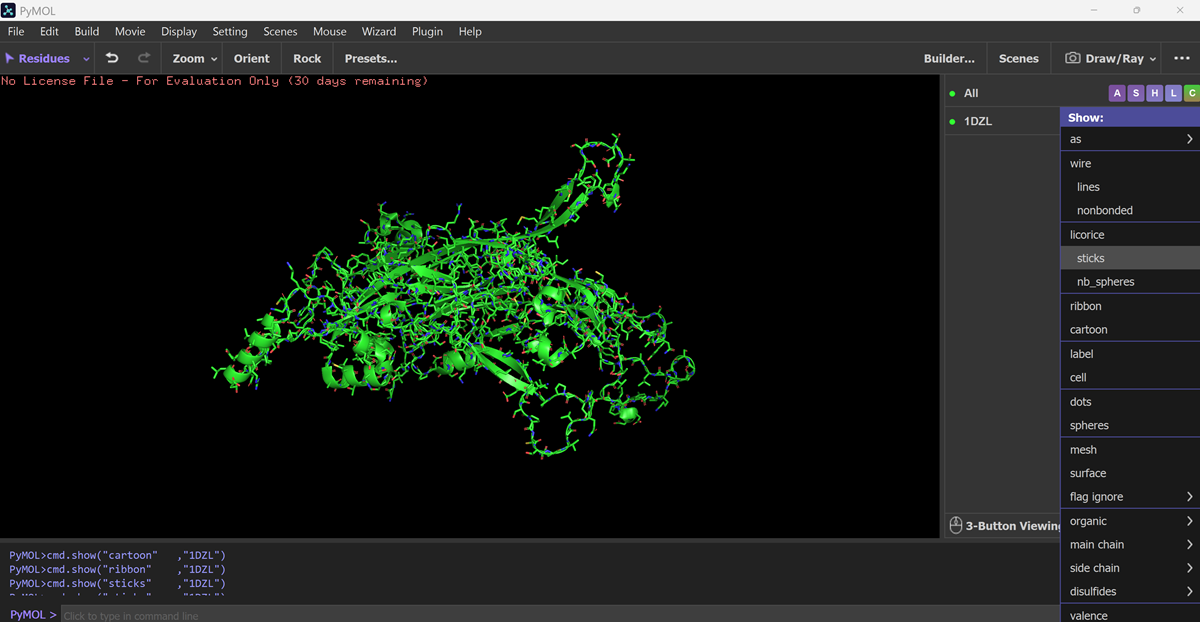
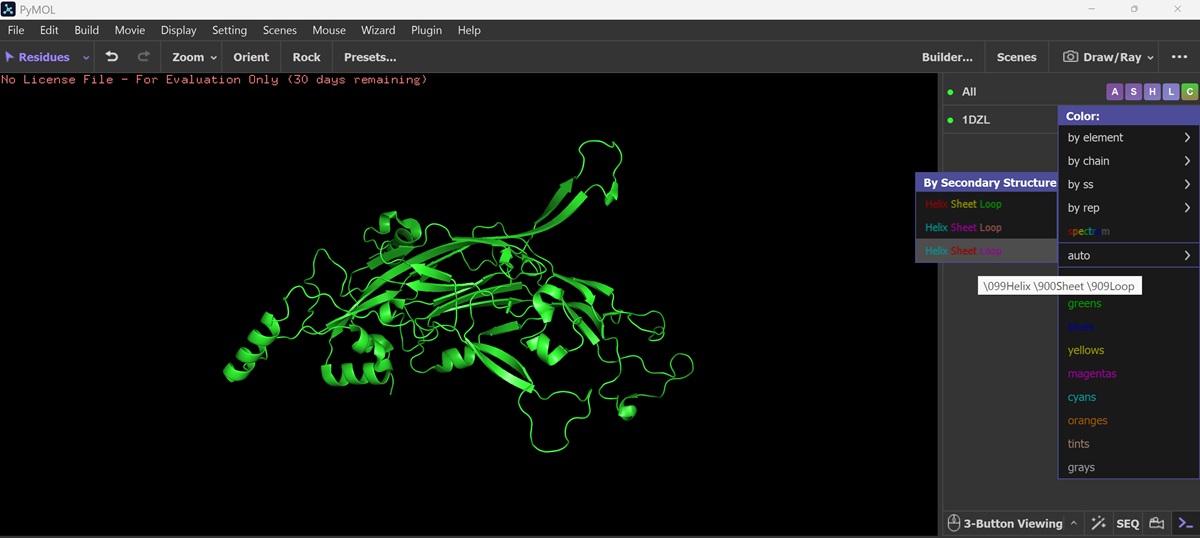
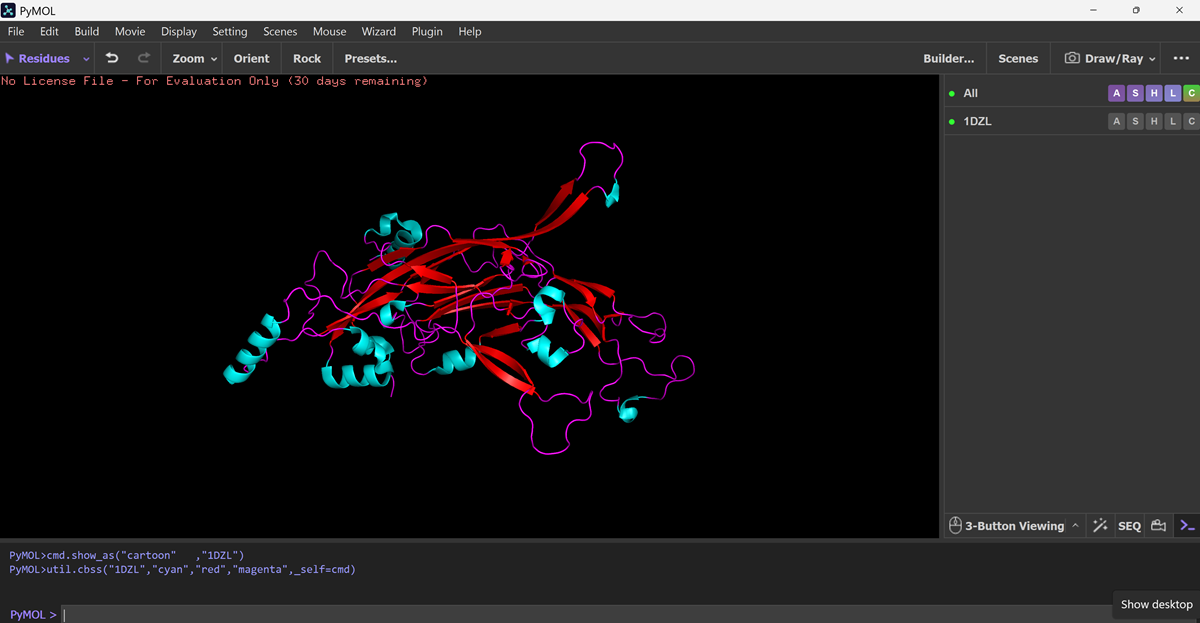
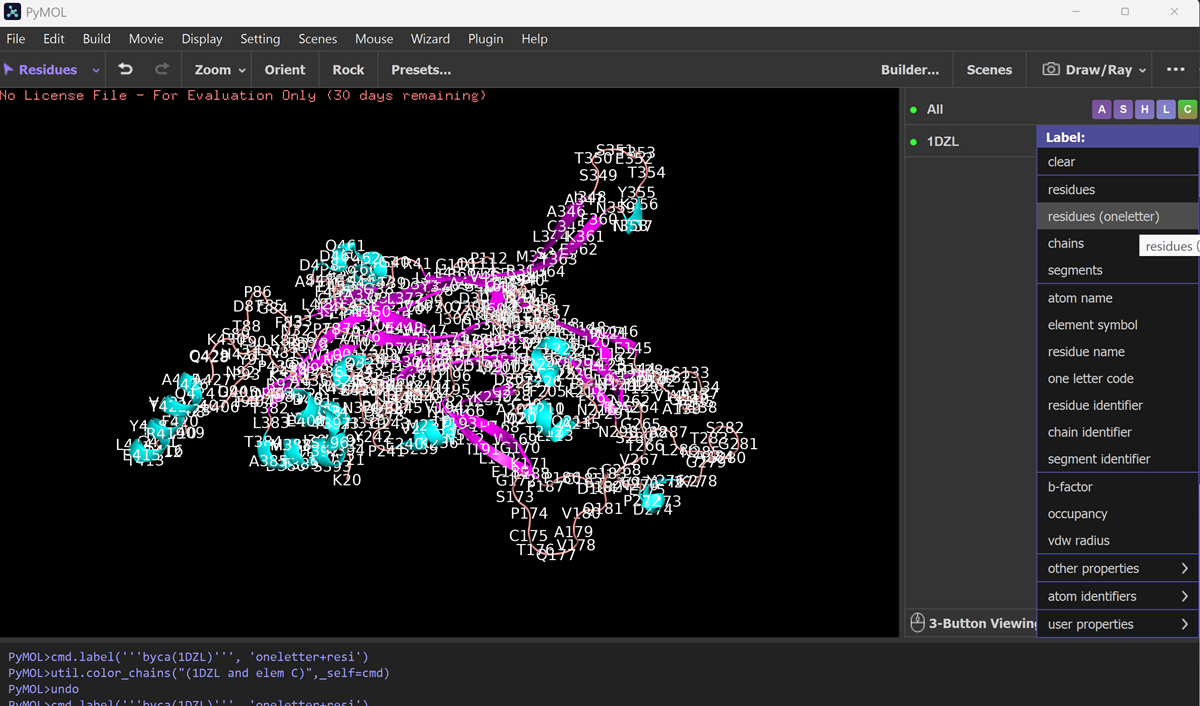
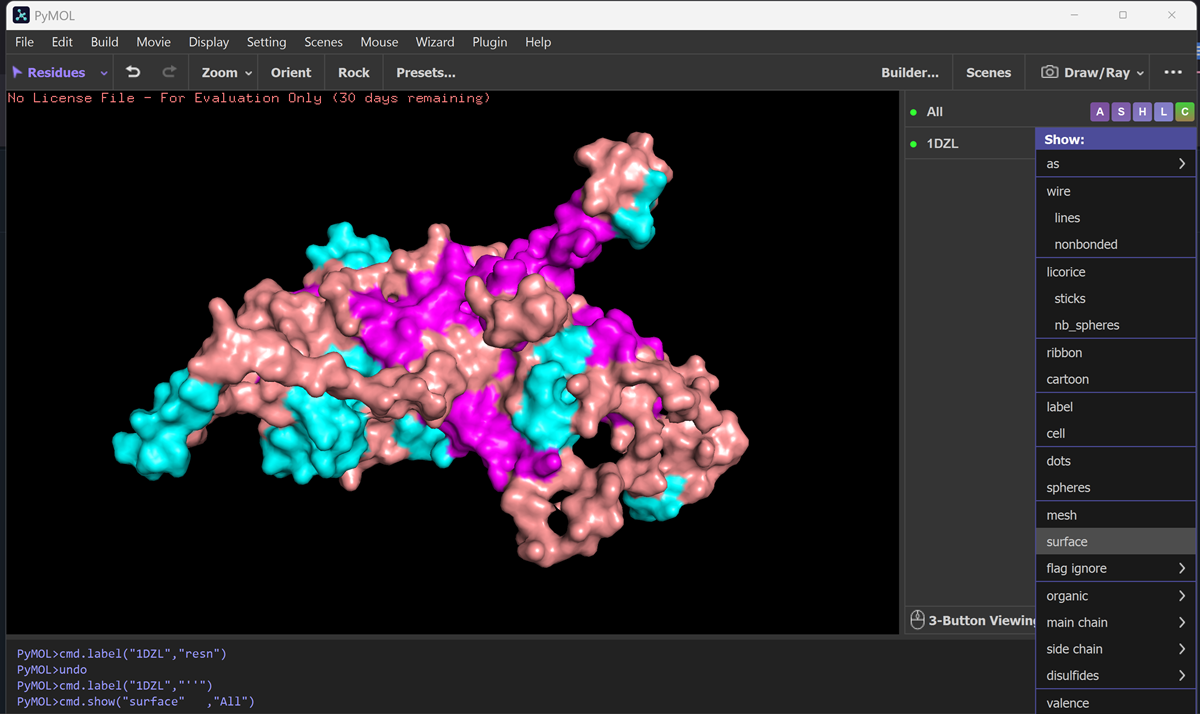
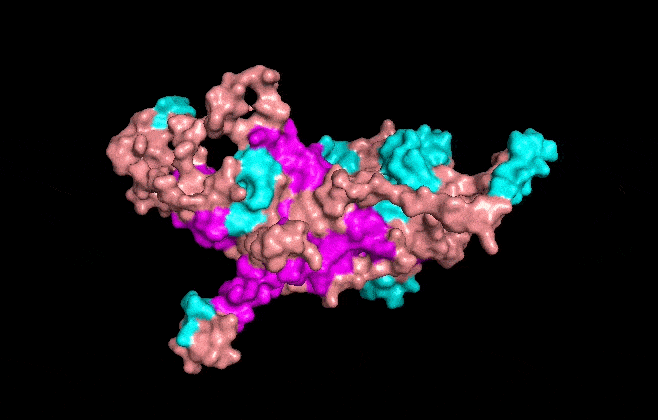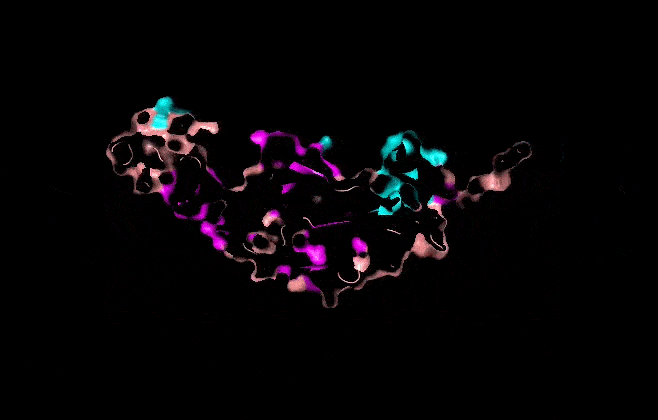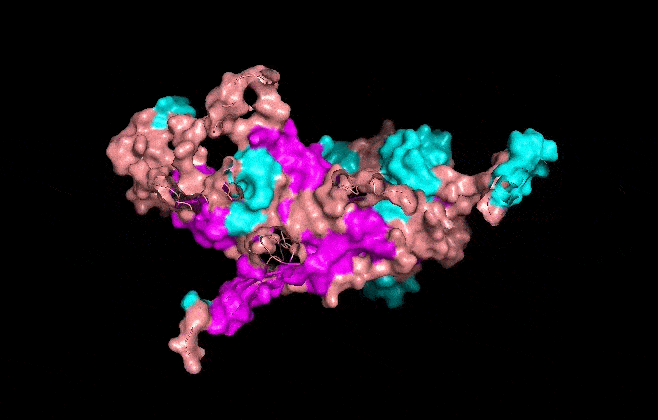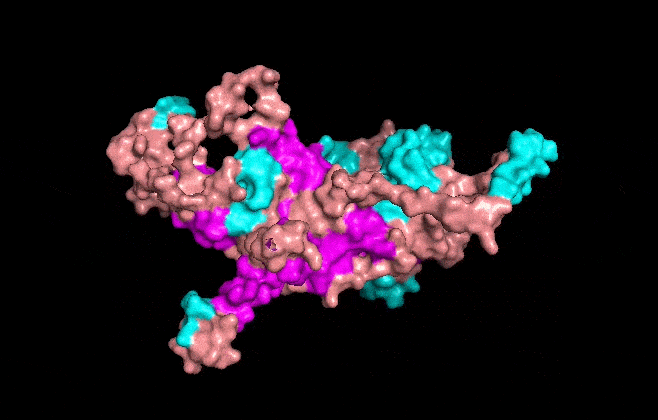
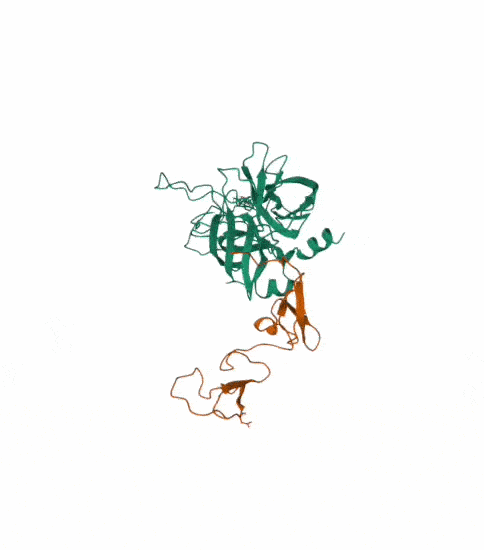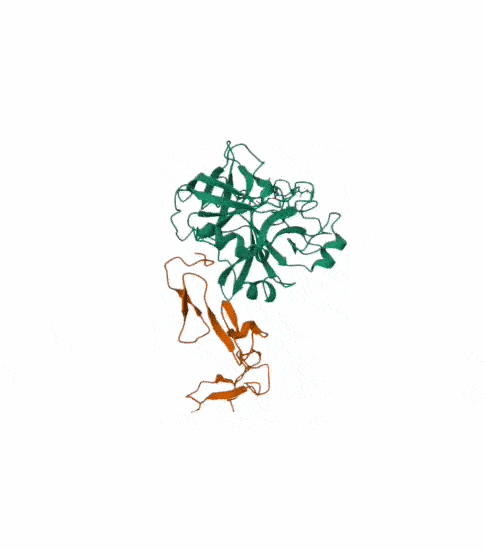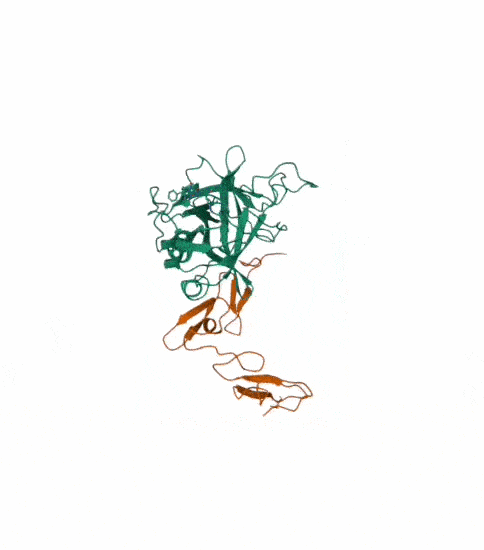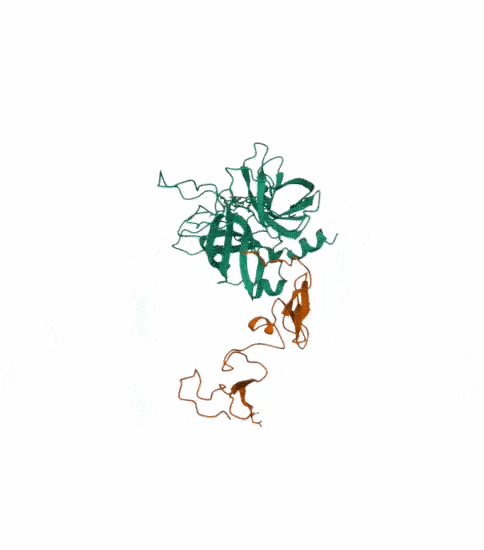

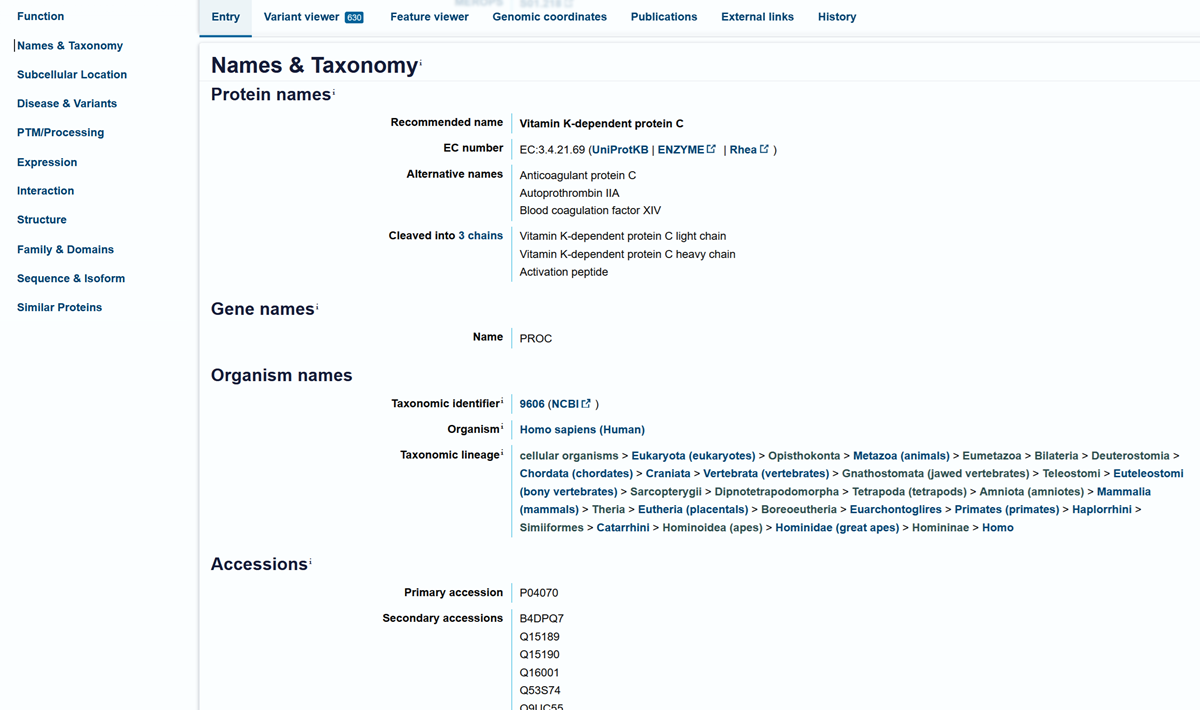
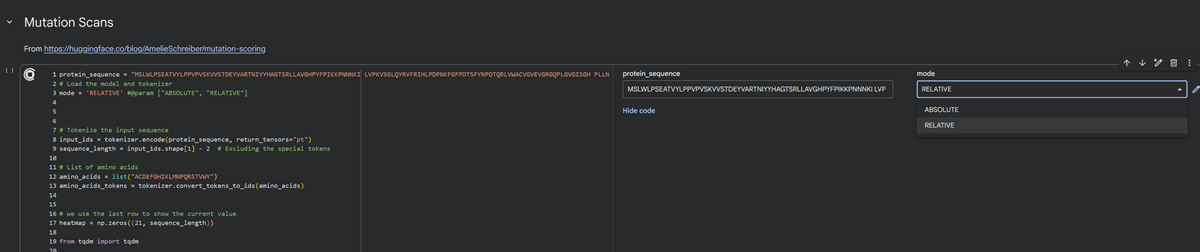
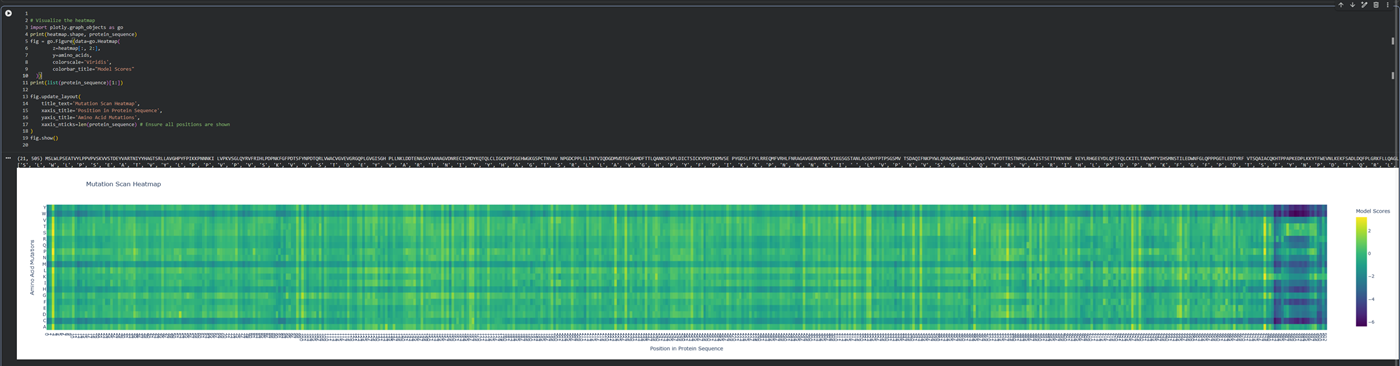
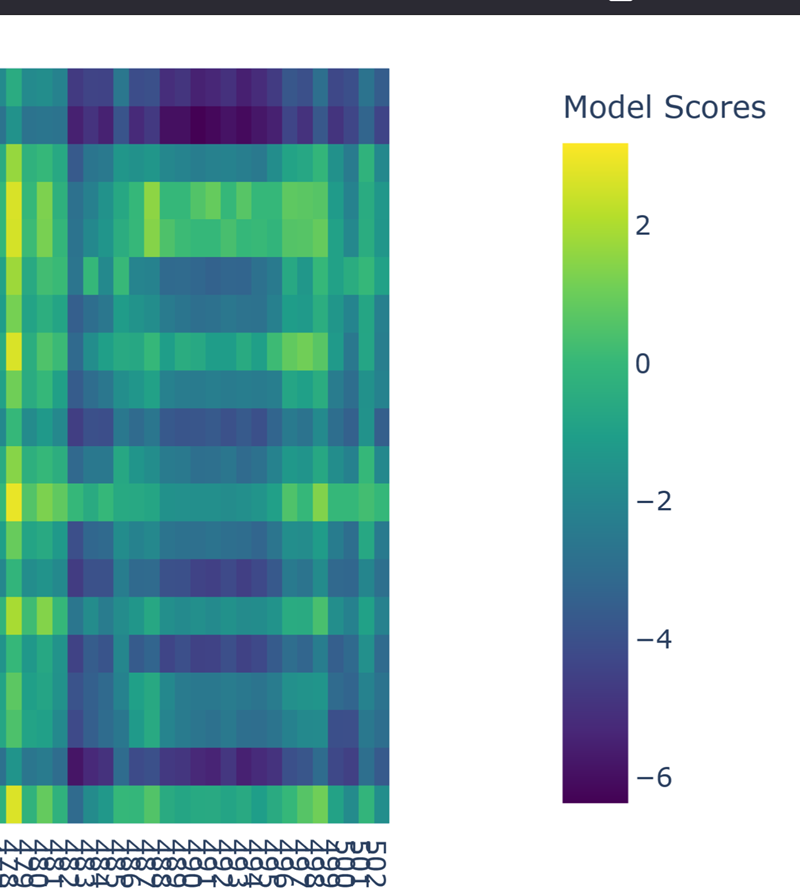
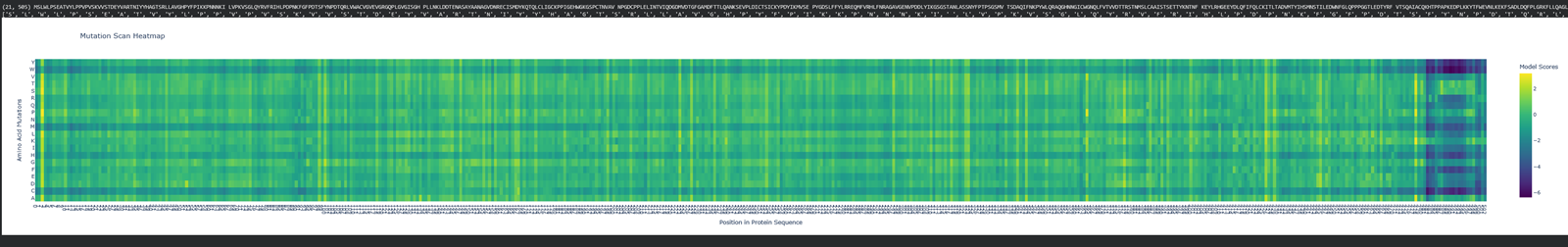
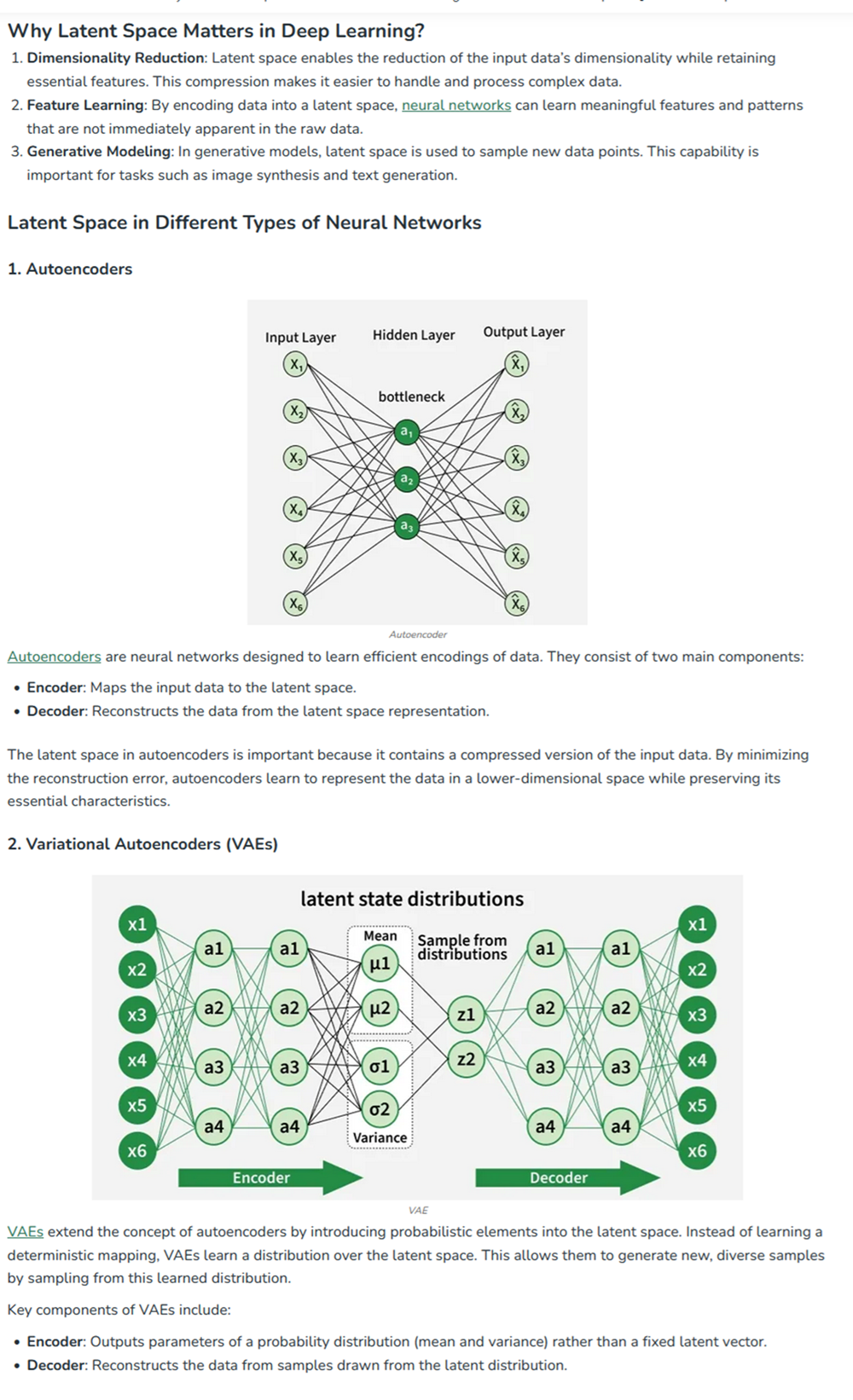
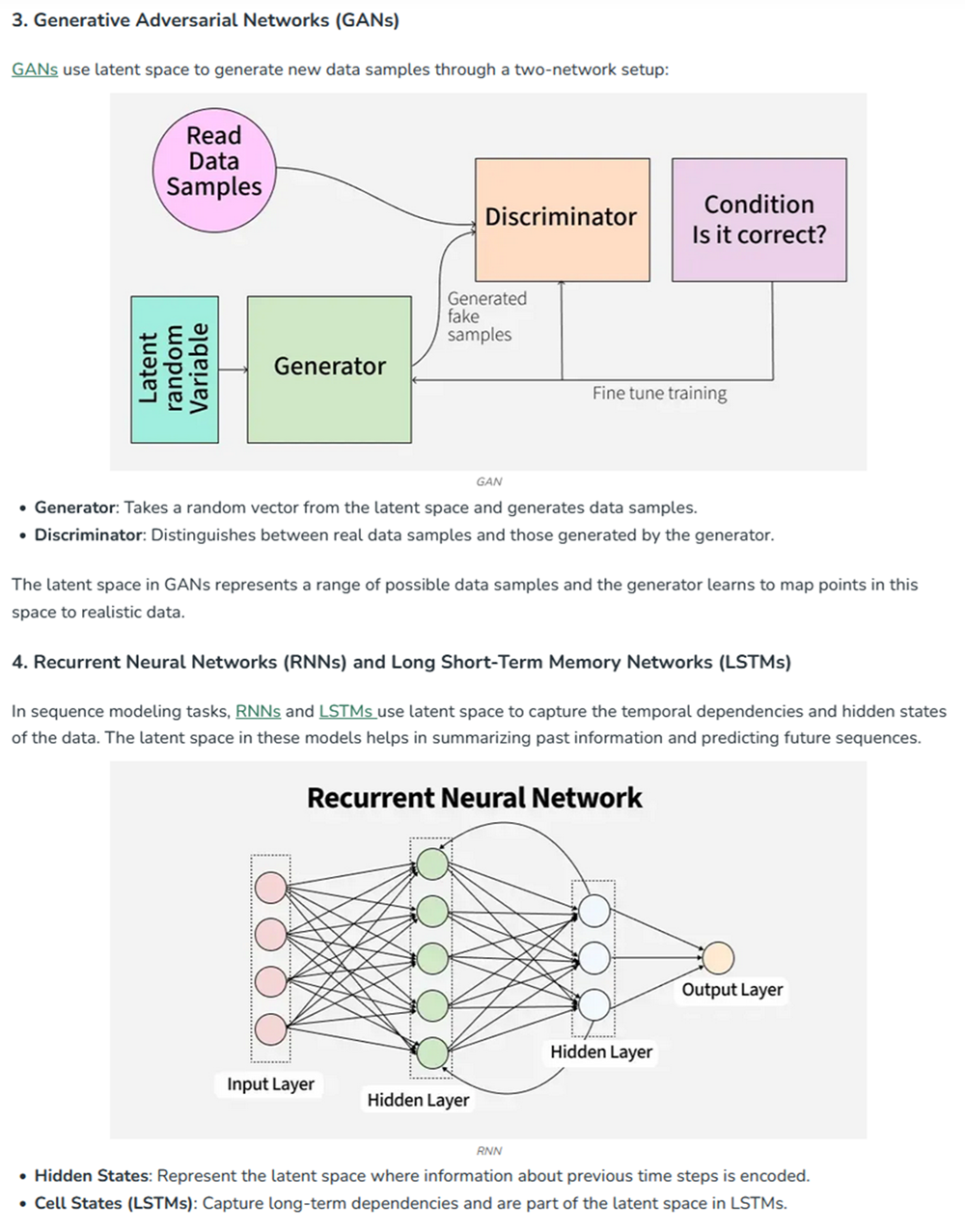
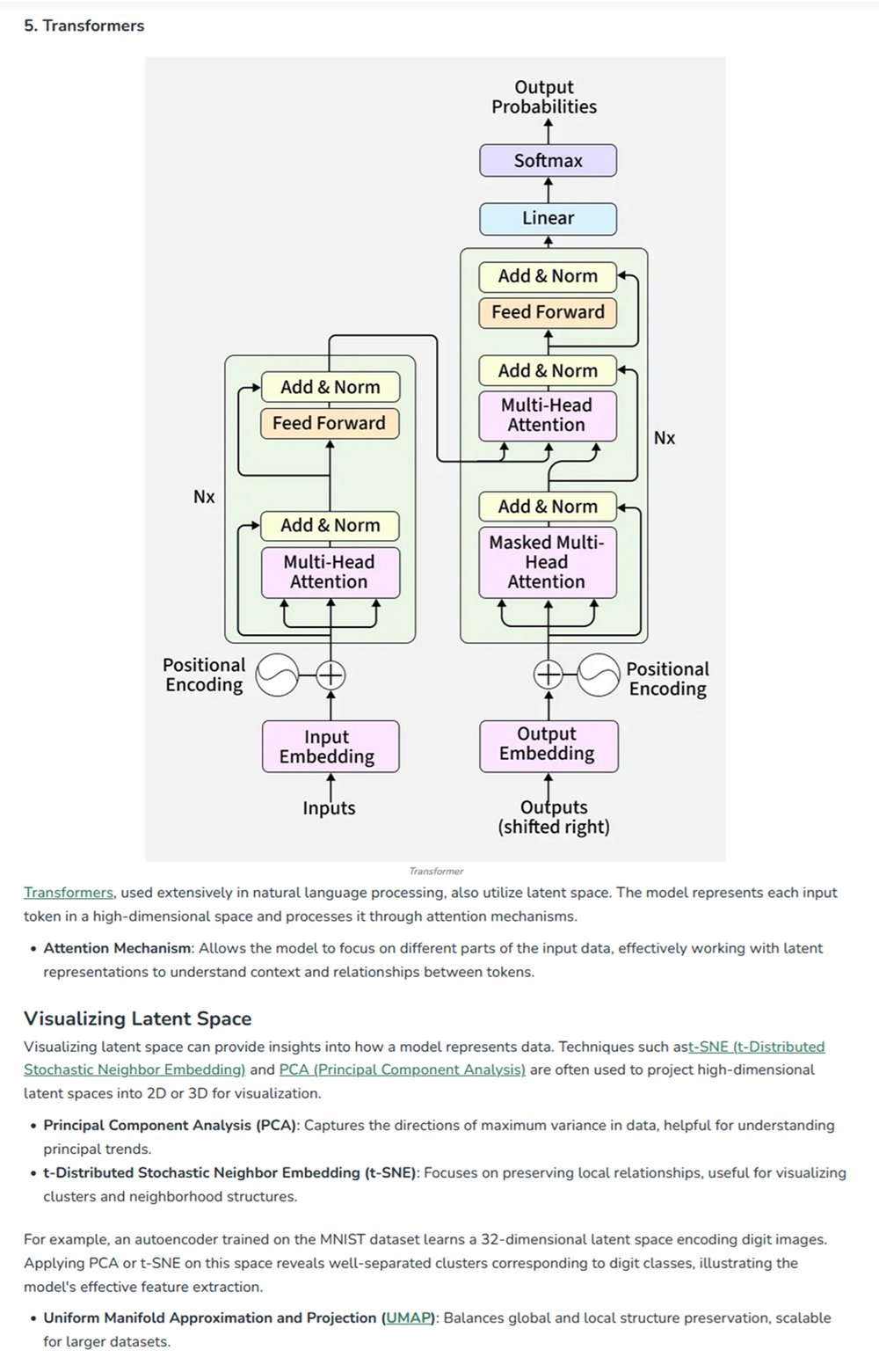

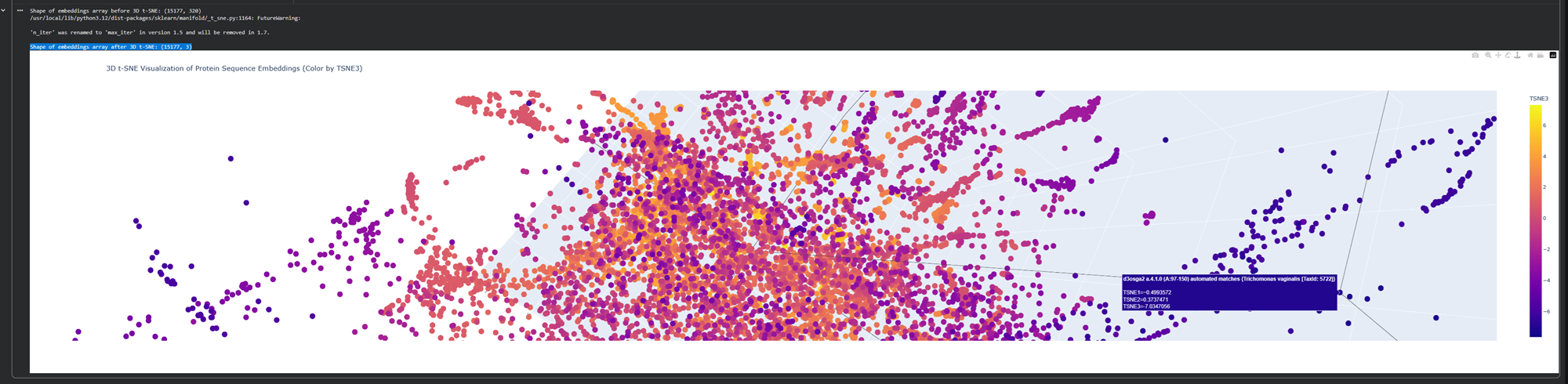
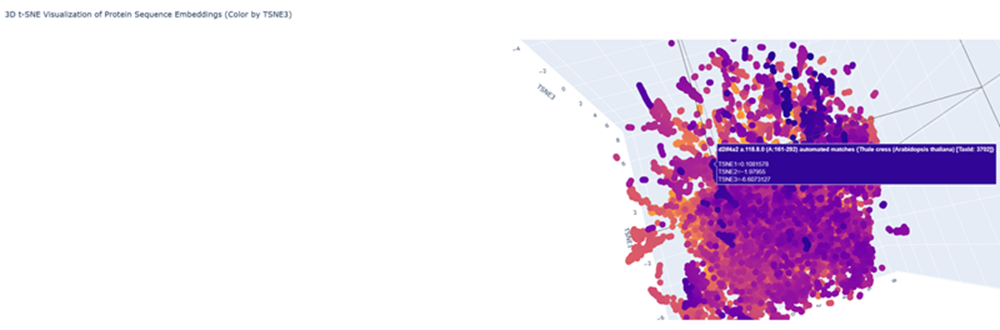
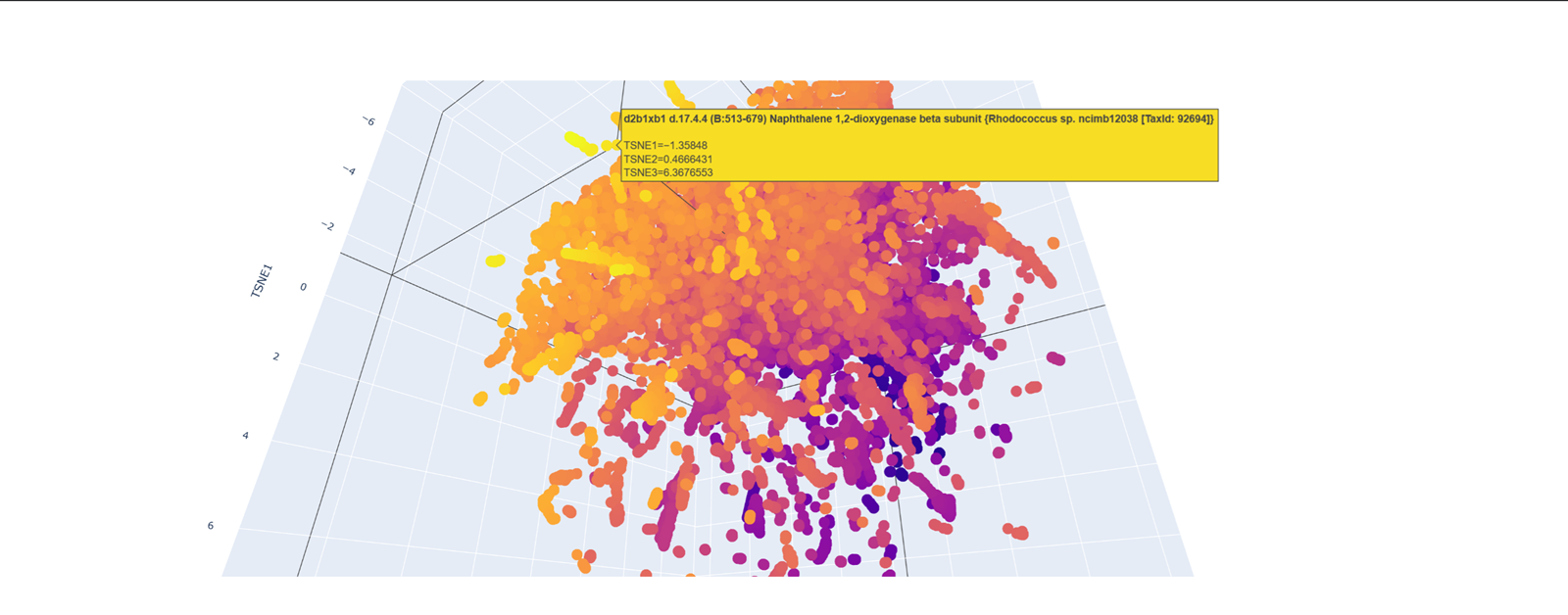
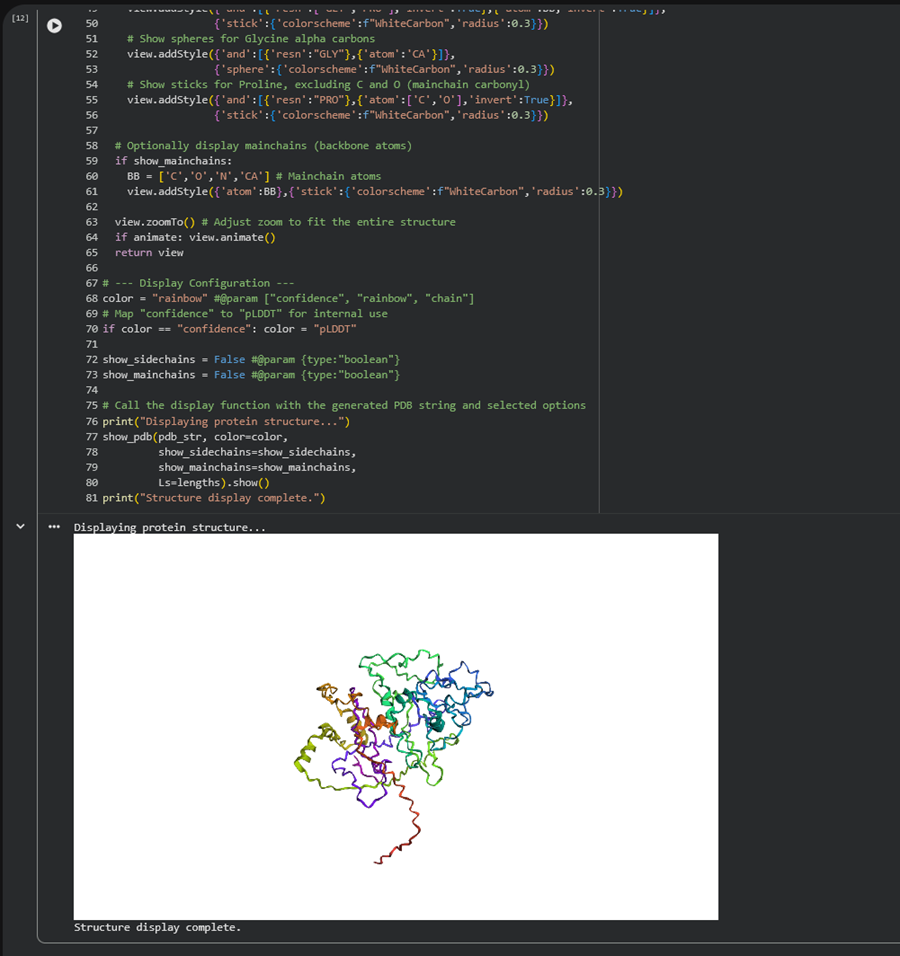

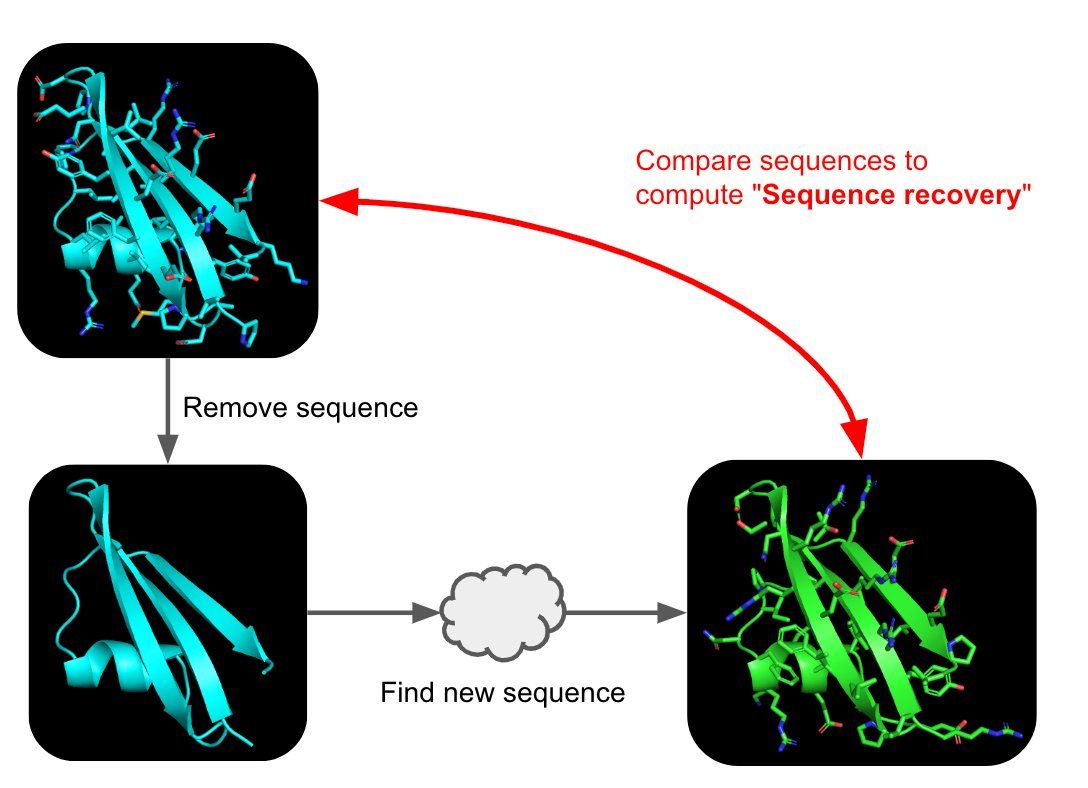
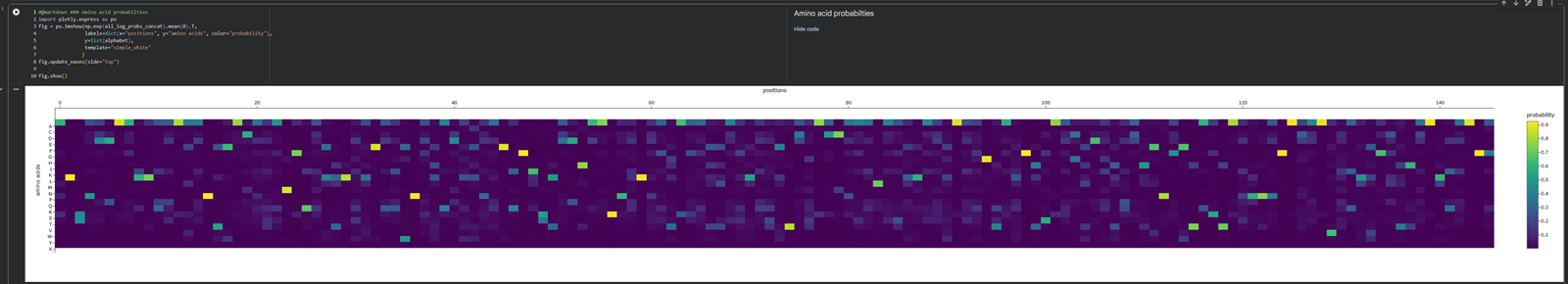
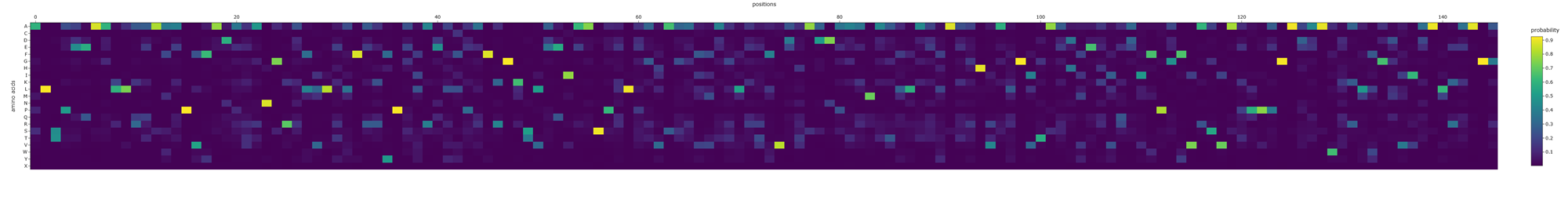
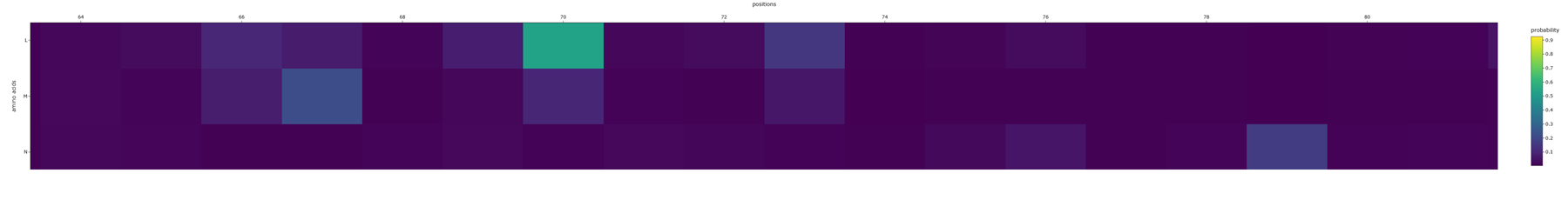
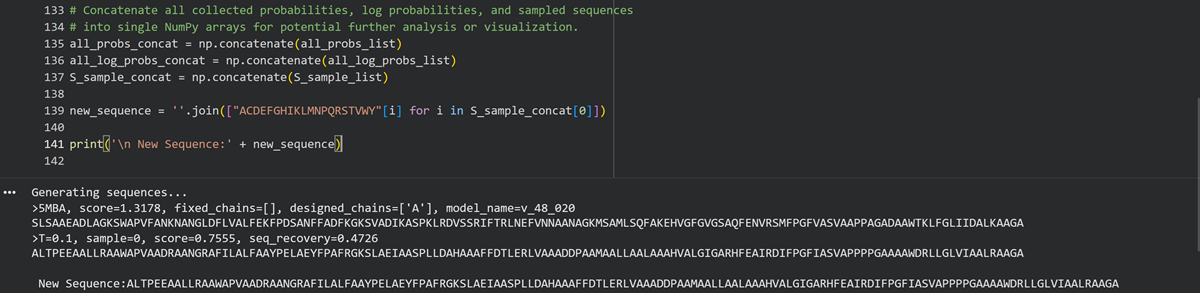
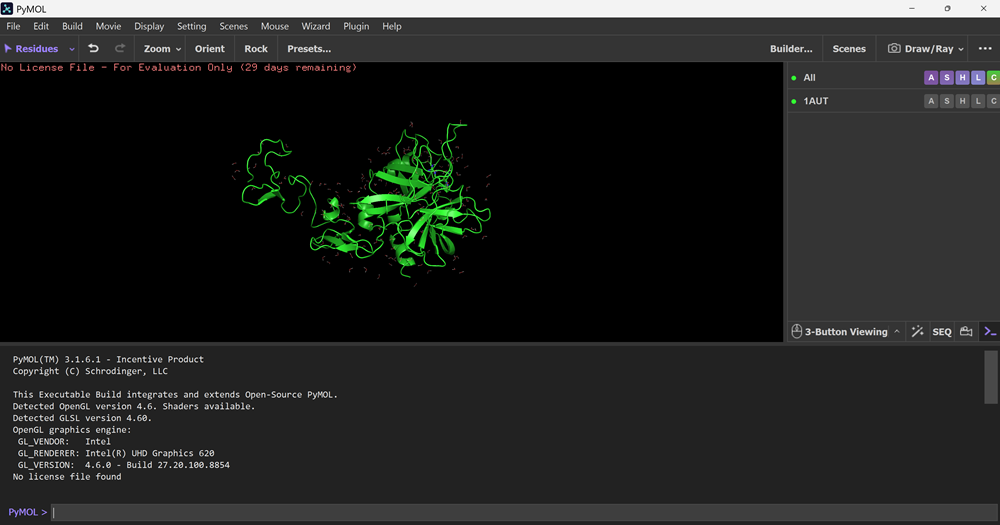
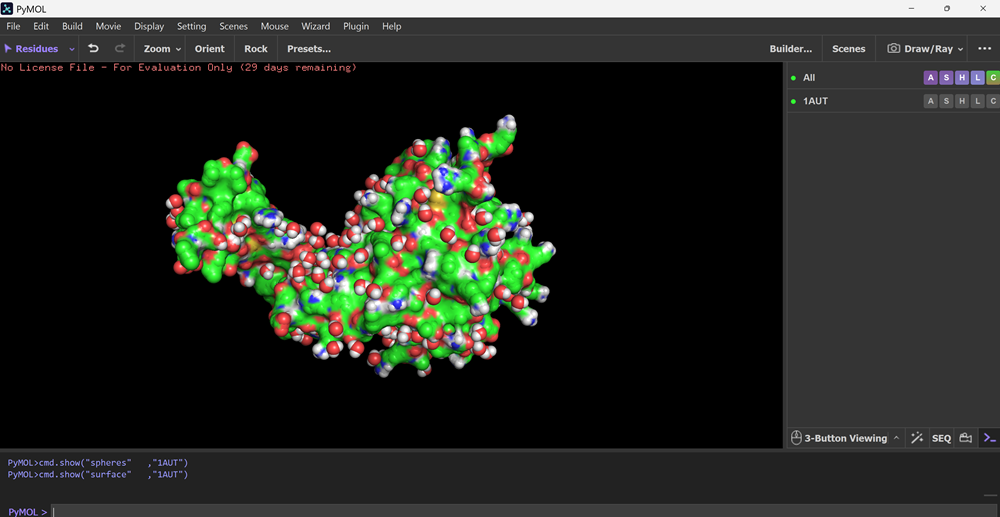


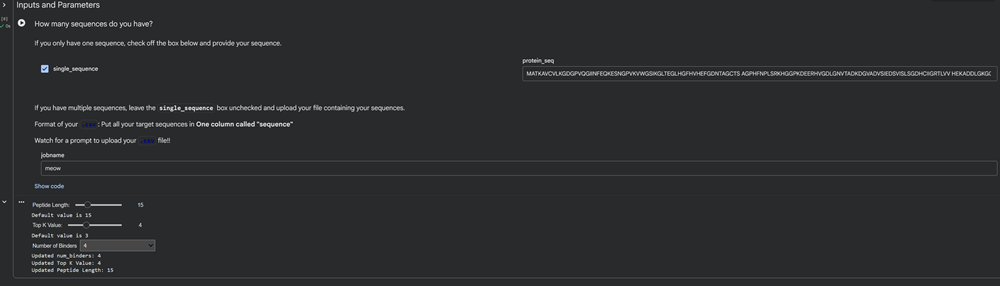
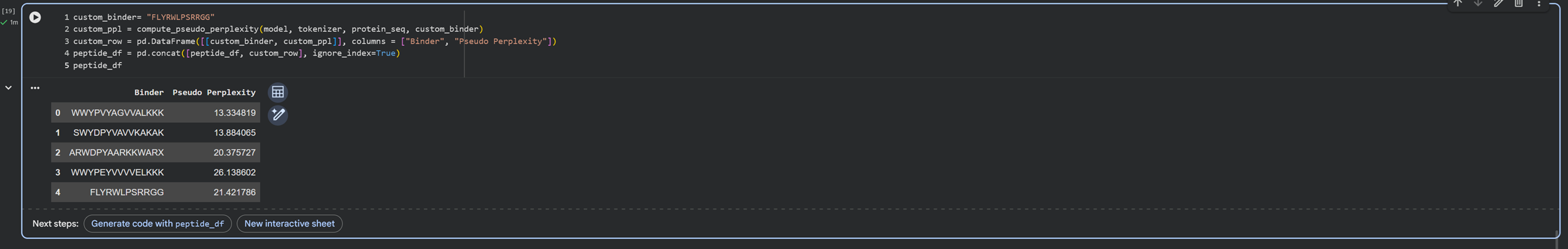
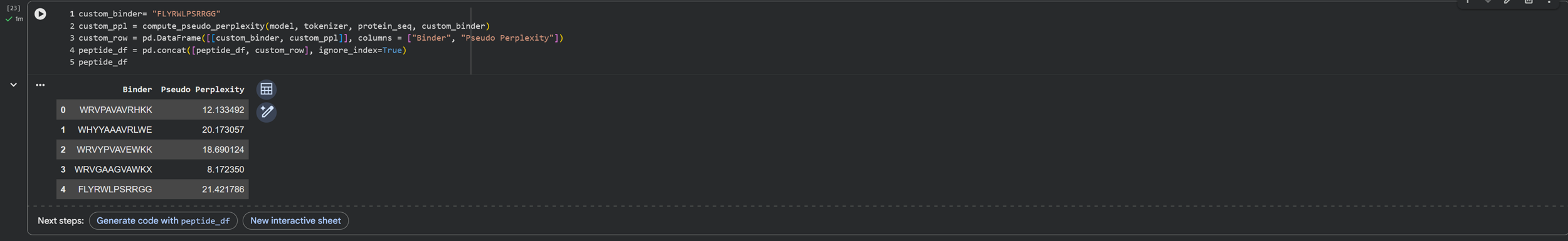
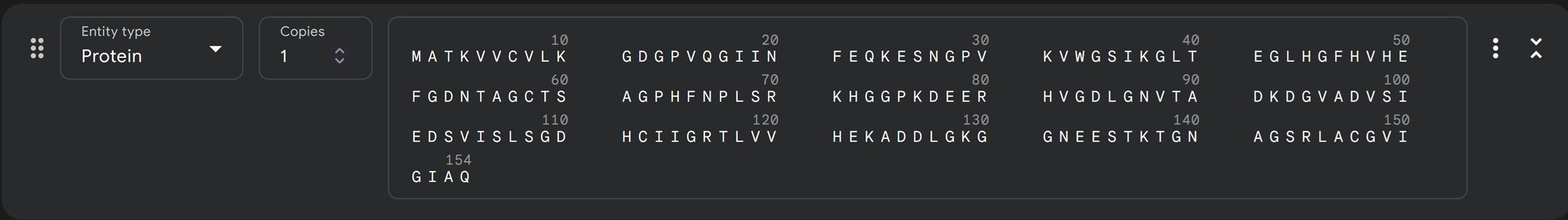

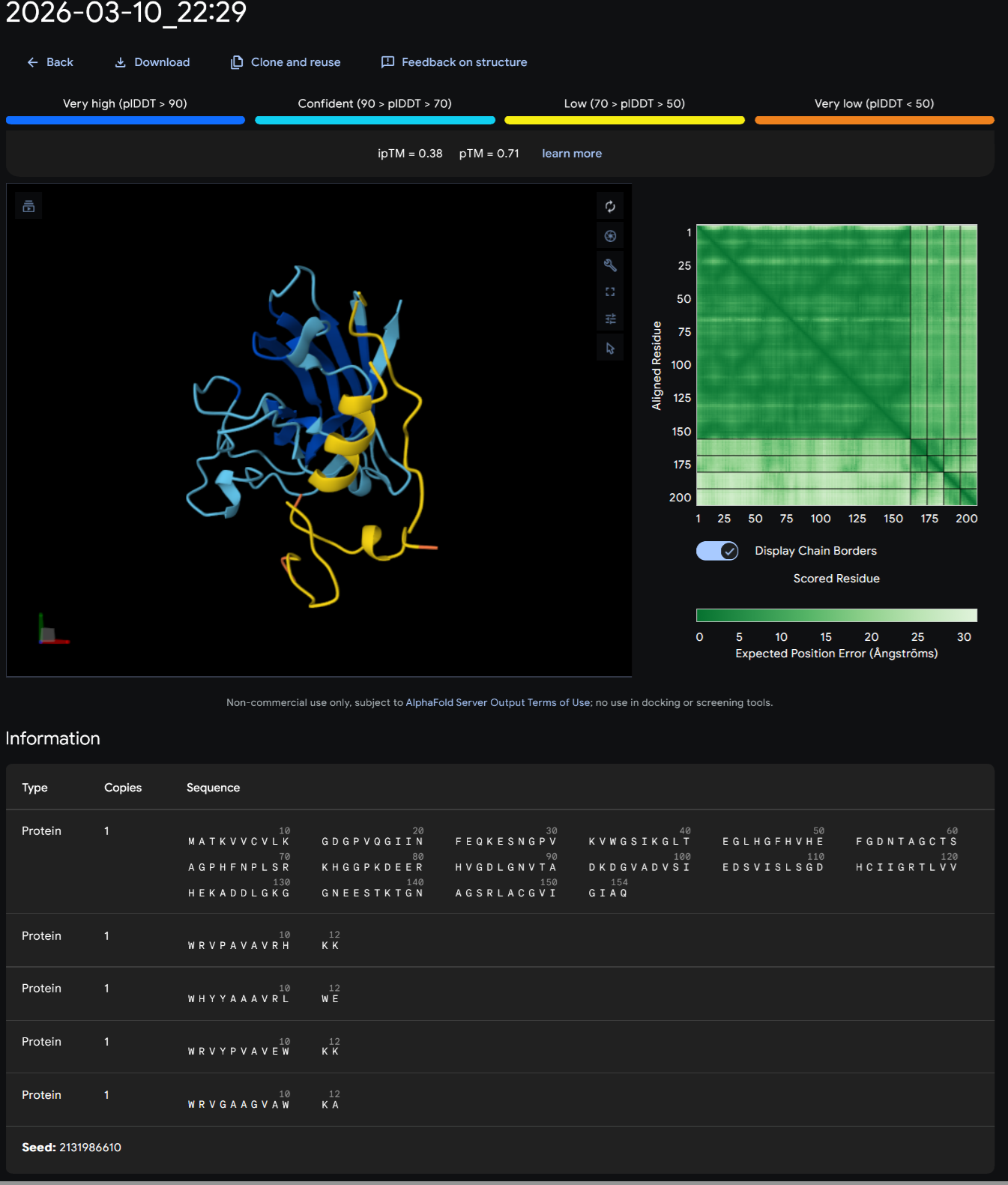
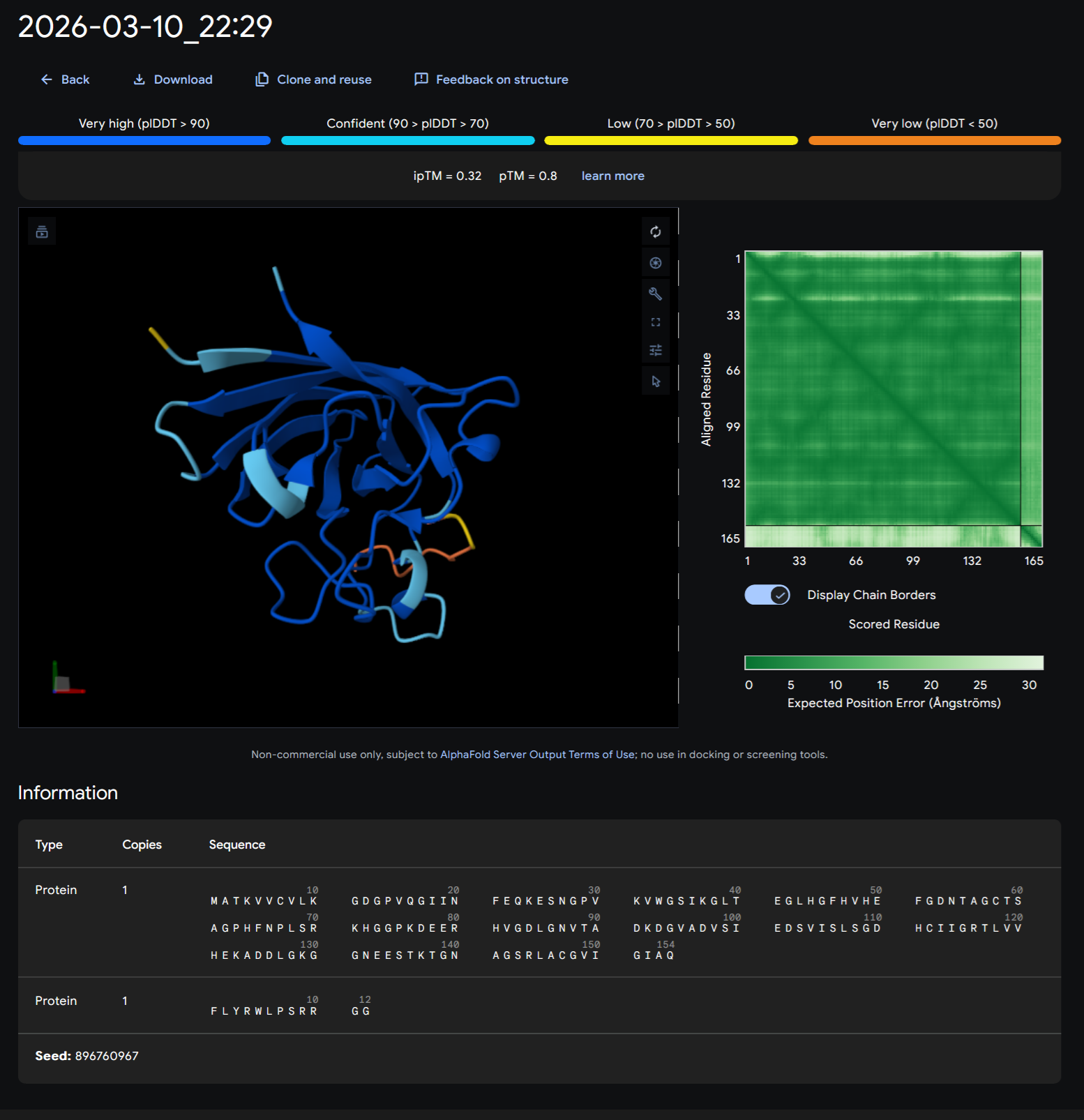
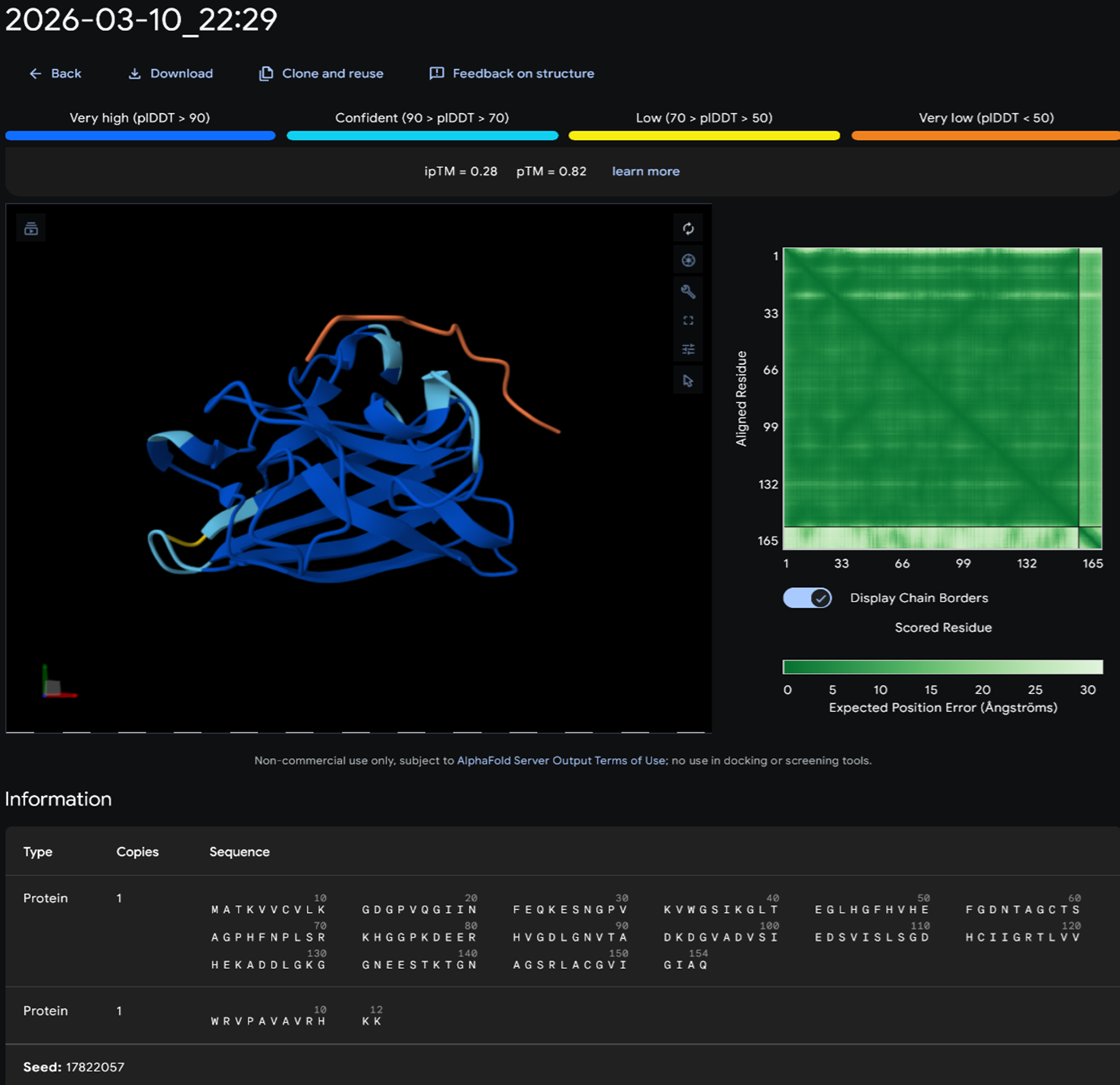
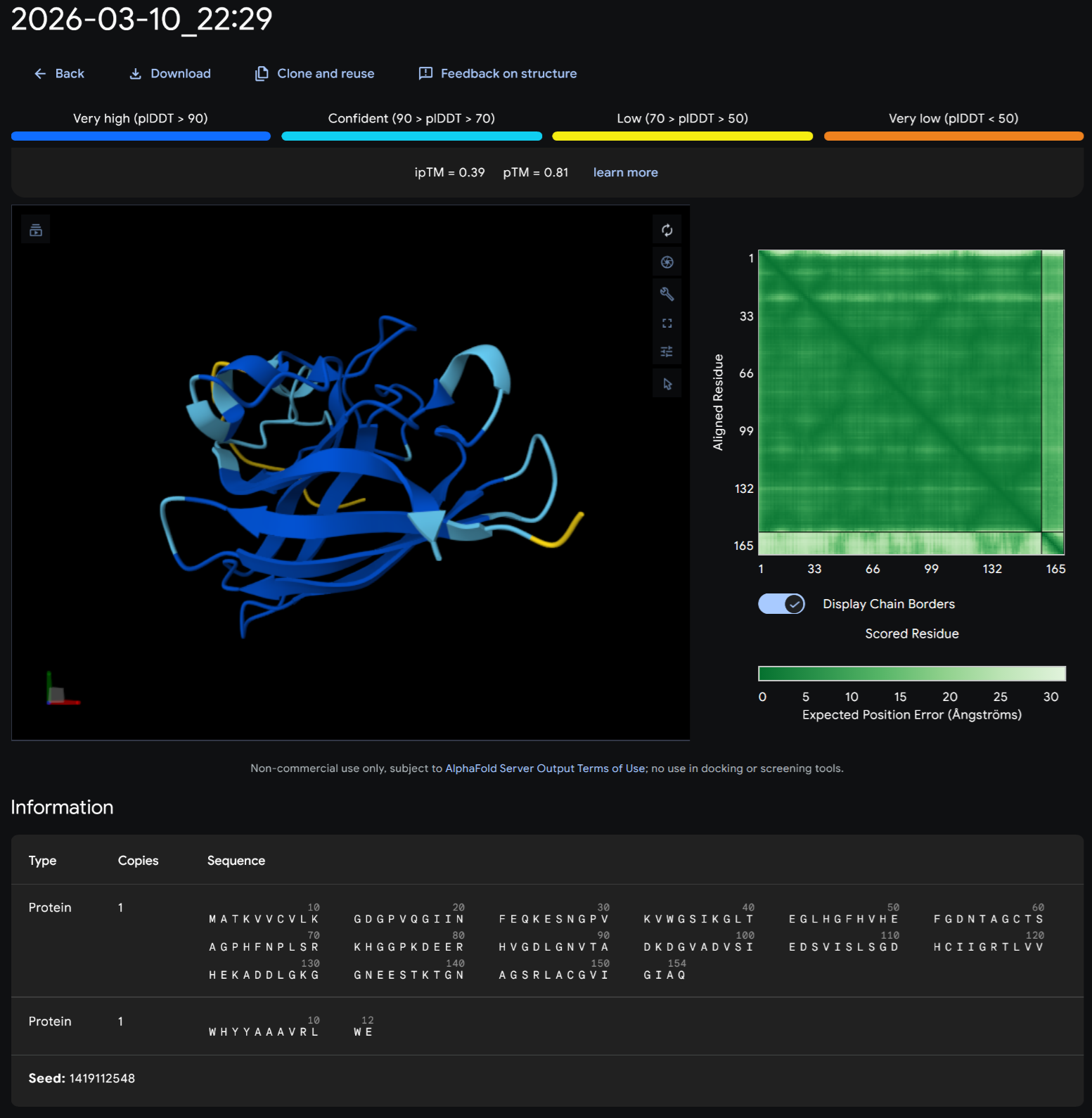
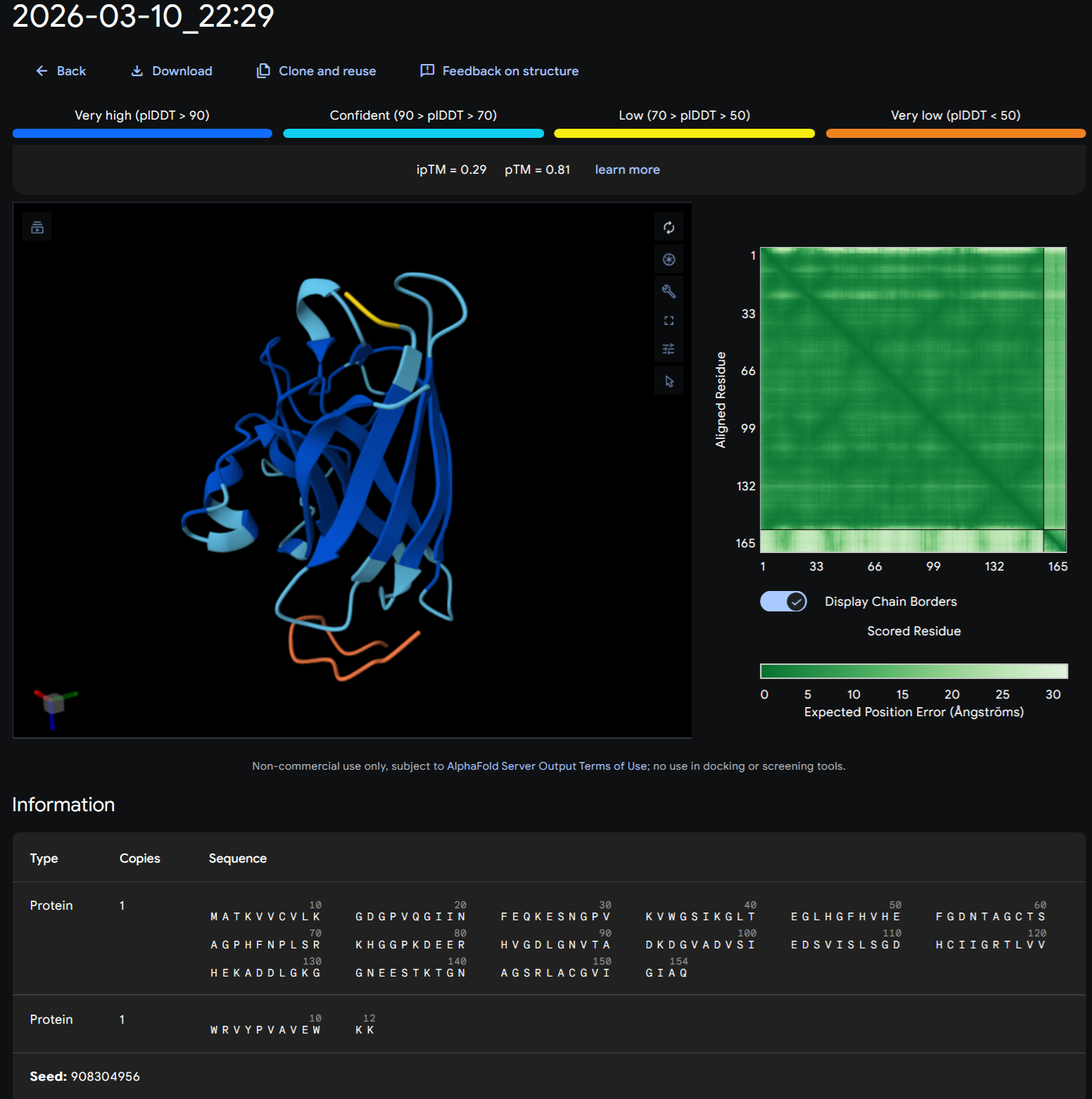
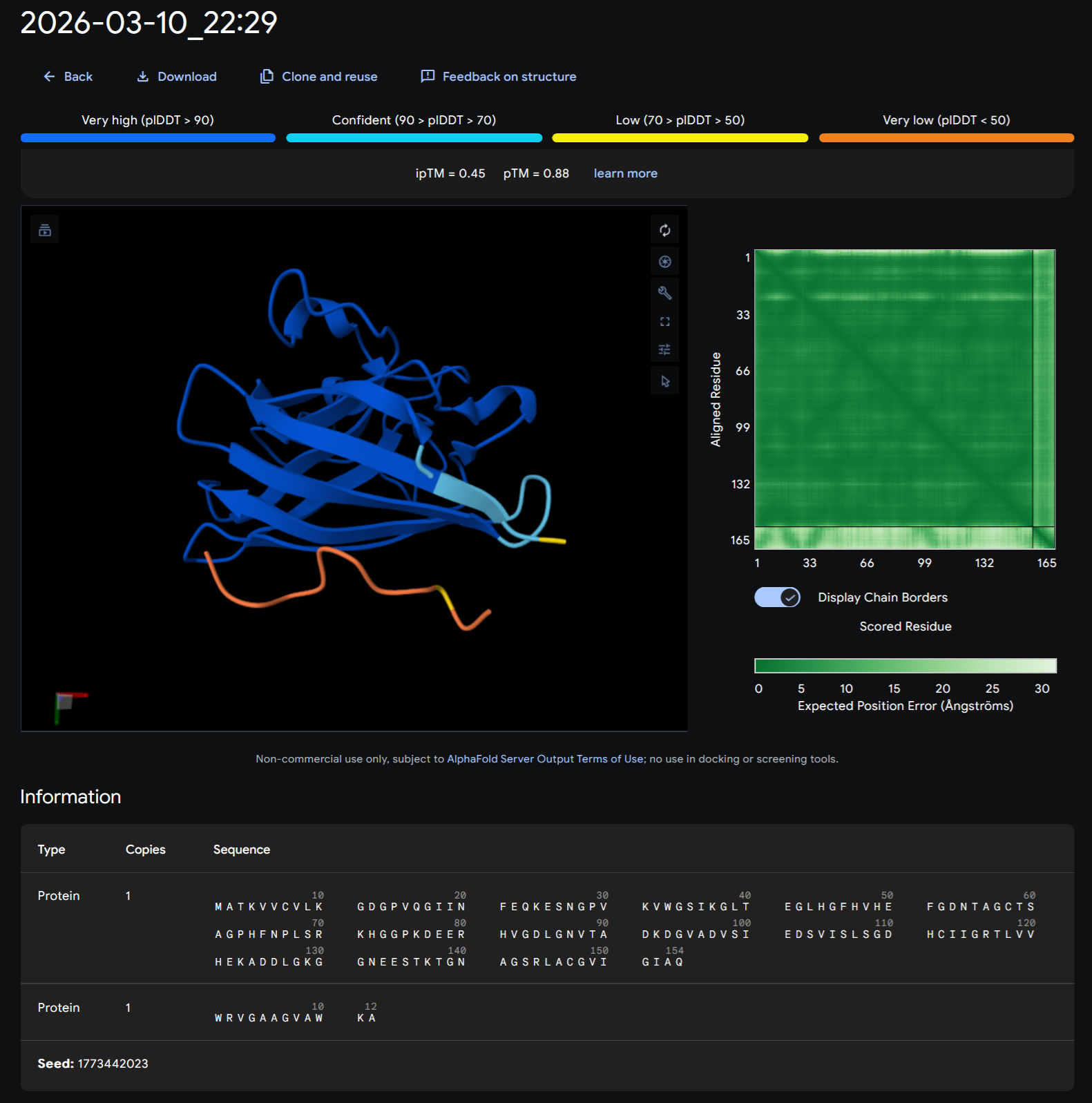
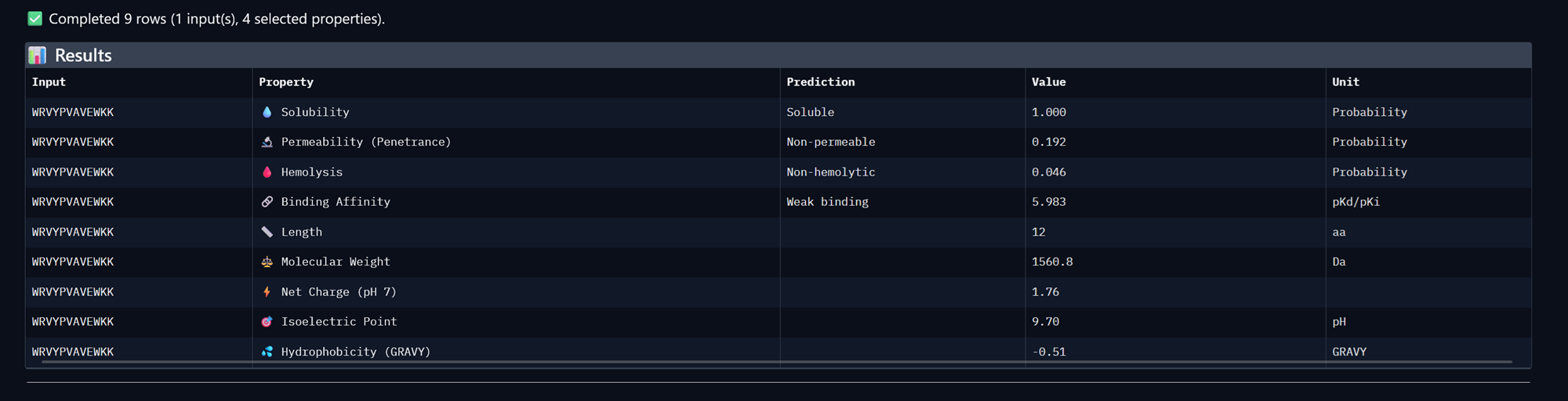
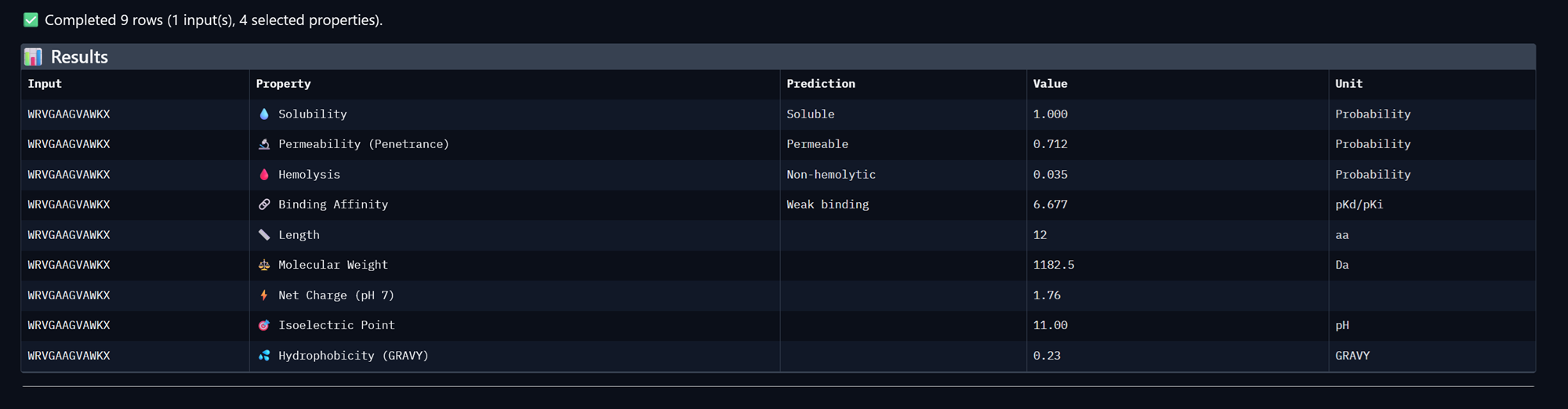

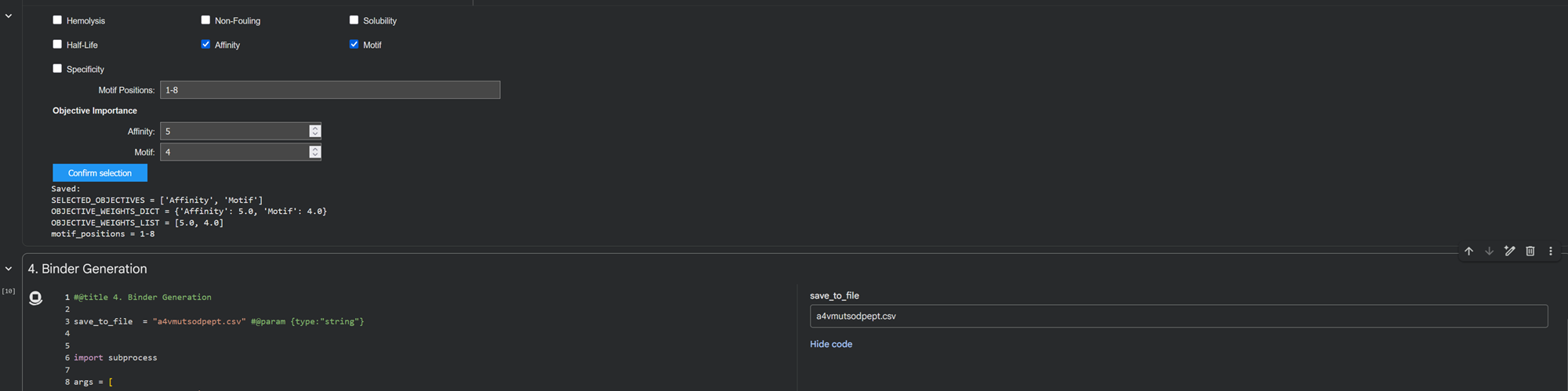
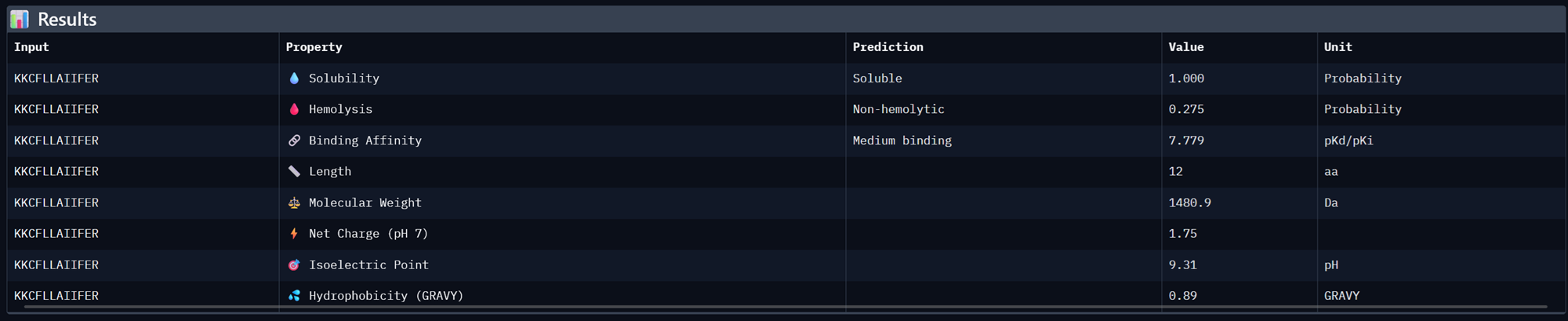
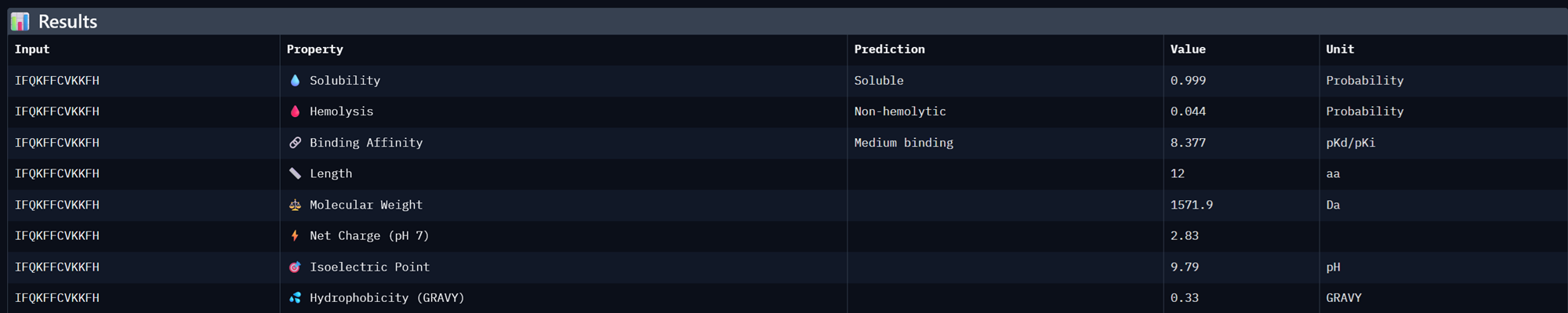
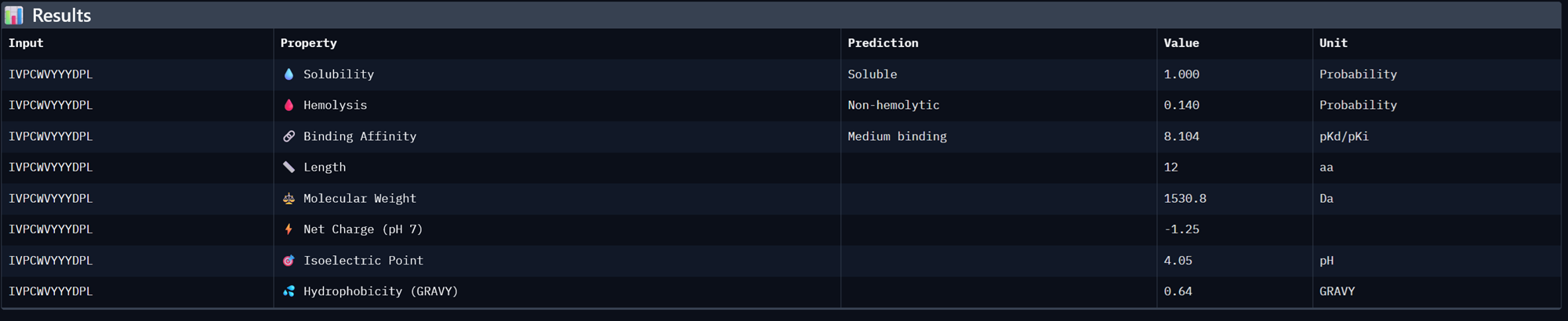
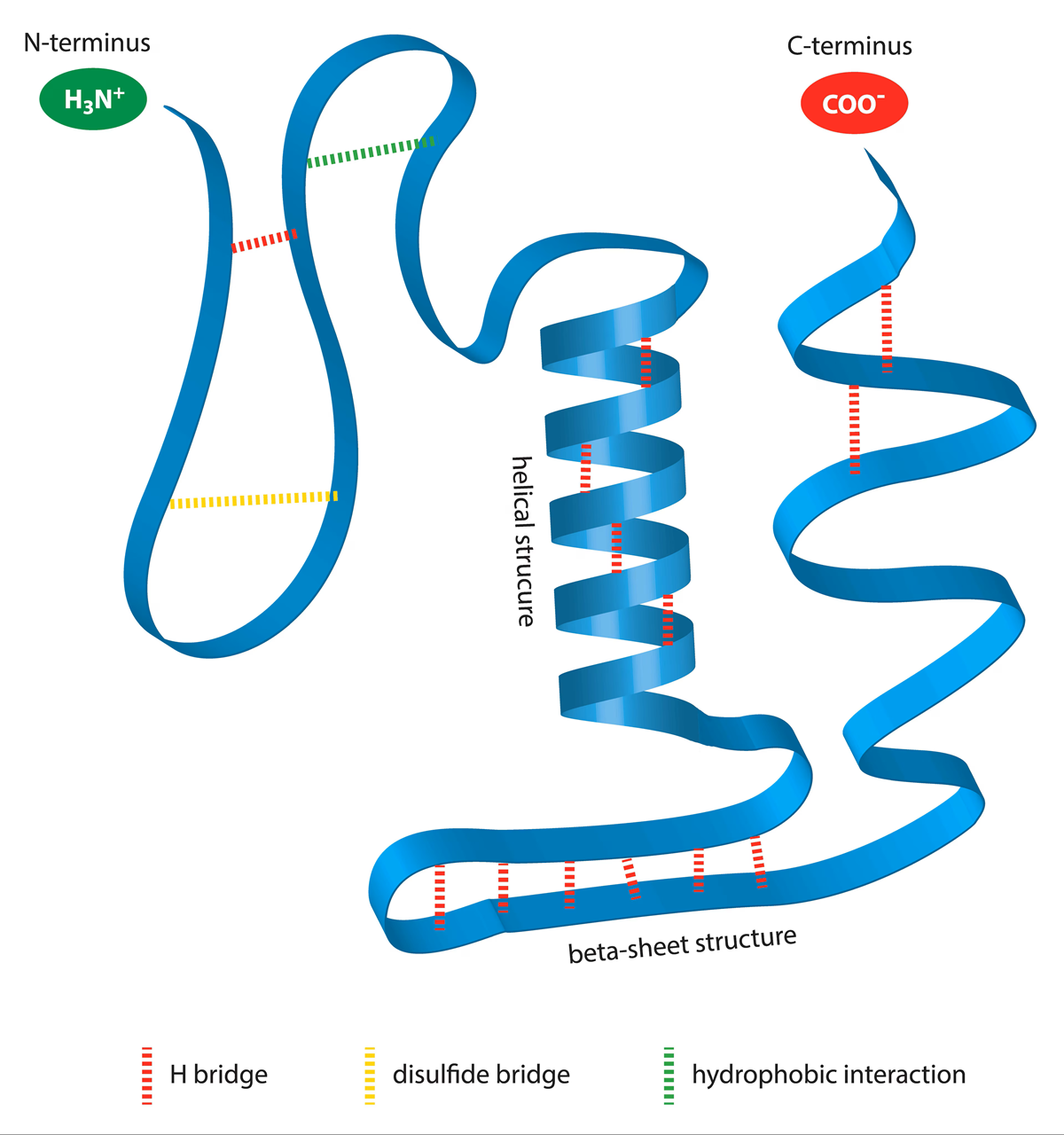
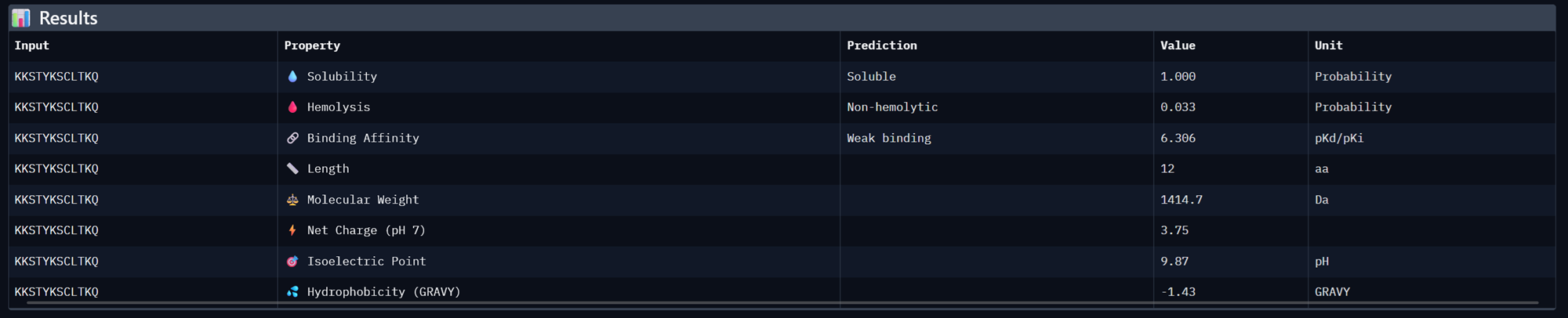
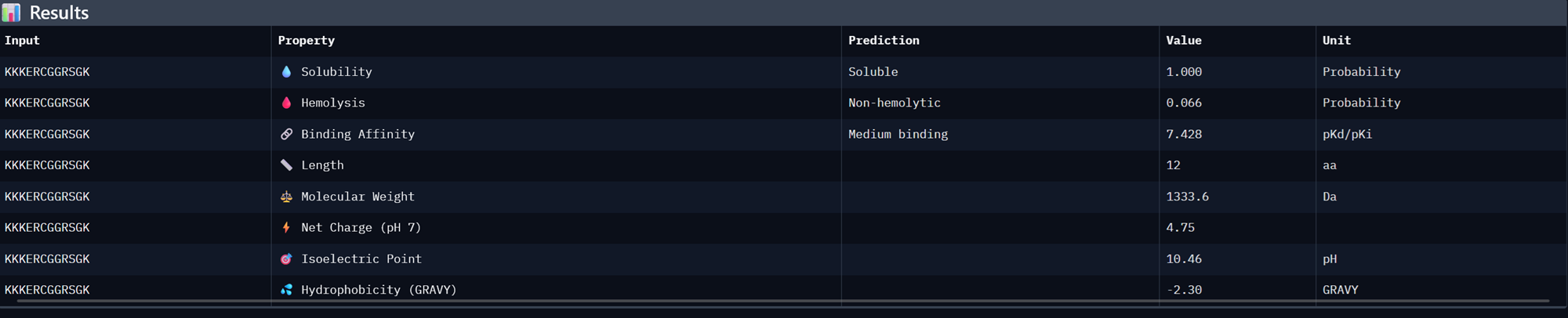
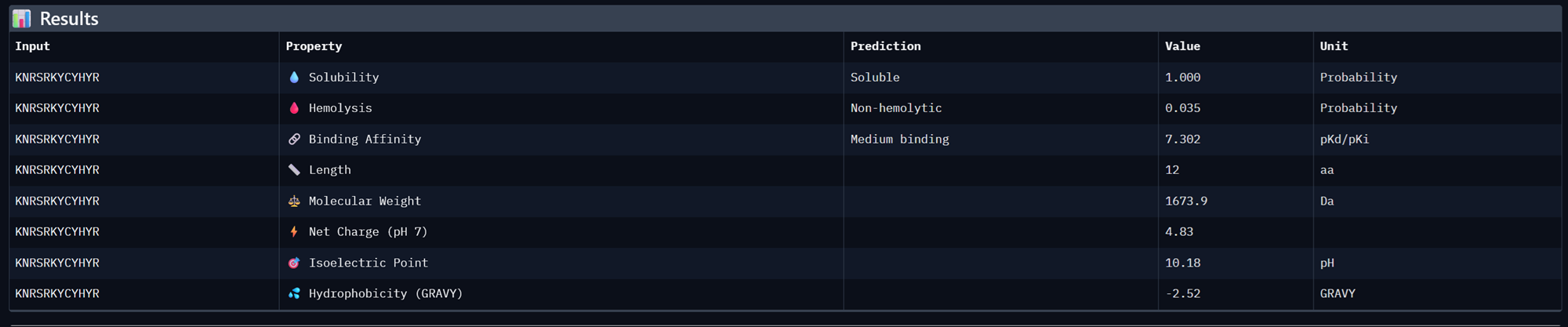
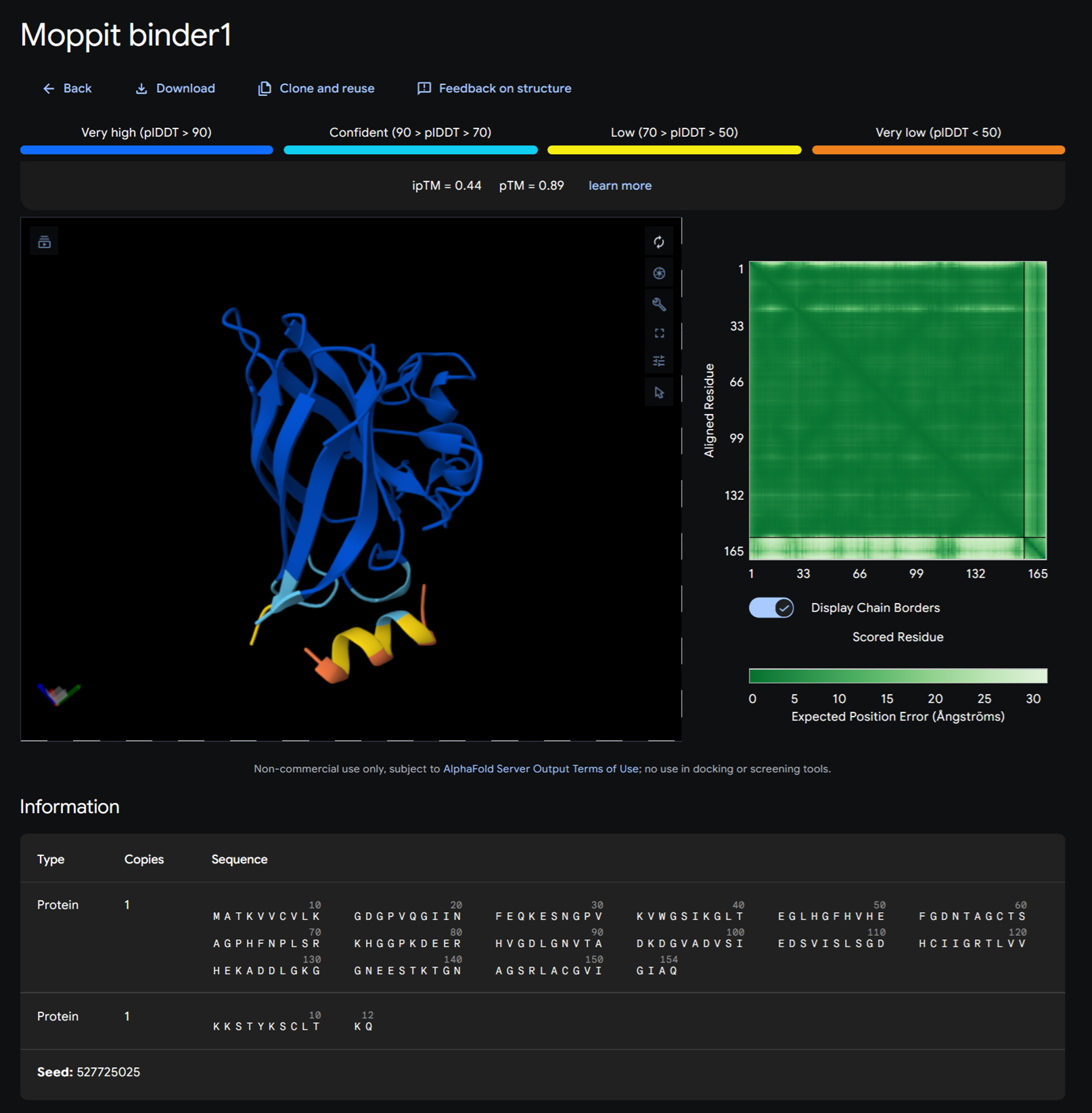
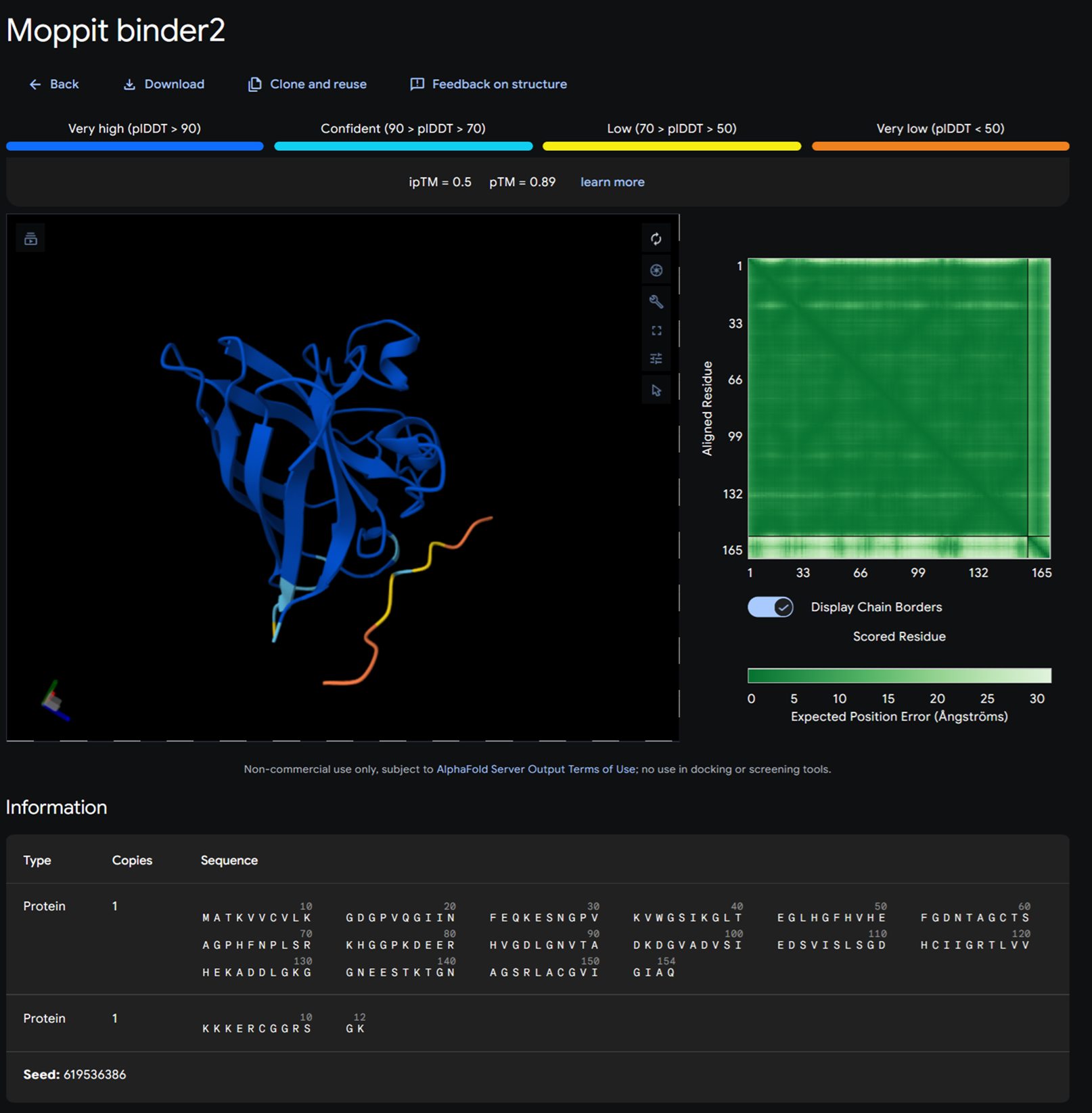
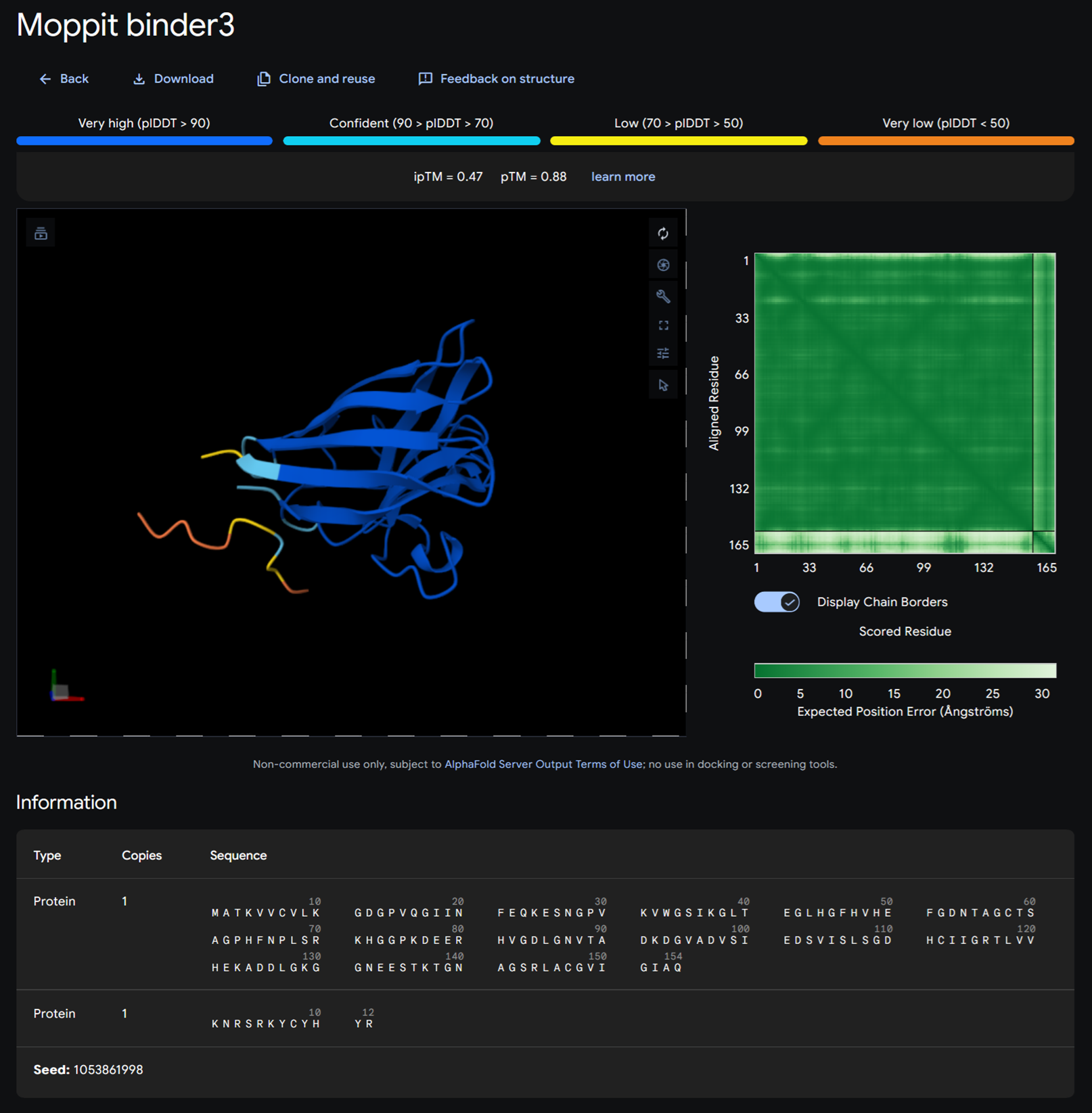

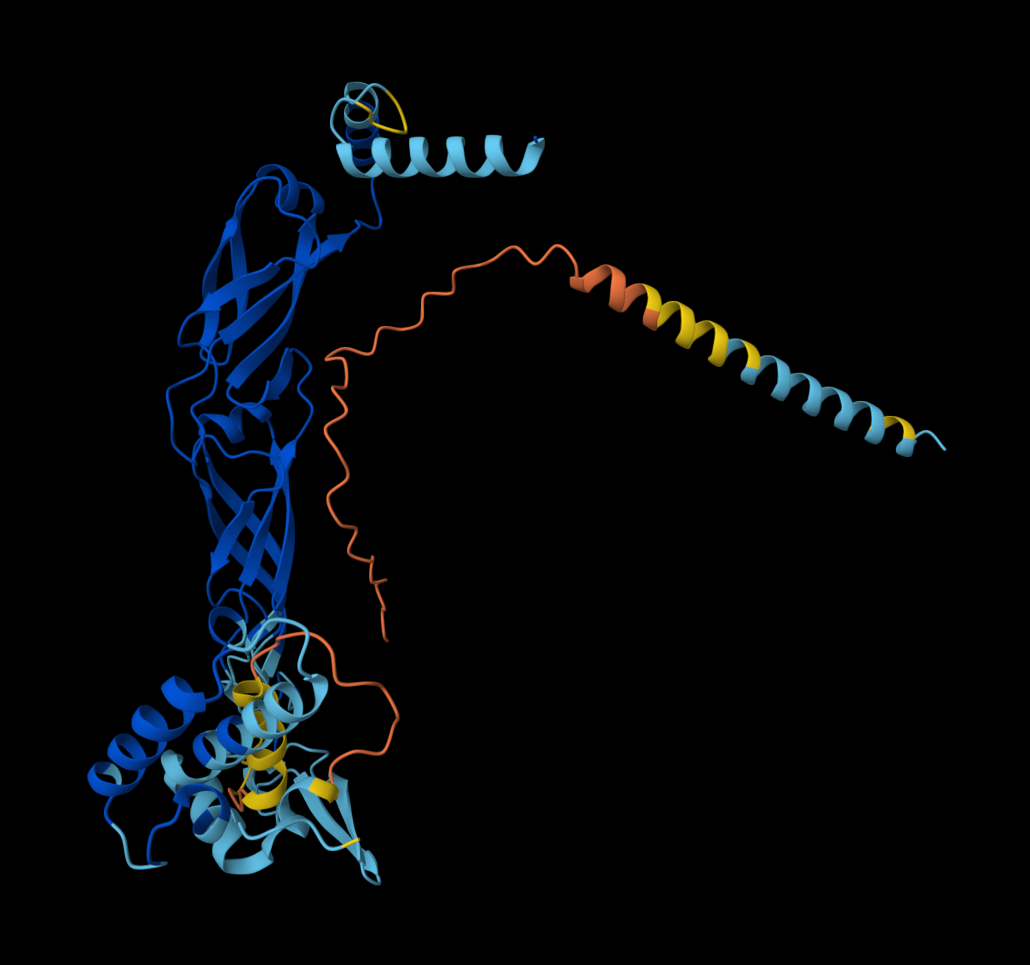

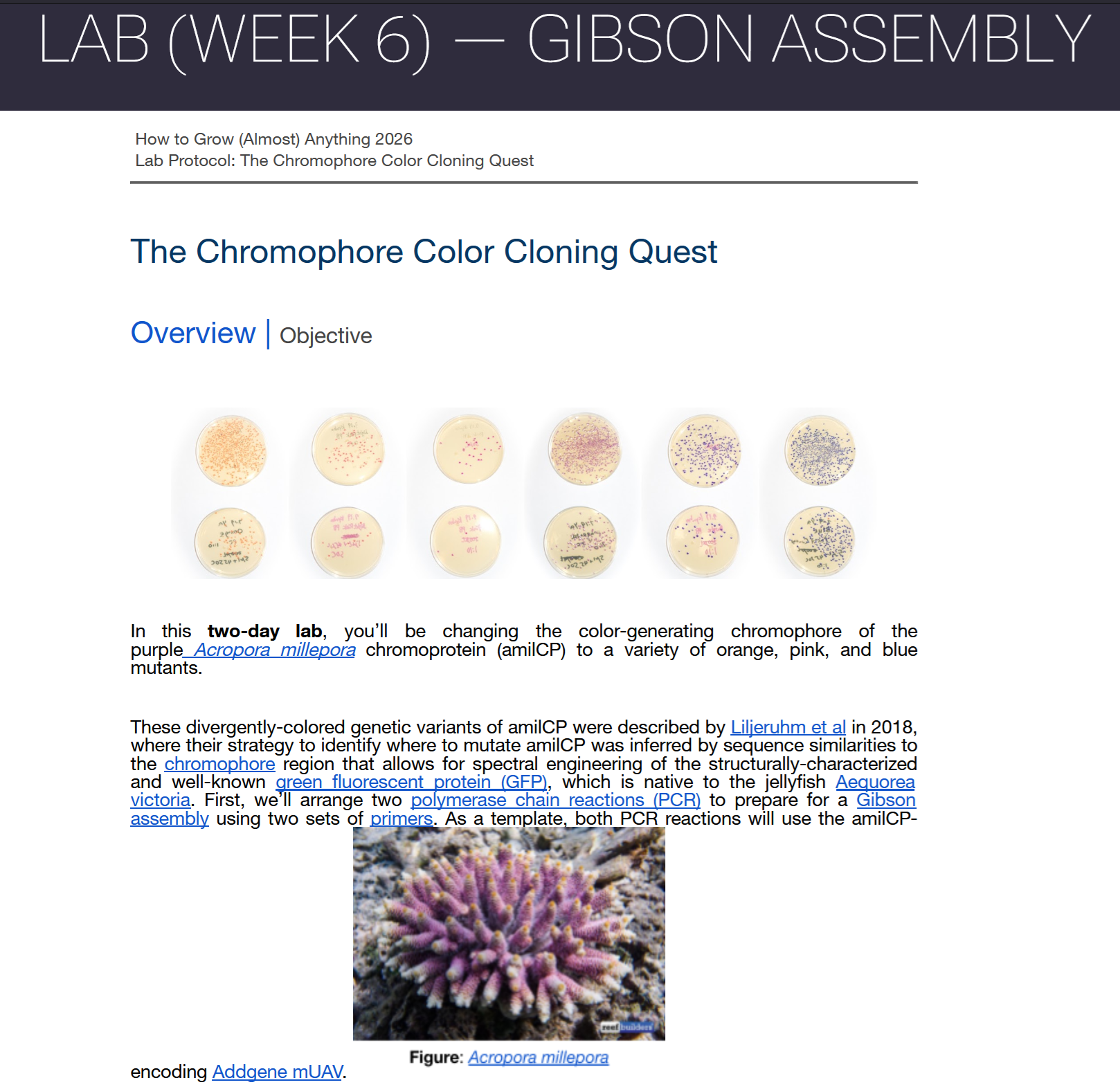

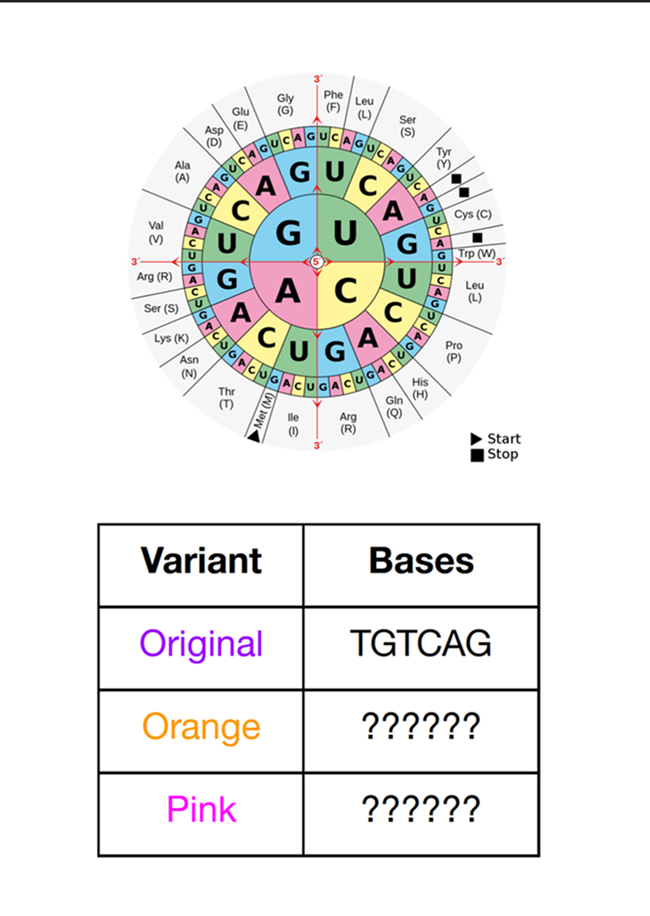

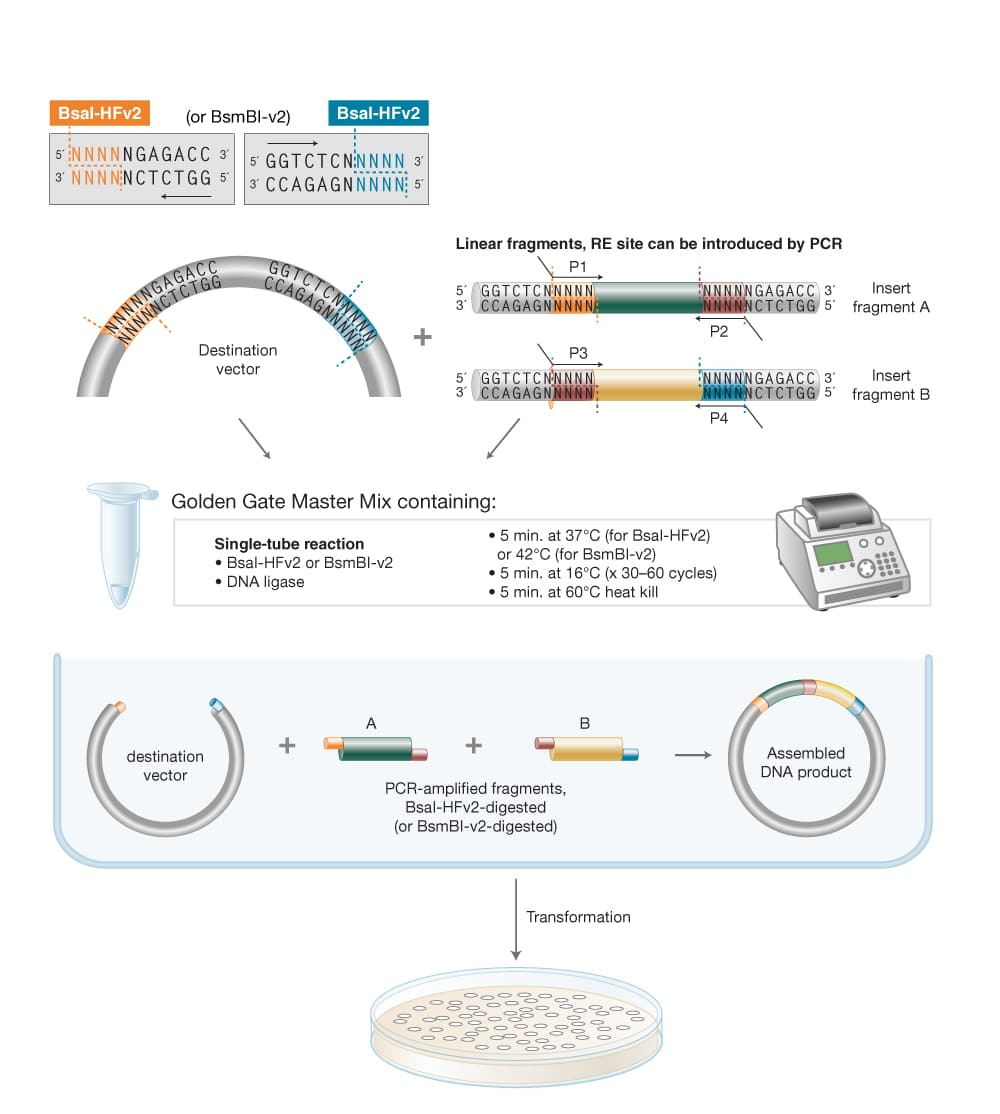
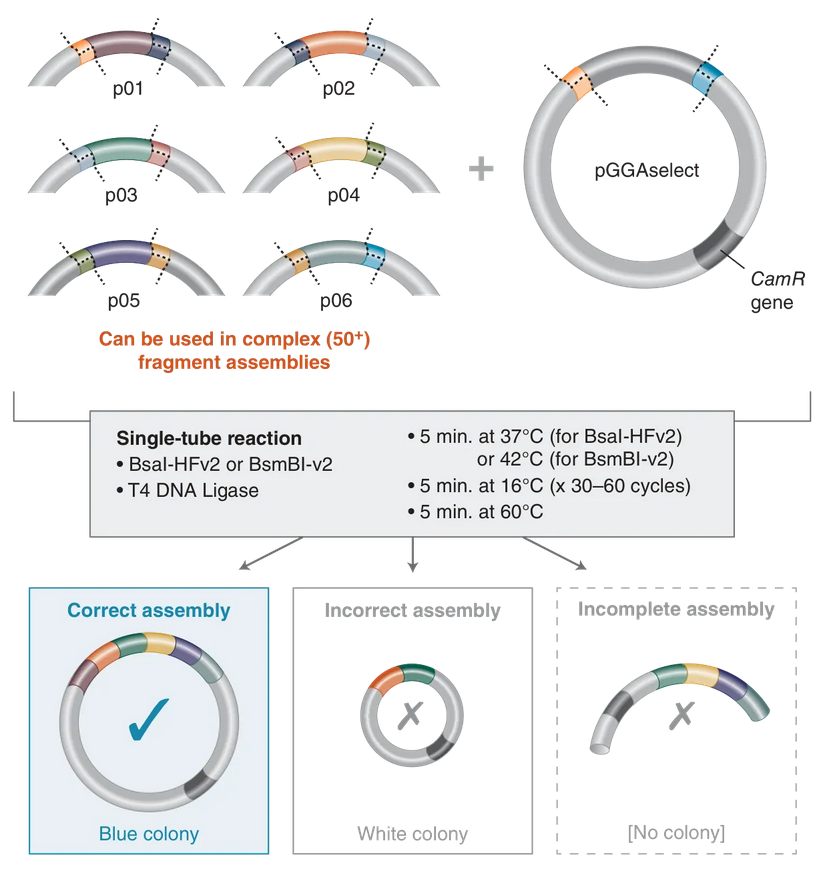
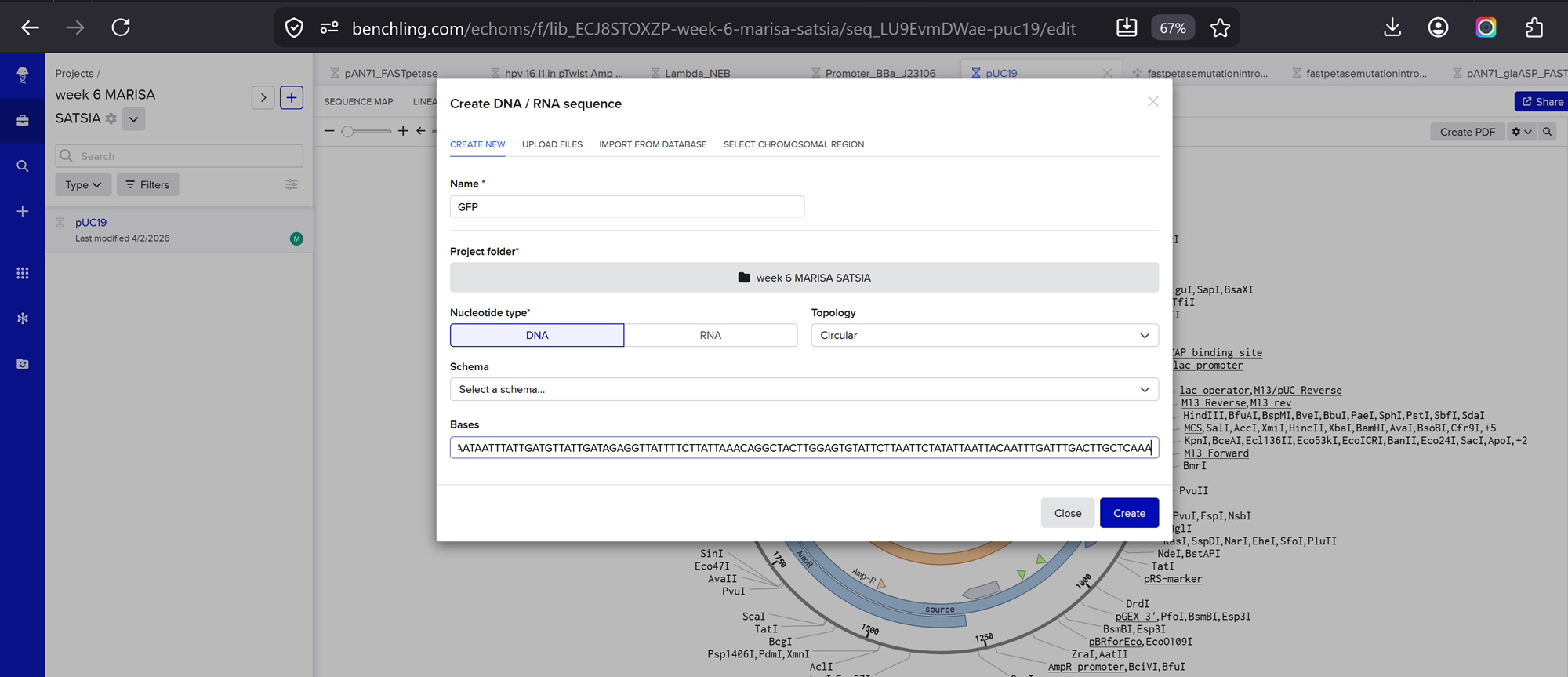
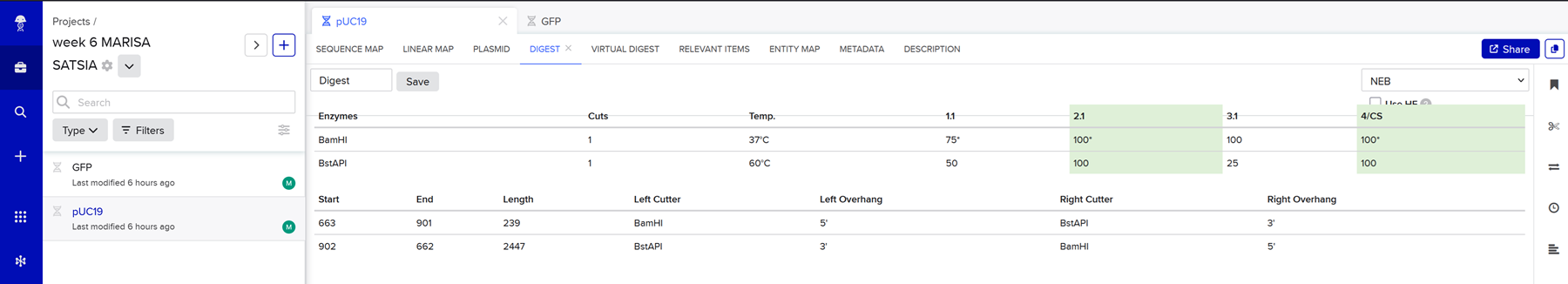
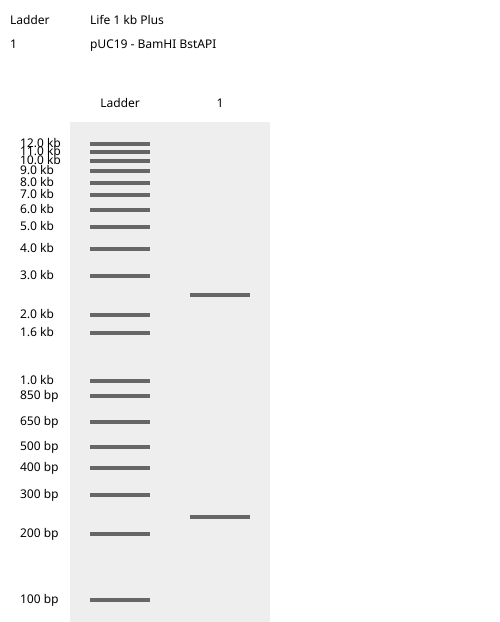
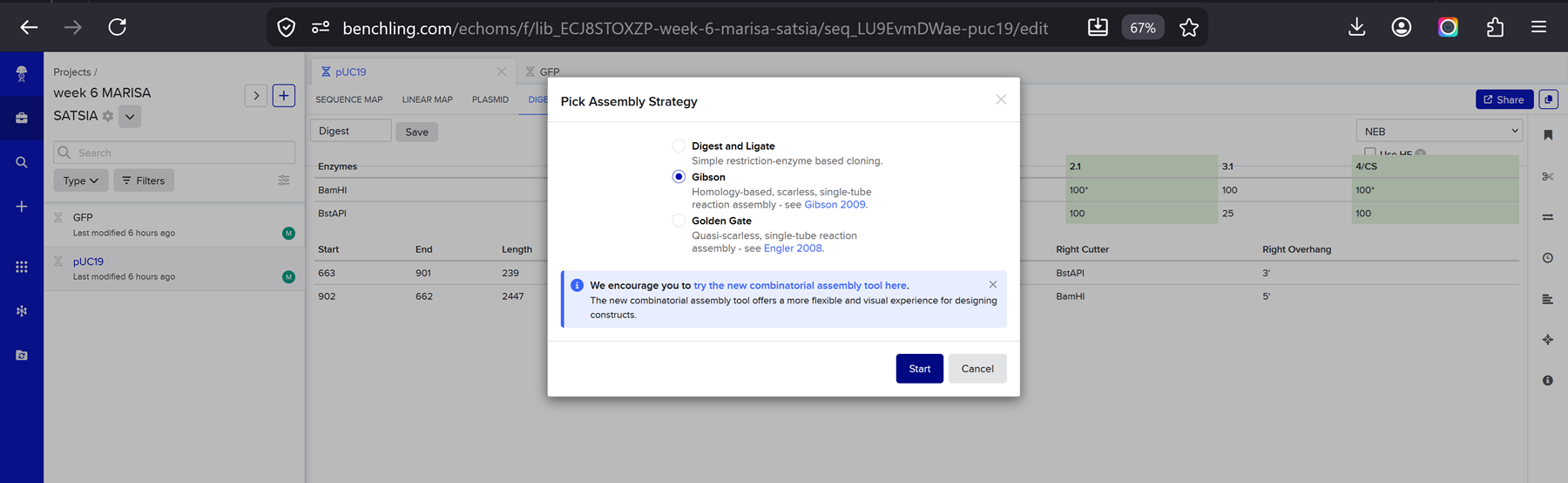
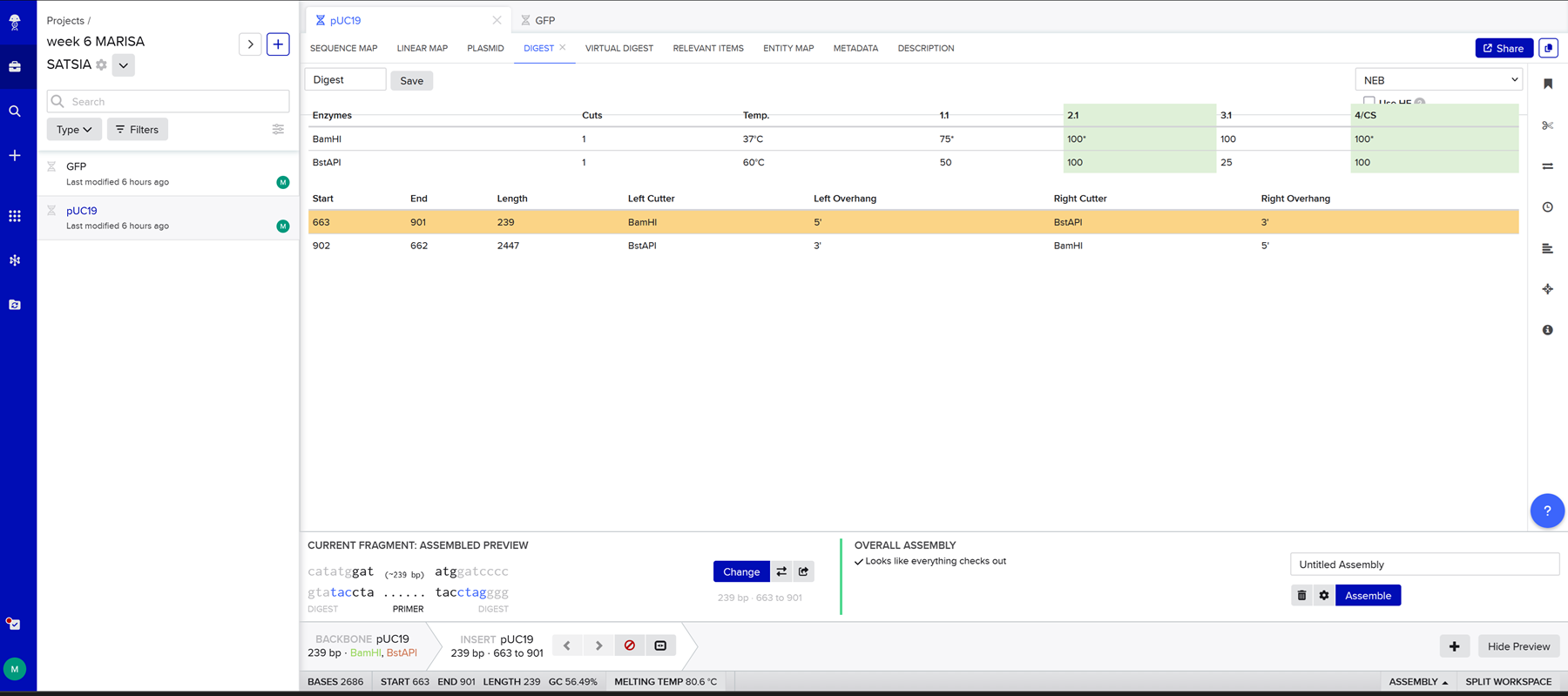
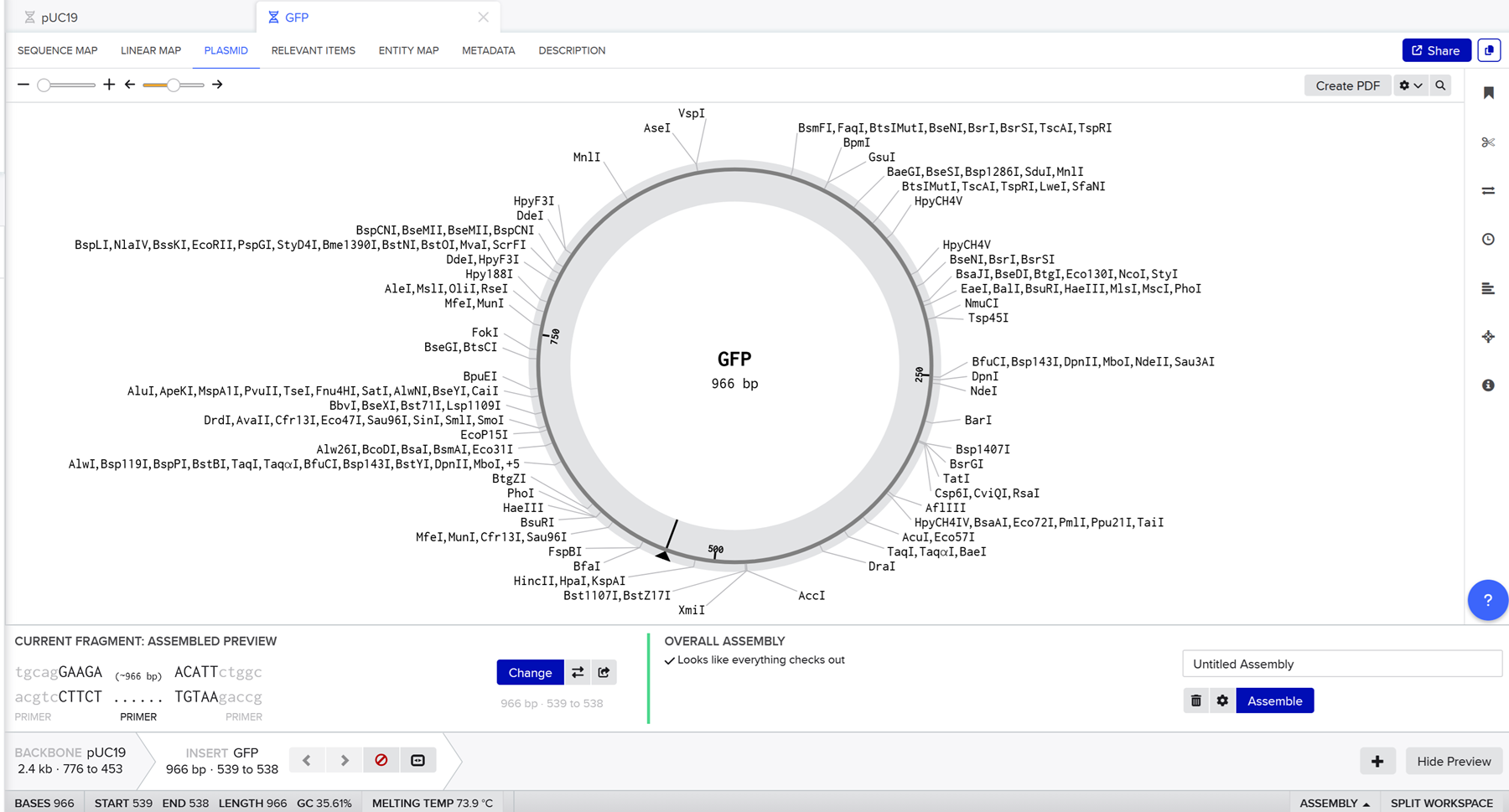
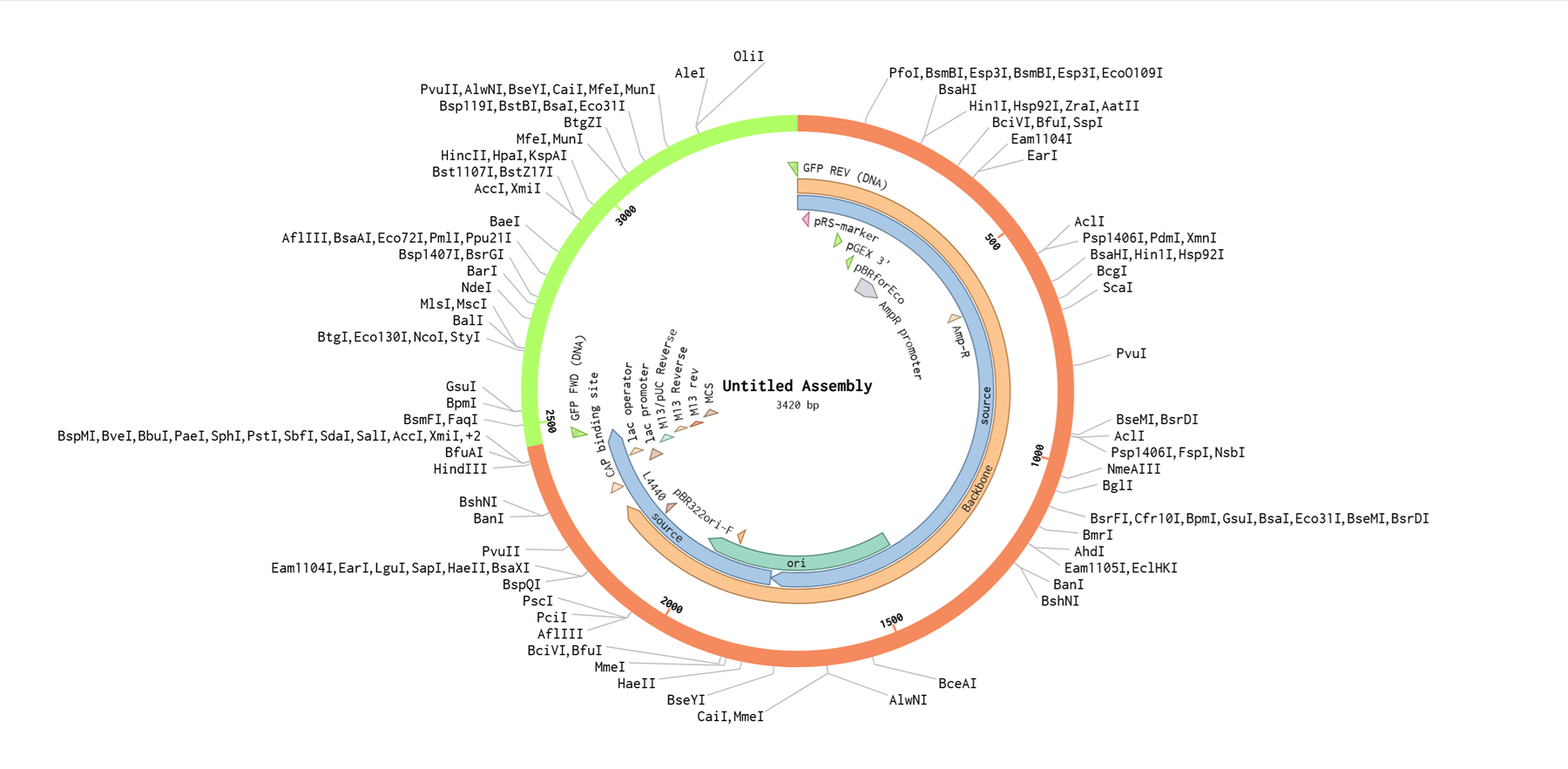
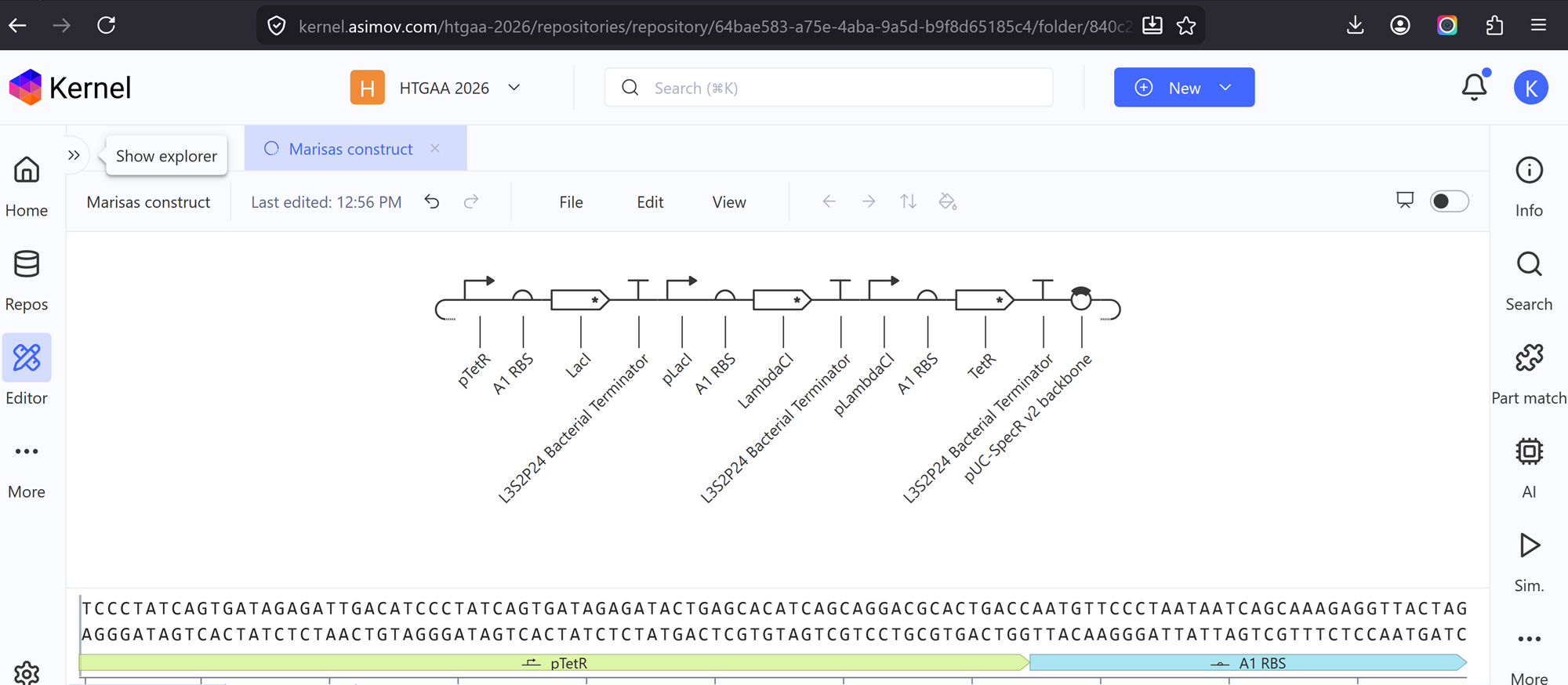
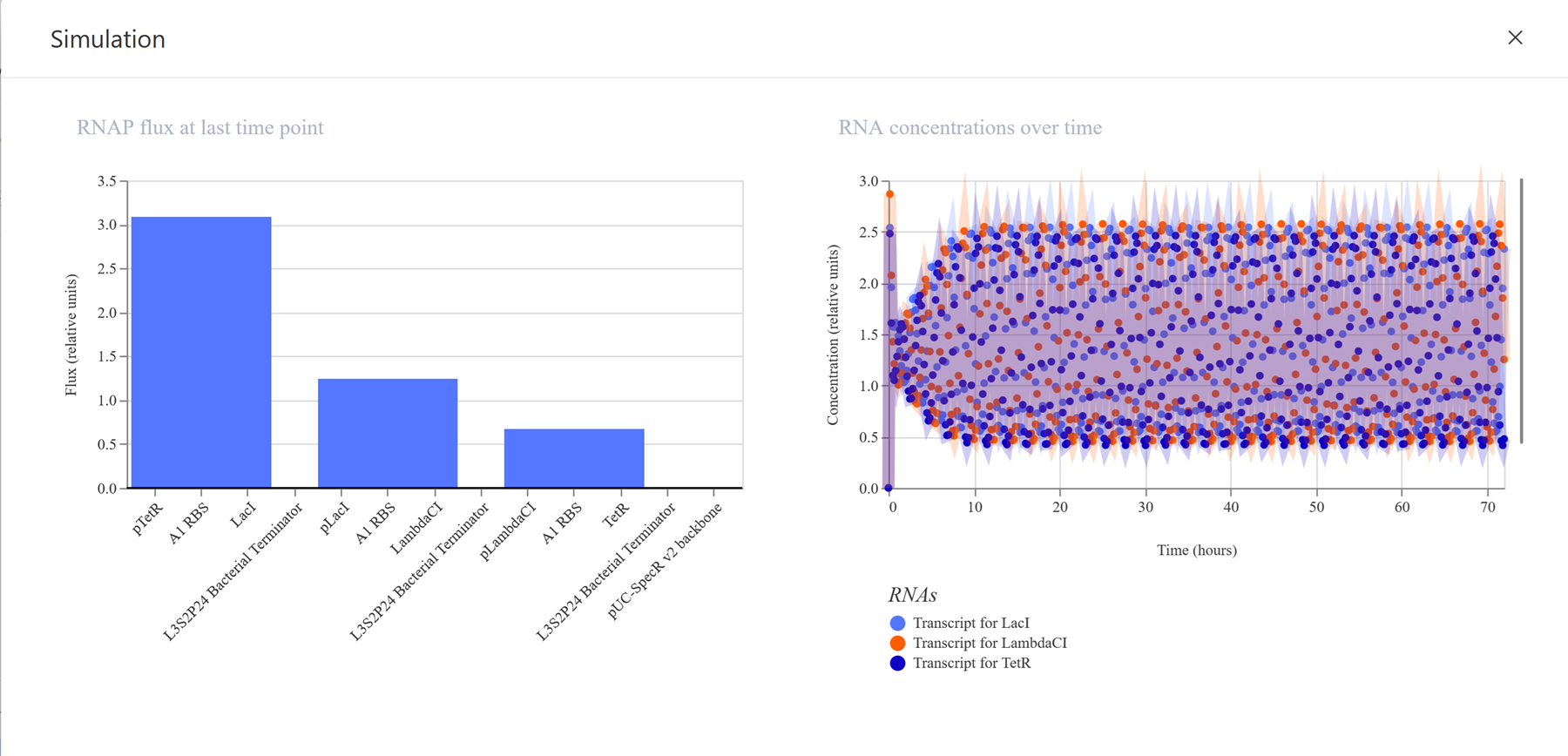
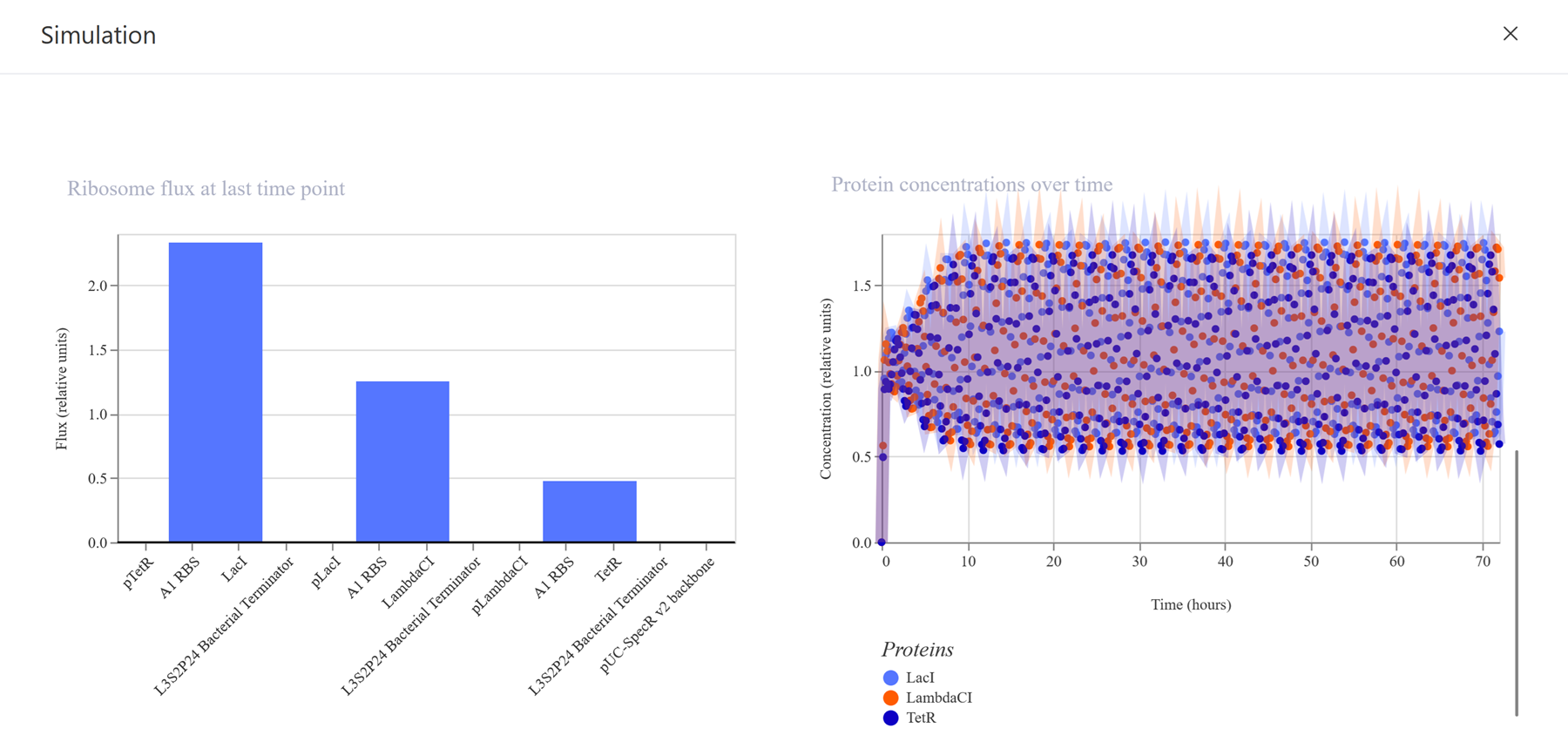
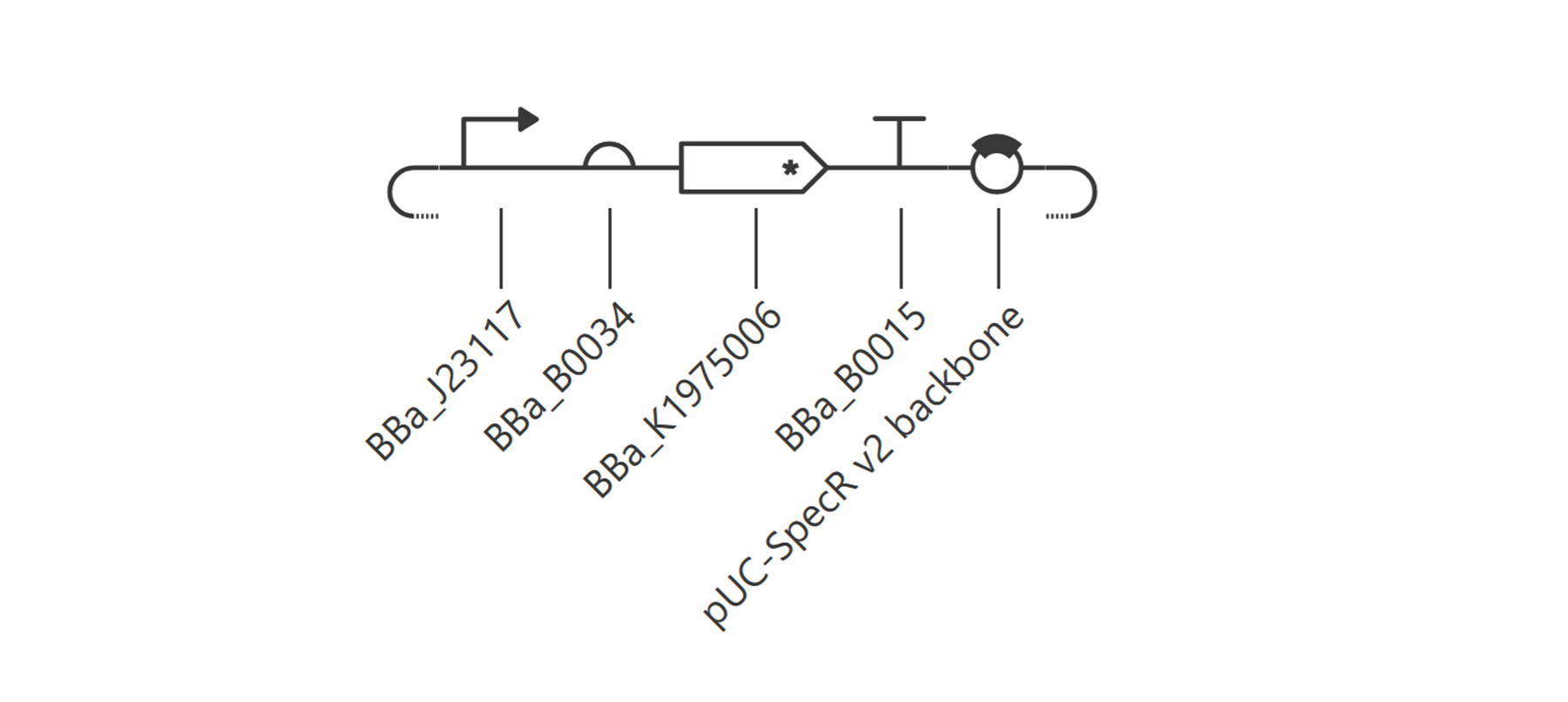
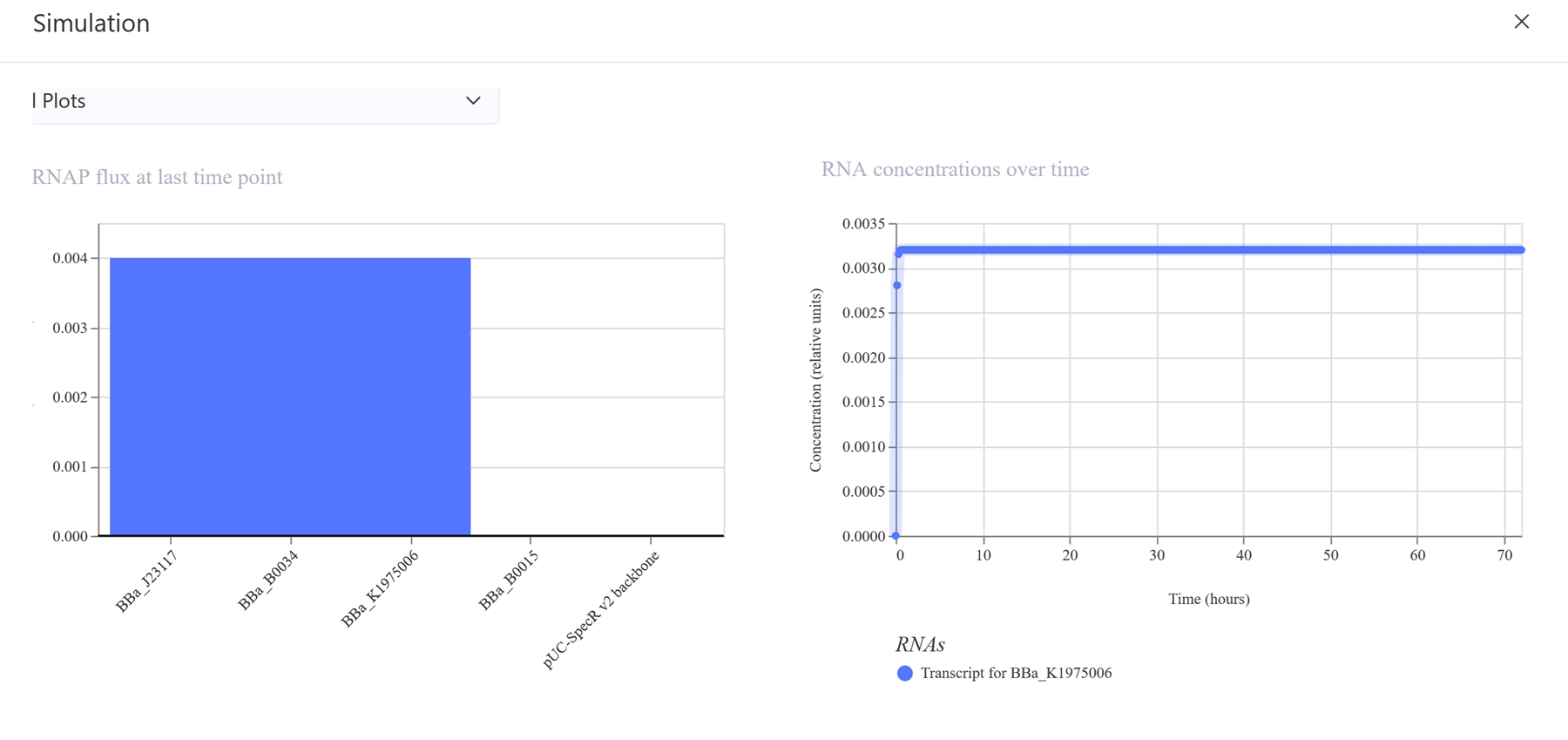
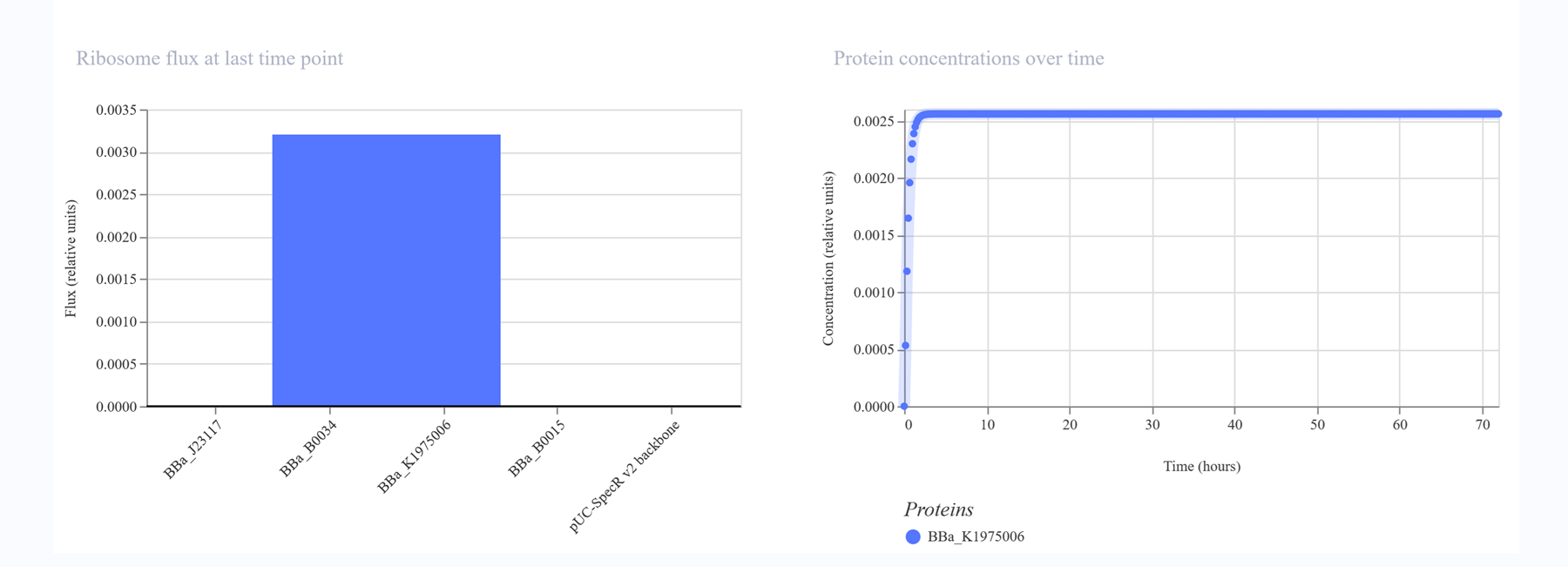
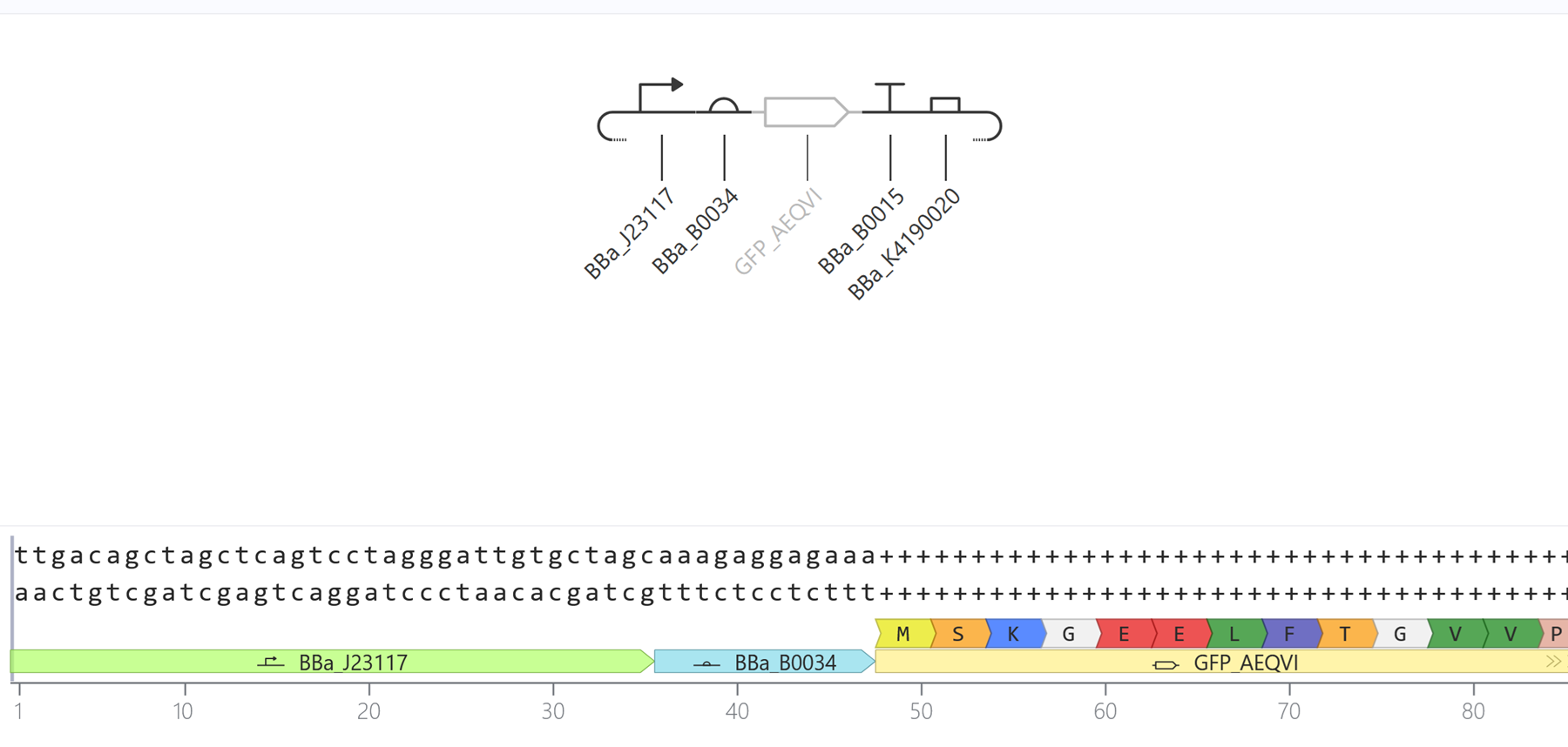
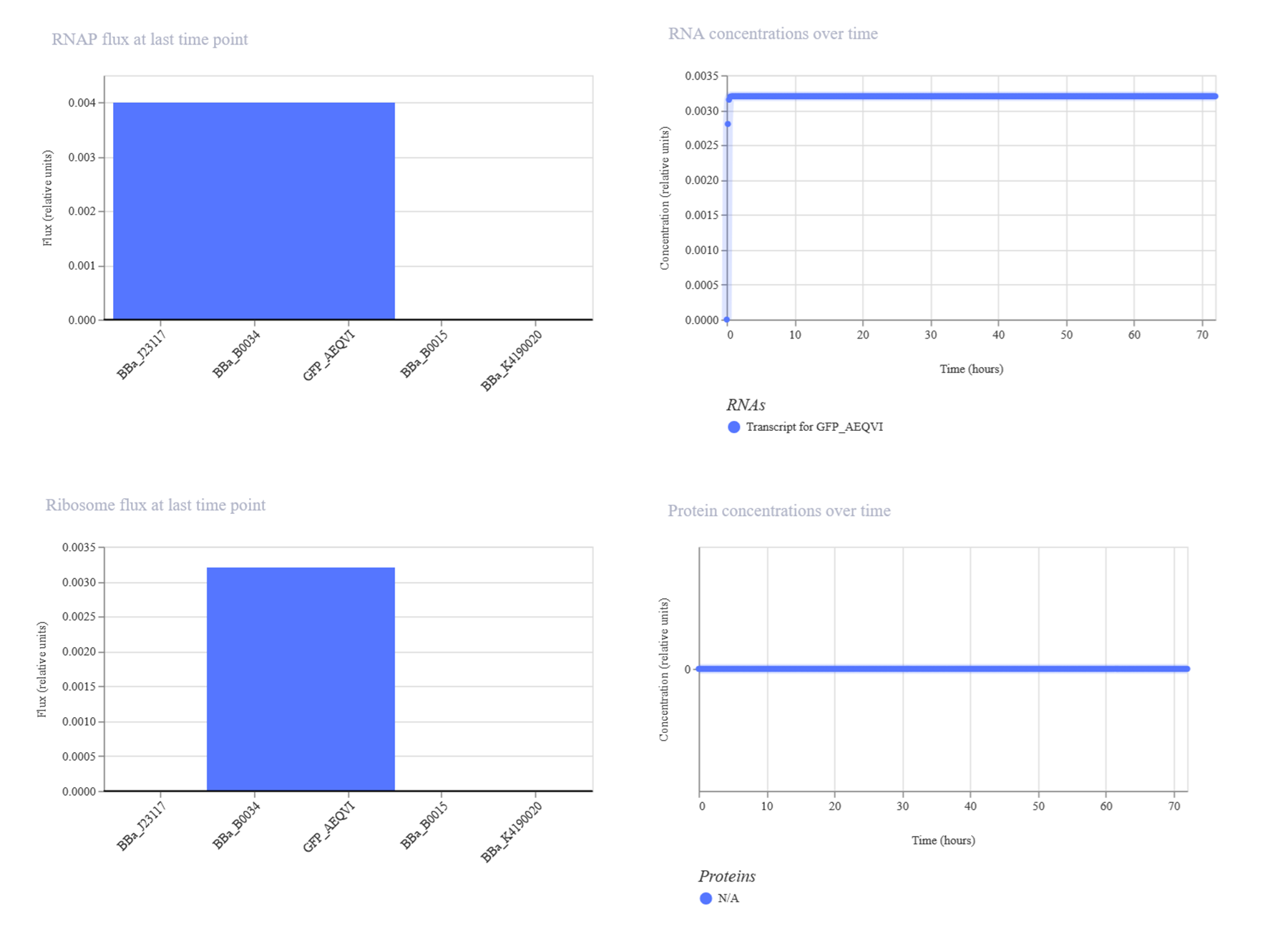

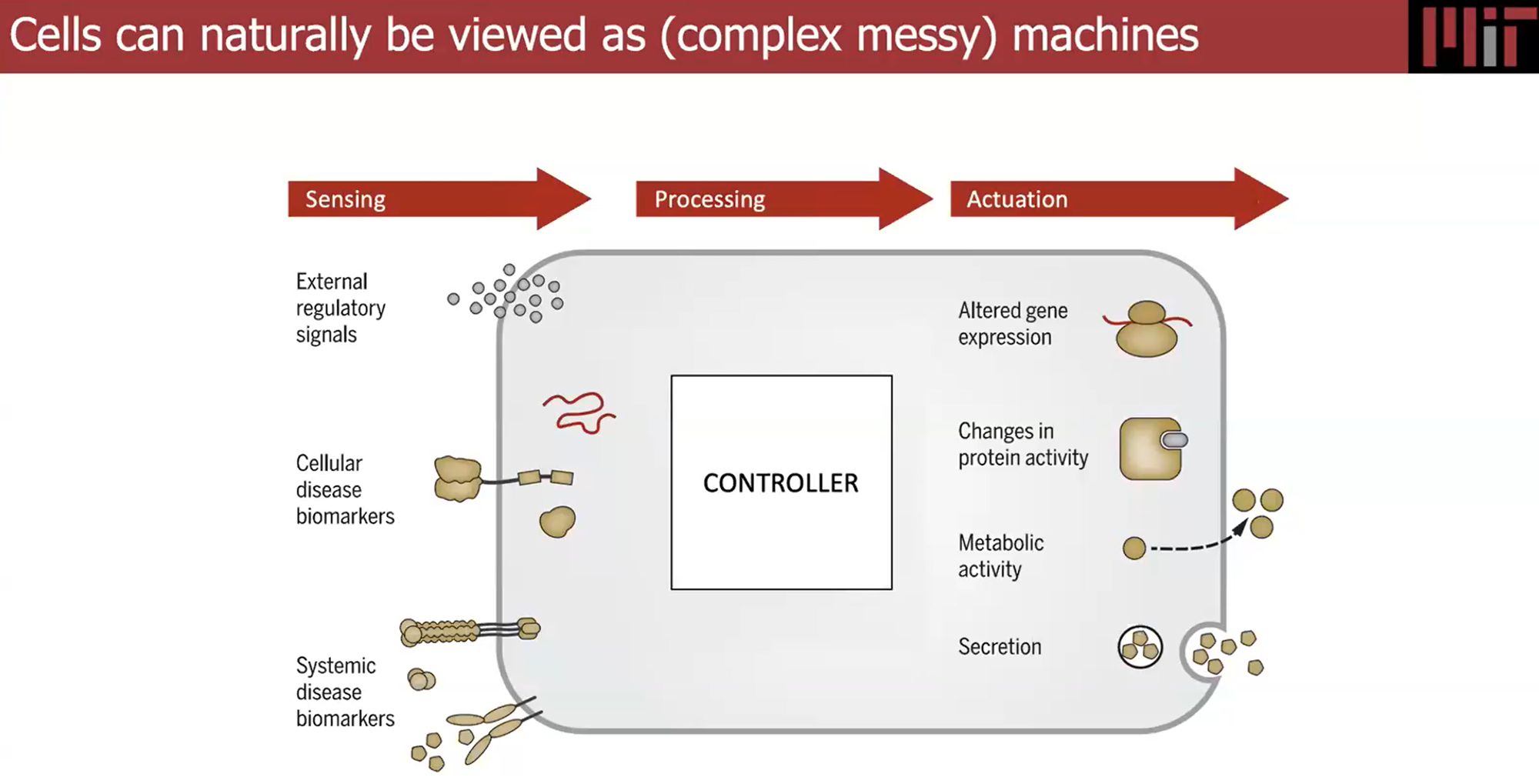

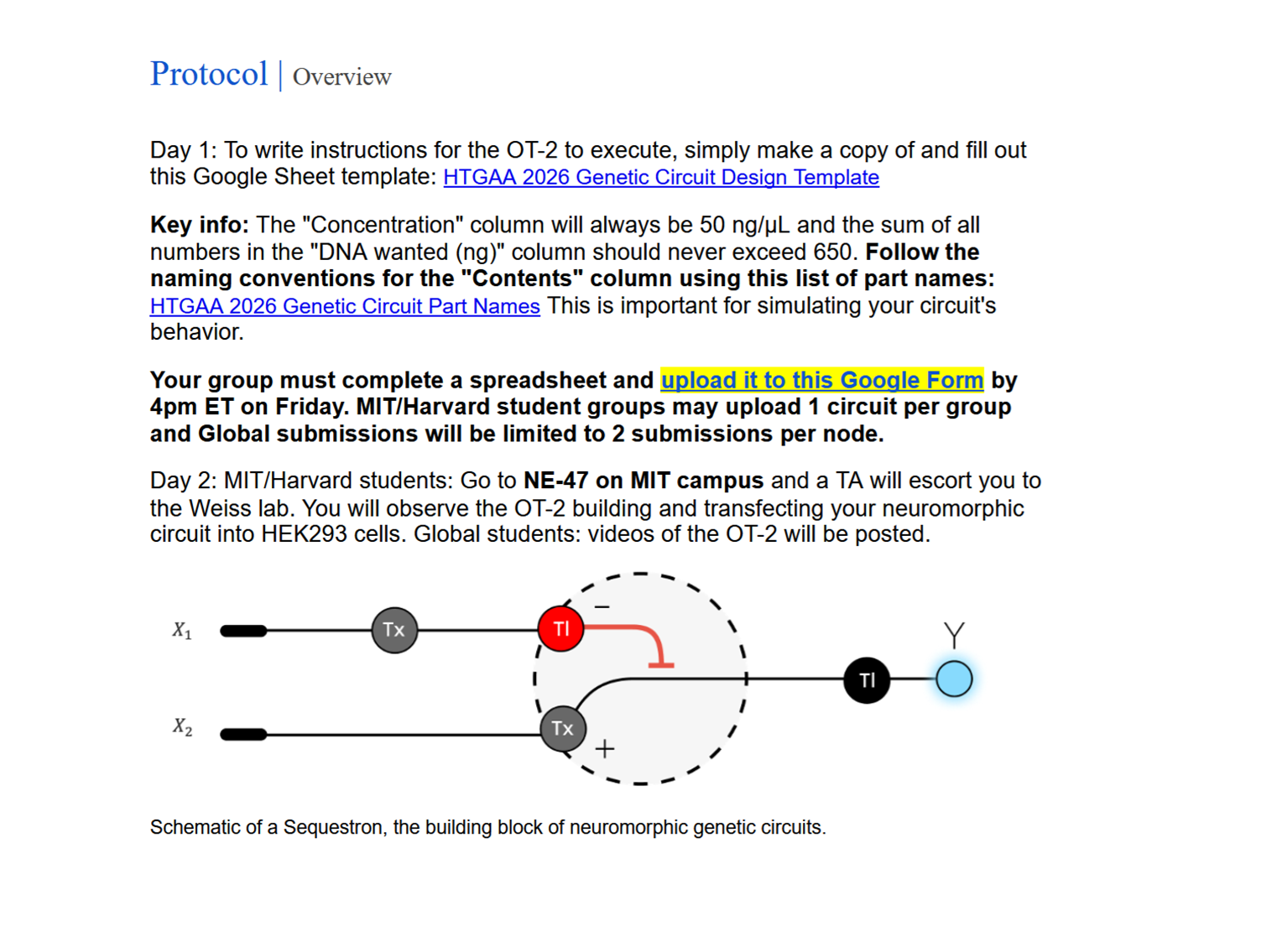
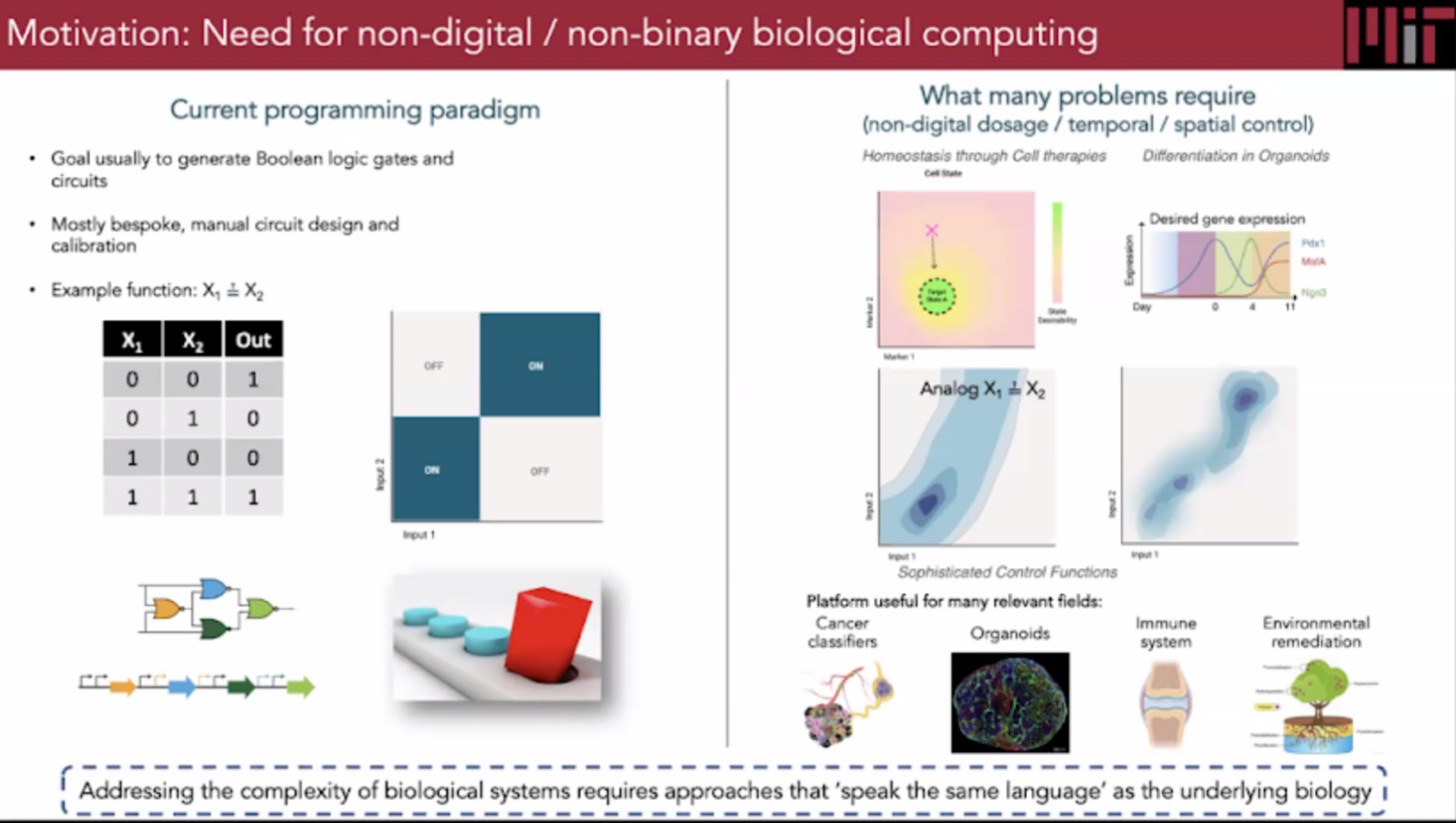
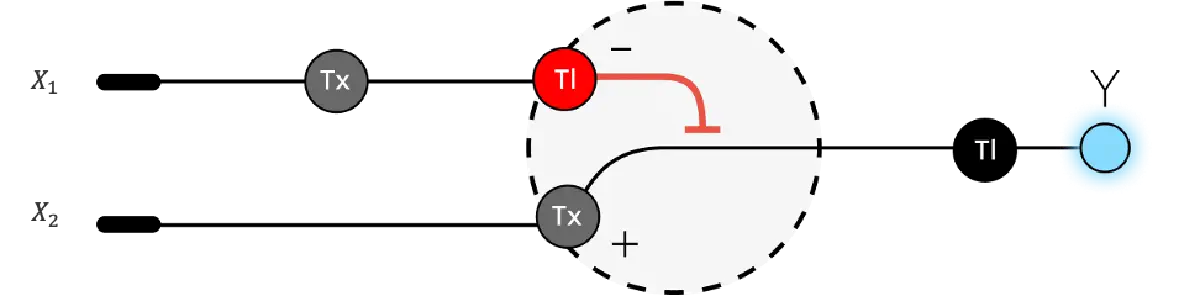
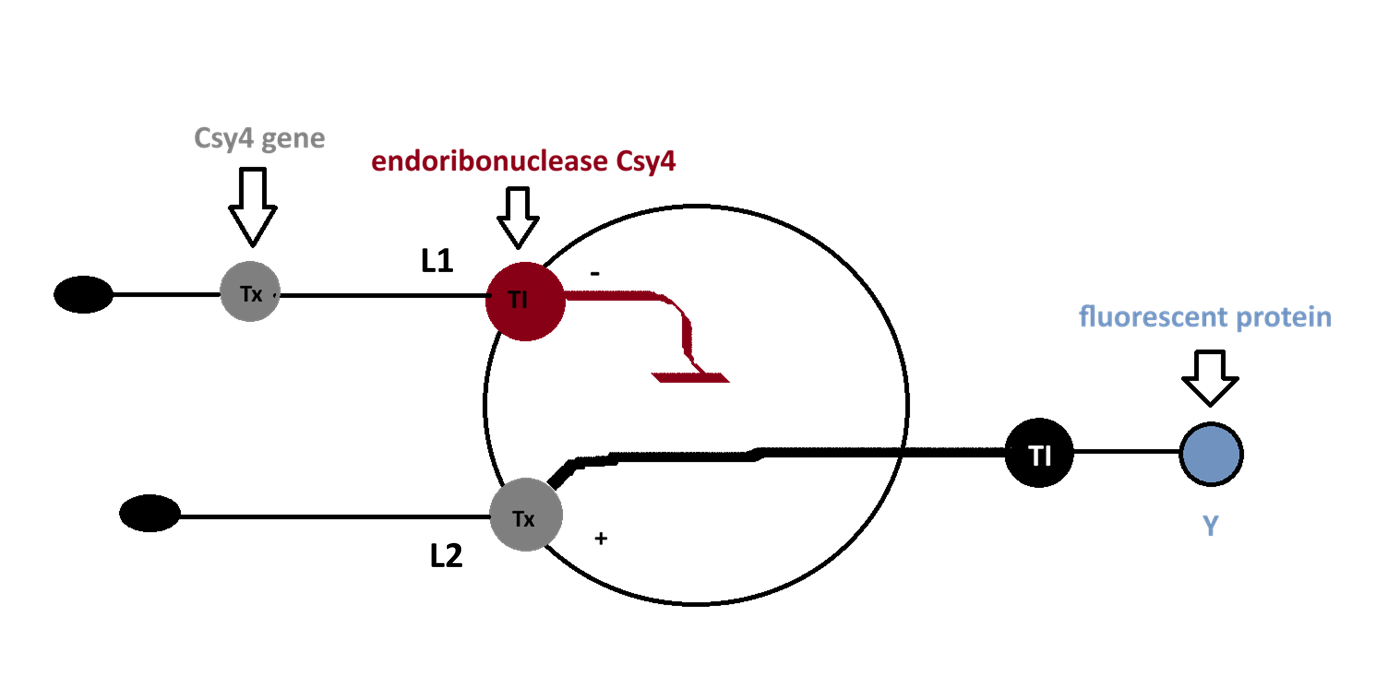



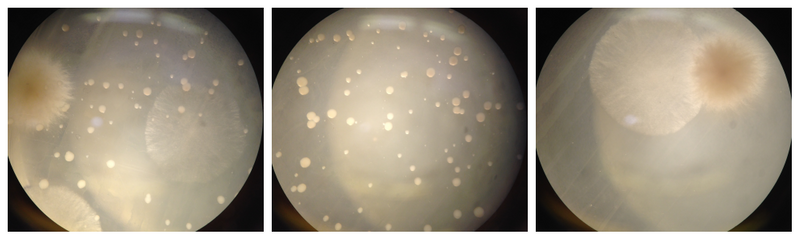
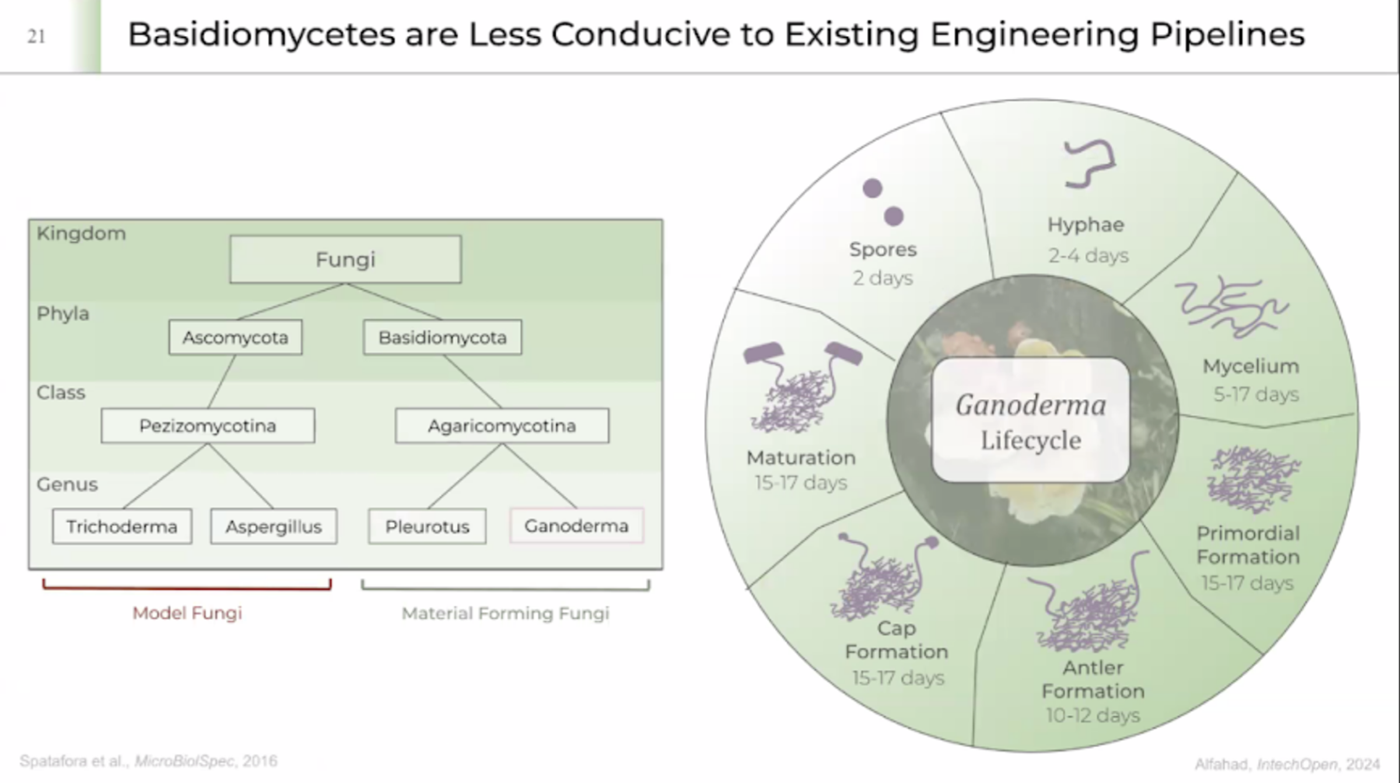
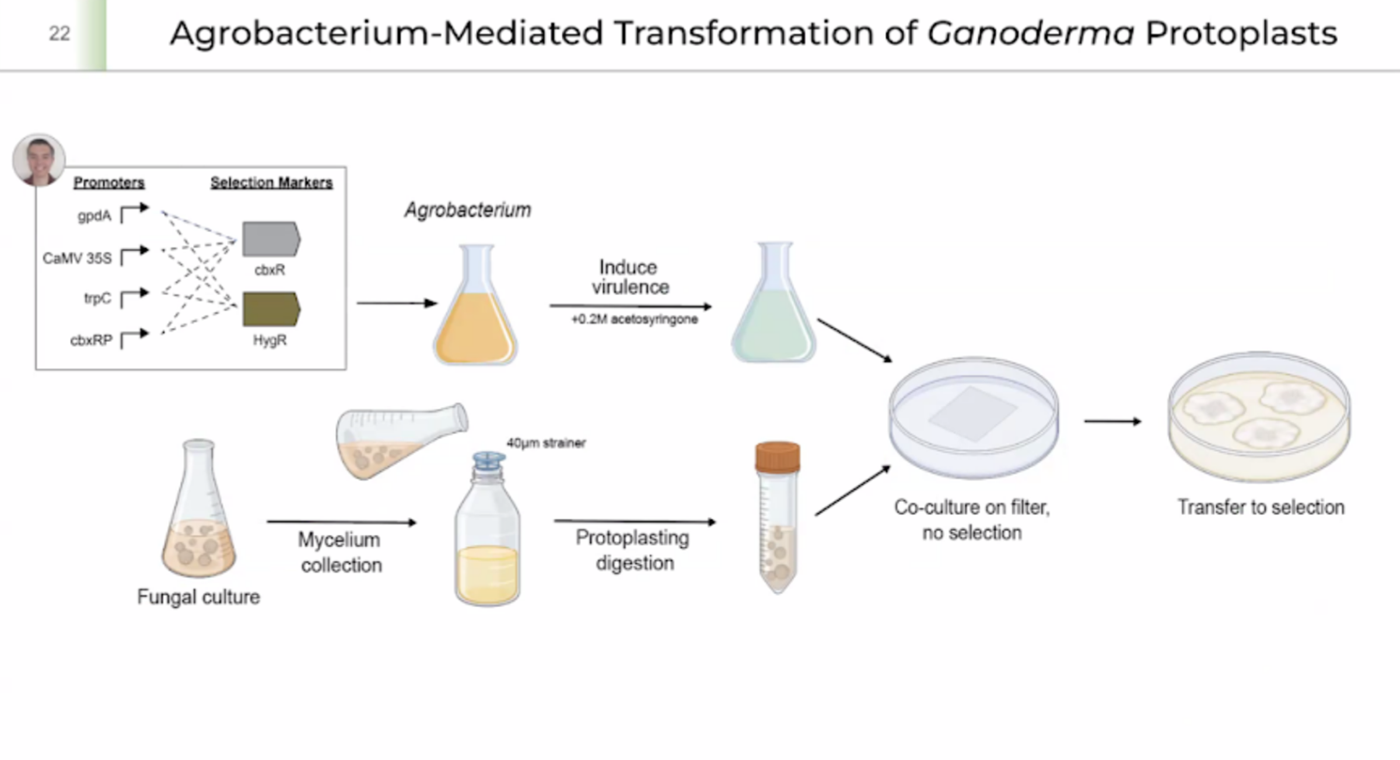
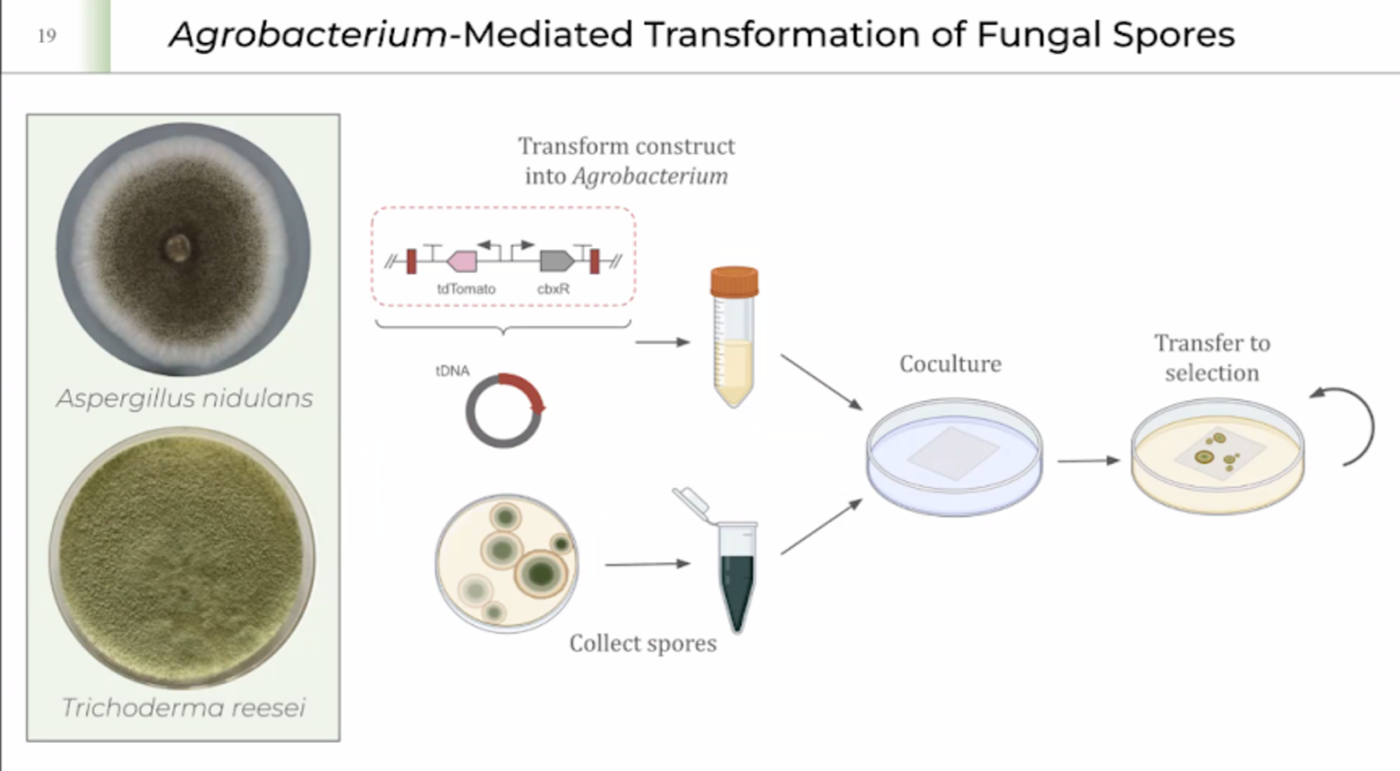
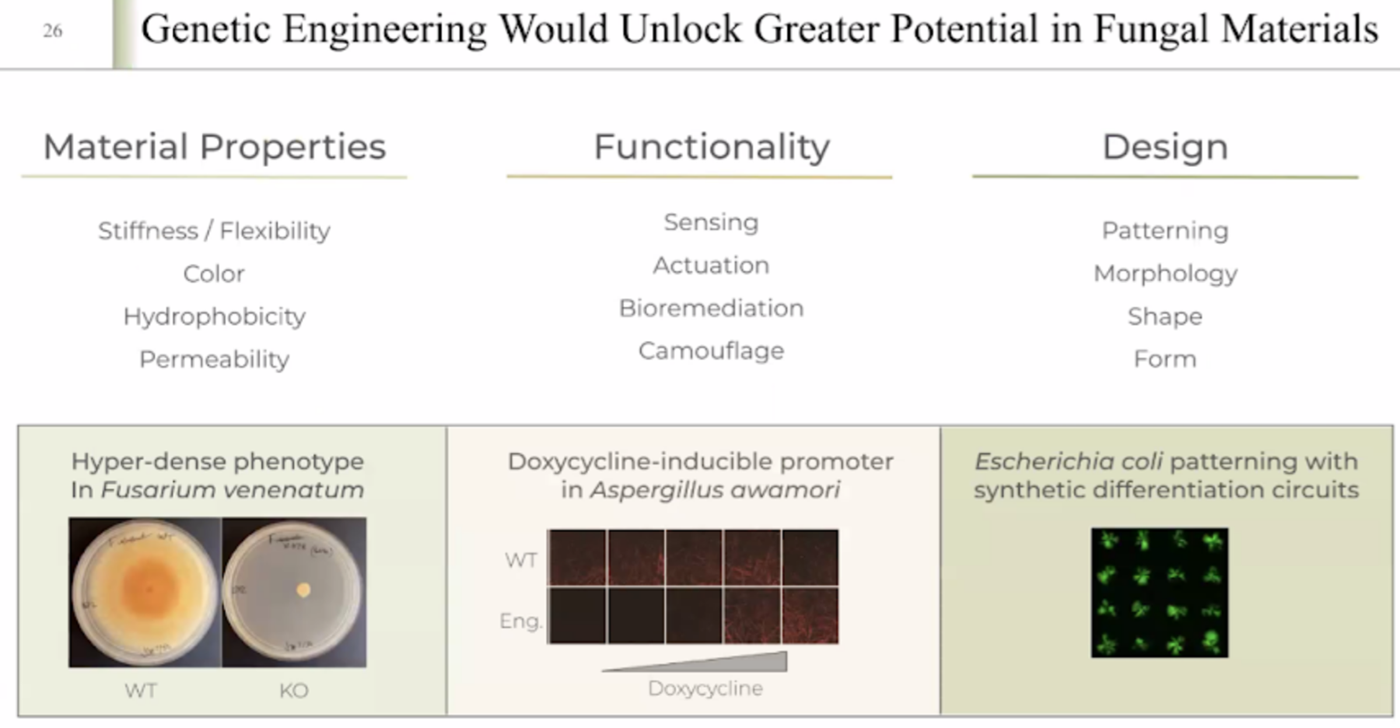




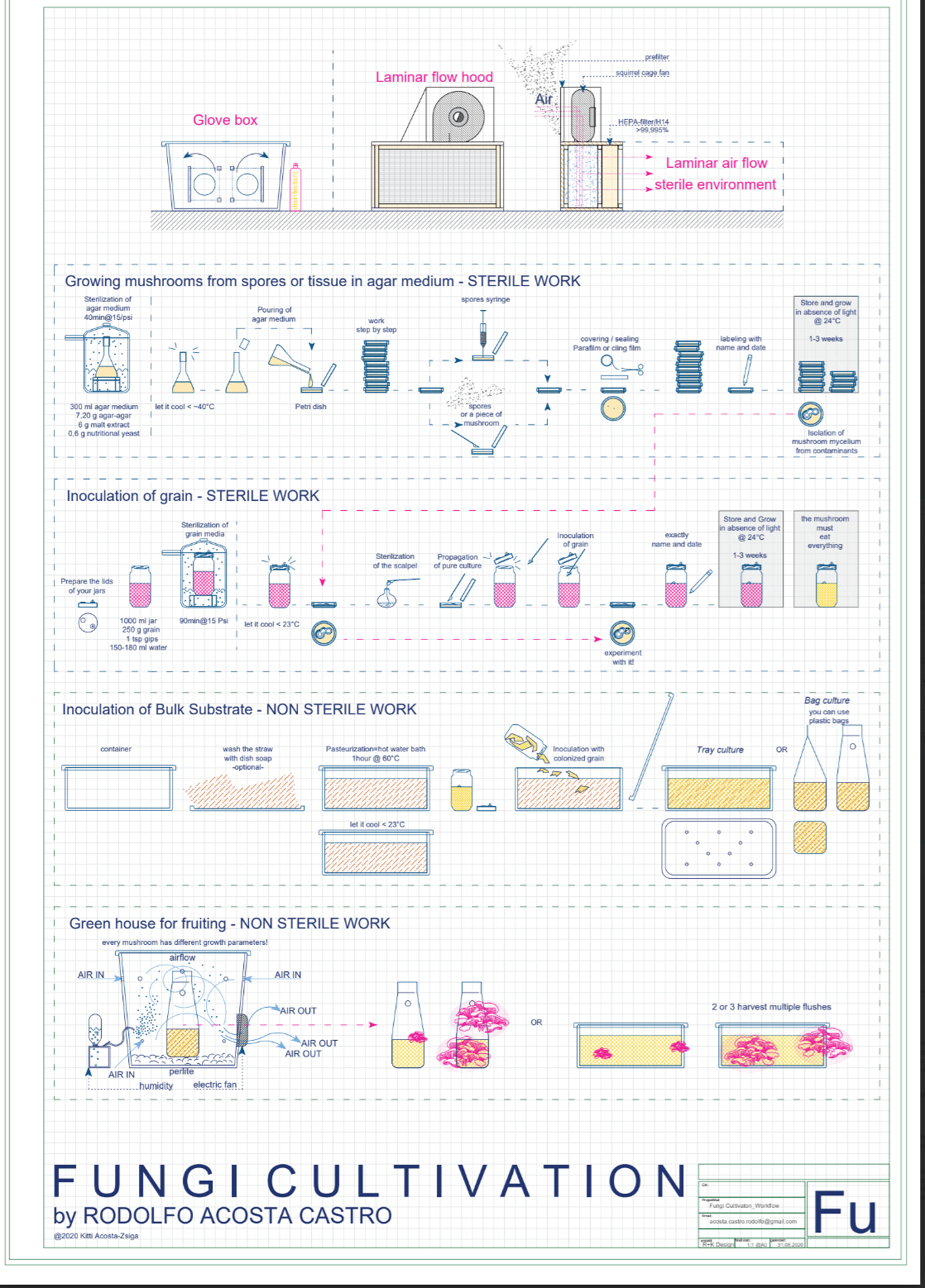


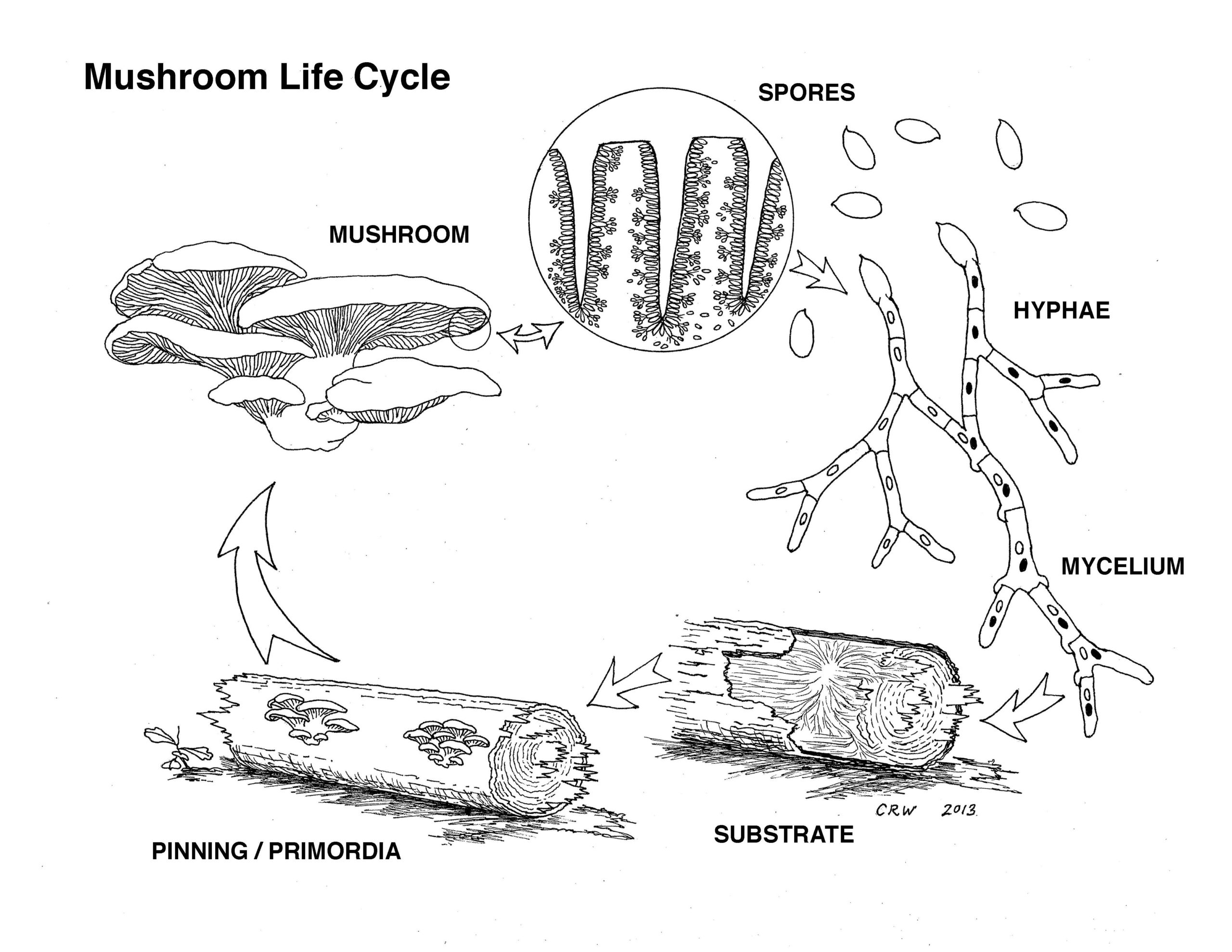
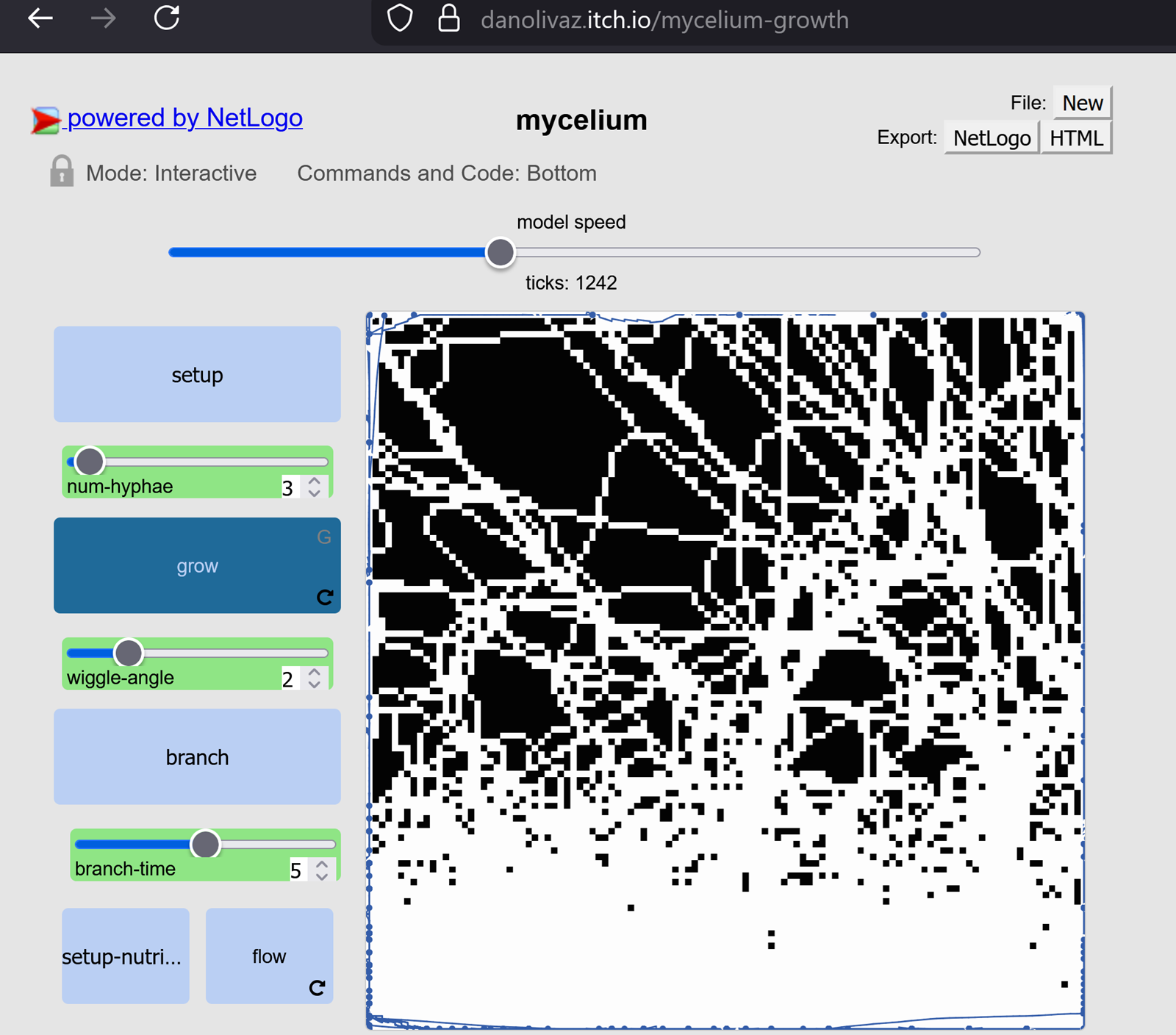

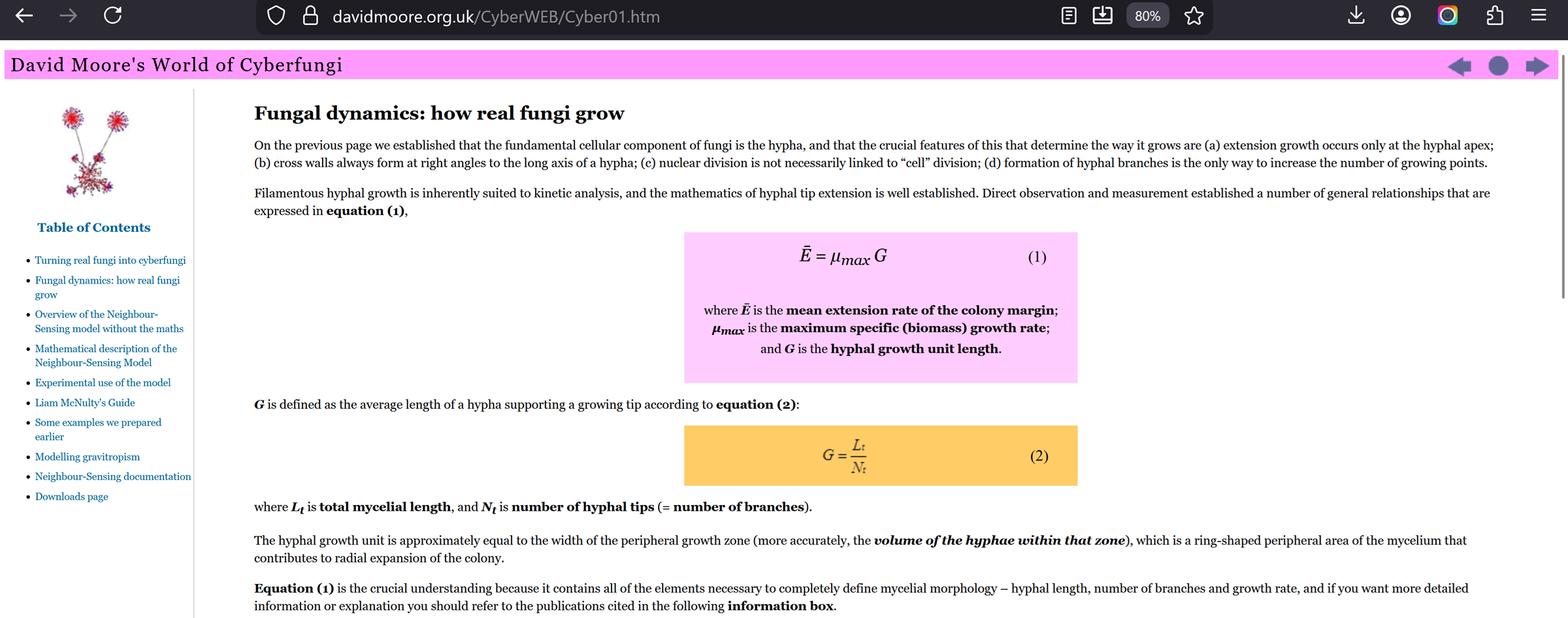



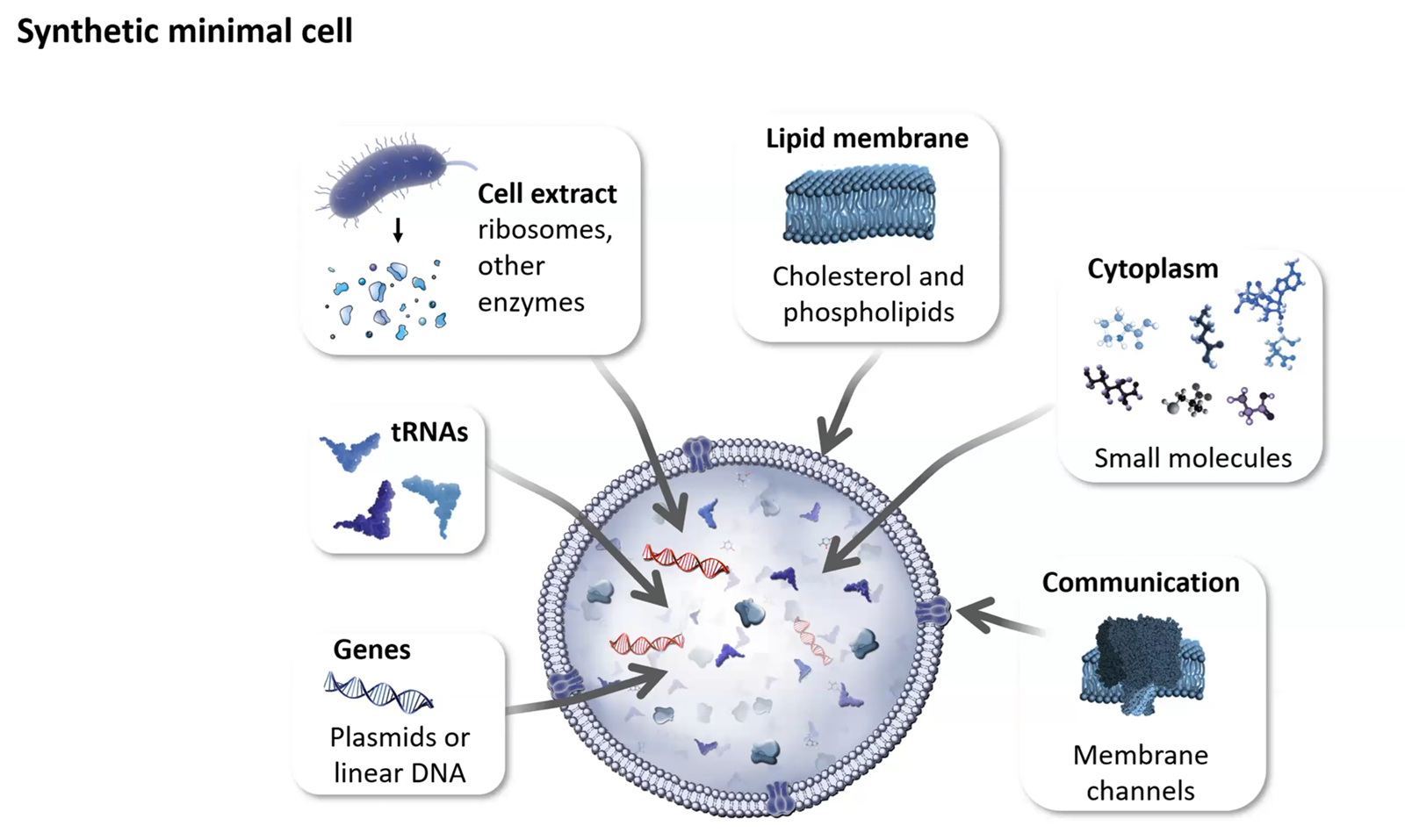
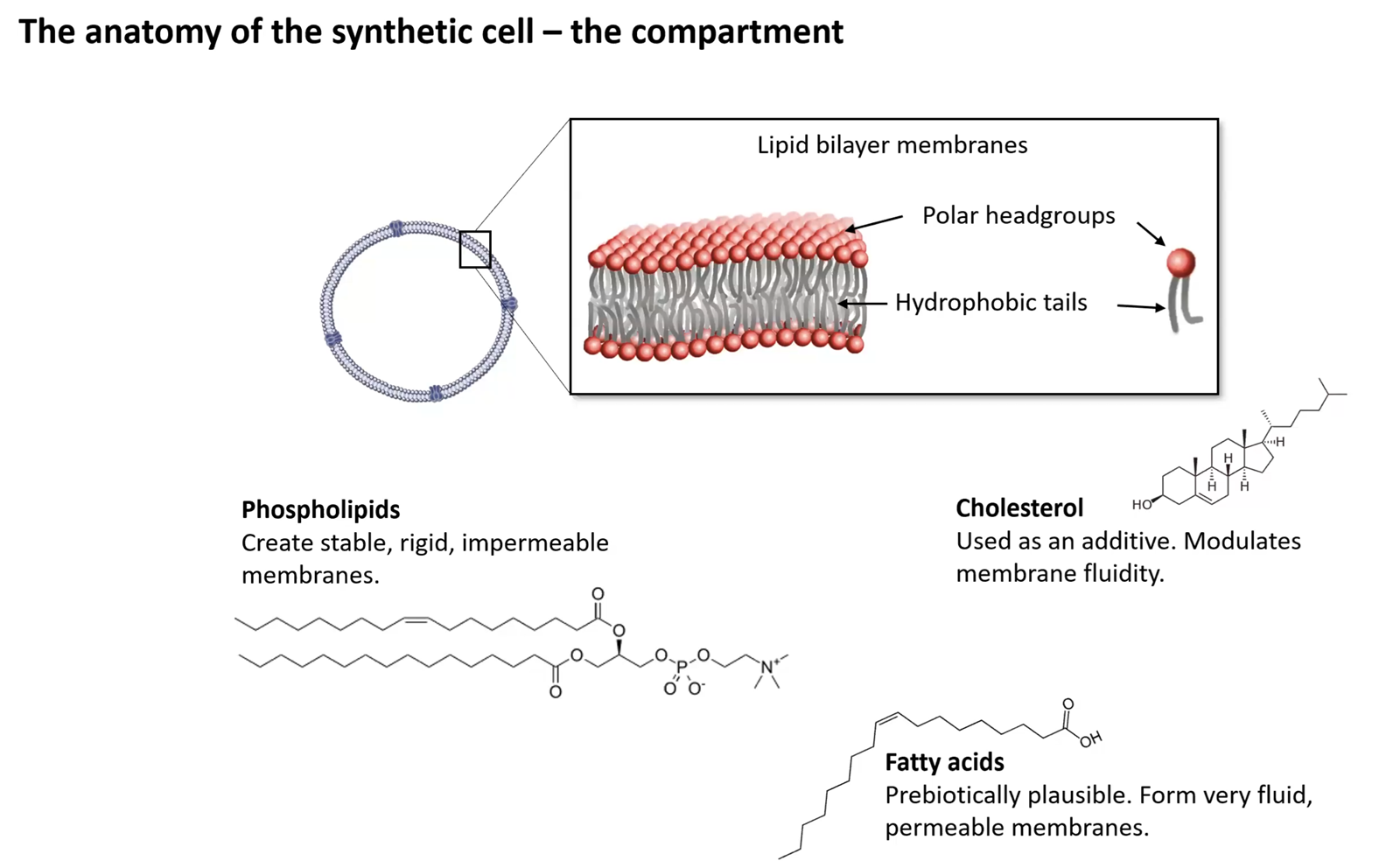
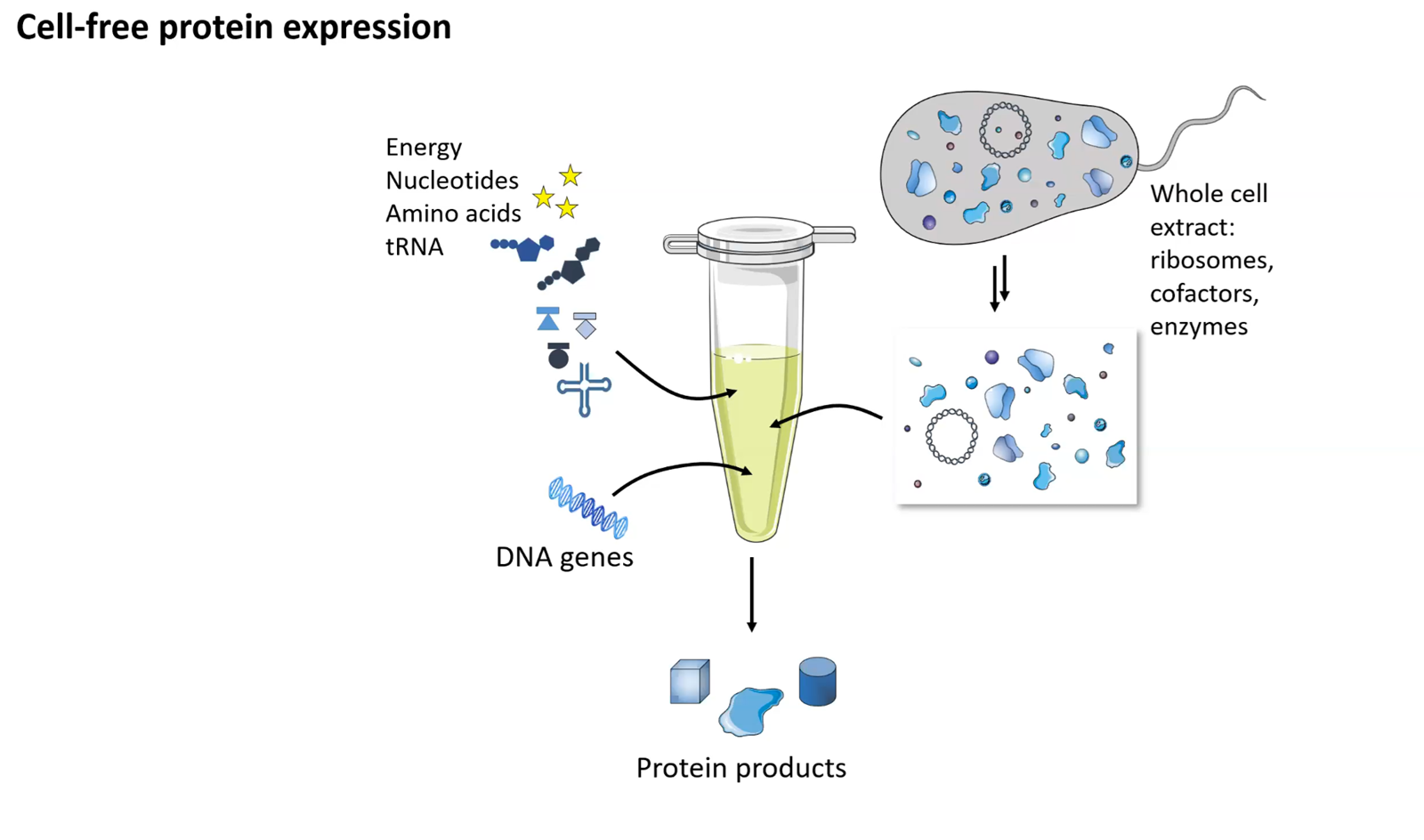

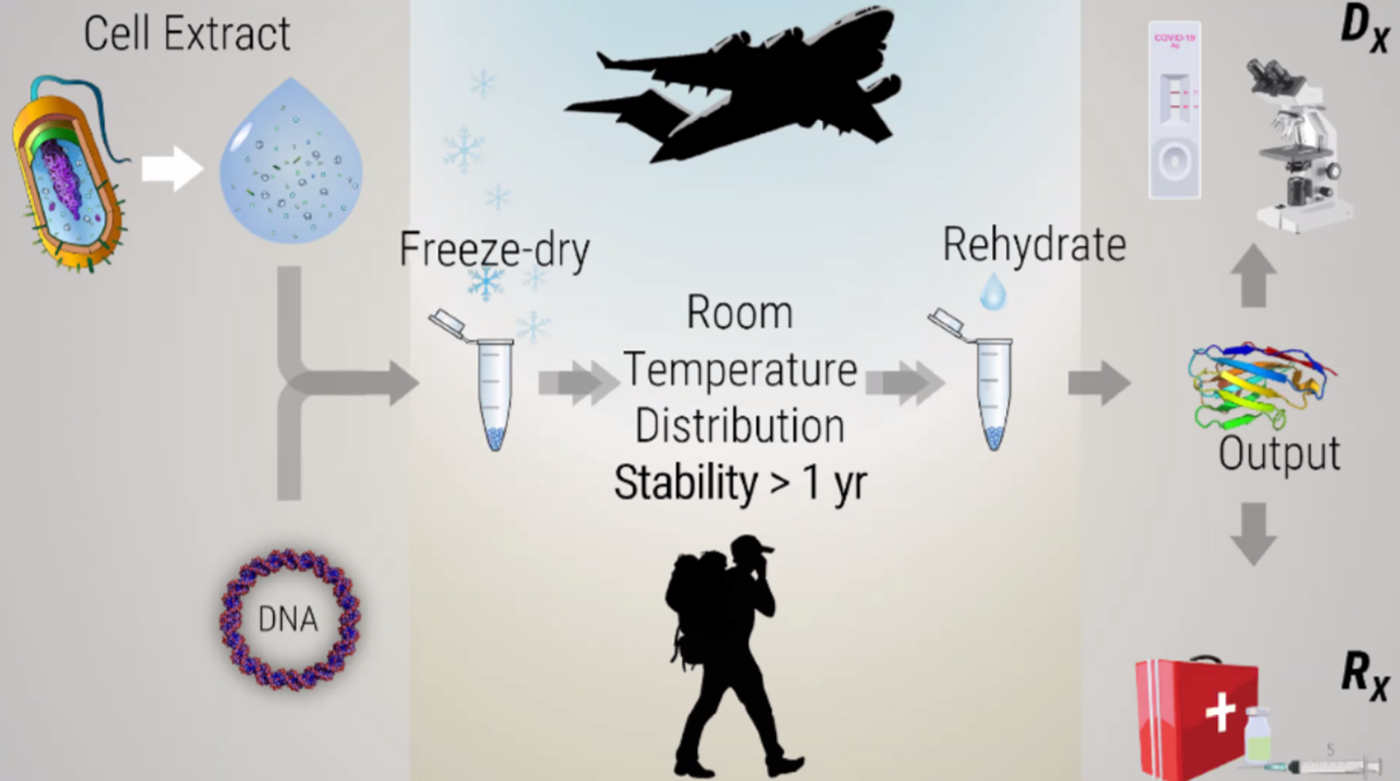
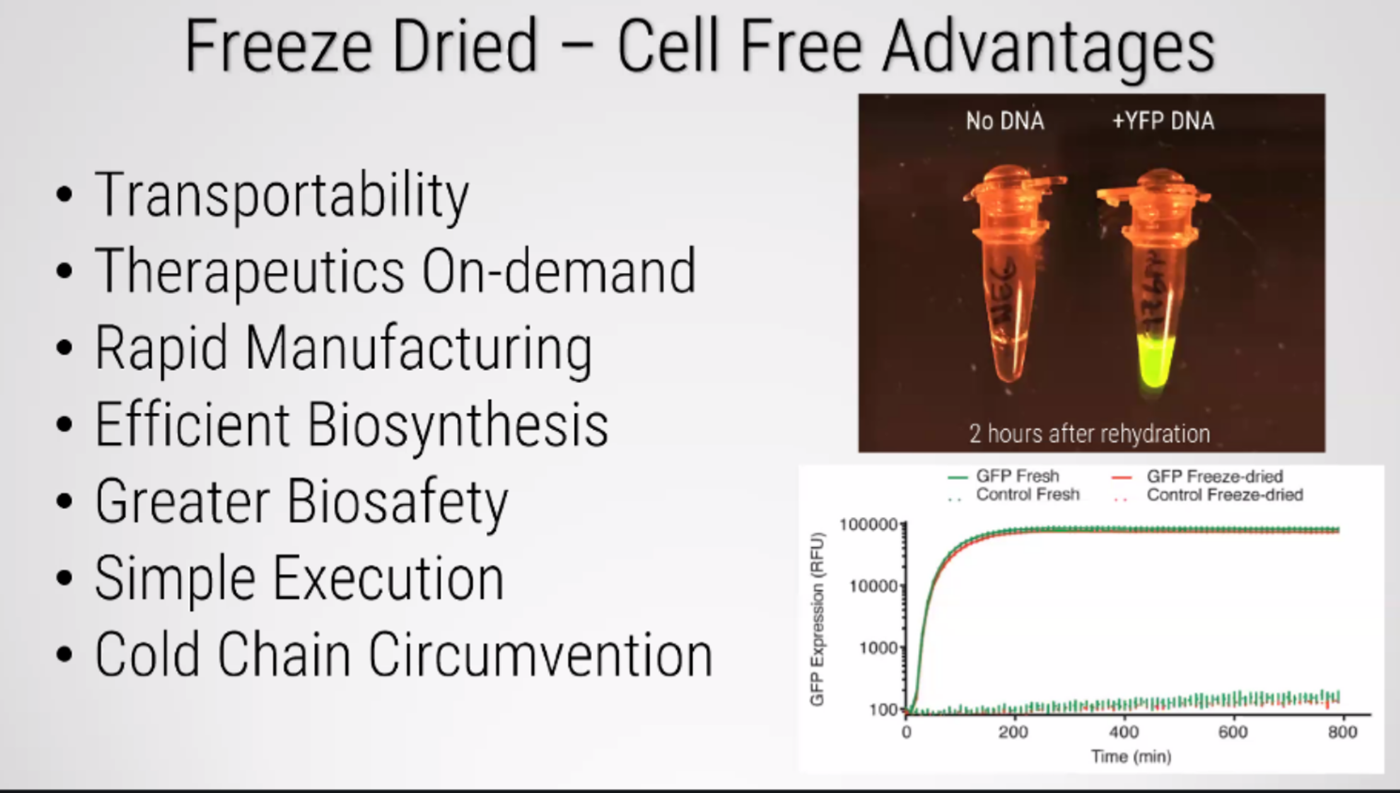

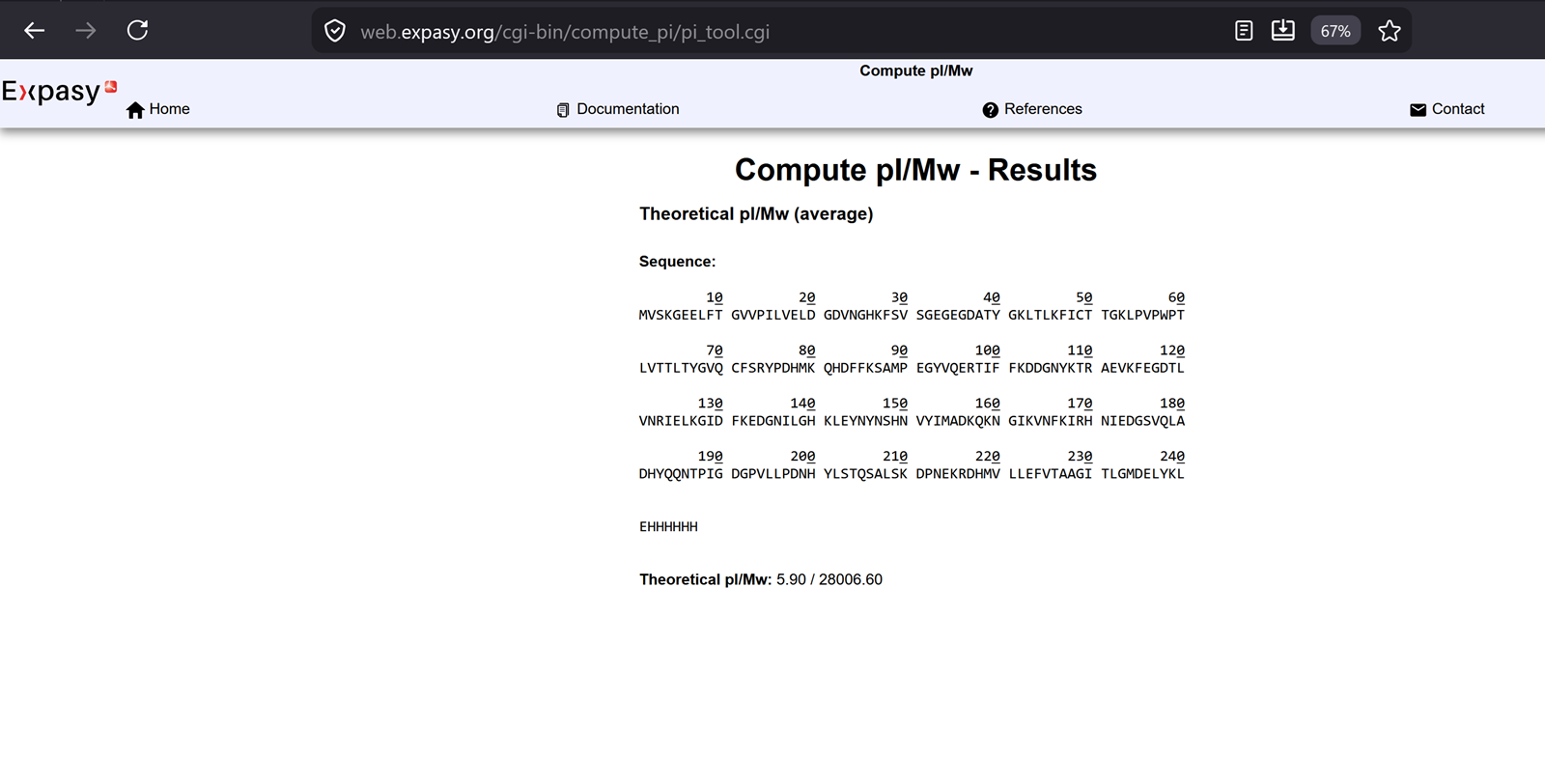
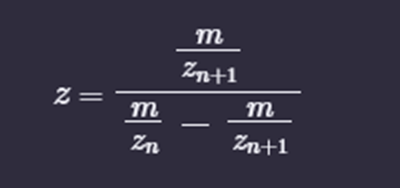

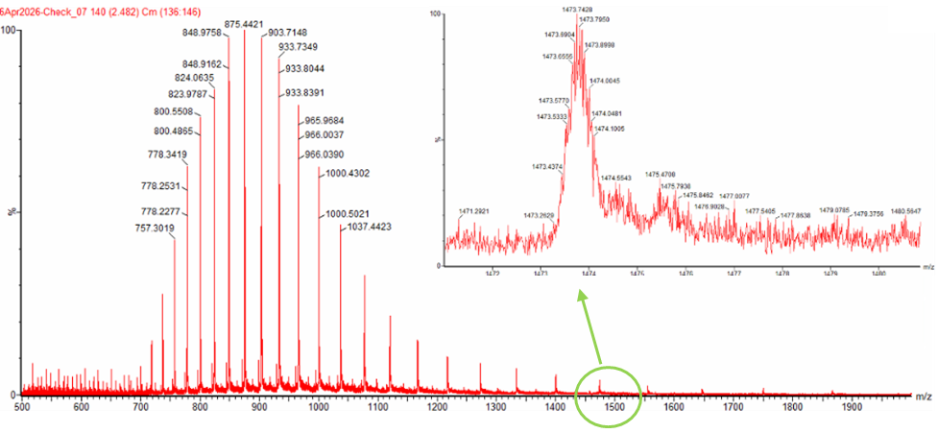
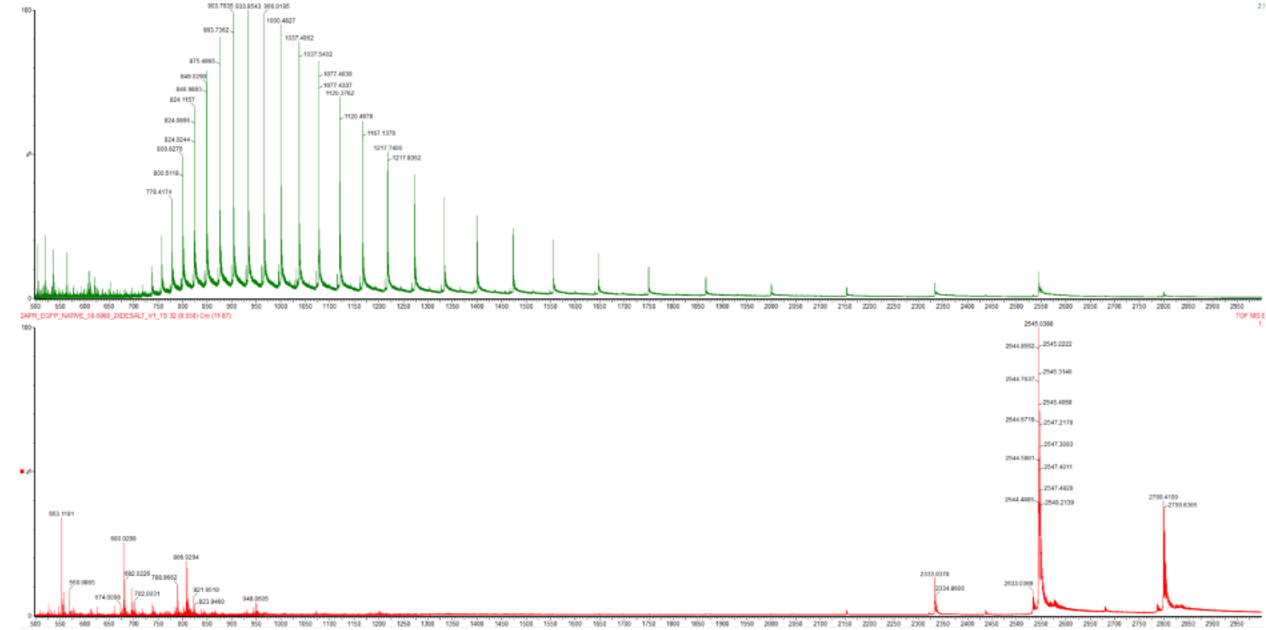
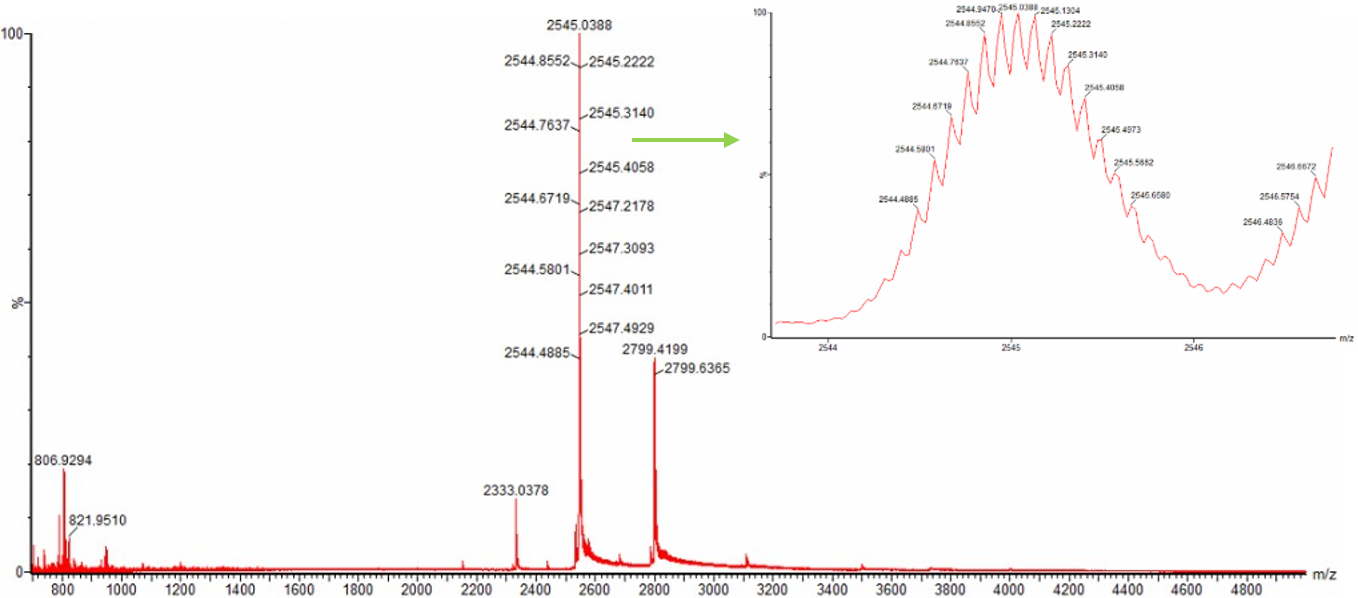
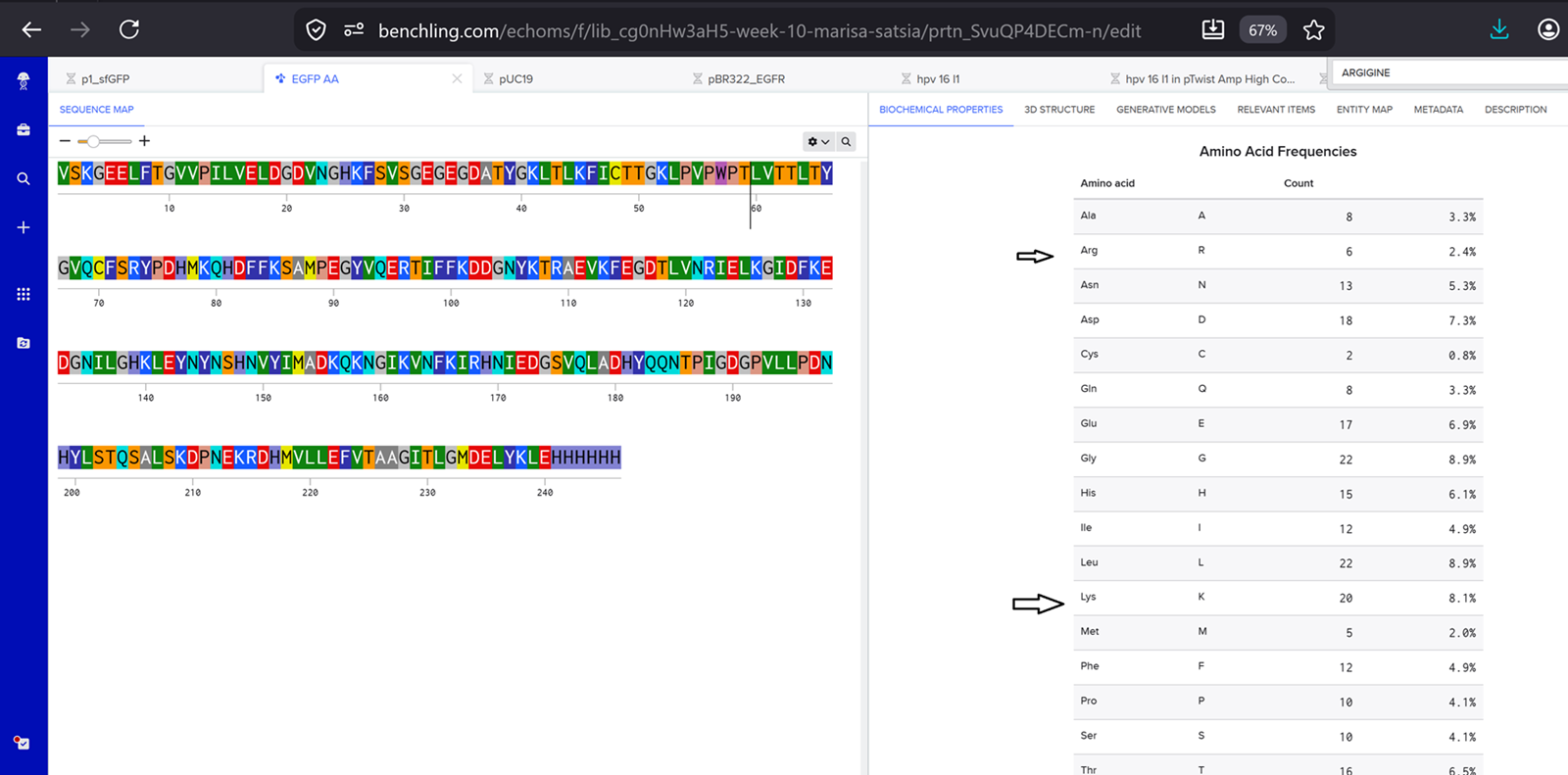
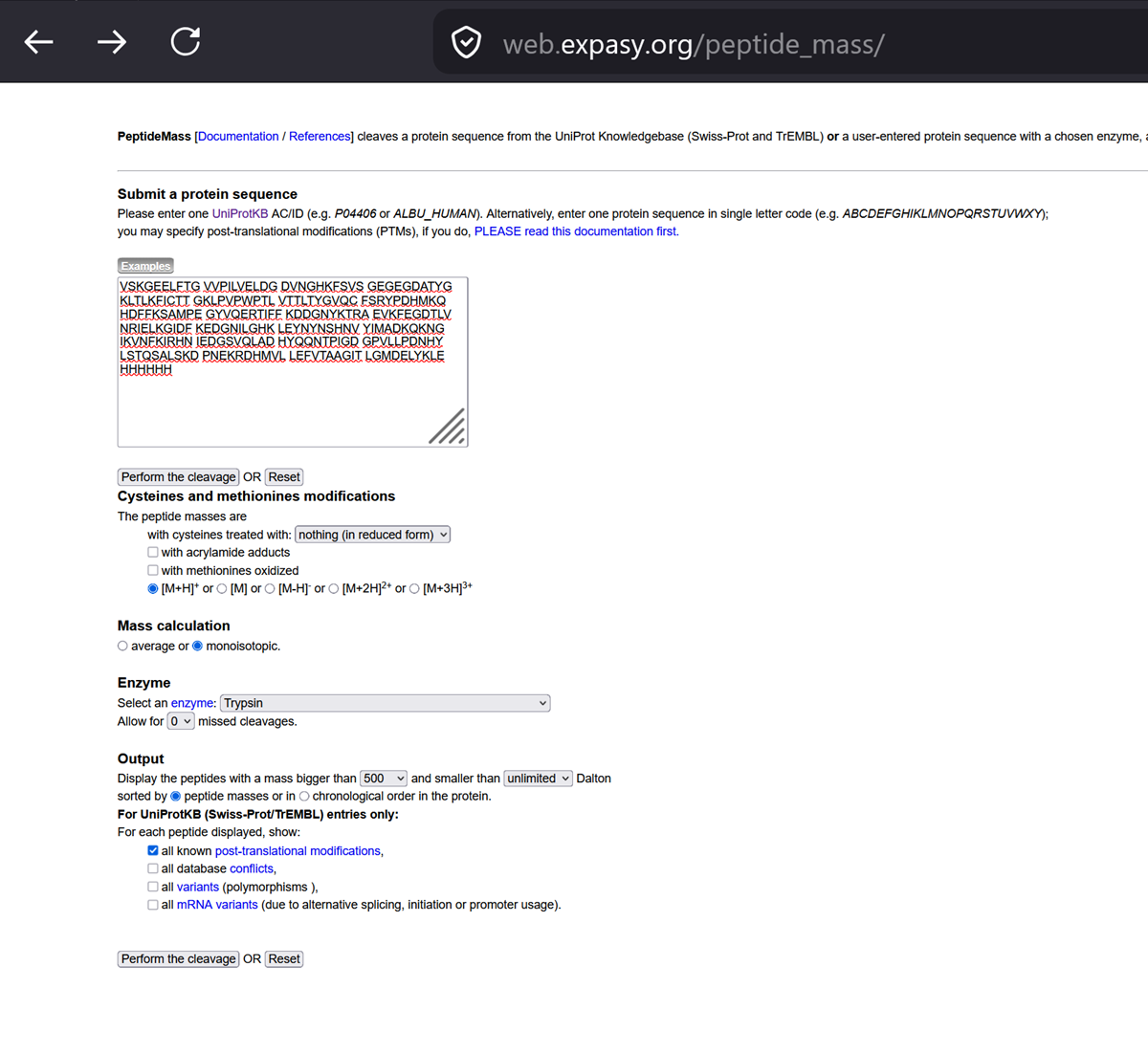
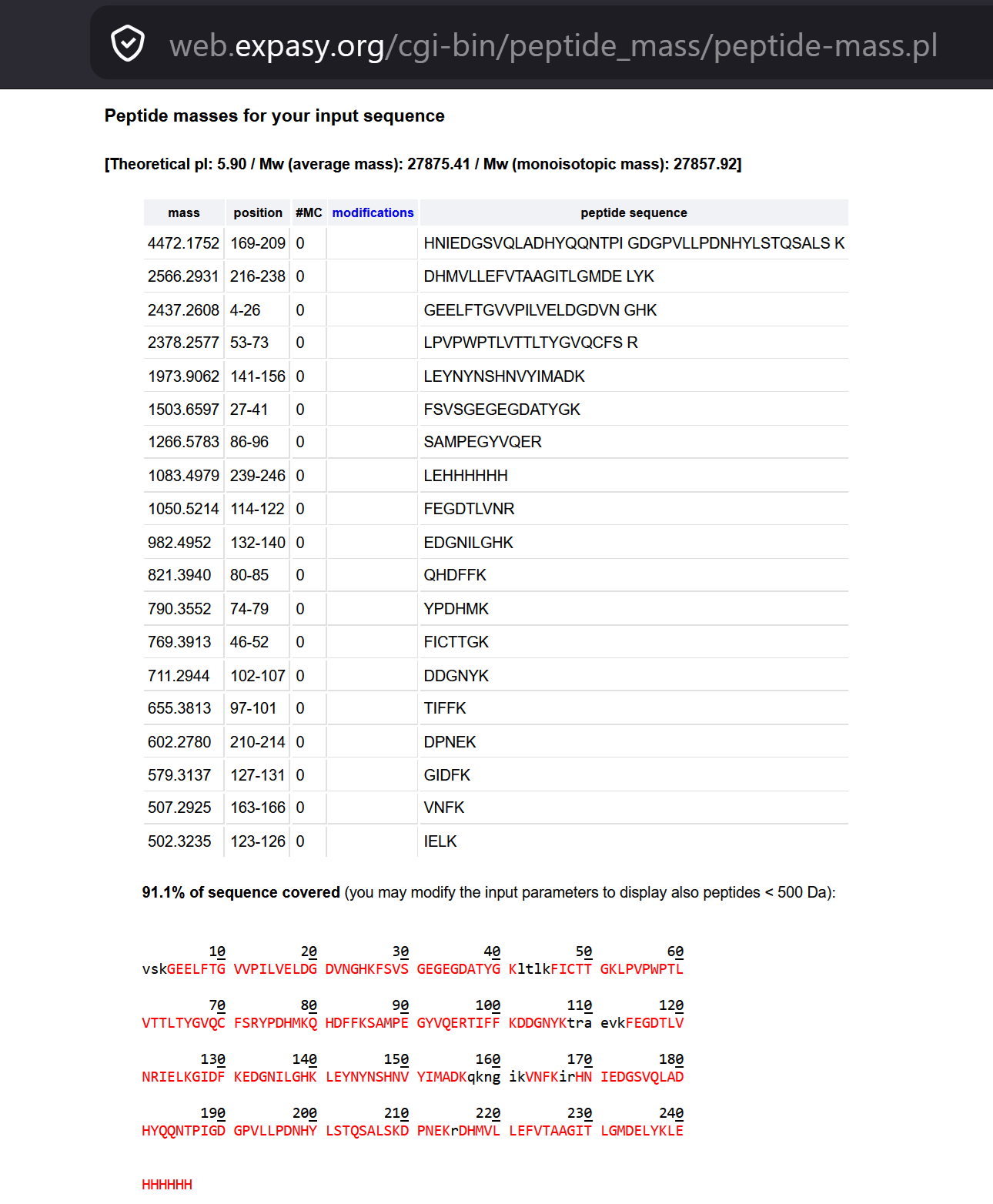
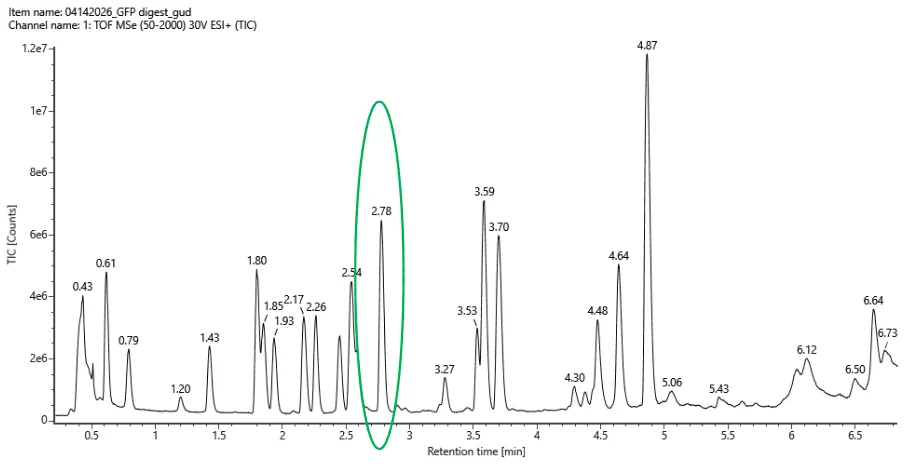
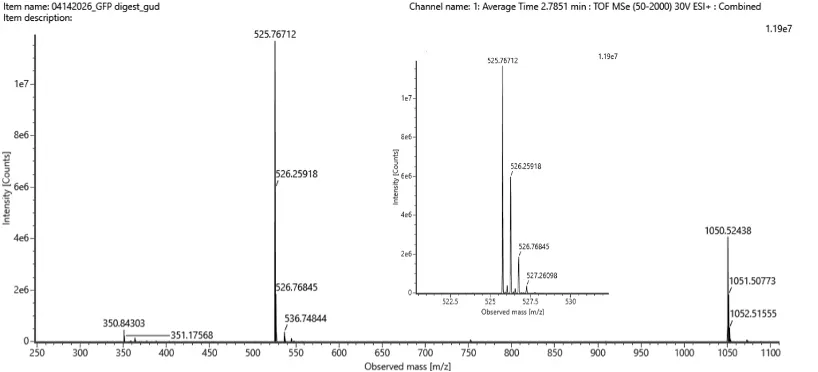
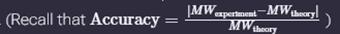
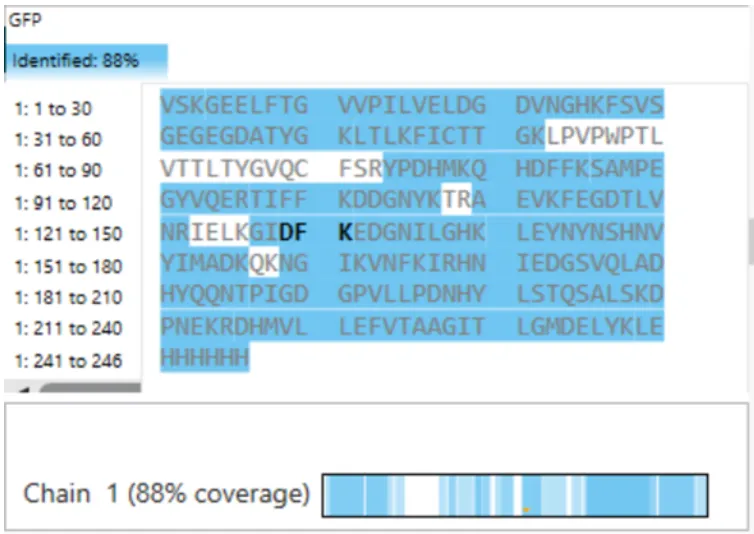
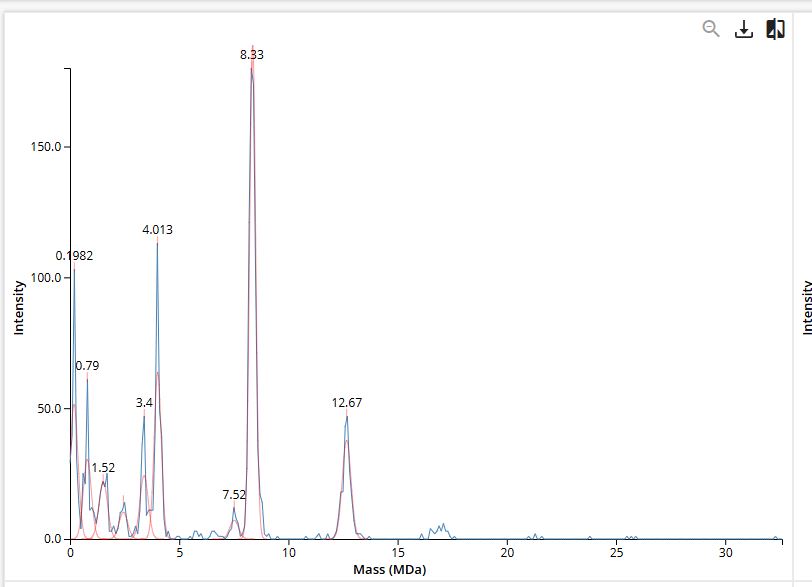





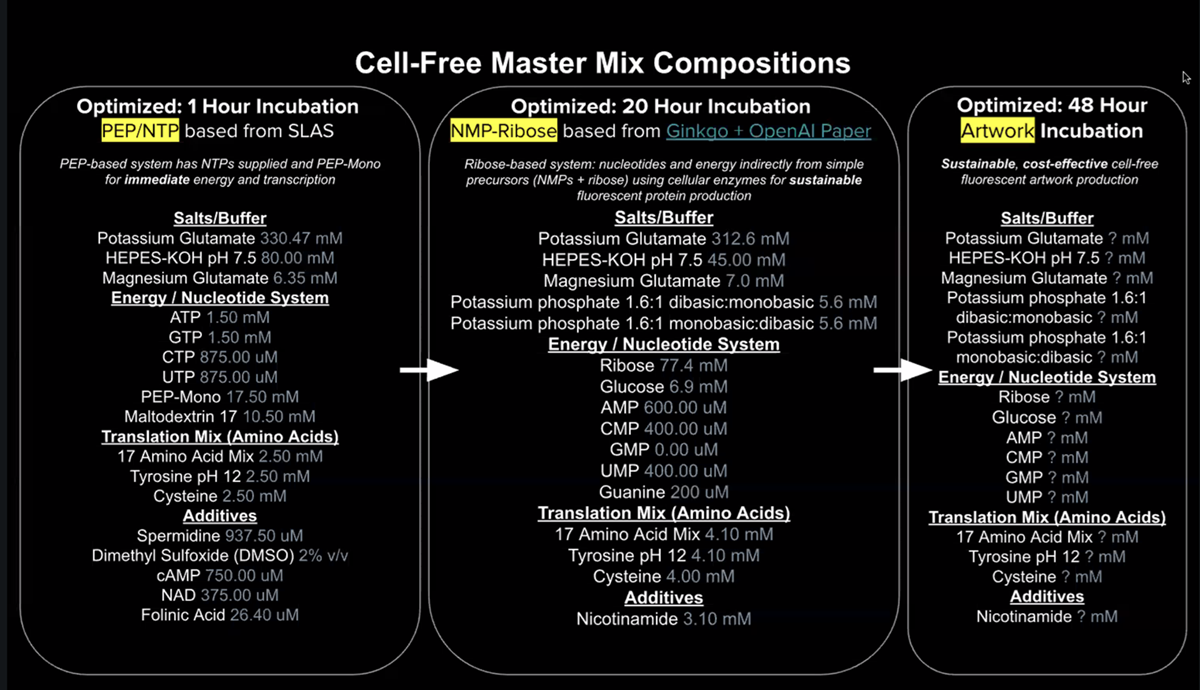

{kind=link}