Week 4 HW: Protein design- PART I
What is protein design?
Objective:
Learn basic concepts:
+amino acid structure
+3D protein visualization
+the variety of ML-based design tools
Brainstorm as a group how to apply these tools to engineer a better bacteriophage (setting the stage for the final project).

Part A. INTUITIVE PART OF THE HOMEWORK!
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
I read the website of a previous htgaa student and found this!
Assuming that the average molecular weight of each amino acid (M) is 100 Daltons or g/mol and mass (m) is 1g, leads to n = m/M = 0.01 mol of amino acids. Taking Avogadro’s constant 1 mol = ~6.022𝐸23 mol(-1) then, X = 0.01 mol * 6.022𝐸23 mol(-1) = 6.022𝐸21 amino acids. Considering that 100 g serving of red meat provides around 28g of protein, this approximates to 3.011E26 amino acid molecules.
- Why do humans eat beef but do not become a cow, eat fish but do not become fish?
We have the ability to digest, break down and transform proteins from other species without becoming what we eat.
- Why are there only 20 natural amino acids?
While only 20 are used in protein synthesis, others exist for specific metabolic roles. These 20 have proven sufficient to build all known life on Earth. The reason isn’t that 20 is a “magic number,” but rather a combination of biological efficiency, evolutionary history, and chemical necessity. One of the most prominent theories (proposed by Francis Crick) suggests that the genetic code became “frozen” early in evolution. Once early life forms settled on these 20 amino acids, any mutation that tried to add a 21st or swap one out would have been lethal. Because every protein in the organism would have changed simultaneously, the organism likely wouldn’t survive. Therefore, we are stuck with the “standard kit” that worked for our most ancient ancestors.
- Can you make other non-natural amino acids? Design some new amino acids.
Ofcourse you can! You can make new building blocks and more synthetic aminoacids :) There are a few different methods to design and create new synthetic aminoacids!
According to this paper these are the methods for reprogramming natural proteins using unnatural amino acids-
Methods for Creating Non-Natural Amino Acids
Chemical Synthesis: This is the most common approach, utilizing organic chemistry reactions such as asymmetric synthesis and transition metal-catalyzed reactions to produce diverse amino acids.
- Gold Catalysis: Recent breakthroughs include using gold catalysis to create amino acids ready for peptide assembly, which simplifies the process of making complex peptides.
- Modifications: This includes modifying natural amino acids by adding, for example, fluorine atoms to enhance stability or altering the backbone through methylation.
Biological Synthesis (Biosynthesis): Engineered microorganisms, such as E. coli, are designed to produce specialized amino acids through metabolic engineering.
Enzyme Catalysis: Enzymes can be used to convert substrates into amino acids with high specificity and, often, in an environmentally friendlier way.
Genetic Code Expansion: Scientists reprogram the cellular machinery to incorporate UAAs into proteins during translation (in vivo).
Applications of these synthetic amino acids are crucial in:
Drug Discovery: Developing more stable and effective peptide-based drugs.
Protein Engineering: Creating enzymes with enhanced activity and stability under harsh conditions.
Antibody-Drug Conjugates (ADCs): Creating unique linkages to bind drugs to antibodies for targeted cancer therapy.
- Where did amino acids come from before enzymes that make them, and before life started?
I have a feeling I have to dive into astrobiology for this, haha!
Overall, the findings of the study indicate that slight differences in the conditions present during aqueous alteration on planetesimals can have big effects on the end abundances of amino acids. Some amino acids can be destroyed and others created and this in turn will affect the availability of amino acids at the origin of life on Earth.
- If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
A a helix made of D-amino acids will form a left-handed helix. Because D-amino acids are mirror images of the naturally occurring L-amino acids (which form right-handed helices), their sterically favored conformation results in an enantiomeric, left-handed structure.
L-Amino Acids: Form Right-Handed helices.
D-Amino Acids: Form Left-Handed helices.
Mechanism: The reversal occurs because the steric hindrance caused by side chains is mirrored in D-amino acids, favoring the opposite twist.
- Can you discover additional helices in proteins?
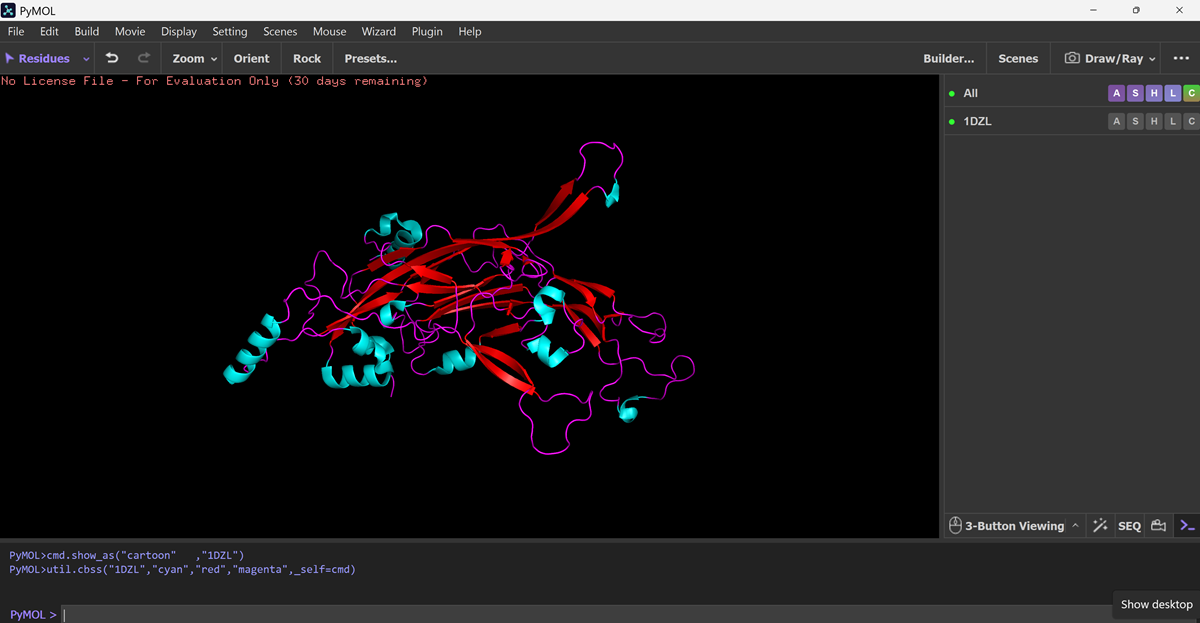
Yes, additional helices in proteins can be discovered and analyzed beyond the common helix, as modern structural biology, computational methods, and tools like AlphaFold continue to reveal more complex and rare structural motifs. While helices and sheets constitute the majority of secondary structures, other helices, such as a helices and b helices, are frequently identified in protein structures
- Why are most molecular helices right-handed?
Most molecular helices, such as DNA and alpha-helices in proteins, are right-handed because this configuration is energetically more stable and allows for tighter packing without steric hindrance (clashing) between atoms. This structural preference is rooted in the intrinsic chirality of their building blocks—left-handed amino acids in proteins and right-handed sugars in DNA.
- Why do β-sheets tend to aggregate?
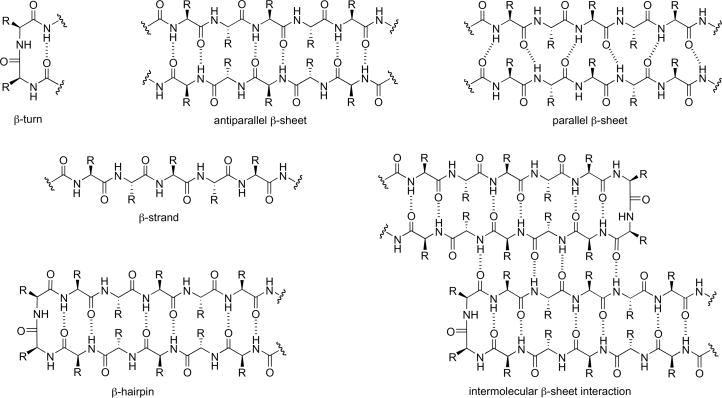
The edges of completely regular β-sheets or β-sandwiches are inherently aggregation-prone, because they are already set up to form further β H-bonding with any other β strands they encounter, Natural β-sheet proteins use negative design to avoid edge-to-edge aggregation.
β-sheets have a natural, intrinsic tendency to aggregate due to their structural properties, which make them ideal building blocks for stable, intermolecular arrangements known as “cross-β” spines or amyloid fibrils. They are prone to aggregating when “edge strands” of a sheet—which contain open hydrogen bond donors/acceptors—encounter other strands, facilitating the growth of intermolecular hydrogen-bonded networks.
Protein β-sheet structures are formed by the lateral alignment of β-strands in parallel or antiparallel orientations and are stabilized by hydrogen bonding, hydrophobic interactions, and other forces Figure 1.
- What is the driving force for β-sheet aggregation?
The driving force for sheet aggregation—commonly referred to as amyloid fibril formation is a complex, multi-faceted process primarily driven by the hydrophobic effect and the maximization of intermolecular hydrogen bonding. These forces allow, often unstructured or partially folded, peptide monomers to overcome the energetic penalty of unfolding and rearrange into highly stable, stacked sheet structures.
- Why do many amyloid diseases form β-sheets?
Amyloid diseases form sheets because this structure represents a highly stable, low-energy, and thermodynamically favorable conformational state for misfolded proteins to aggregate. The sheets form “cross-” spines, where peptide strands align perpendicularly to the fiber axis, allowing for tight"dry" (water-free) interlocking of amino acid side chains (steric zippers) and maximum hydrogen bonding.
More specifically, here and here I found more answers-
Here is why amyloid diseases specifically form sheets:
Thermodynamic Stability: The sheet conformation is extremely stable, allowing misfolded, often disordered proteins (e.g., Alzheimer’s A) to aggregate into insoluble amyloid fibrils.
Dry Steric Zippers: Amyloid fibrils are held together by a “dry steric zipper” where two-sheets interdigitate, leaving no room for water between them, creating a very strong, stable structure.
Maximum Hydrogen Bonding: The in-register sheets maximize inter-strand hydrogen bonding, which stabilizes the fibrous structure.
“Generic” Protein Property: Many proteins have the potential to form sheets if subjected to specific conditions (e.g., PH changes, denaturation), meaning it is a common, almost universal"default" misfolding state.
Templated Growth: The sheet structure allows for an “indefinitely repeating” pattern, which enables the rapid recruitment of more monomers to the growing fiber.
Can you use amyloid β-sheets as materials?
Yes!! Amyloids are Building Blocks for Macroscopic Functional Materials. Amyloid sheets are used as highly stable, versatile building blocks for advanced functional materials, including hydrogels, nanofibrils, and conductive coatings. Due to their ordered cross-structure, they are engineered for applications like 3D cell culture, tissue engineering, water filtration, and drug delivery, exploiting their remarkable strength and resistance to environmental degradation.
Design a β-sheet motif that forms a well-ordered structure.
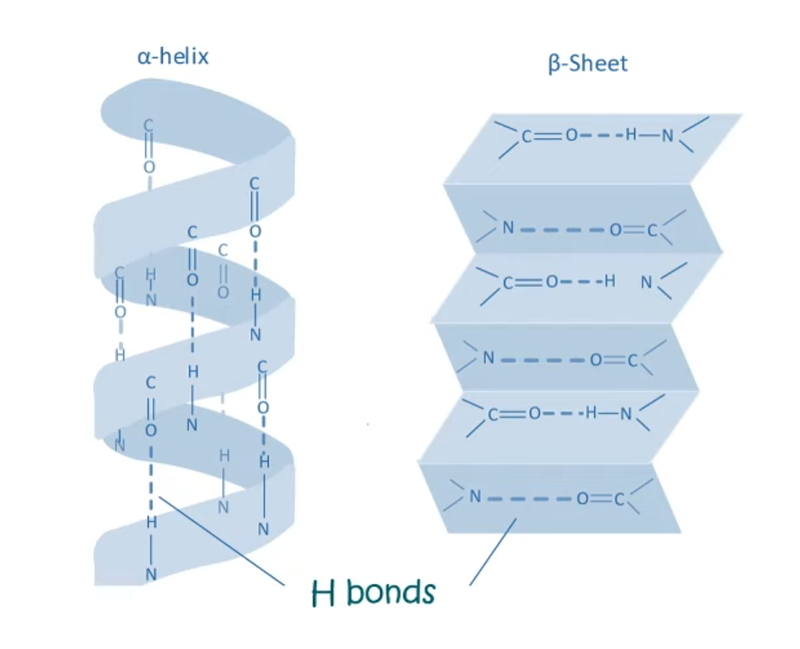
Secondary structural features- b pleated sheet. N-H groups in the backbone of one strand of aminoacids form HYDROGEN BONDS with C=O groups in the backbone of adjacent strands. Less stearic constraint compared to alpha helices. 2 orientations-> parallel and antiparallel.
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
NGLViewer: NGL Viewer is a collection of tools for web-based molecular graphics. WebGL is employed to display molecules like proteins and DNA/RNA with a variety of representations.
PyMOL: PyMOL is a user-sponsored molecular visualization system on an open-source foundation, maintained and distributed by Schrödinger.
Chimera: A highly extensible program for interactive visualization and analysis of molecular structures and related data, including density maps, supramolecular assemblies, sequence alignments, docking results, trajectories, and conformational ensembles.
Briefly describe the protein you selected and why you selected it. I chose hpv 16 L1 protein because I used it during the second week for reading, writing and editing DNA.
Identify the amino acid sequence of your protein.
MYVWSLQRLQ KVPVKGLGYI GIGQHPYYNY KELVPVTSTN YITKSEPEIG NGLYQISVKL KKLSTGVLSA YIHTSDEVLS VPFNKILVQS PVGQNIFRFP LGTVLADTVT STNMSLGTIV PYVLSTSVPT SMVSSGSCIS APNKIPAPGY IYMRTVASDV SKVNTSVLYN SKSLPVTSIS VNKGRFTTNS LTQTNLTVES NTNTVLSIDG FAPGTNLGIT VQPPVGSGNY IFRIPITKQP GSTVVRIVDD TSTYNSLGSL LHPNDNIPLT AGVGEVVISE DTDTSSTSQT PVSSGVNFSN TGLDNYITQT NVTVTQDTGD TYLSNGLKFD NQGLTPLTVQ PVTGRTSLVS GYVTTSNKTV TDASTTYSLS NPTAGNPNSG IYISNGTIGT GGVTSVTPGT NTSVSQTVPG GGTDYNILQA TLTAVEDSTT VVTNVFAGNV NPTSTSPNTS SLVTTPSSGT YISNTSSATS TNTSAVSATP TSSAVSLSTS TTSSISSLTS TVTAVSSSTT AAKPKFTLGK RKATPTTSST STTAKRKK
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
It is a 531 amino acid, highly conserved major capsid structural protein that self-assembles into virus-like particles (VLPs). The prototype sequence, often derived from HPV-16 reference genomes (e.g.UniProt P03101), features key neutralizing epitopes, particularly within surface-exposed loops. Common variants show minor mutations (e.g., H76Y, T176N, T266A) depending on geographical lineage. Serine (S) is generally identified as the most frequently occurring amino acid in the L1 major capsid.
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs. Here is my search for HPV 16 L1 PROTEIN HOMOLOGS. I have 181 holomogs. On the map red indicated close relation to other families
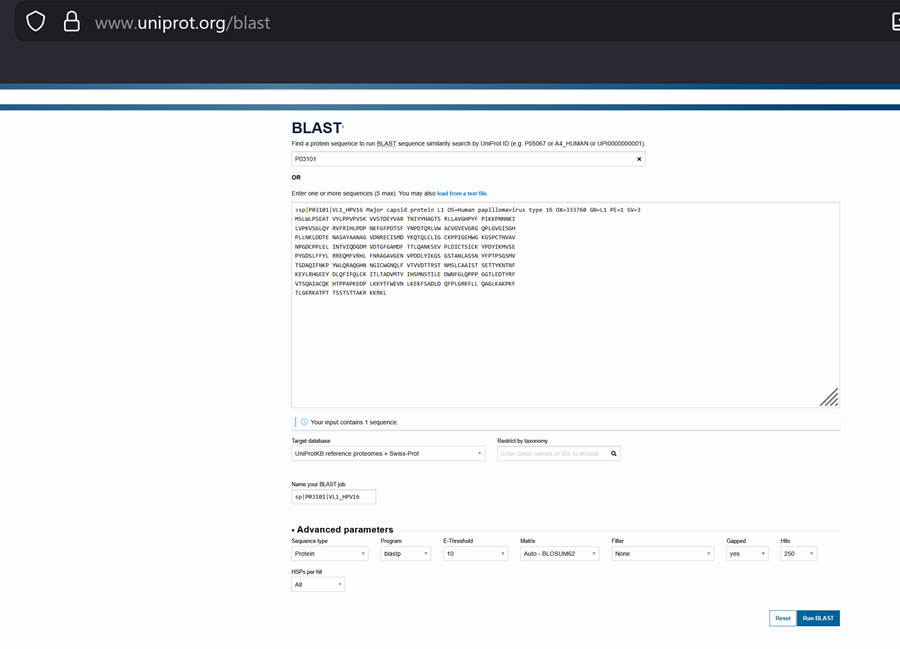
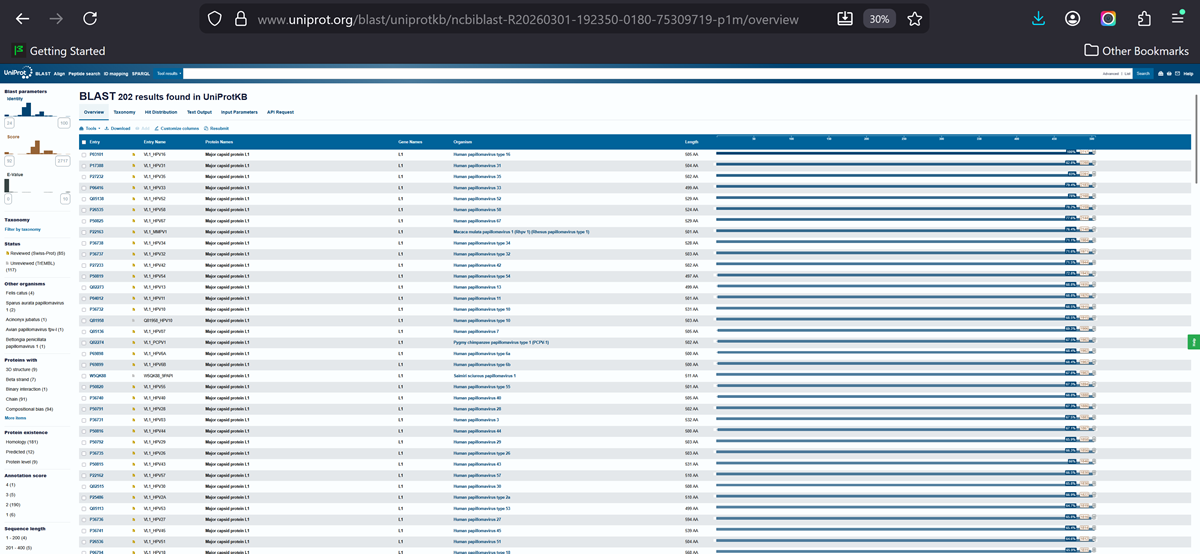
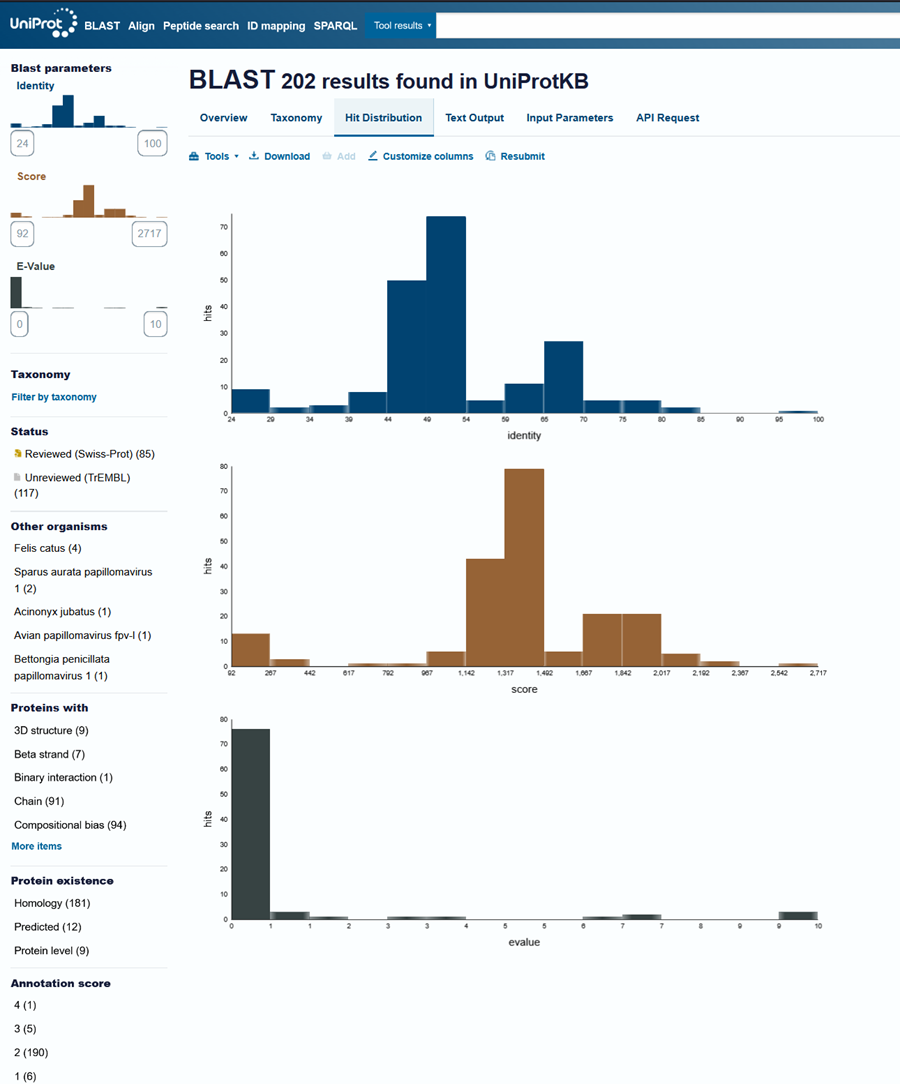
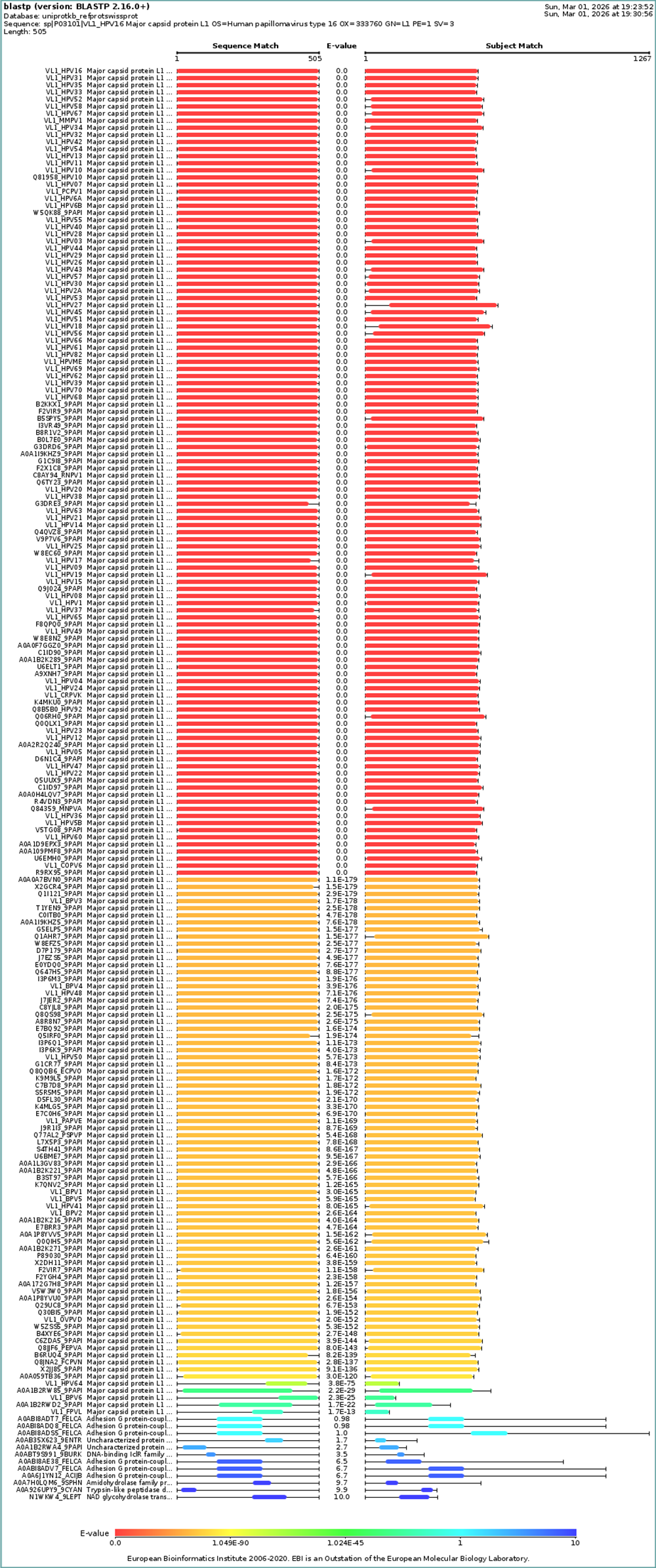
Does your protein belong to any protein family? It is a part of the papillomaviridae L1 protein family.
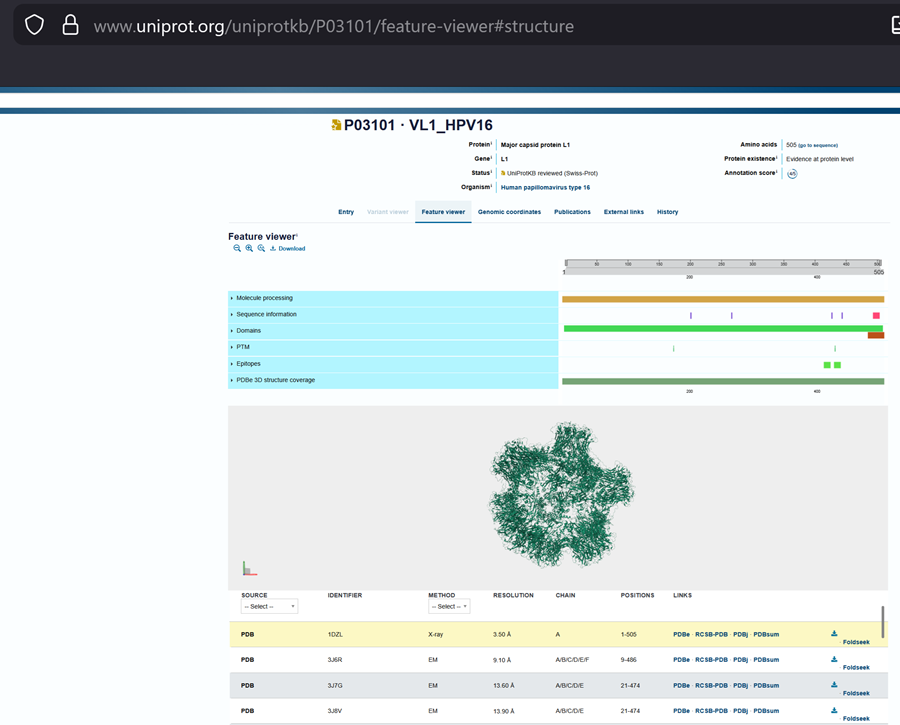
- Identify the structure page of your protein in RCSB
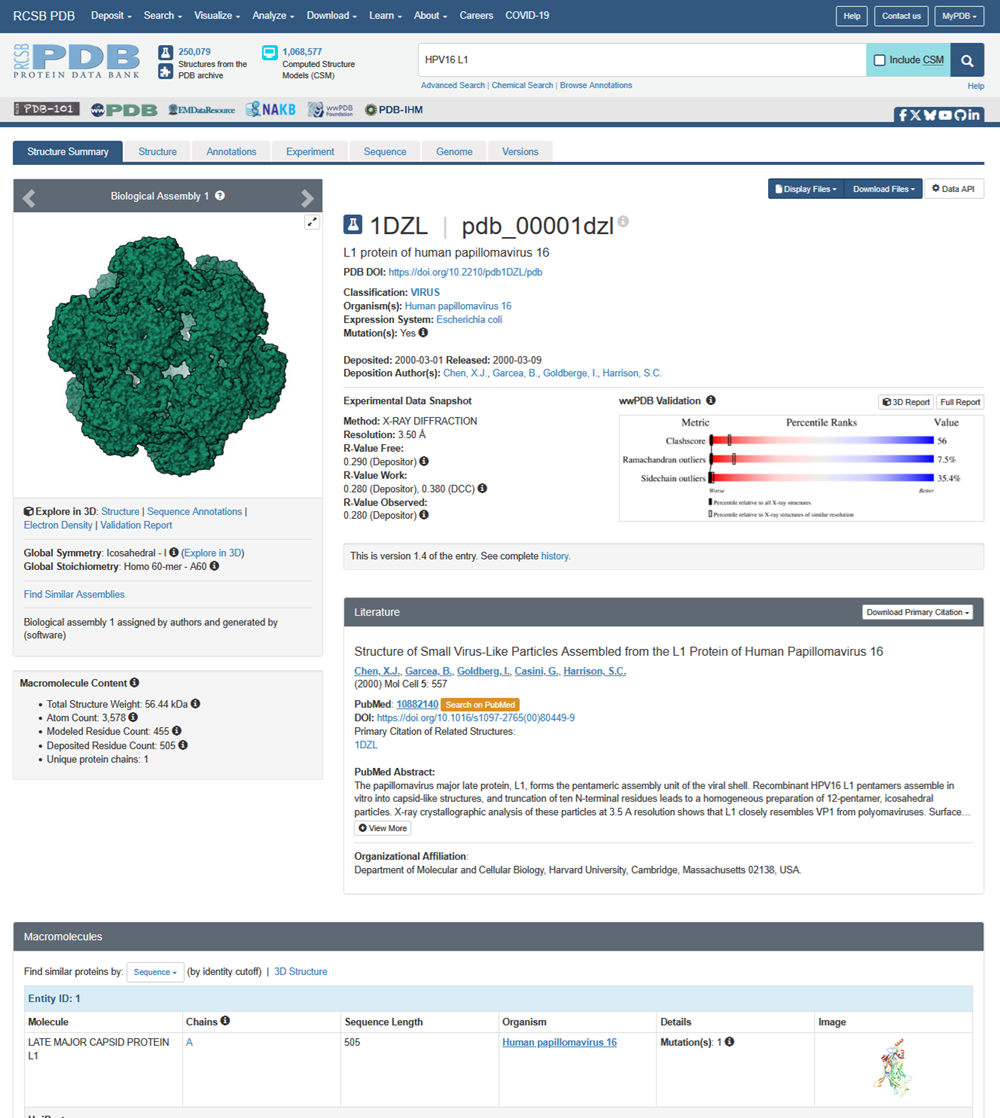
Here is the DOI of the protein on RCSB. I went to the homepage and searched in the top right for my protein and this is what i got!
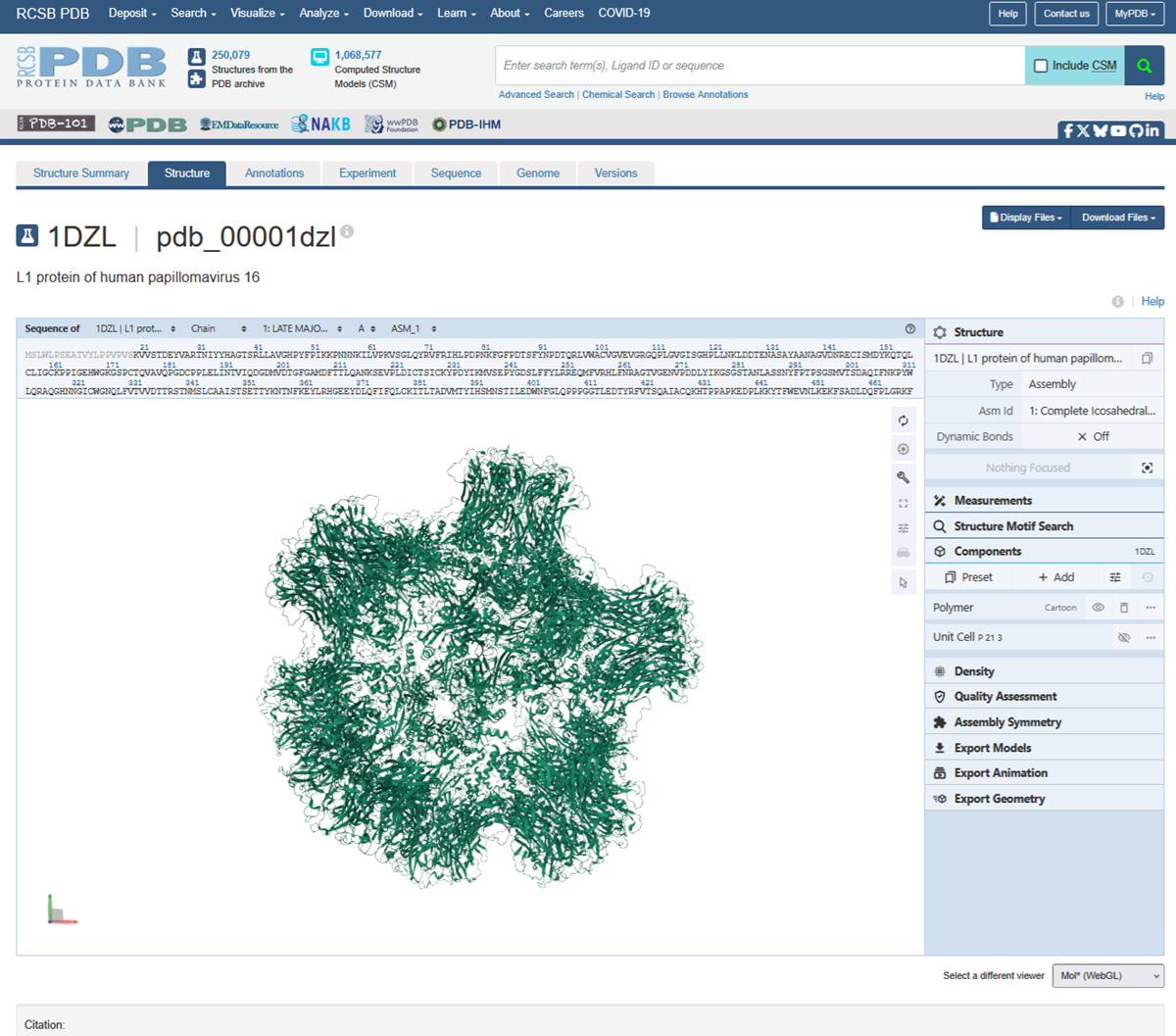
Then to explore the 3D structure I clicked under the image on the left.

I also made an animation of the unwind assembly of the HPV16 L1 PROTEIN.
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
L1 protein of human papillomavirus 16
PDB DOI: https://doi.org/10.2210/pdb1DZL/pdb
Classification: VIRUS
Organism(s): Human papillomavirus 16
Expression System: Escherichia coli
Mutation(s): Yes
Deposited: 2000-03-01 Released: 2000-03-09
Deposition Author(s): Chen, X.J., Garcea, B., Goldberge, I., Harrison, S.C.
Experimental Data Snapshot
Method: X-RAY DIFFRACTION
Resolution: 3.50 Å R-Value Free: 0.290 (Depositor) R-Value Work: 0.280 (Depositor), 0.380 (DCC) R-Value Observed: 0.280 (Depositor)
wwPDB Validation
Are there any other molecules in the solved structure apart from protein?
Nop?
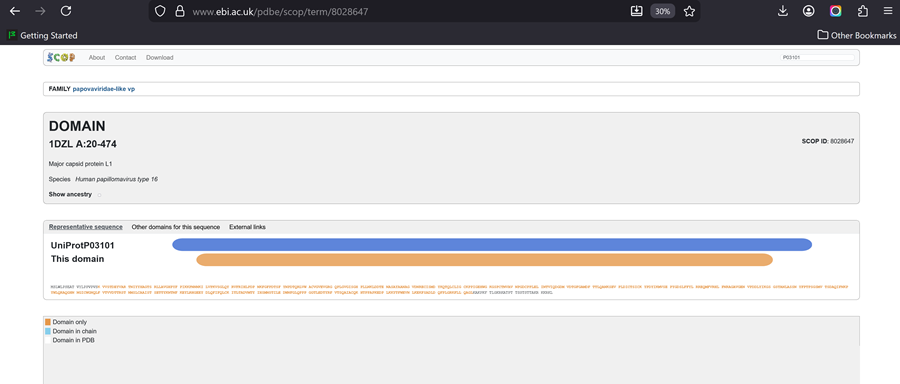
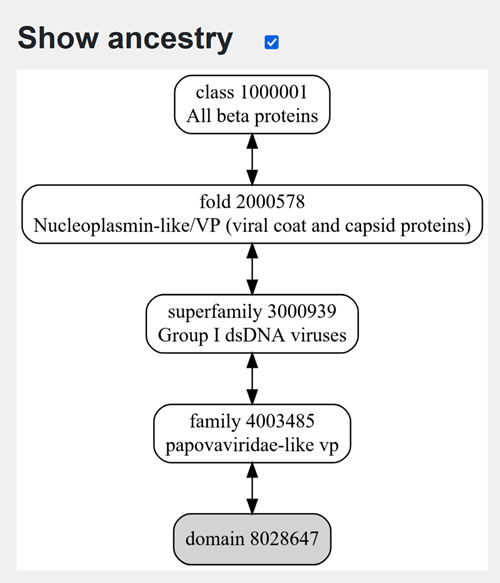
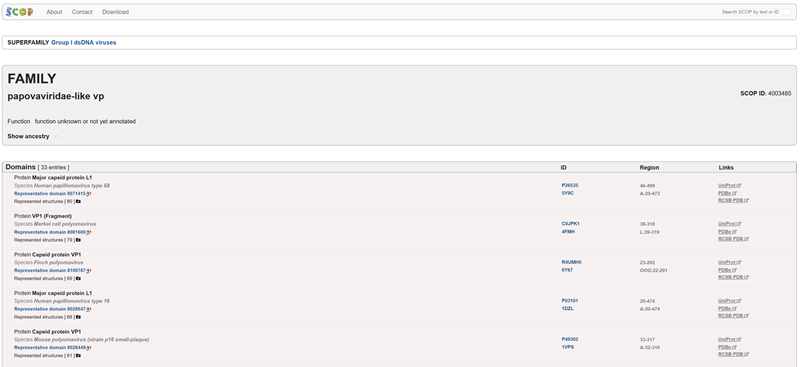
Does your protein belong to any structure classification family?
Family-> papovaviridae-like vp
- Open the structure of your protein in any 3D molecule visualization software
Soooo! I tried to download pymol and as we discussed during bioclub Thursdays myself and Flo had to register to use it!
PyMol Tutorial Here(hint: ChatGPT is good at PyMol commands)
When I opened the PYMOL app on my laptop I got this message and chose skip activation and now I only have 30 day evaluation period -> NO LICENSE FILE
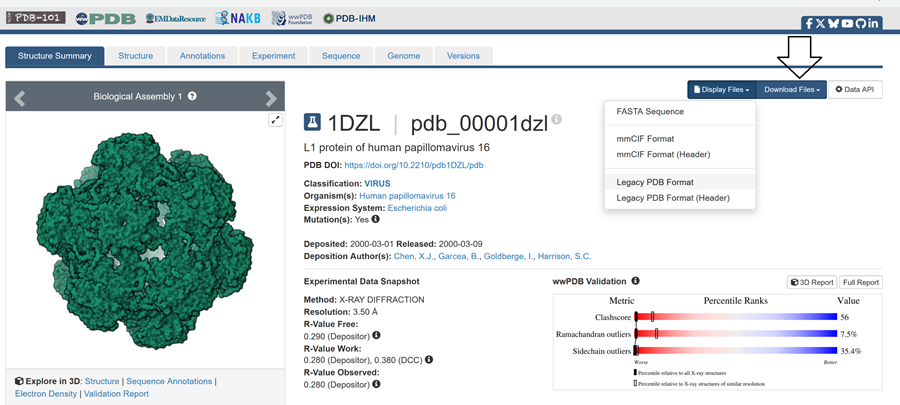
And then I had to download the PDB files for the proten on rcsb.org!
Here I found the PYMOL tutorial from step 1 of the process of visualisation on PYMOL.
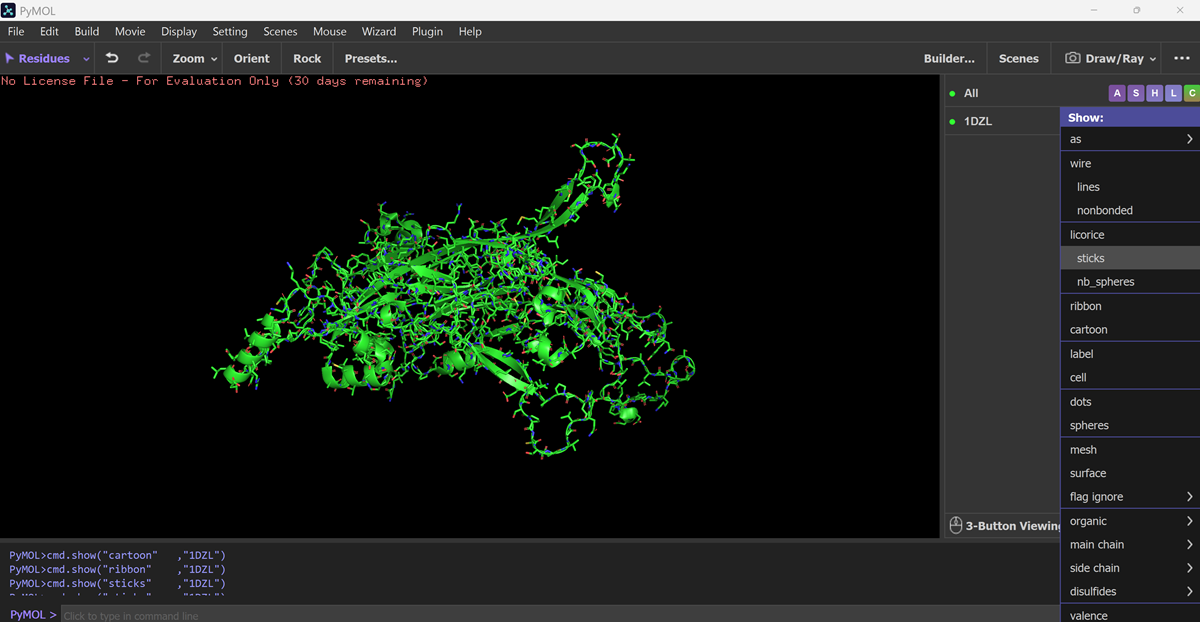
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
I downloadd my pdb files for my protein and went to FILE-OPEN on PYMOL.
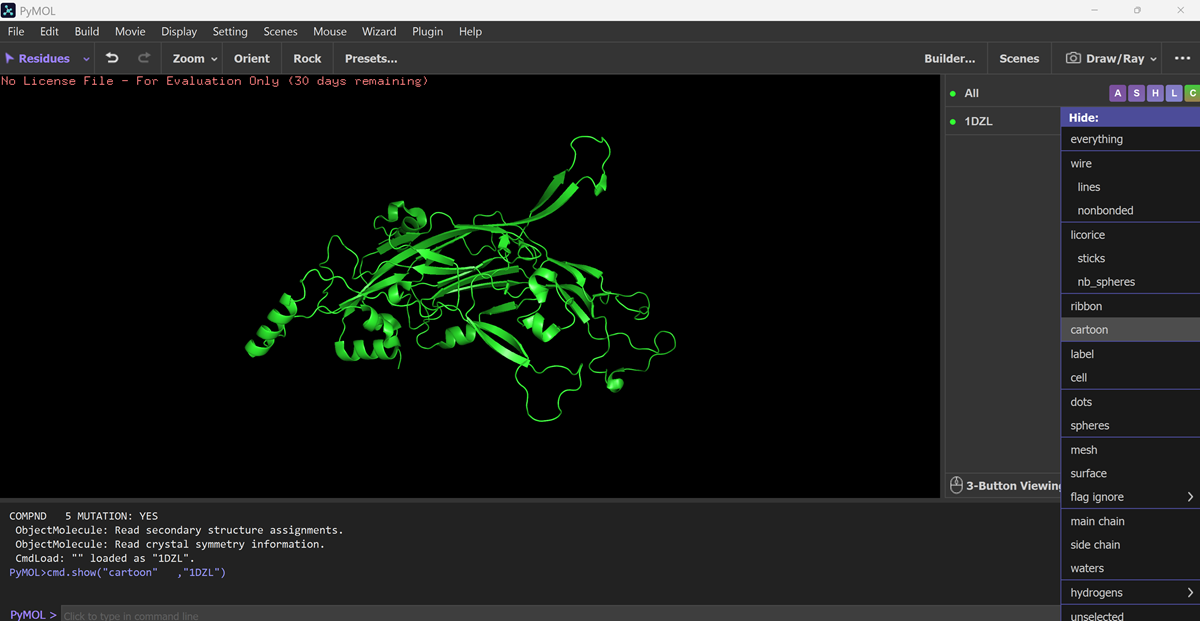
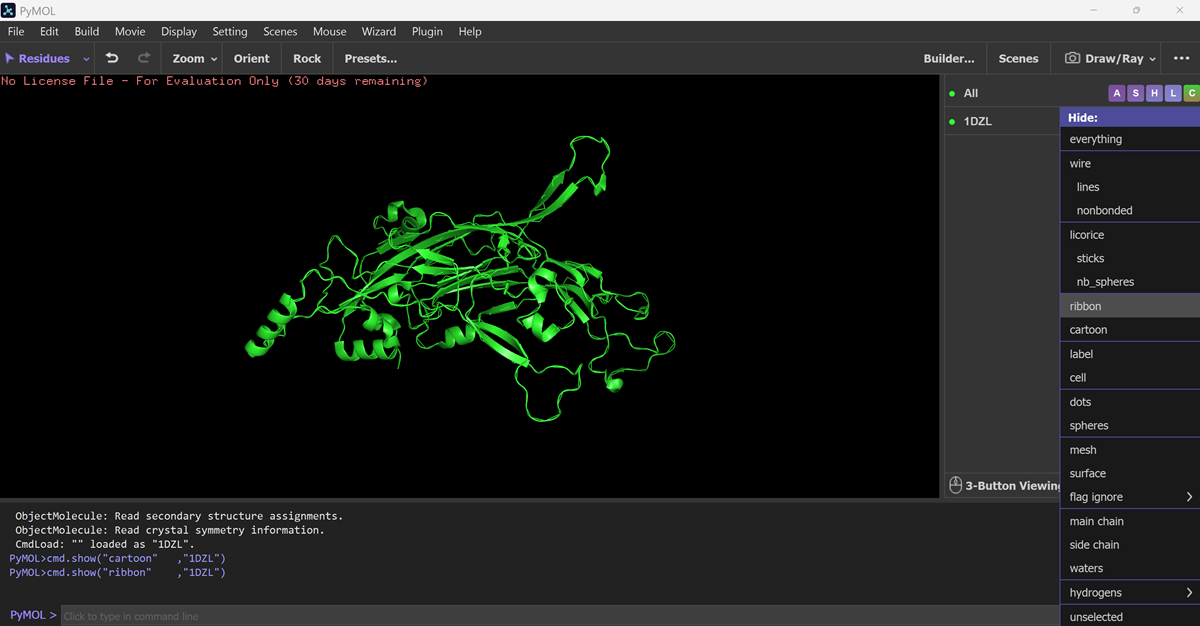
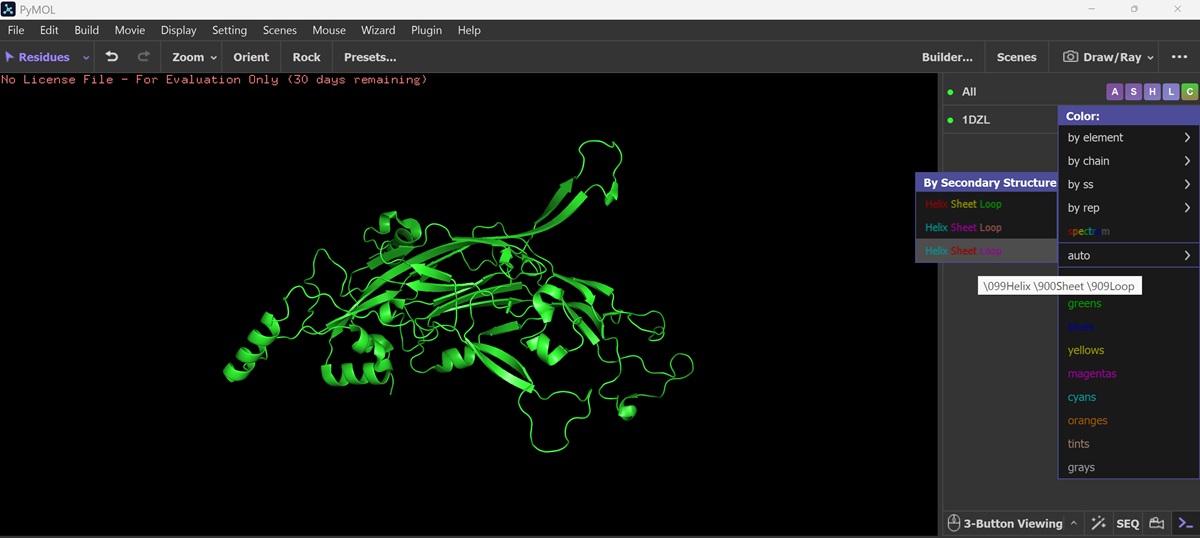
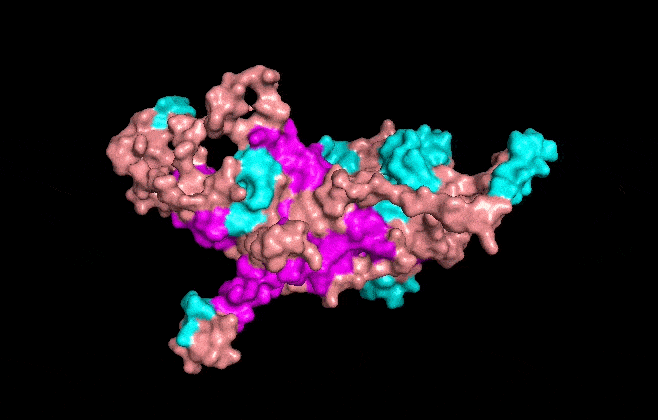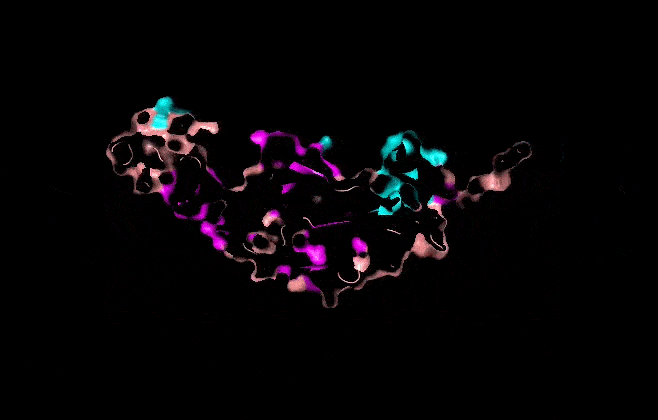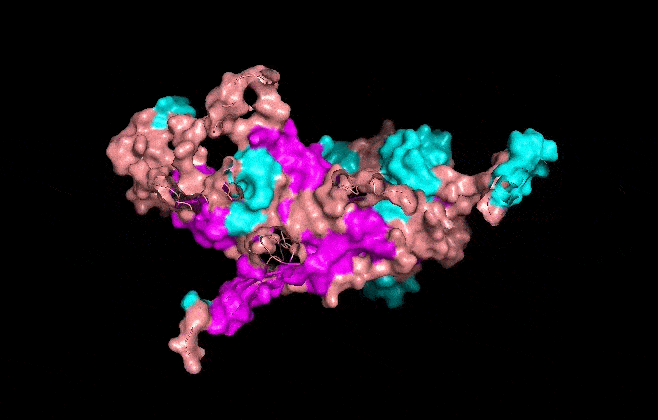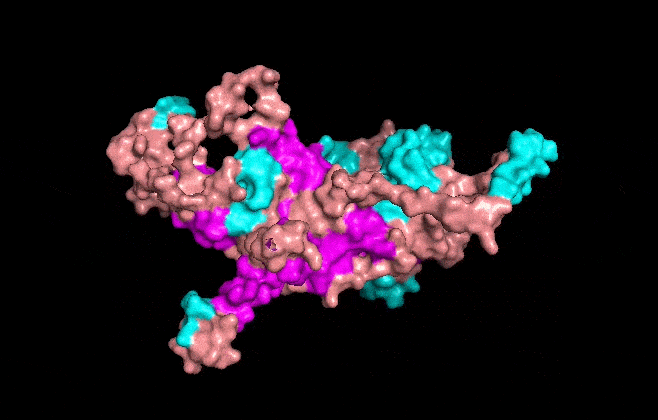
Color the protein by secondary structure. Does it have more helices or sheets?
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
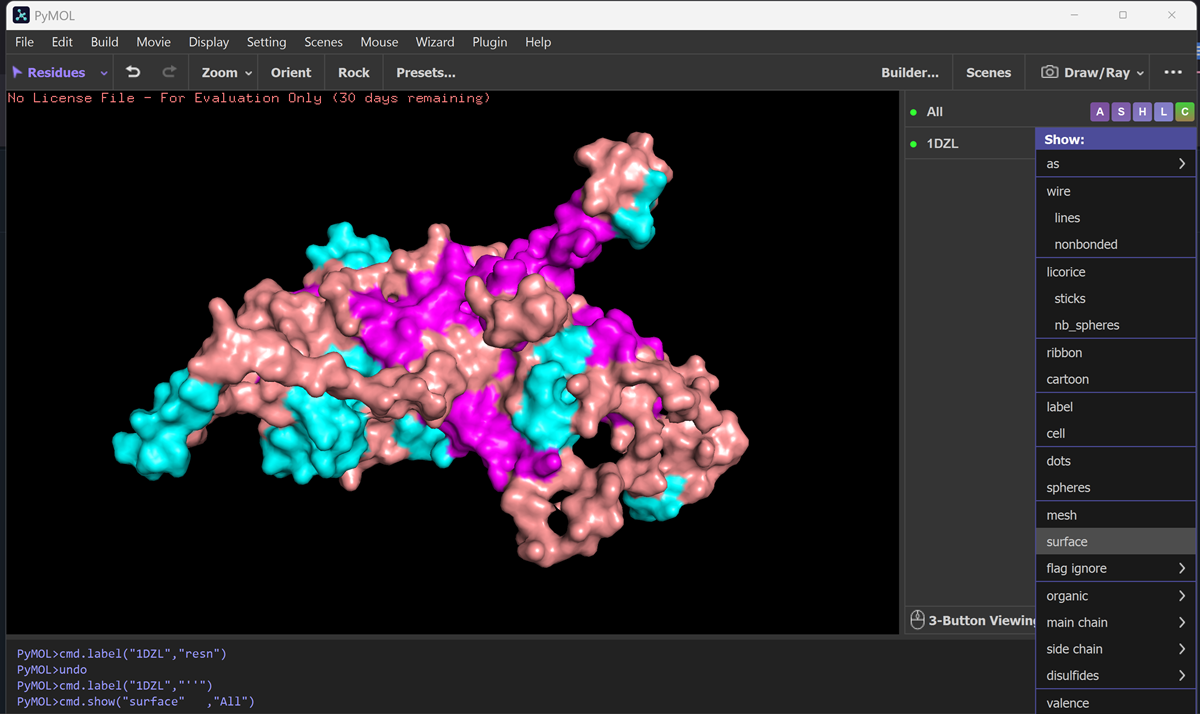
It has some holes aka binding pockets!
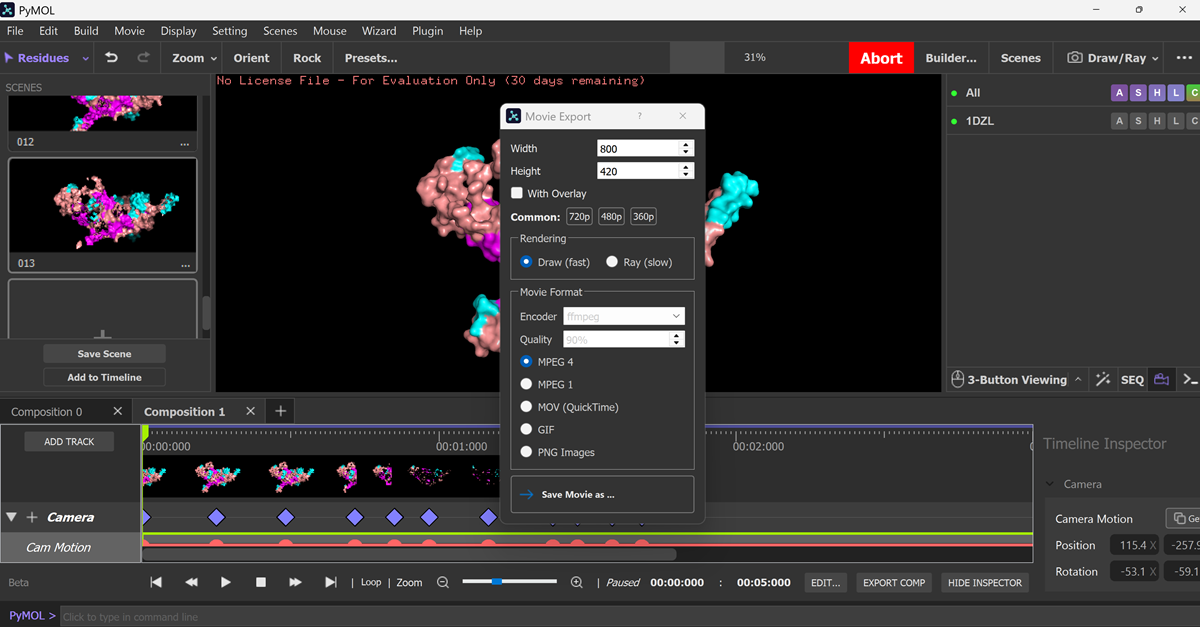
I made an animation using the commands below.
Aaaand this is the mp4 animation I made. I later converted it into a gif and optimized it.
Part C. Using ML-Based Protein Design Tools
In this section, we will learn about the capabilities of modern protein AI models and test some of them in your chosen protein.
Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
Choose your favorite protein from the PDB.
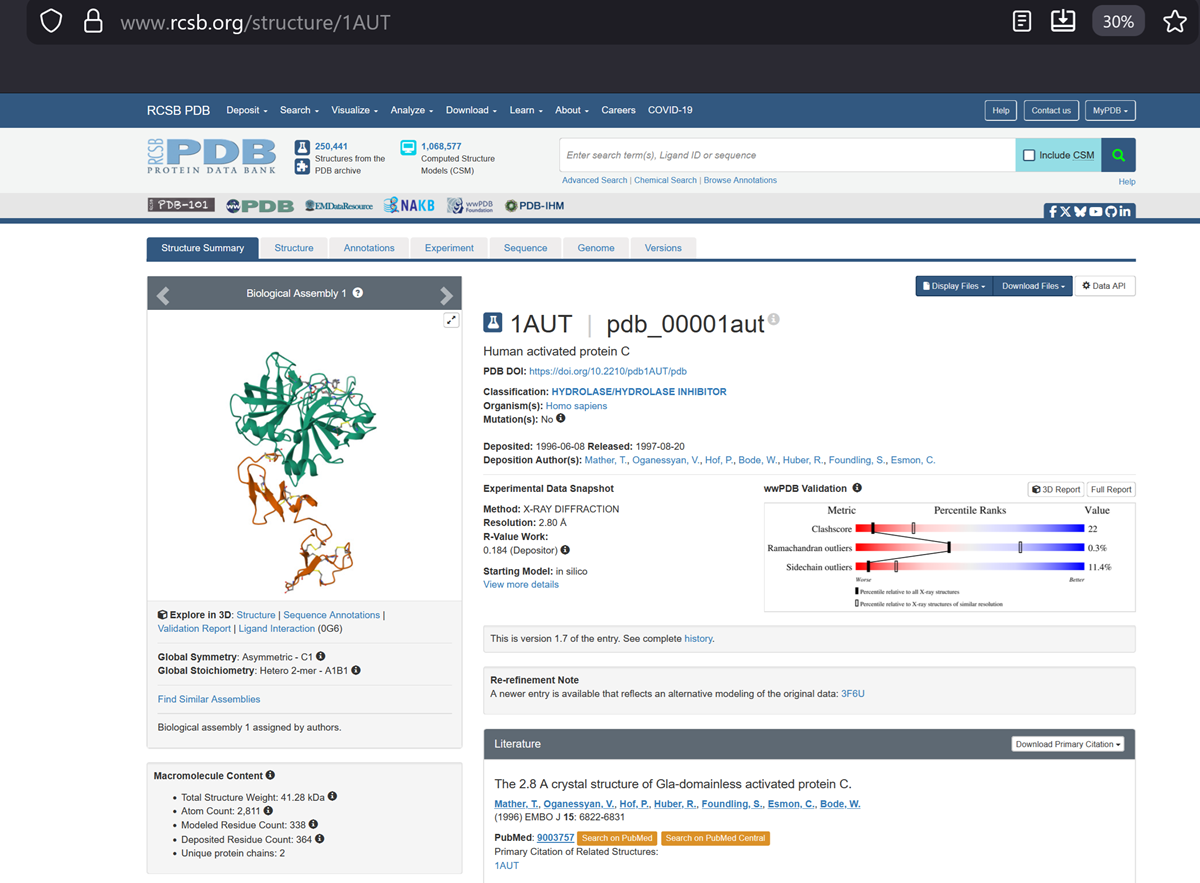
Protein C, also known as autoprothrombin IIA and blood coagulation factor XIV is a zymogen, that is, an inactive enzyme. The activated form plays an important role in regulating anticoagulation, inflammation, and cell death and maintaining the permeability of blood vessel walls in humans and other animals. Activated protein C (APC) performs these operations primarily by proteolytically inactivating proteins Factor Va and Factor VIIIa. APC is classified as a serine protease since it contains a residue of serine in its active site. In humans, protein C is encoded by the PROC gene, which is found on chromosome 2. The zymogenic form of protein C is a vitamin K-dependent glycoprotein that circulates in blood plasma. Its structure is that of a two-chain polypeptide consisting of a light chain and a heavy chain connected by a disulfide bond. The protein C zymogen is activated when it binds to thrombin, another protein heavily involved in coagulation, and protein C’s activation is greatly promoted by the presence of thrombomodulin and endothelial protein C receptors (EPCRs).
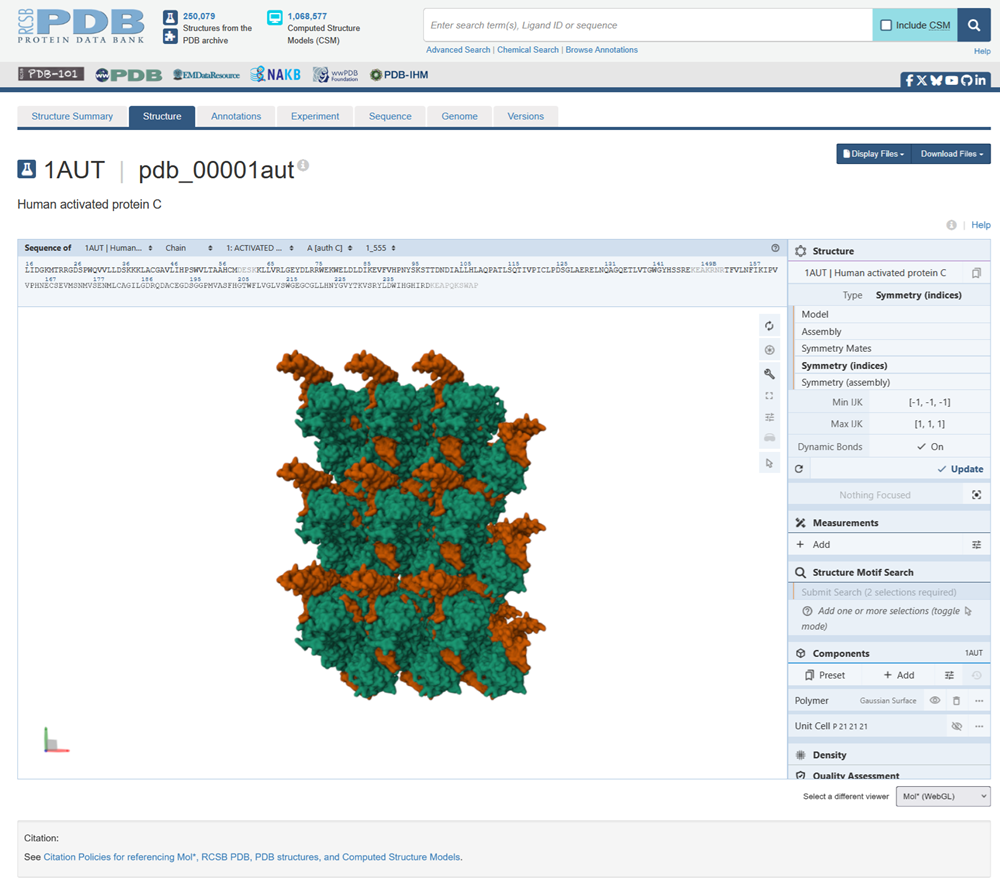
This is the structure with symmetry indices. I exported the animation on the rcsb website and rendered it too. Then i downloaded the mp4 and converted it into a gif.
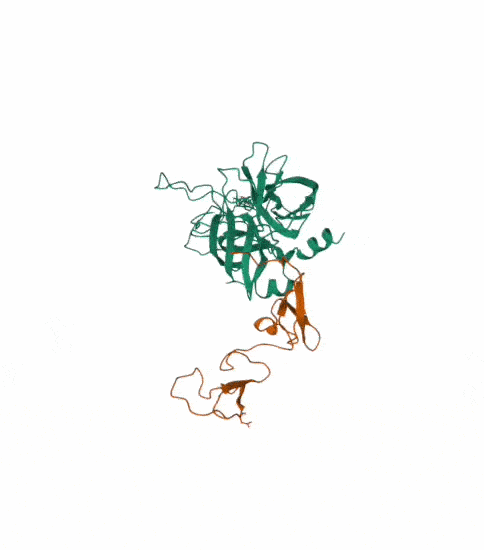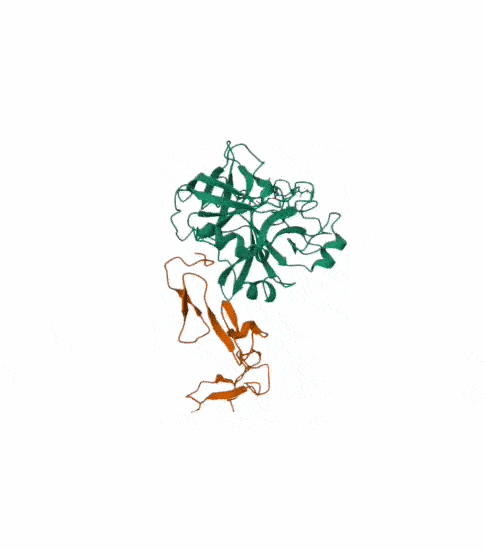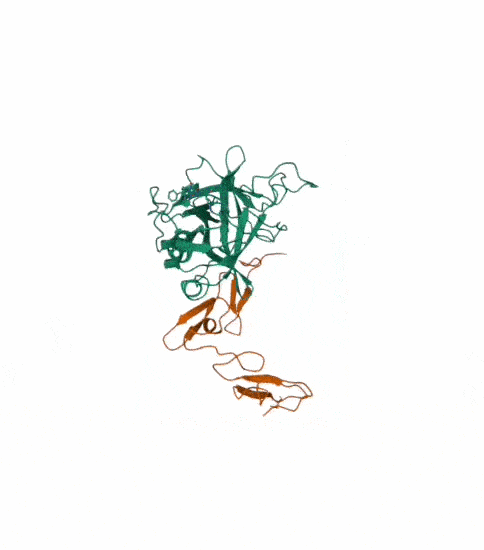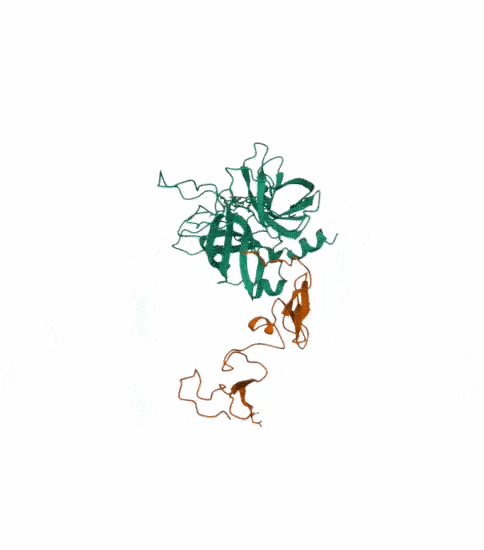
I also made another animation by choosing the unwind assembly under the export animation section on the right hand side.

Some more details about c protein
On uniprots website, Vitamin K-dependent protein C.
And this is the canonical sequence!
sp|P04070|PROC_HUMAN Vitamin K-dependent protein C OS=Homo sapiens OX=9606 GN=PROC PE=1 SV=1
MWQLTSLLLFVATWGISGTPAPLDSVFSSSERAHQVLRIRKRANSFLEELRHSSLERECI EEICDFEEAKEIFQNVDDTLAFWSKHVDGDQCLVLPLEHPCASLCCGHGTCIDGIGSFSC DCRSGWEGRFCQREVSFLNCSLDNGGCTHYCLEEVGWRRCSCAPGYKLGDDLLQCHPAVK FPCGRPWKRMEKKRSHLKRDTEDQEDQVDPRLIDGKMTRRGDSPWQVVLLDSKKKLACGA VLIHPSWVLTAAHCMDESKKLLVRLGEYDLRRWEKWELDLDIKEVFVHPNYSKSTTDNDI ALLHLAQPATLSQTIVPICLPDSGLAERELNQAGQETLVTGWGYHSSREKEAKRNRTFVL NFIKIPVVPHNECSEVMSNMVSENMLCAGILGDRQDACEGDSGGPMVASFHGTWFLVGLV SWGEGCGLLHNYGVYTKVSRYLDWIHGHIRDKEAPQKSWAP
Part C1. Protein Language Modeling
I will use both the sequence of HPV 16 L1 protein for this and the C PROTEIN (let’s see if i manage! 0.0). I am having a lot of issues with my internet in the last 10 days due to the situation in Iran, Lebanon and here in Cyprus because of drone attacks on the british military bases.
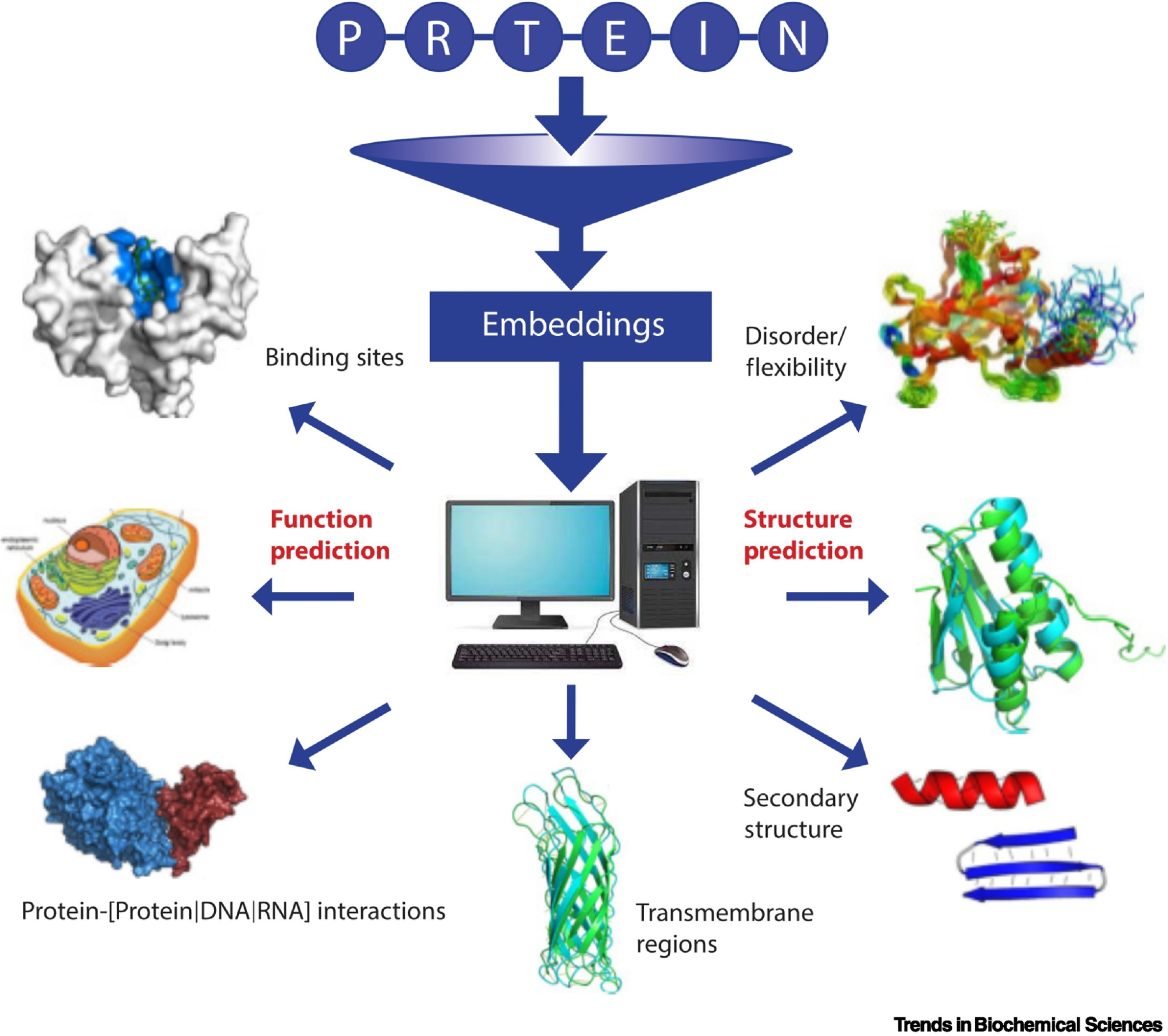
Picture Source: Bordin, Nicola et al (2023). Novel machine learning approaches revolutionize protein knowledge. Trends in Biochemical Sciences, Volume 48, Issue 4, 345 - 359
Here you can find my colab notebook!
Deep Mutational Scans
Deep mutational scan of hpv 16 l1.
Here is again the dna seq of hpv 16 l1 -
MSLWLPSEATVYLPPVPVSKVVSTDEYVARTNIYYHAGTSRLLAVGHPYFPIKKPNNNKILVPKVSGLQYRVFRIHLPDPNKFGFPDTSFYNPDTQRLVWACVGVEVGRGQPLGVGISGHPLLNKLDDTENASAYAANAGVDNRECISMDYKQTQLCLIGCKPPIGEHWGKGSPCTNVAVNPGDCPPLELINTVIQDGDMVDTGFGAMDFTTLQANKSEVPLDICTSICKYPDYIKMVSEPYGDSLFFYLRREQMFVRHLFNRAGAVGENVPDDLYIKGSGSTANLASSNYFPTPSGSMVTSDAQIFNKPYWLQRAQGHNNGICWGNQLFVTVVDTTRSTNMSLCAAISTSETTYKNTNFKEYLRHGEEYDLQFIFQLCKITLTADVMTYIHSMNSTILEDWNFGLQPPPGGTLEDTYRFVTSQAIACQKHTPPAPKEDPLKKYTFWEVNLKEKFSADLDQFPLGRKFLLQAGLKAKPKFTLGKRKATPTTSSTSTTAKRKKRKL
I am waiting for aaaaaages for the cells to excute -.-
I did run all and added the sequence in the sections like MUTATION SCANS and RUN ESM FOLD and then I got an error about hugging face. I did take a screnshot and forgot to save it (no comment!). It was something like get a token from hugging face to continue and something about a secret tab. No idea! I then signed up on hugging face because that was the response of the 1st or 2nd cell in the googlecolab file.
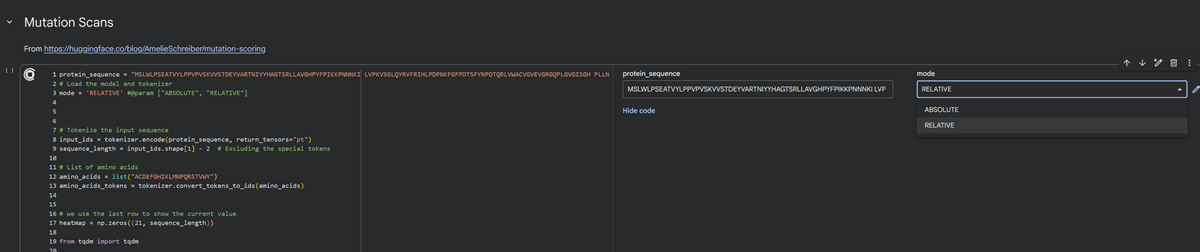
I have 2 modes to choose from for the deep mutational scan for my protein sequence- RELATIVE or ABSOLUTE. I chose RELATIVE.
Amelie Schreiber writes about Predicting the Effects of Mutations on Protein Function with ESM-2.
- Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
I noticed there are different models I can choose from. After reading a lot about latent space and encoding and transformers I can tell you that ESM stands for Evolutionary Scale Modeling are protein language models designed to design and predict 3d structures based on evolutionary information! We are using ESM2 models that are transformer based models that learn representation from massive protein sequence databases, which can be used to predict structure (ESMFold).
I used the model esm2_t6_8M_UR50D.
- Can you explain any particular pattern? (choose a residue and a mutation that stands out)
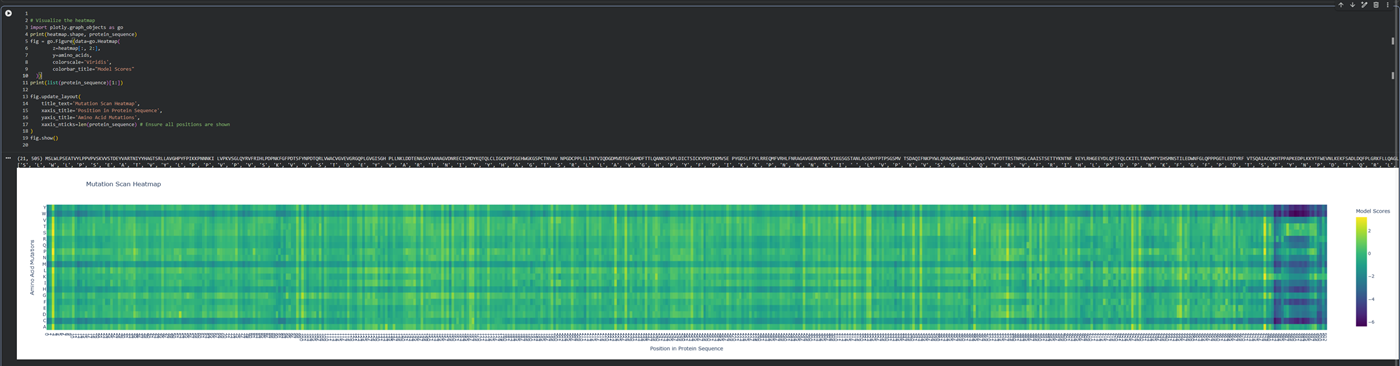
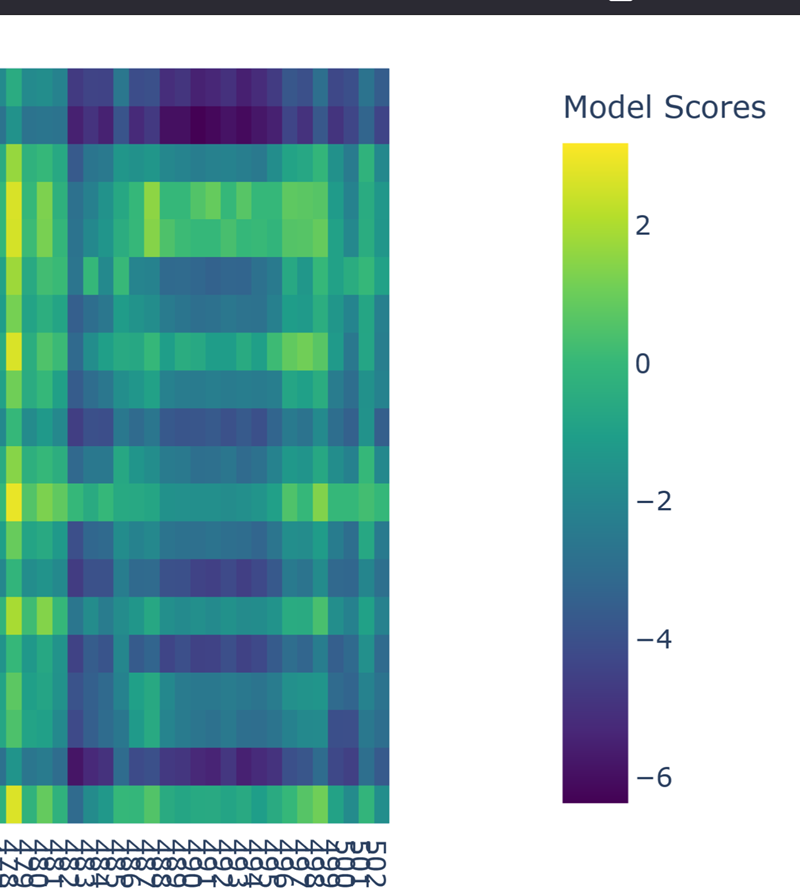
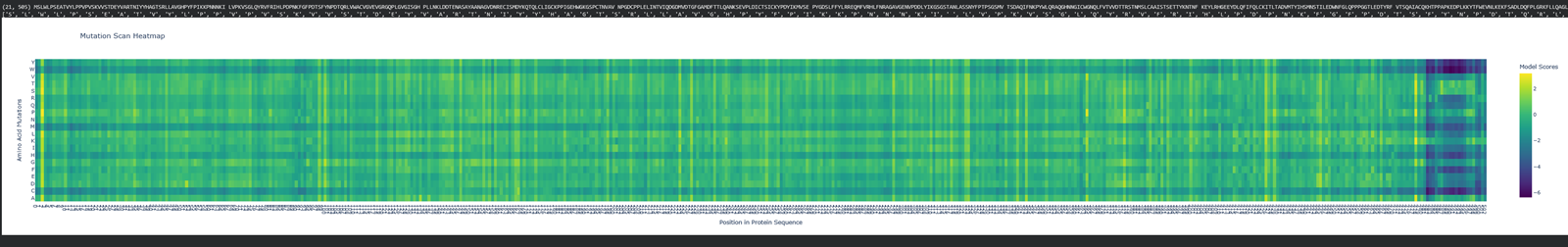
- (Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
I will see if I have extra time to answer this in the following day!
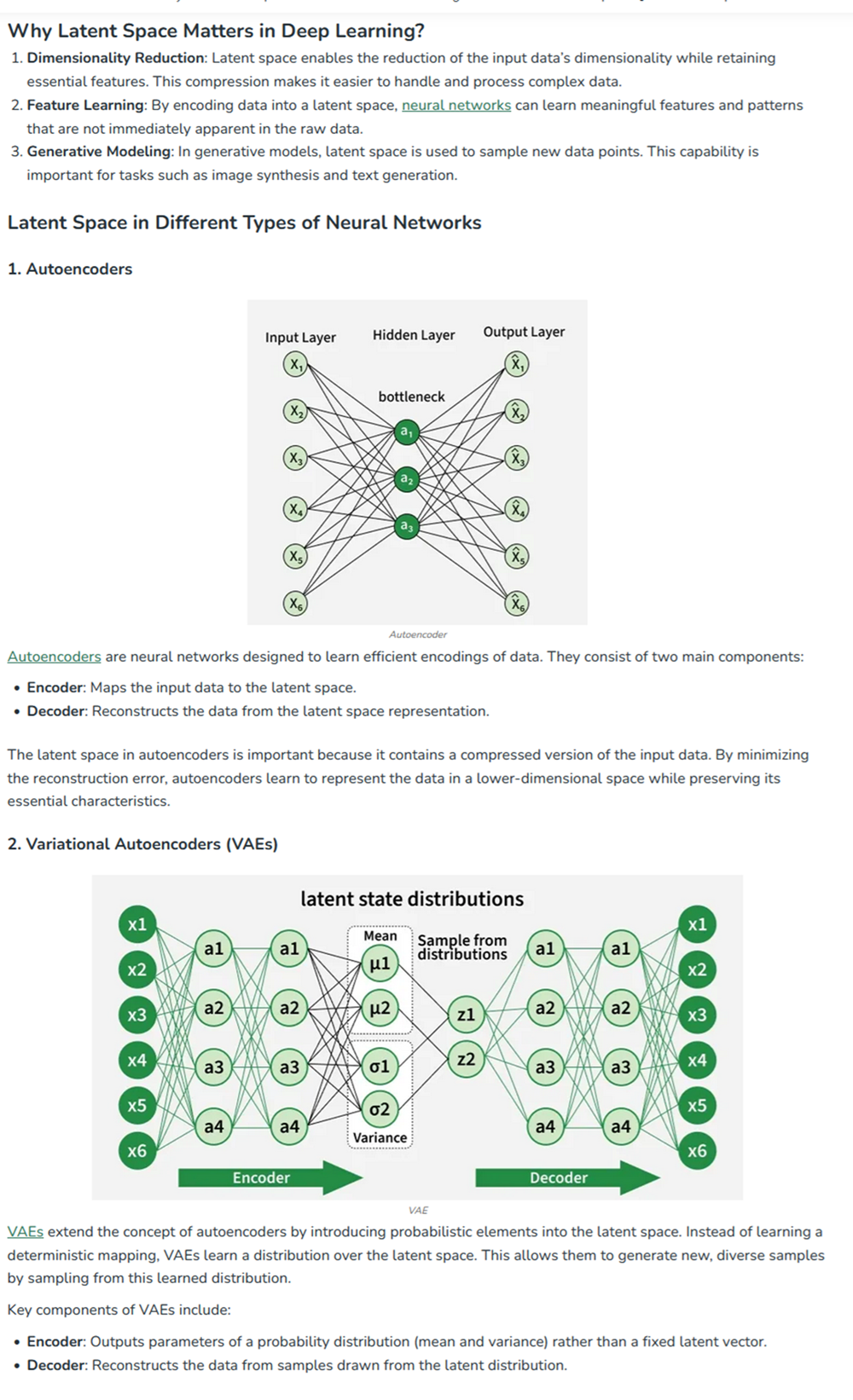
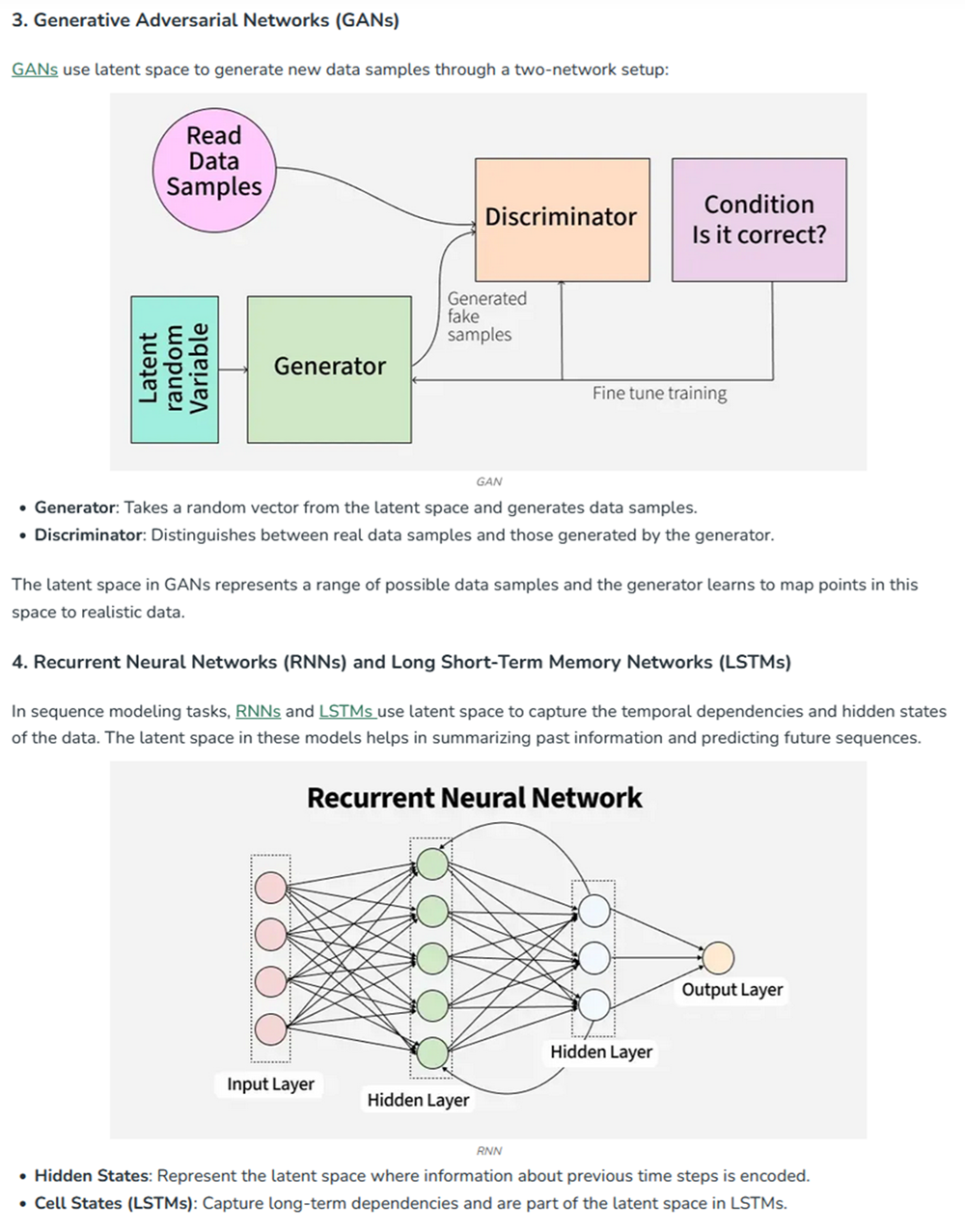
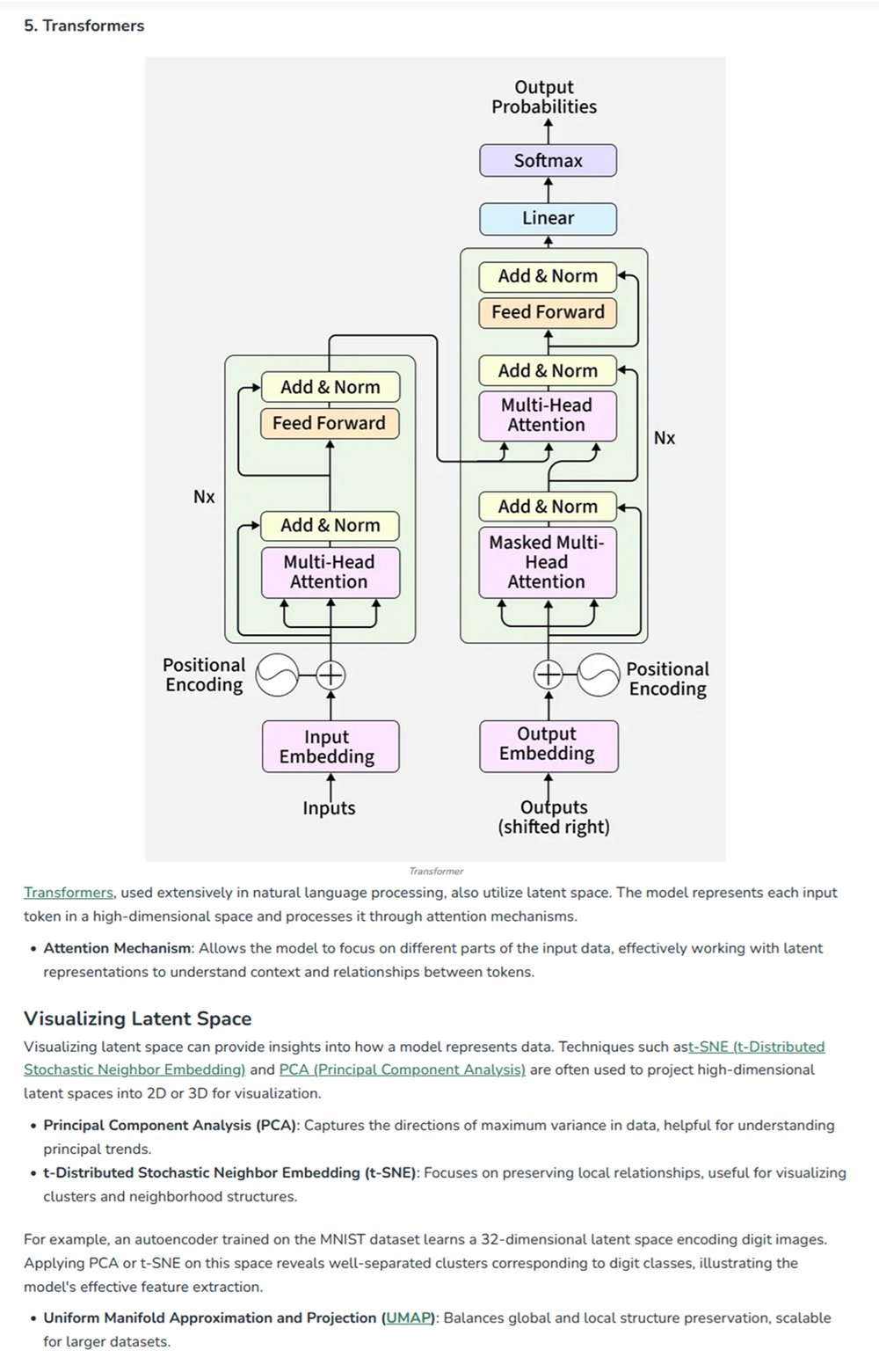
Latent Space Analysis
I did some ML and AI in 2020 in Berlin. I remember learning about latent space analysis and through HTGAA I am remembering things I learned in the past but connecting them to bio stuff, which is something that I honestly enjoy a lot but I feel I need a bit more time to experiment more and get used to these concepts again. I did some phython too as well as algorithmic botany. <3
I found this website that explains latent space in deep learning also.

At the end of cell 10 while its developing the 3d figure to show I got this message -> Shape of embeddings array before 3D t-SNE: (15177, 320) /usr/local/lib/python3.12/dist-packages/sklearn/manifold/_t_sne.py:1164: FutureWarning:’n_iter’ was renamed to ‘max_iter’ in version 1.5 and will be removed in 1.7.
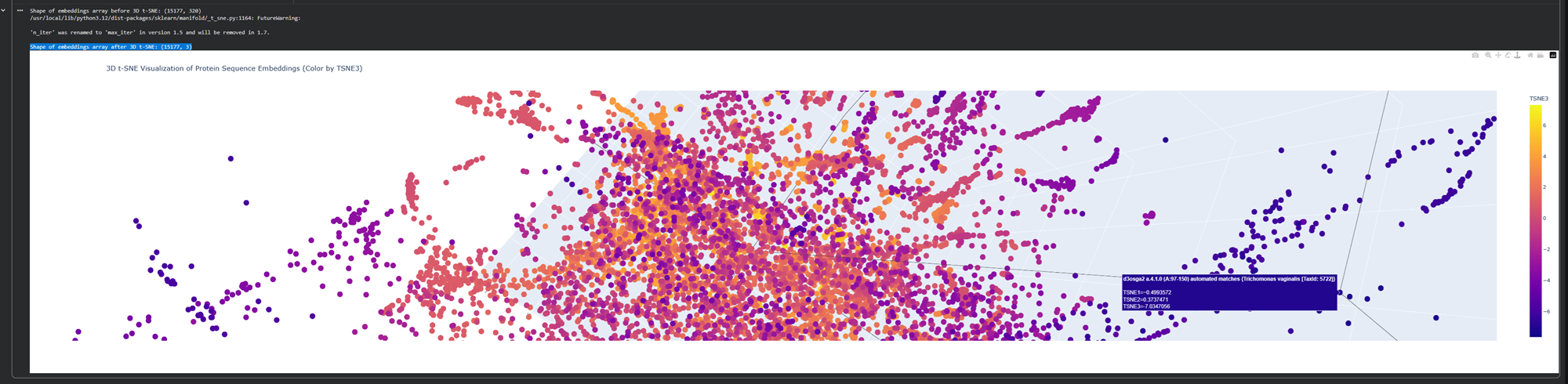
Shape of embeddings array after 3D t-SNE: (15177, 3)
Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins?
I explored the neighbooring organisms and added screenshots of similar protein structures to less similar. Found some interesting ones!
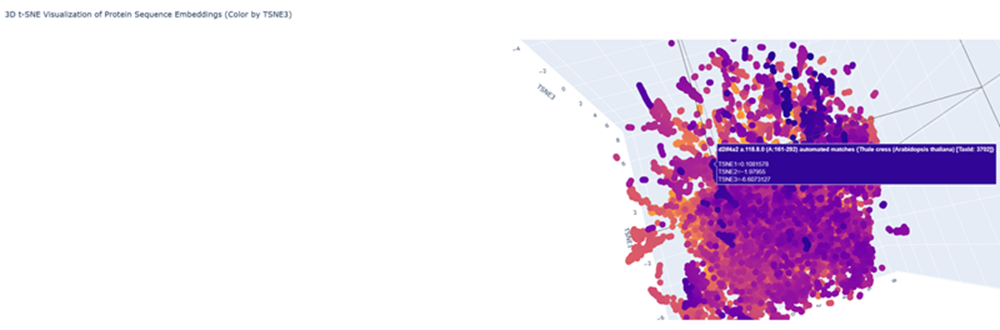
Here is an examples with less similar proteins!
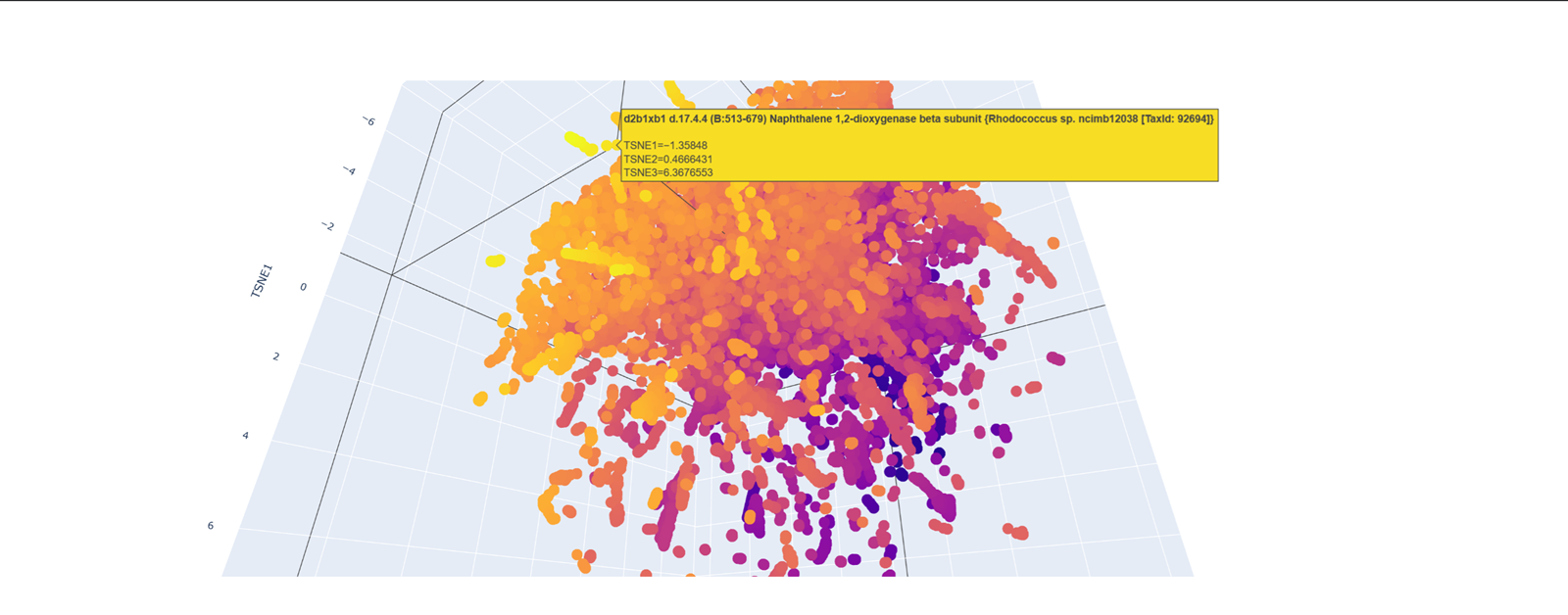
- Place your protein in the resulting map and explain its position and similarity to its neighbors.
Part C2. Protein Folding
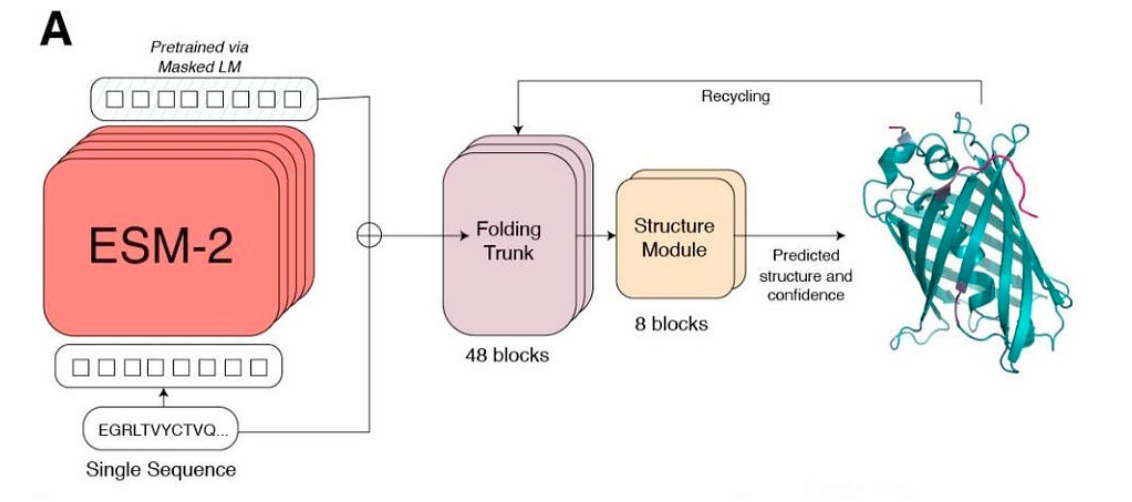
Picture Source: Lin et al (2023). Evolutionary-scale prediction of atomic-level protein structure with a language model.
Folding a protein
- Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
I managed to figure it out, I did not have enough space and was using the wrong memory option.
Total sequence length: 505
Running ESMFold inference for sequence with length 505...
Prediction complete. ptm: 0.120 plddt: 28.209
Results saved to test_18a92/
CPU times: user 1min 24s, sys: 8.67 s, total: 1min 33s
Wall time: 2min 10s
Display (optional)
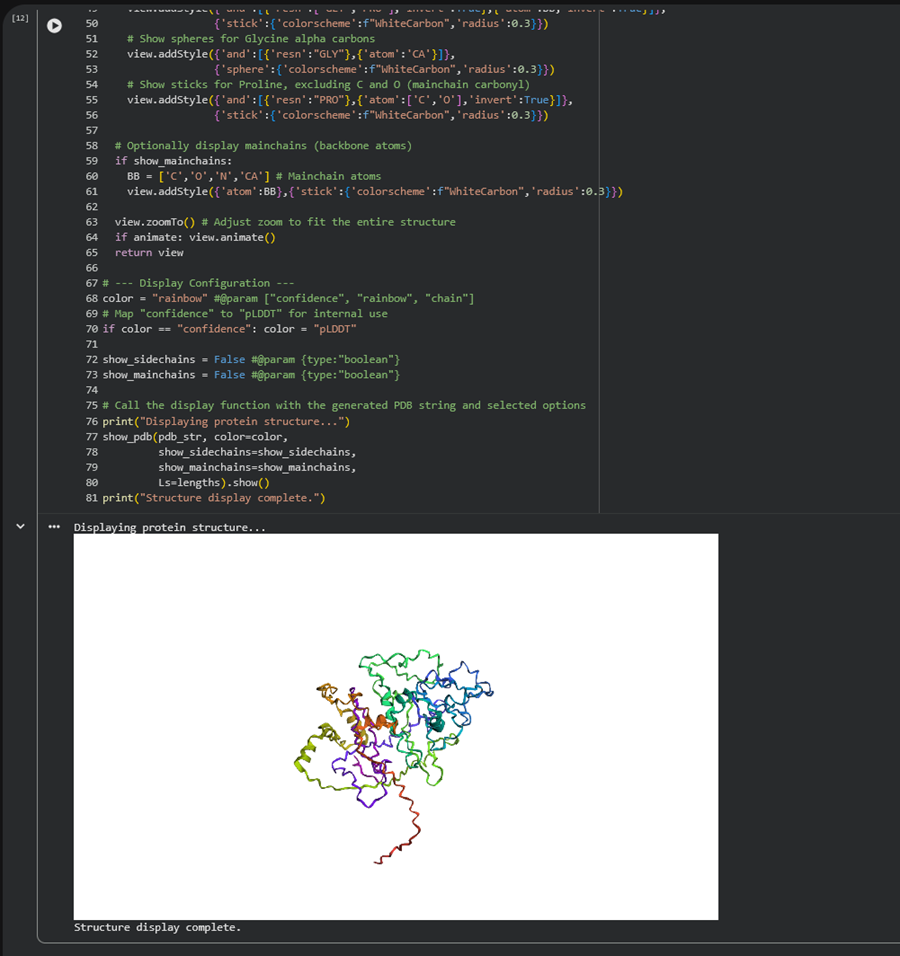

I made this gif by getting screenshots of my 3d model and then using photoshop I turned the pngs into a sequence and made an mp4. I then went to this website to turn the mp4 into a gif an optimize it!
- Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
Part C3. Protein Generation
Picture Source: 1. Post from Sergey Ovchinnikov 2. Roney, Ovchinnikov et al (2022). State-of-the-art estimation of protein model accuracy using AlphaFold. Phys. Rev. Lett. 129, 238101
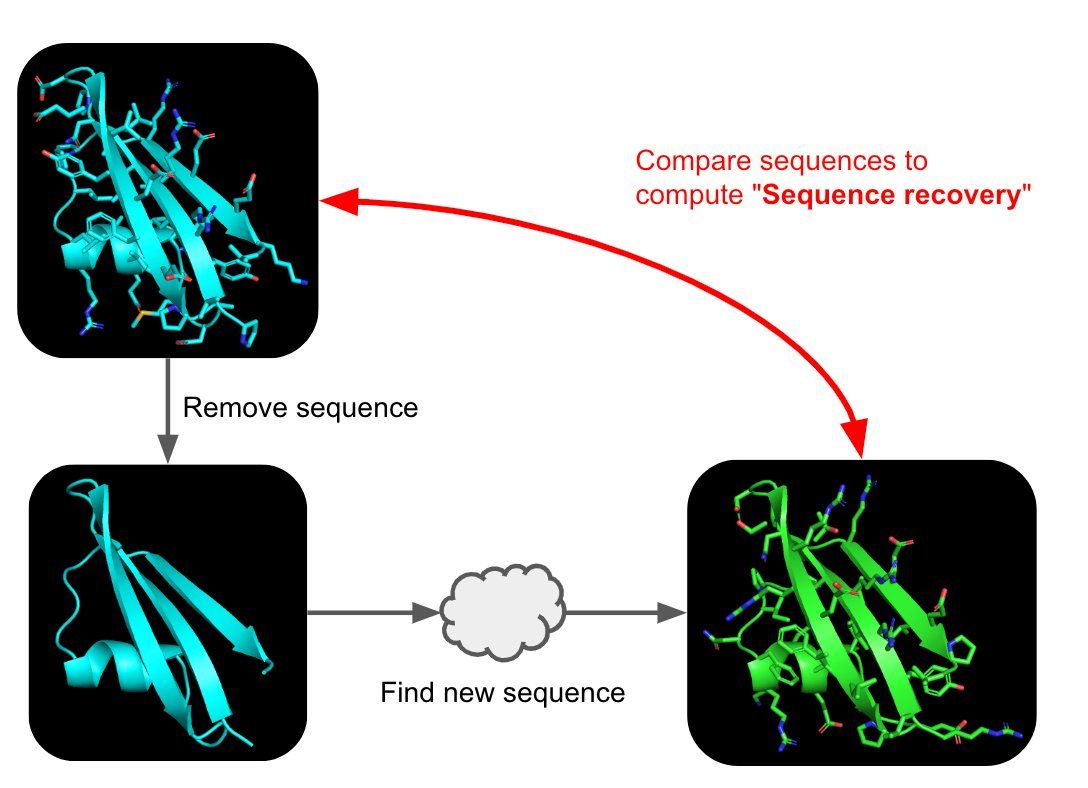
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
- Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
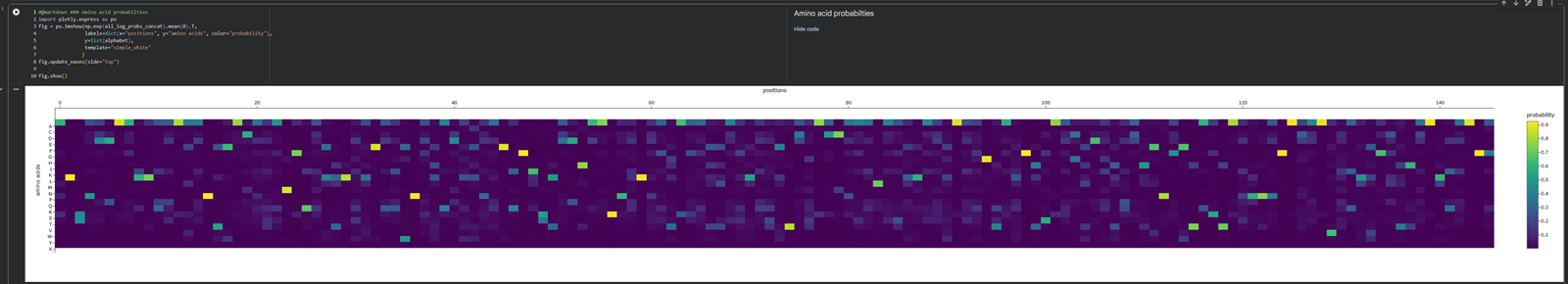
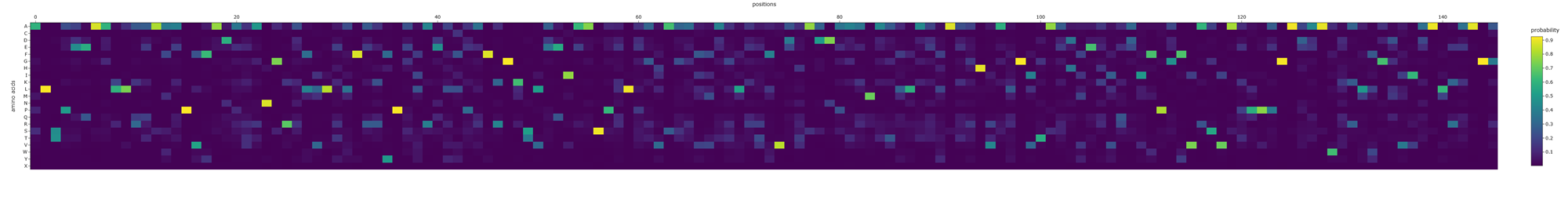
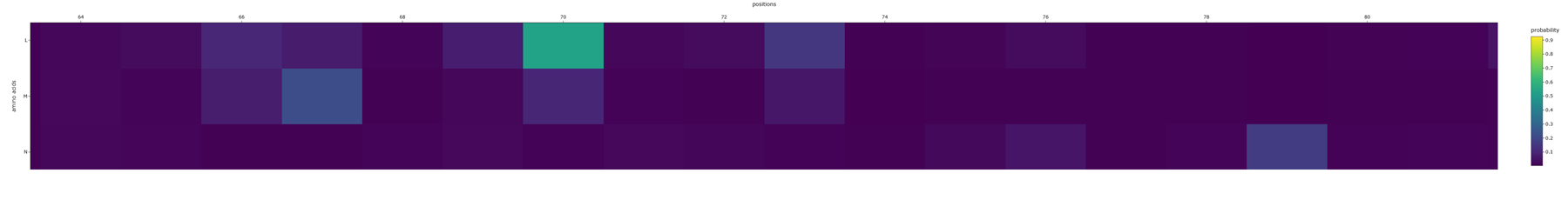
Aaaaand here is a better picture showing the position of the aminoacids and their probability :)
- Input this sequence into ESMFold and compare the predicted structure to your original.
EXTRA PYMOL PLAY FOR C PROTEIN
Ribbon
Surface
AAAnnnnnd I made another animation in pymol that I then converted to an optimized gif!
Part D. Group Brainstorm on Bacteriophage Engineering
1. Find a group of ~3–4 students
I formed a group with the following people in our node :)
and weee have a new member!
2. Read through the Phage Reading material listed under “Reading & Resources” below.
3. Review the Bacteriophage Final Project Goals for engineering the L Protein:
Increased stability (easiest)
Higher titers (medium)
Higher toxicity of lysis protein (hard)
4. Brainstorm Session
+ Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”).
+ Write a 1-page proposal (bullet points or short paragraphs) describing:
Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
+ Why do you think those tools might help solve your chosen sub-problem?
+ Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”).
+ Include a schematic of your pipeline.
+ Each individually put your plan on your HTGAA website.
+ Include your group’s short plan for engineering a bacteriophage
This resource may be useful: HTGAA Protein Engineering Tools.
5. Each individually put your plan on your HTGAA website
+ Include your group’s short plan for engineering a bacteriophage
