Week 5 HW: PROTEIN DESIGN PART II

Part A: SOD1 Binder Peptide Design (From Pranam)
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Your challenge:
Design short peptides that bind mutant SOD1.
Then decide which ones are worth advancing toward therapy.
You will use three models developed in our lab:
PepMLM: target sequence-conditioned peptide generation via masked language modeling.
PeptiVerse: therapeutic property prediction.
moPPIt: motif-specific multi-objective peptide design using Multi-Objective Guided Discrete Flow Matching (MOG-DFM).
Part 1: Generate Binders with PepMLM
Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
You can find the uniprot link here!
>sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS
AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV
HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
So in the mutant, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
SOD1SEQ WITH HIGHLIGHTED ALANINE A4V
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
V Mutated SOD1 Seq
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
Here you can find my colabnotebook!
- Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
| Binder | Pseudo Perplexity |
|---|---|
| SWYDVVAGVYKAAAK | 8.742062391716917 |
| WWYGELVAVVKARAX | 14.3777905642455 |
| WRWPVYAGVKAAARK | 7.102400937372668 |
| SWWPELAGRKKWRRK | 21.326166626669973 |
Unfortunately my connection was timed out and I had to run the cells again and I generated another 4 binders so I am going to be using these to compare the custom binder below as control!
| Binder | Pseudo Perplexity |
|---|---|
| WWYPVYAGVVALKKK | 13.334819029505825 |
| SWYDPYVAVVKAKAK | 13.884064880953778 |
| ARWDPYAARKKWARX | 20.375727062709792 |
| WWYPEYVVVVELKKK | 26.13860234017482 |
- To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
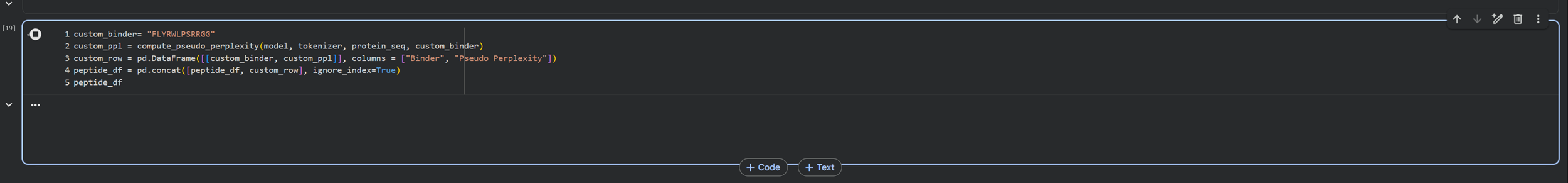
So I assume we are using FLYRWLPSRRGG as control of our experiment. So we need to find a way to add to the code and add our peptide to the list and to get the perplexity score.
I checked out Diogos documentation for and I noticed he asked Gemini to help him with this and i did the same!
Here is the code that gemini helped me write in order to introduce a custom binder into the mix! In all honesty, Diogos documentation was right haha.
And we have generated the custom binder and a table for comparison :)
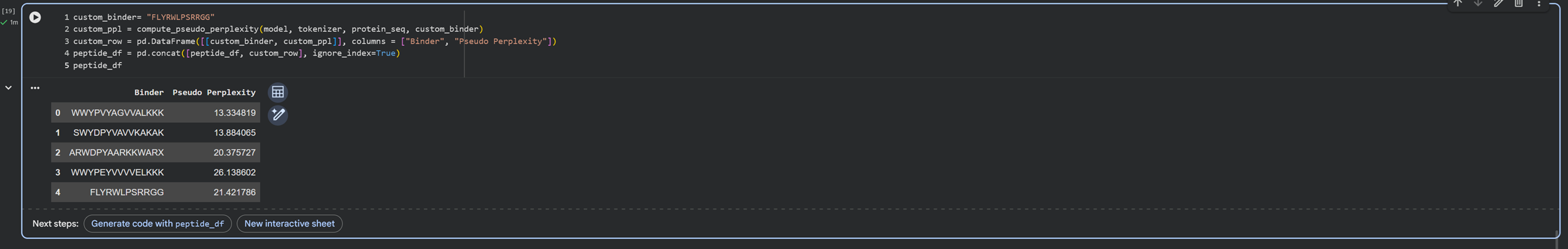
Buuuuut I notice that the additional binder only has 12 peptides instead of 15 like the binders I chose to generate. So I will run this again so I can generate the same length peptides as the control/custom binder!
Here we go again -.-
| Binder | Pseudo Perplexity |
|---|---|
| WRVPAVAVRHKK | 12.133492 |
| WHYYAAAVRLWE | 20.173057 |
| WRVYPVAVEWKK | 18.690124 |
| WRVGAAGVAWKX | 8.172350 |
I run the last code cell I made again to get the value of the custom binder in relation to the binders generated previously.
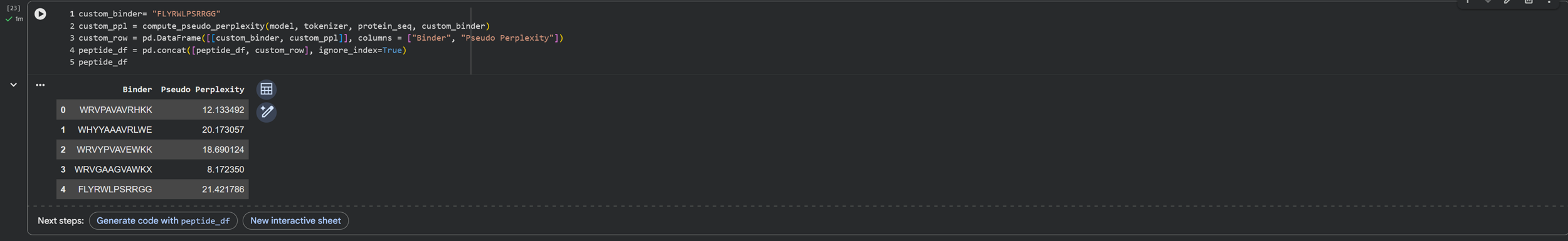
- Record the perplexity scores that indicate PepMLM’s confidence in the binders.
HuggingFace PepMLM-650M MODEL CARD HERE.
Part 2: Evaluate Binders with AlphaFold3
+Navigate to the AlphaFold Server: alphafoldserver.com
+For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
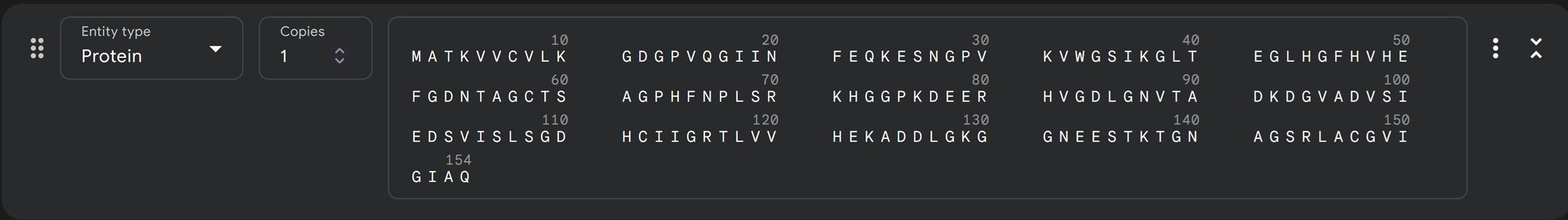
I added the mutant SOD1 sequence and now I am attempting to add the separate chains to model the protein-peptide complex!
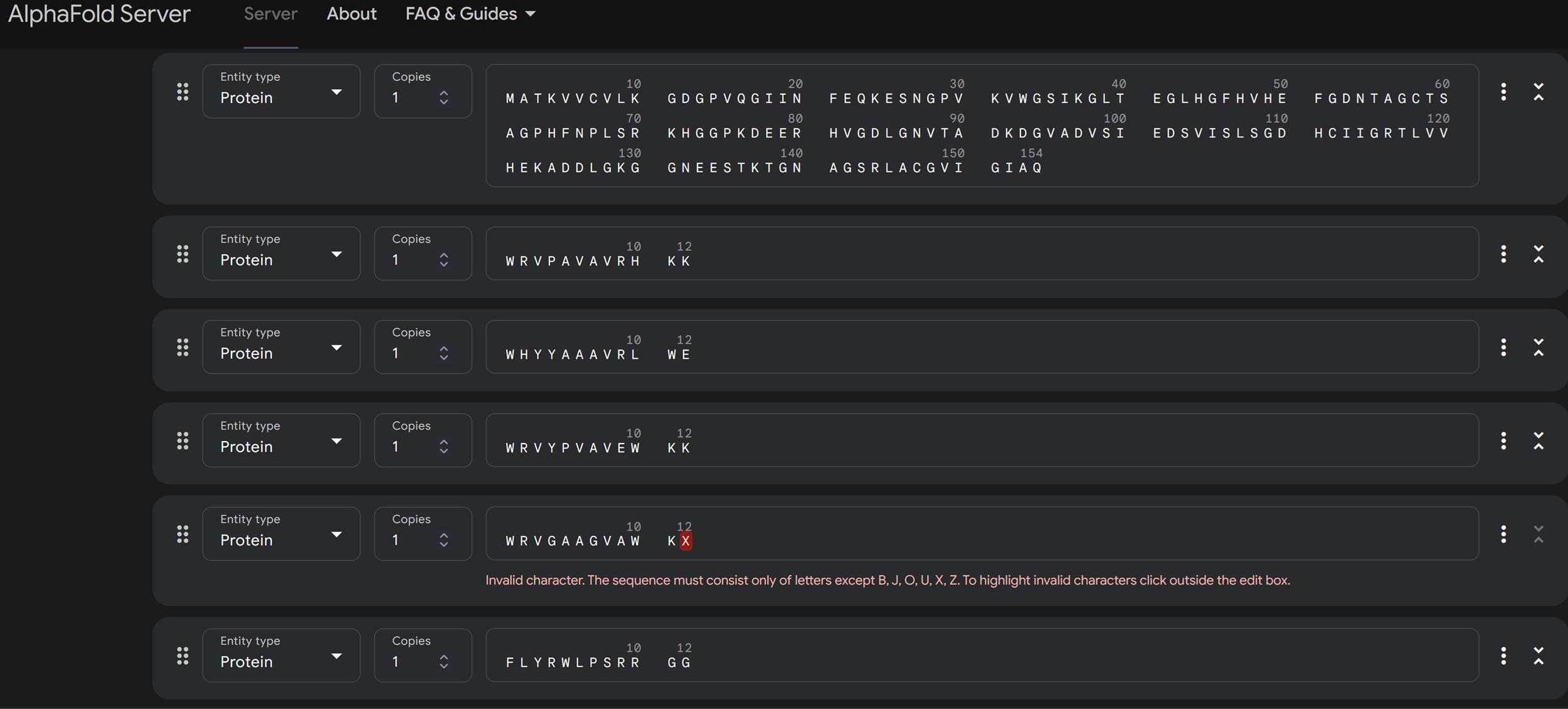
I have no idea if I am doing this right and I already got an error as you can see in the image above regarding the peptide residue X of the peptide generated WRVGAAGVAWKX. I then scrolled below or above and read the FAQ’s.
What should I do if I have unknown residues or nucleotides in my protein, DNA or RNA sequence? AlphaFold Server was not designed to model unknown residues or nucleotides (e.g. X for the unknown residues and N for unknown nucleotides). Please substitute by one of the standard residues or nucleotides that is appropriate for your particular case. In general, consider following substitutions:
-Proteins: replace unknown protein residues with alanine (A)
-DNA: replace unknown nucleotides by poly-T (T), but other nucleotides are also suitable
-RNA: replace unknown nucleotides by poly-U (U), but other nucleotides are also suitable
Based on this information i substitute X with A -> WRVGAAGVAWKX to WRVGAAGVAWKA!

Next steps!
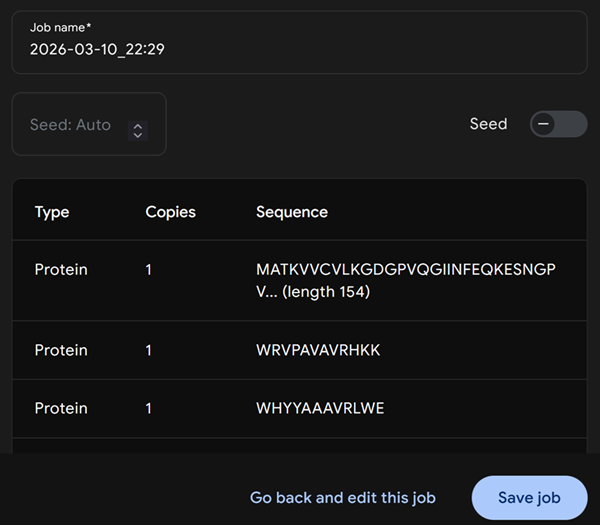
Then continue and preview job but this is what I got.
I then realised I have to run the mutant sod1 sequence and each peptide separately and run each job separately otherwise when i run all peptides together with the sequence this is what i get.
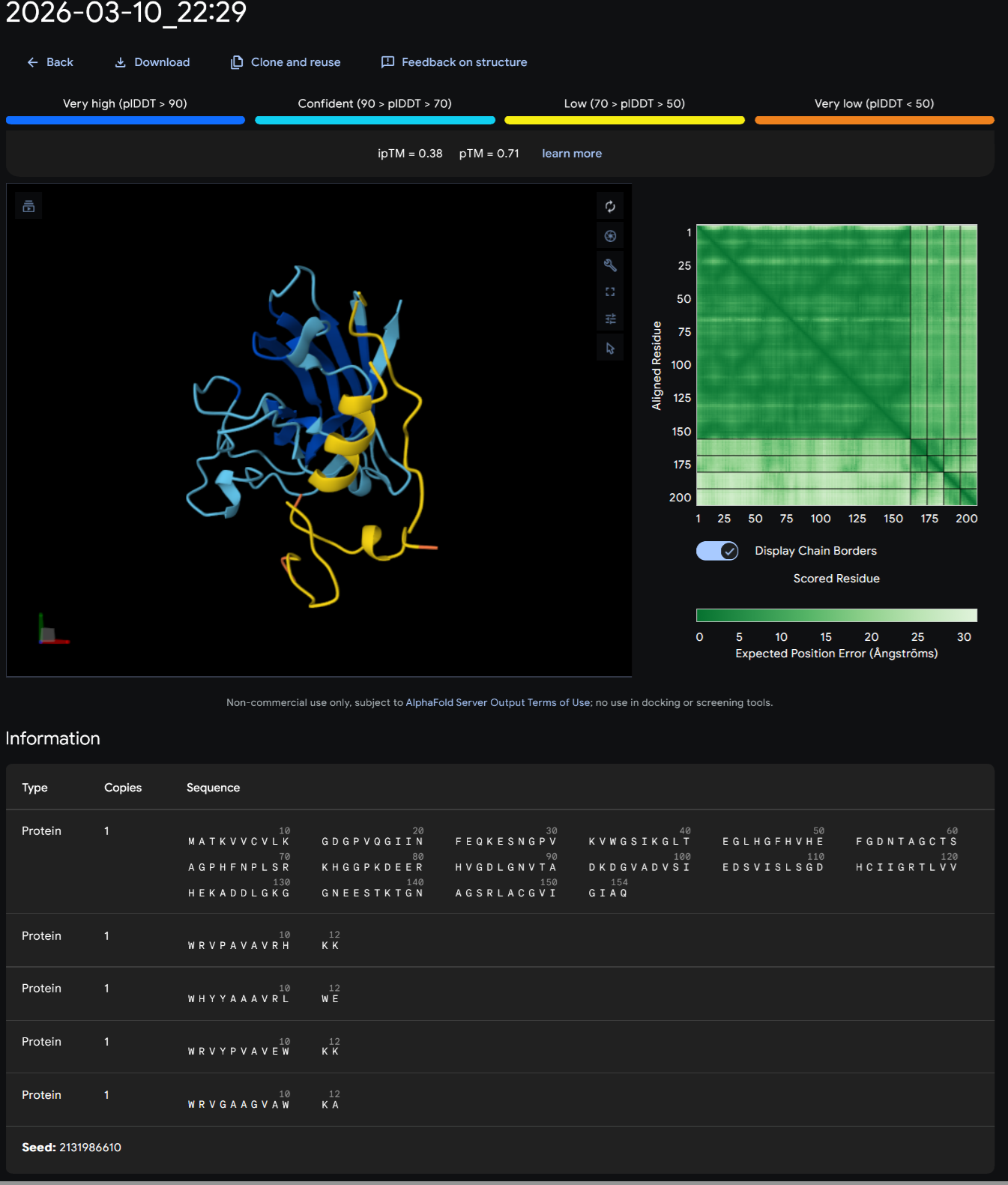
CONTROL PEPTIDE- FLYRWLPSRRGG
I continued by running 5 different jobs for all my different peptides and I started from the control peptide FLYRWLPSRRGG.
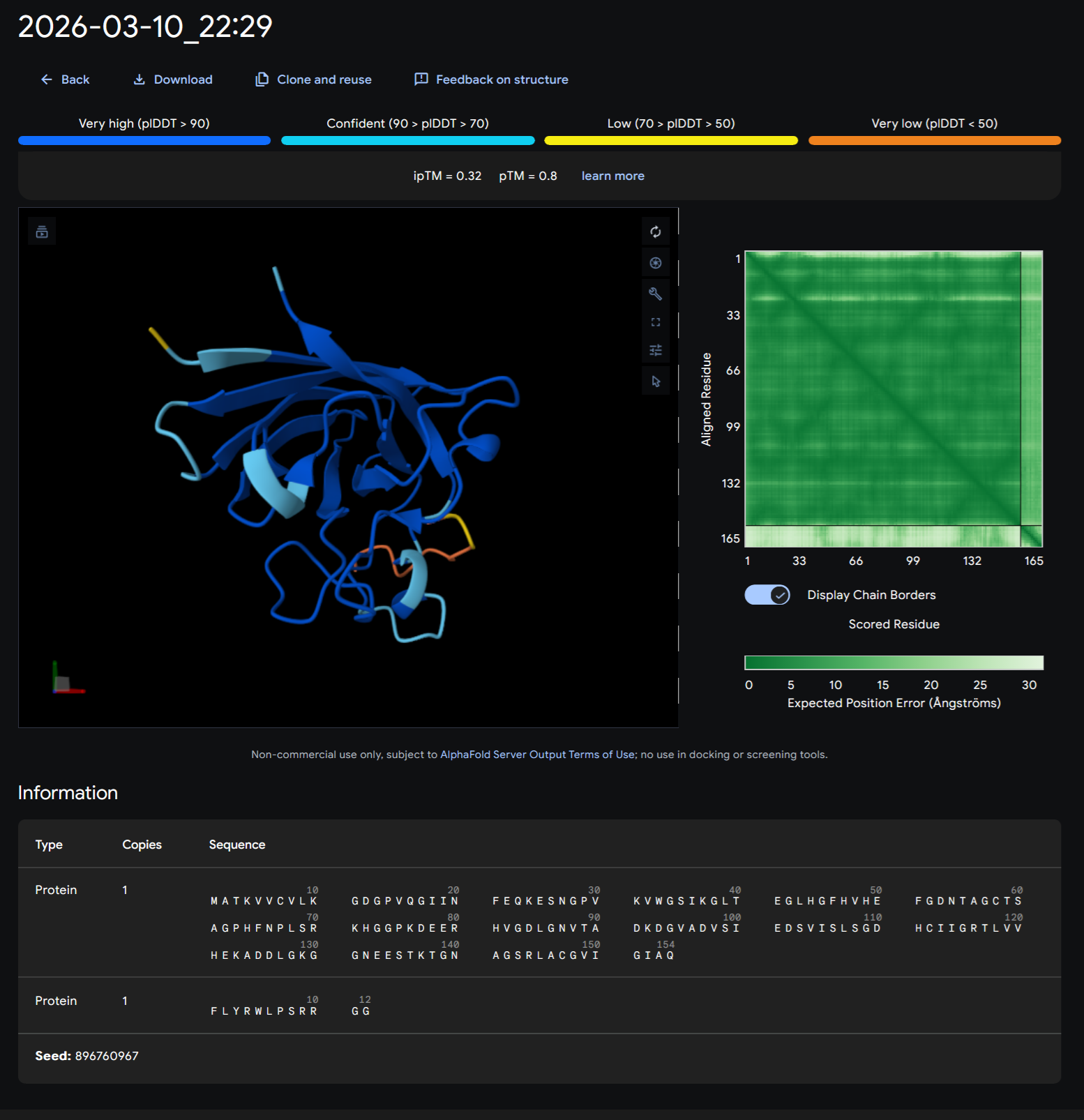
PEPTIDE 1-WRVPAVAVRHKK
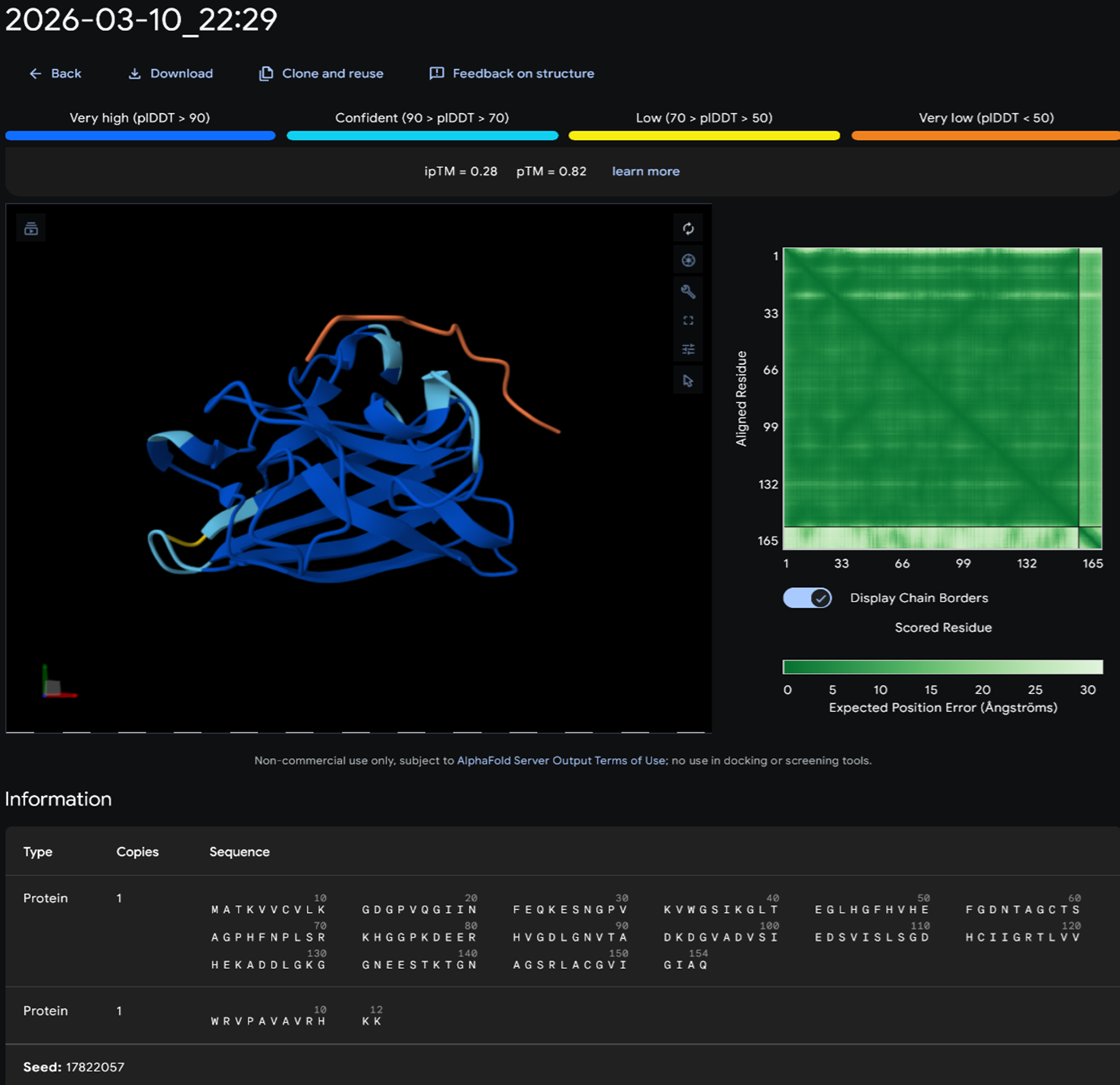
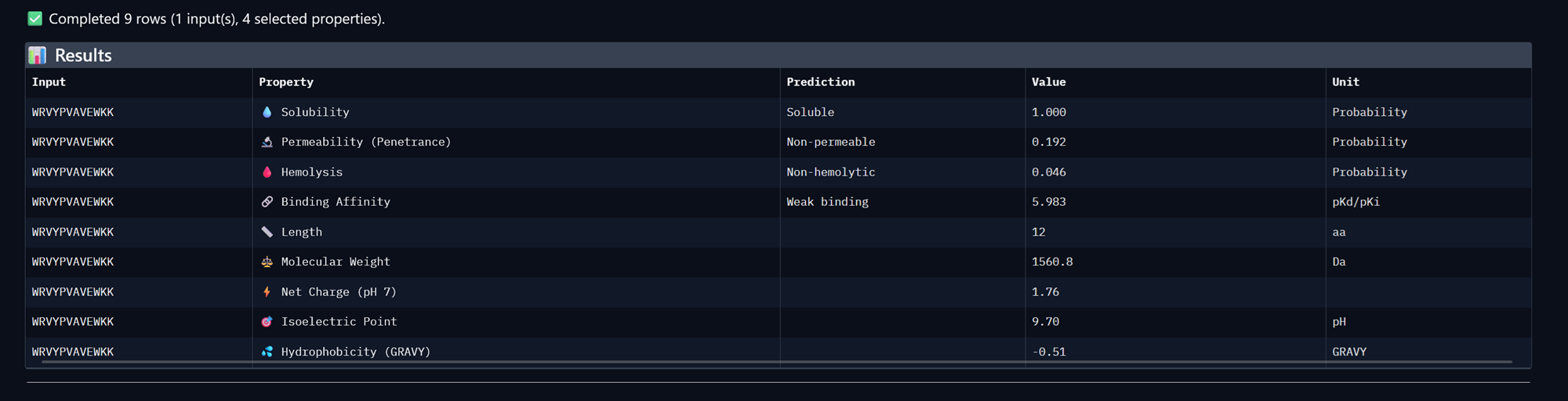
PEPTIDE 2-WHYYAAAVRLWE
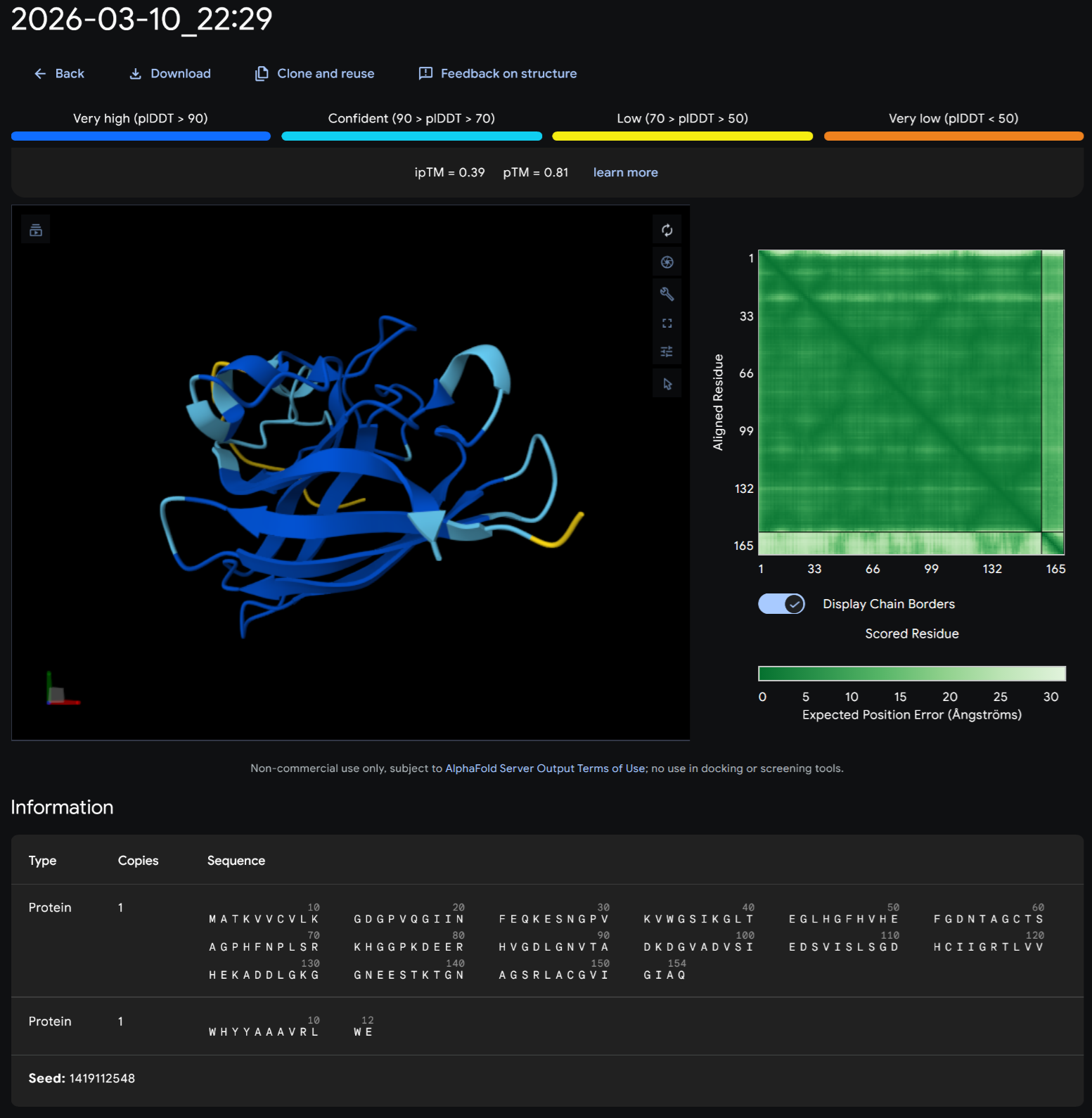
PEPTIDE 3-WRVYPVAVEWKK
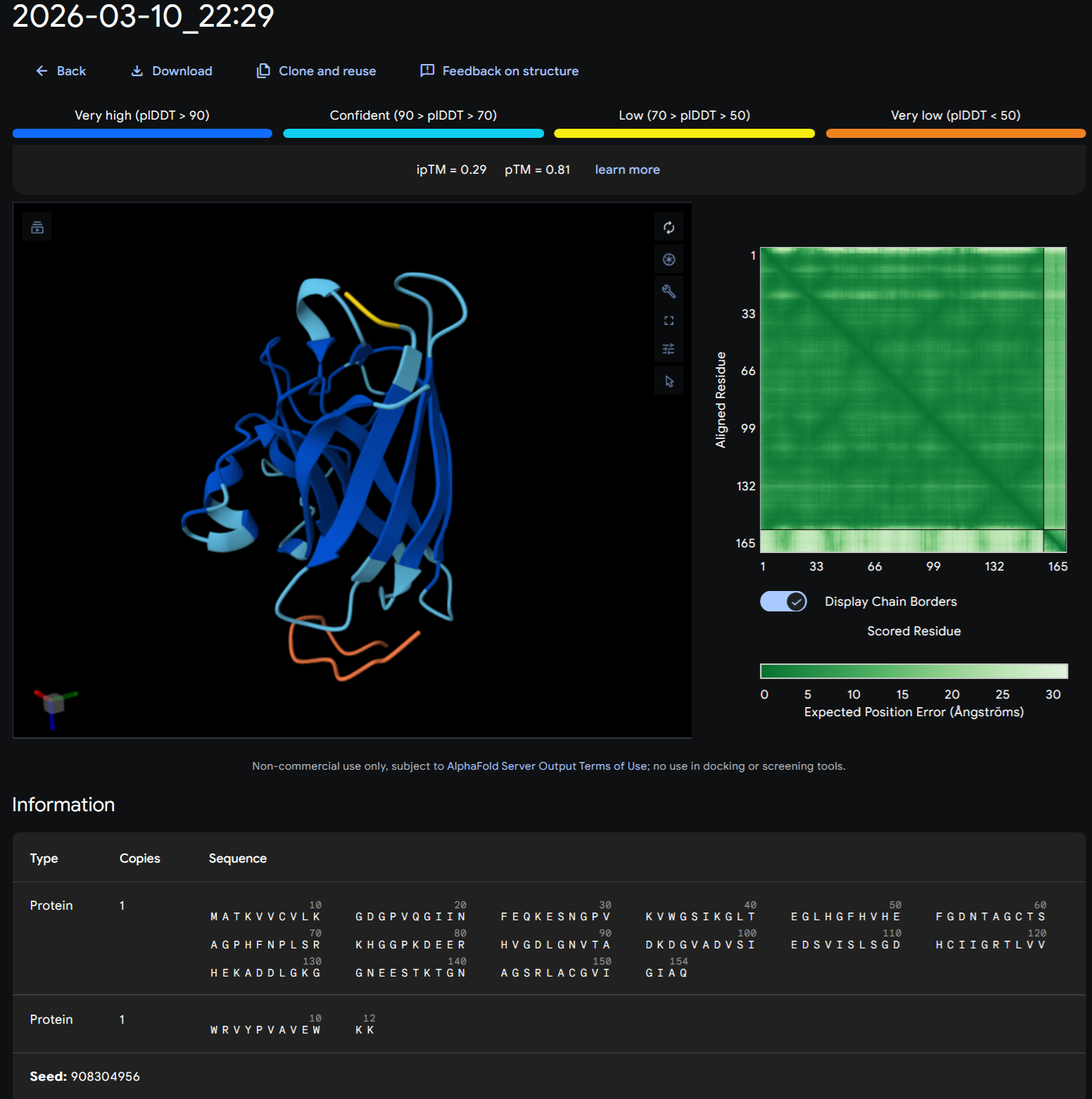
PEPTIDE 4-WRVGAAGVAWKX to WRVGAAGVAWKA
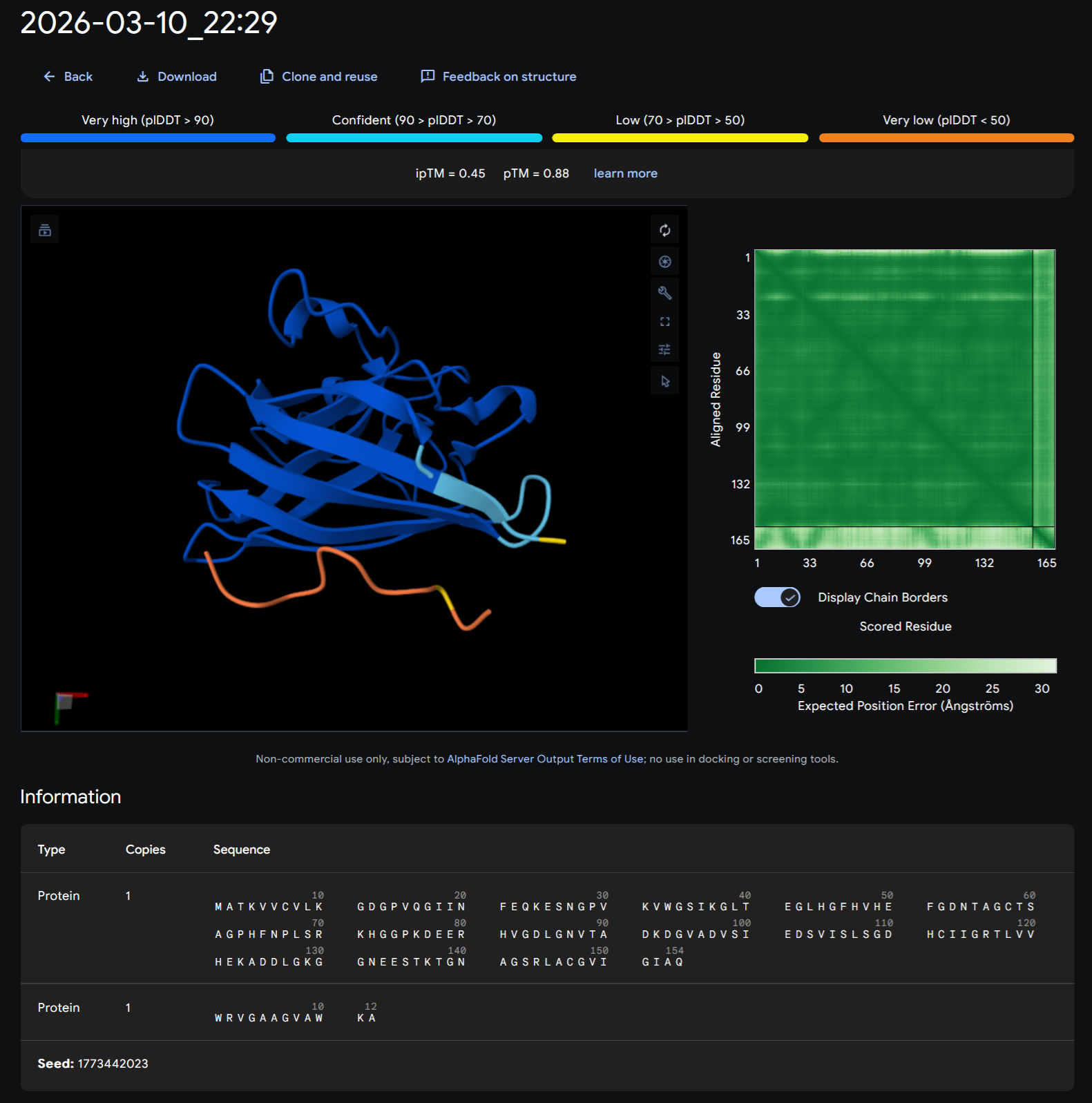
+Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
About the ipTM score:
From ebi.ac.uk.
Here are the scores:
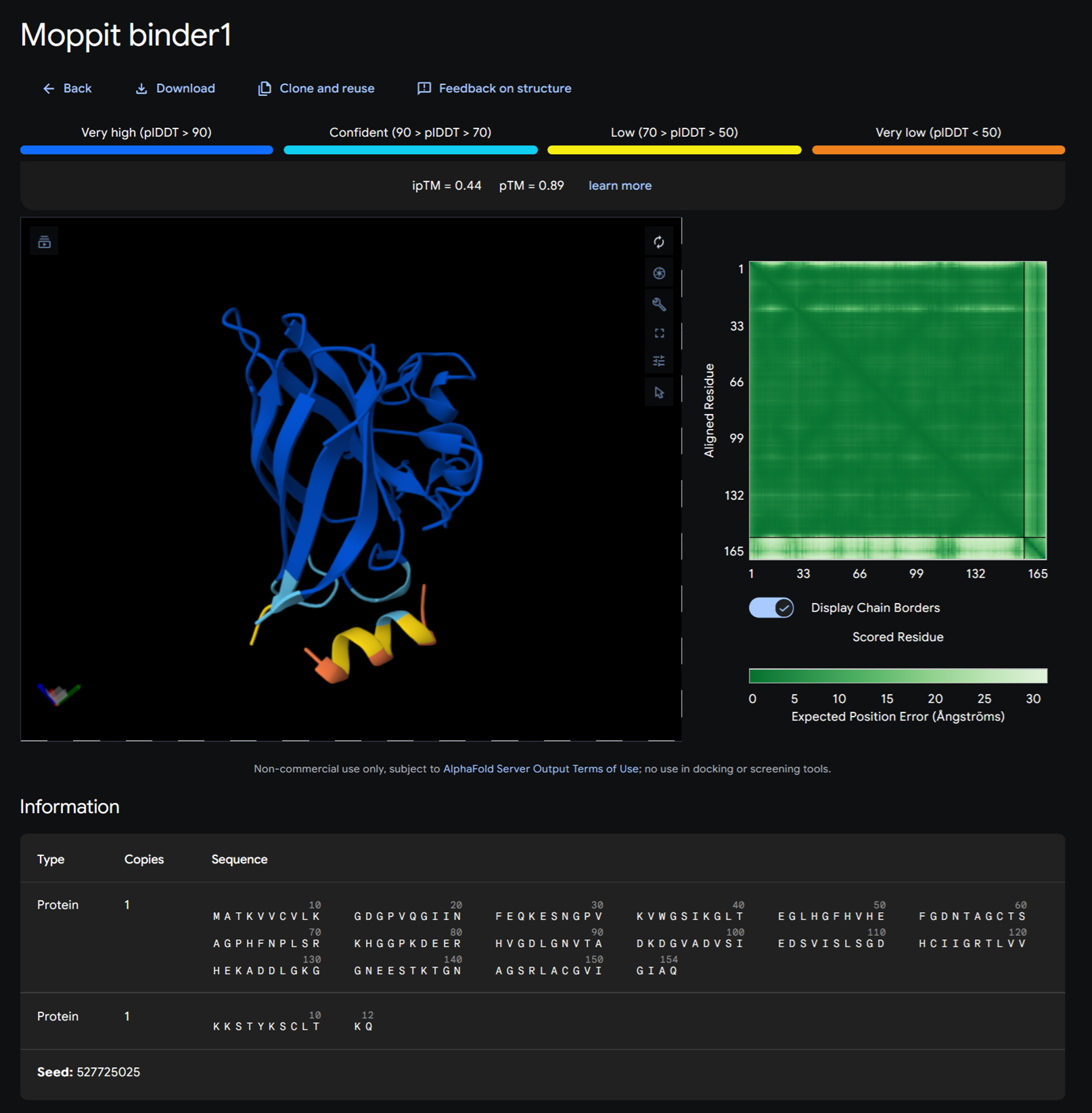
| # | Peptide | iptM |
|---|---|---|
| 0 | FLYRWLPSRRGG | 0.32 |
| 1 | WRVPAVAVRHKK | 0.28 |
| 2 | WHYYAAAVRLWE | 0.39 |
| 3 | WRVYPVAVEWKK | 0.29 |
| 4 | WRVGAAGVAWKA | 0.45 |
As you can see from the visuals generated above in alphafold and the iptM scores above of the control peptide and generated peptides have an iptM score under 0.6 that suggests failed prediction.
| Peptide | Binding info |
|---|---|
| FLYRWLPSRRGG | Peptide does not bind-floats away from the protein and not engaging with the N-terminus, the beta barrel or the Dimer Interface |
| WRVPAVAVRHKK | Peptide does not bind-floats away from the protein and not engaging with the N-terminus, the beta barrel or the Dimer Interface |
| WHYYAAAVRLWE | Peptide does not bind-floats away from the protein and not engaging with the N-terminus, the beta barrel or the Dimer Interface |
| WRVYPVAVEWKK | Peptide does not bind-floats away from the protein and not engaging with the N-terminus, the beta barrel or the Dimer Interface |
| WRVGAAGVAWKA | Peptide does not bind-floats away from the protein and not engaging with the N-terminus, the beta barrel or the Dimer Interface |
+In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
WRVGAAGVAWKA with an iptM score of 0.45 is the only peptide generated that exceeds the known binder FLYRWLPSRRGG.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
For Peptiverse go here.
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
+Paste the peptide sequence.
+Paste the A4V mutant SOD1 sequence in the target field.
+Check the boxes
Predicted binding affinity
Solubility
Hemolysis probability
Net charge (pH 7)
Molecular weight
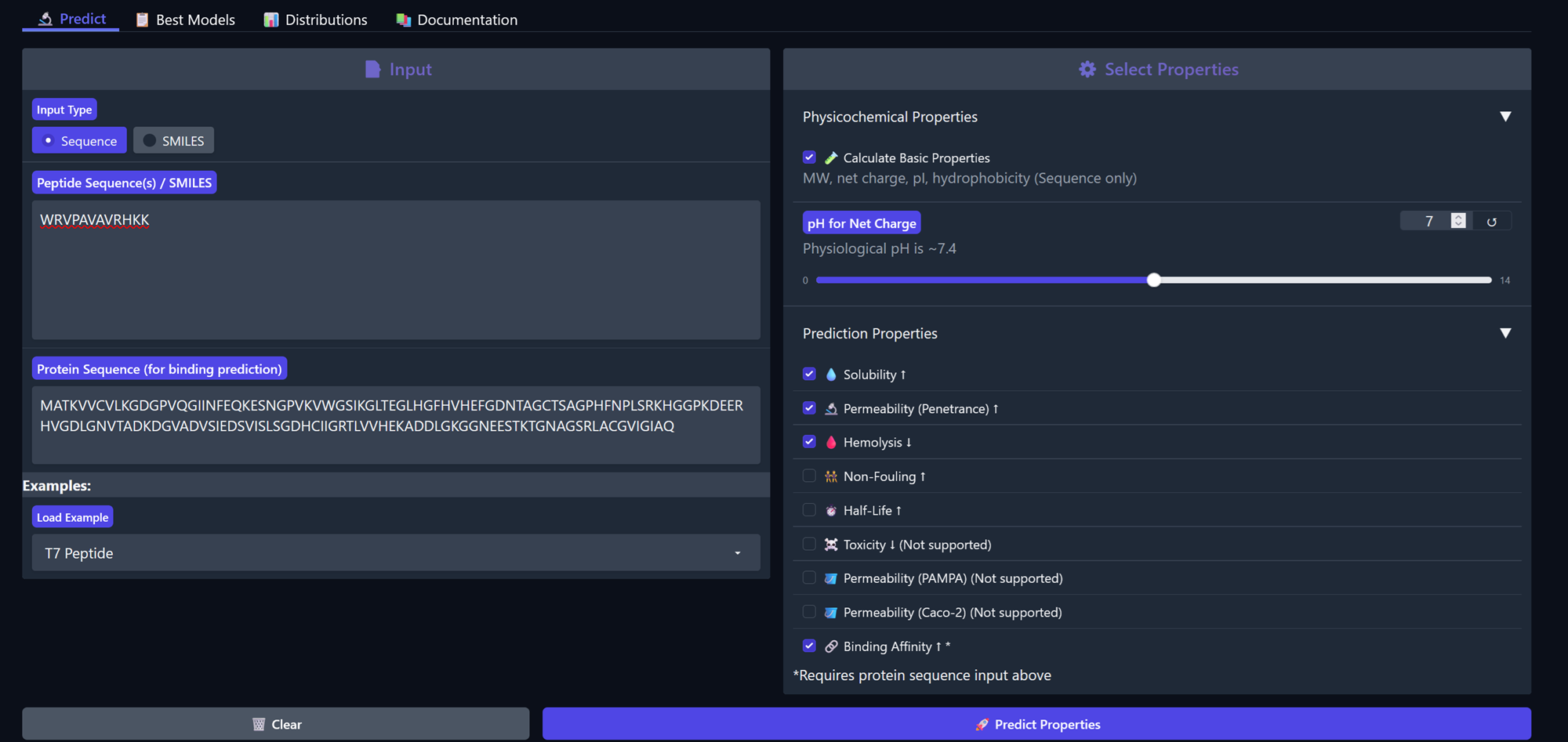
Peptide 1, WRVPAVAVRHKK
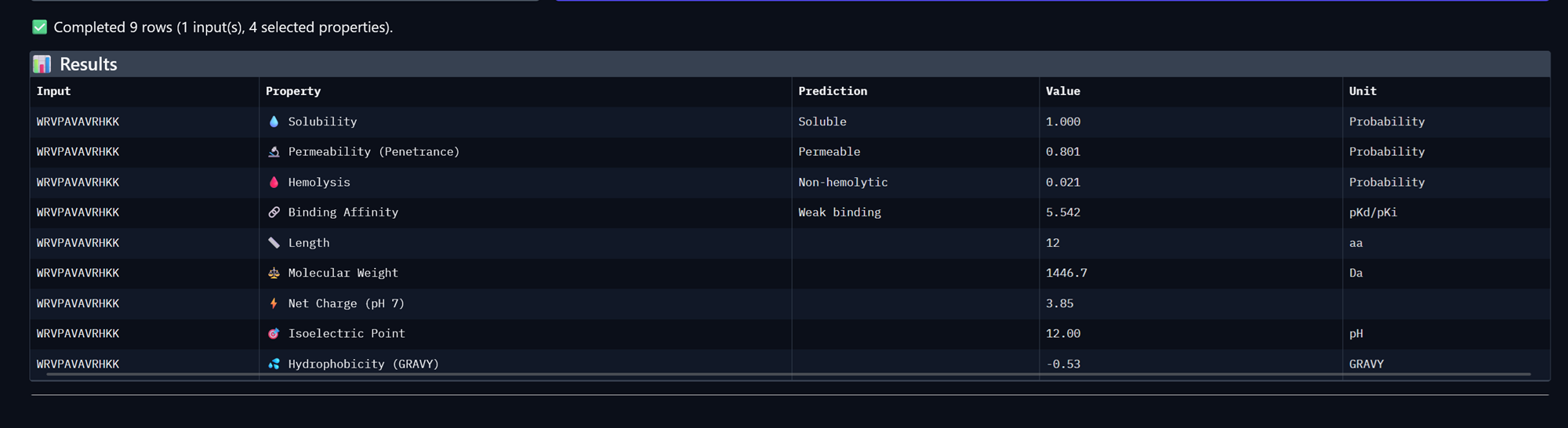
Peptide 2, WHYYAAAVRLWE
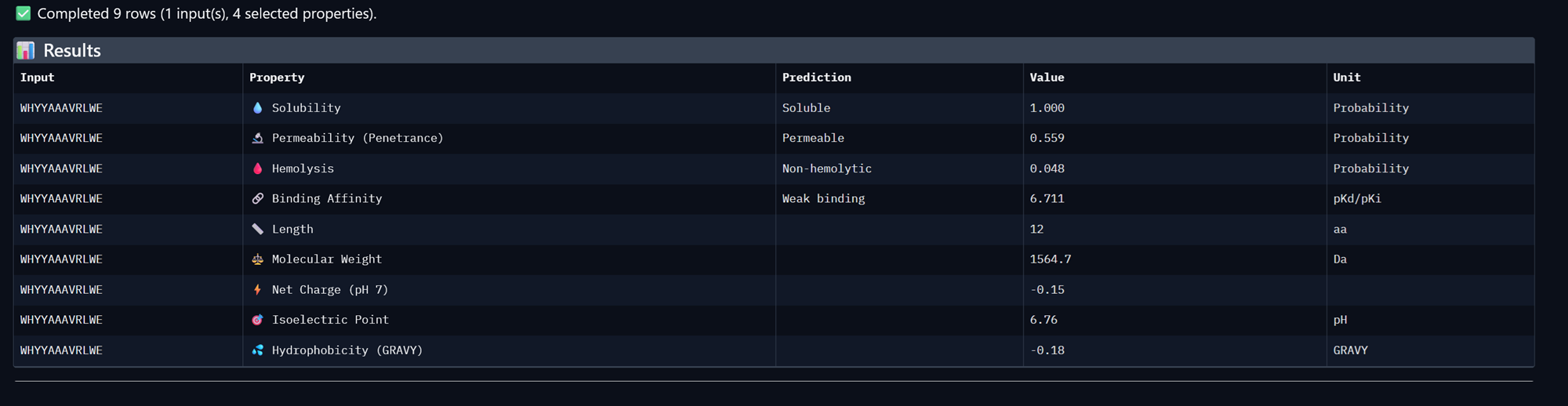
Peptide 3, WRVYPVAVEWKK
Peptide 4, WRVGAAGVAWKA
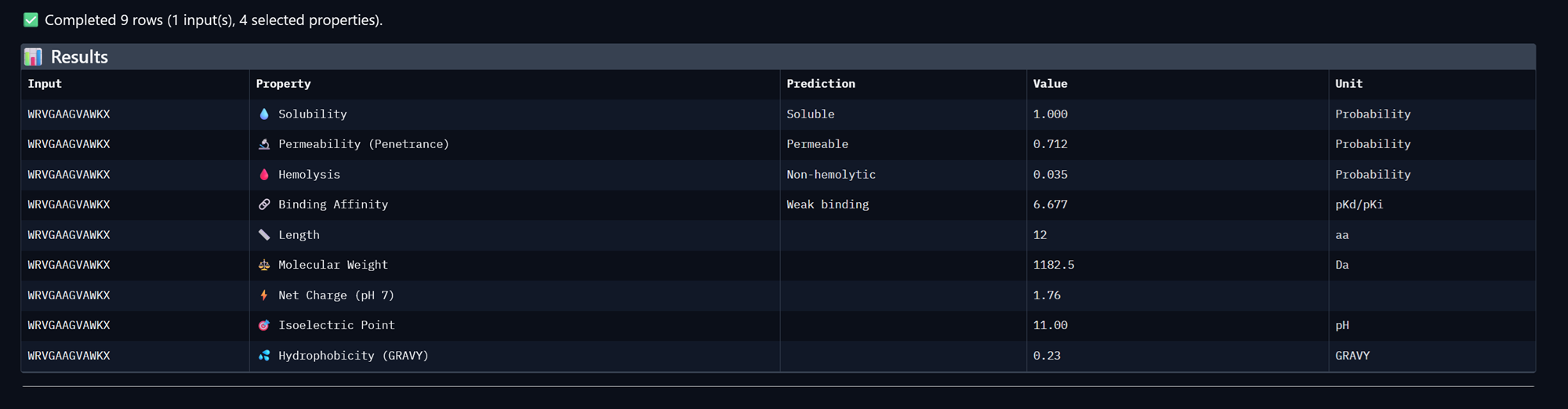
+Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Peptides with higher ipTMP do show stronger predicted affinities as we can see above. All of our binders have weak binding properties and none of them really best balances predicted binding and therapeutic properties.
+Choose one peptide you would advance and justify your decision briefly.
I would choose the peptide with the higher binding affinity and the ones that are permeable and in this case it is peptide WHYYAAAVRLWE.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
- Open the moPPit Colab linked from the HuggingFace moPPIt model card
- Make a copy and switch to a GPU runtime.
- In the notebook: -Paste your A4V mutant SOD1 sequence. -Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch). -Set peptide length to 12 amino acids. -Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
Here is my moPPit colab -.-
I generated 3 peptides for the A4V mutated SOD1 sequence choosing the parameters I am more interested in in order to to optimize the the binding process such as affinity and motif.

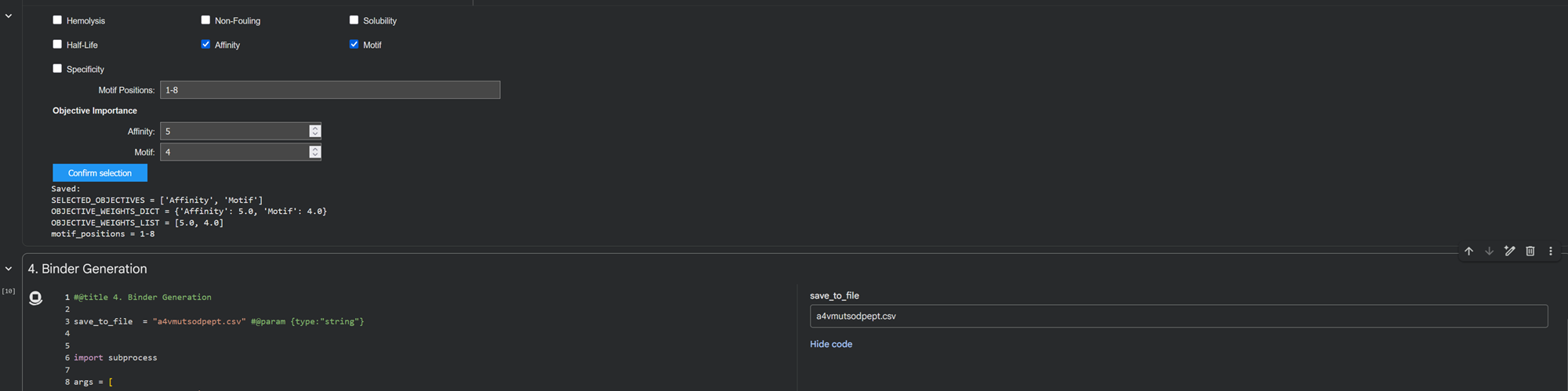
- After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
PepMLM generated peptides for a4v sod1 mutant
The known binder is the first one on the table below.
| Peptide | iptM | ptM |
|---|---|---|
| FLYRWLPSRRGG | 0.32 | 0.80 |
| WRVPAVAVRHKK | 0.28 | 0.82 |
| WHYYAAAVRLWE | 0.39 | 0.81 |
| WRVYPVAVEWKK | 0.29 | 0.81 |
| WRVGAAGVAWKA | 0.45 | 0.88 |
moPPit generated peptides
After the command was executed (after a whiiiiile) I then found my moppit files on the sideback of the colab notebook and downloaded the csv file I saved through the code named-> a4vmutsodpept.csv
| Binder | Affinity | Motif |
|---|---|---|
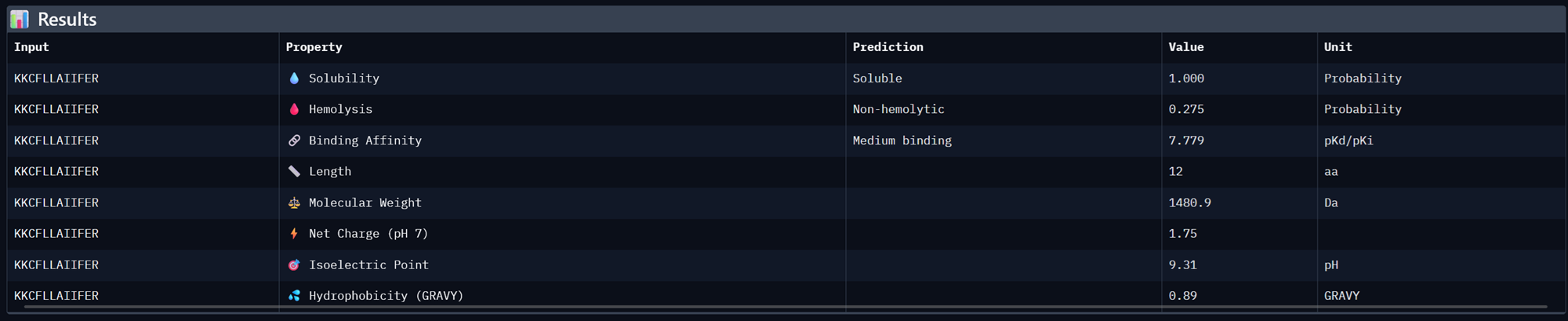
| KKCFLLAIIFER | 7.779302597045898 | 0.7270036339759827 |
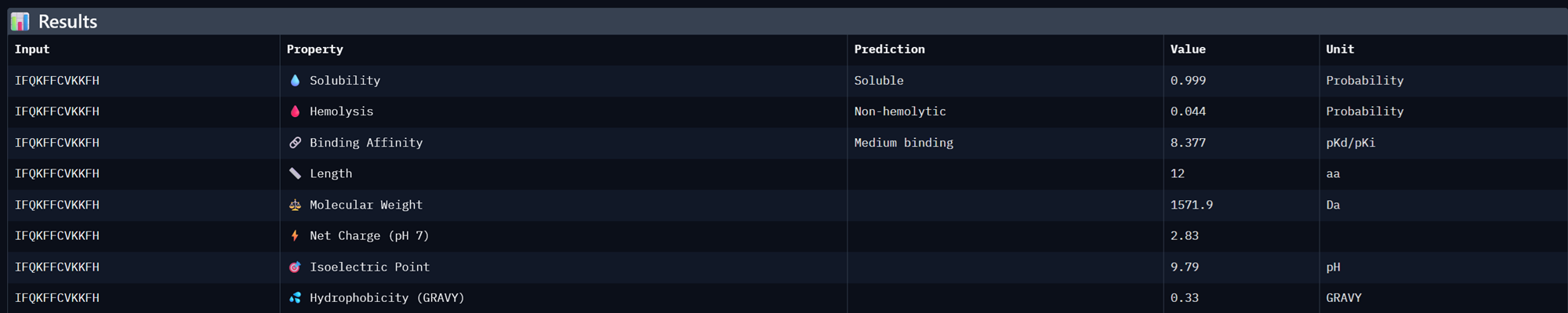
| IFQKFFCVKKFH | 8.377154350280762 | 0.7129143476486206 |
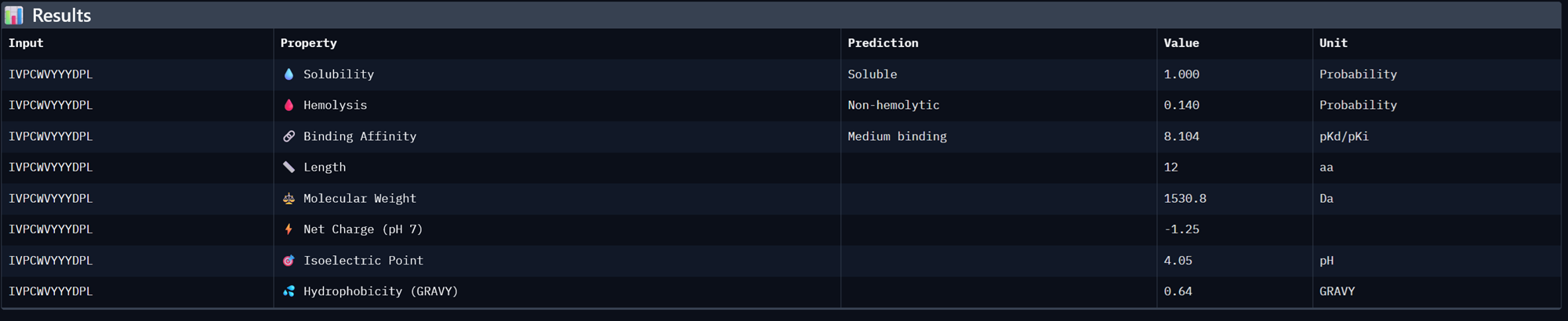
| IVPCWVYYYDPL | 8.104171752929688 | 0.695409893989563 |
I took the 3 moPPit peptides generated from the colab notebook above and analysed their properies in Peptiverse <3
Binder 1- KKCFLLAIIFER
Binder 2- IFQKFFCVKKFH
Binder 3- IVPCWVYYYDPL
I then generated another 3 moPPit peptides by checking more properties in the colab notebook such as solubility, hemolysis and specificity.
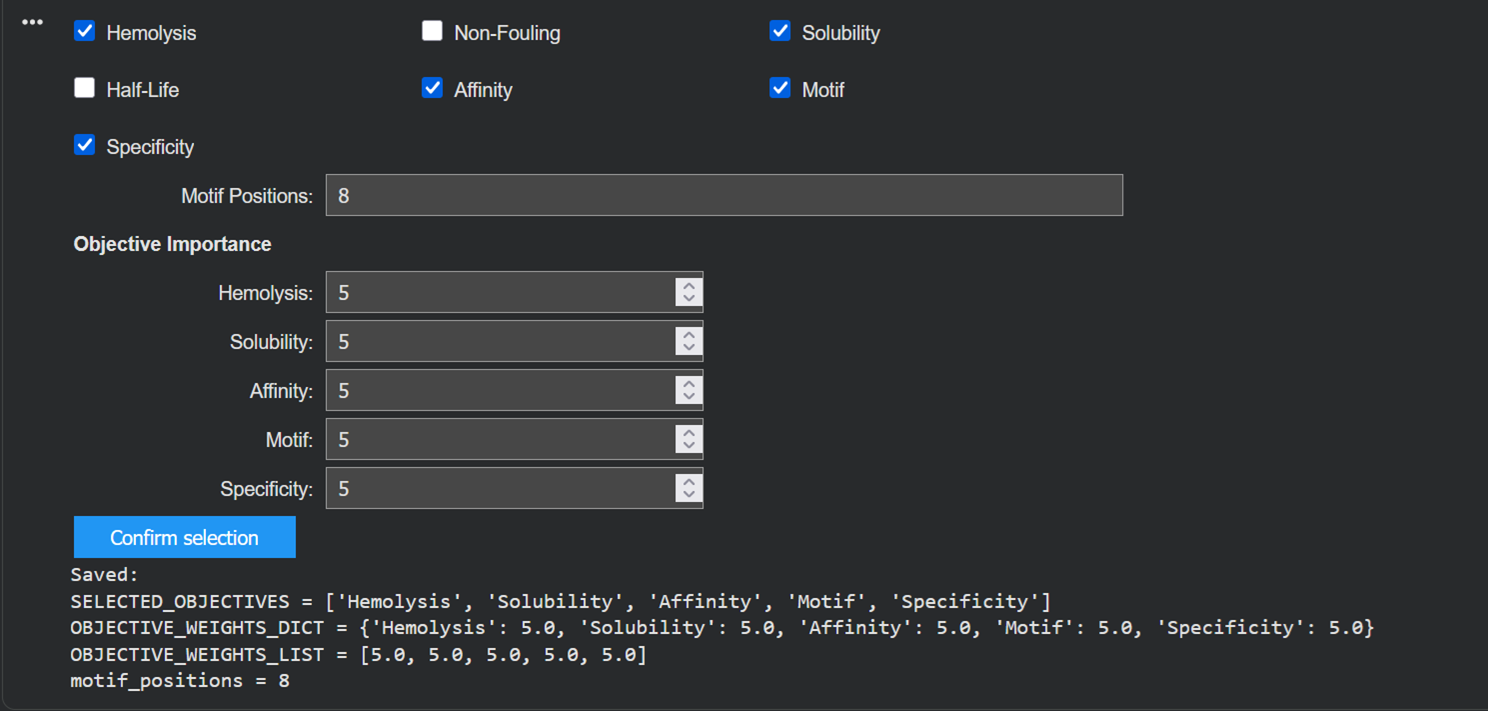
Here is the table:
| Binder | Hemolysis | Solubility | Affinity | Motif | Specificity |
|---|---|---|---|---|---|
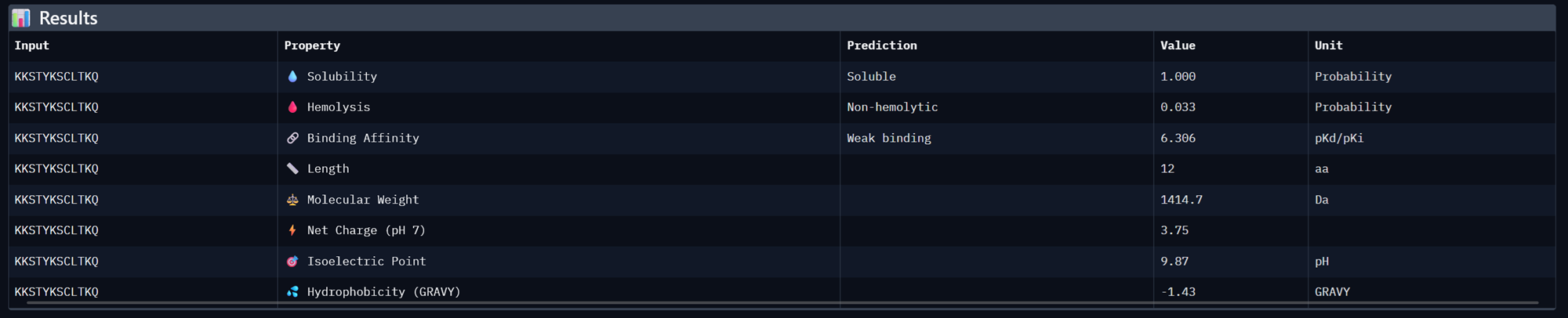
| KKSTYKSCLTKQ | 0.9520842246711254 | 0.9166666865348816 | 6.306060791015625 | 0.3722824454307556 | 0.6474359035491943 |
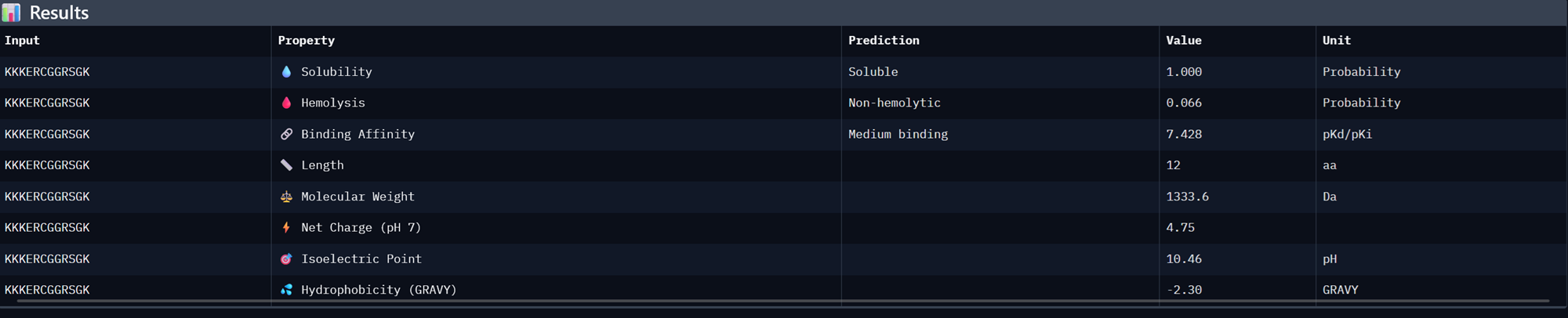
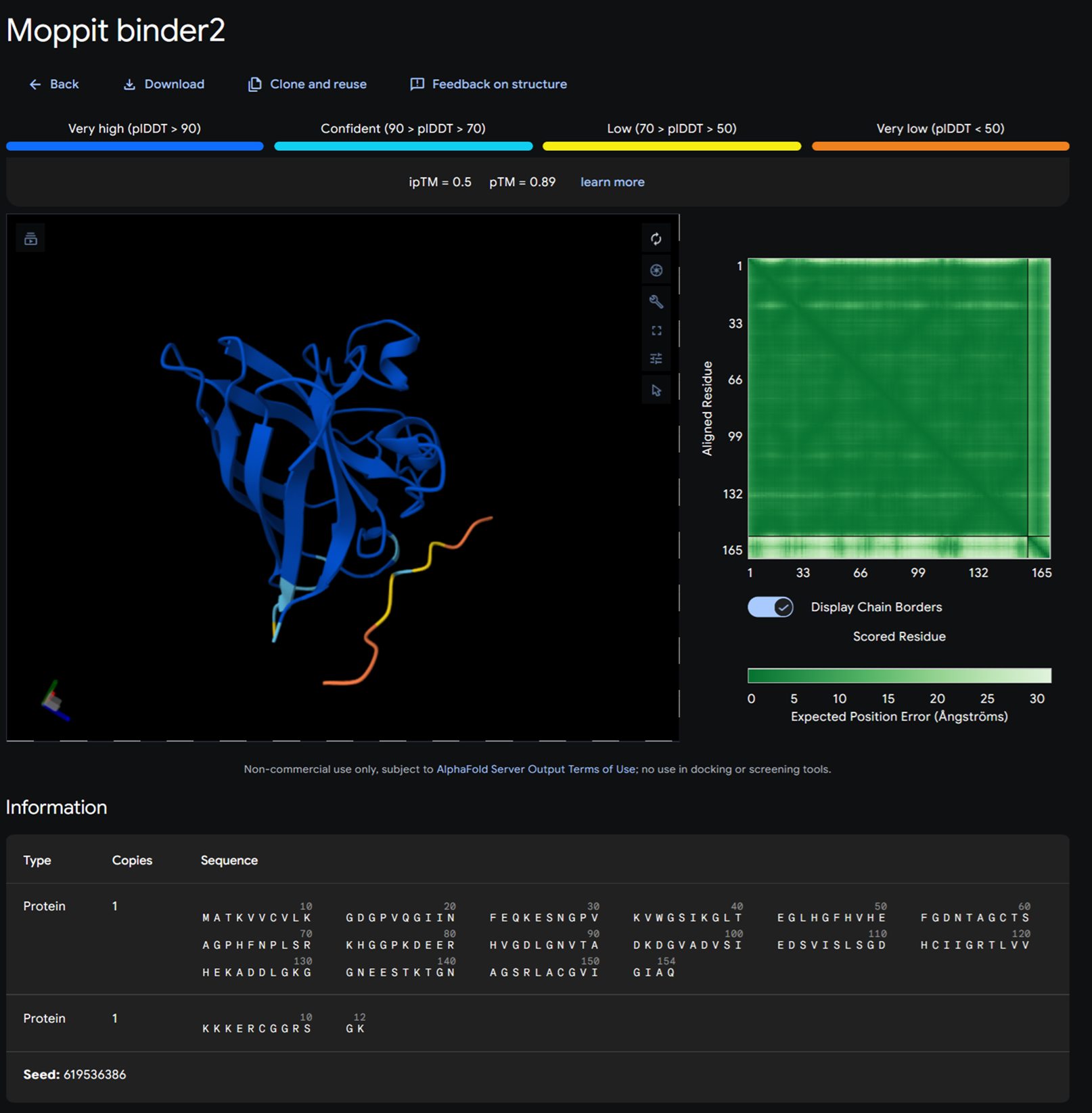
| KKKERCGGRSGK | 0.967436034232378 | 1.0 | 7.427951812744141 | 0.018683239817619324 | 0.942307710647583 |
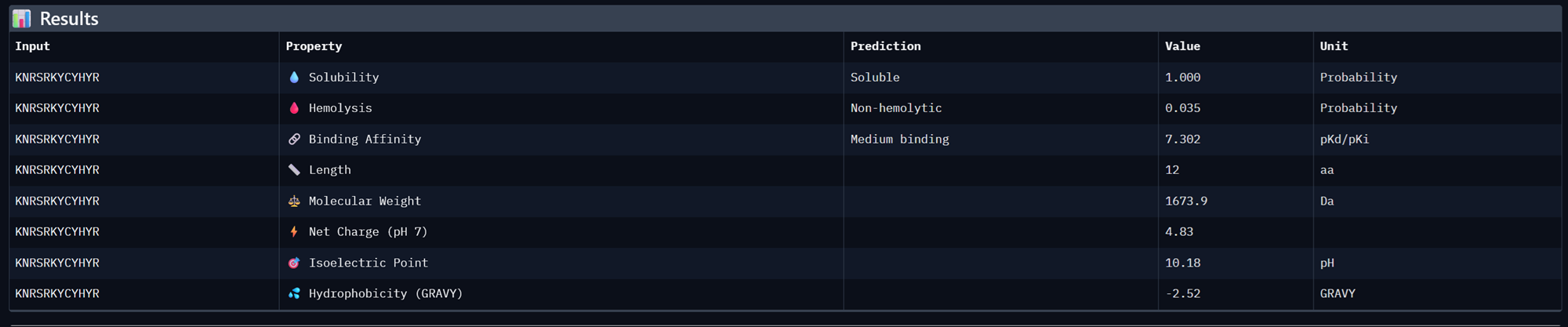
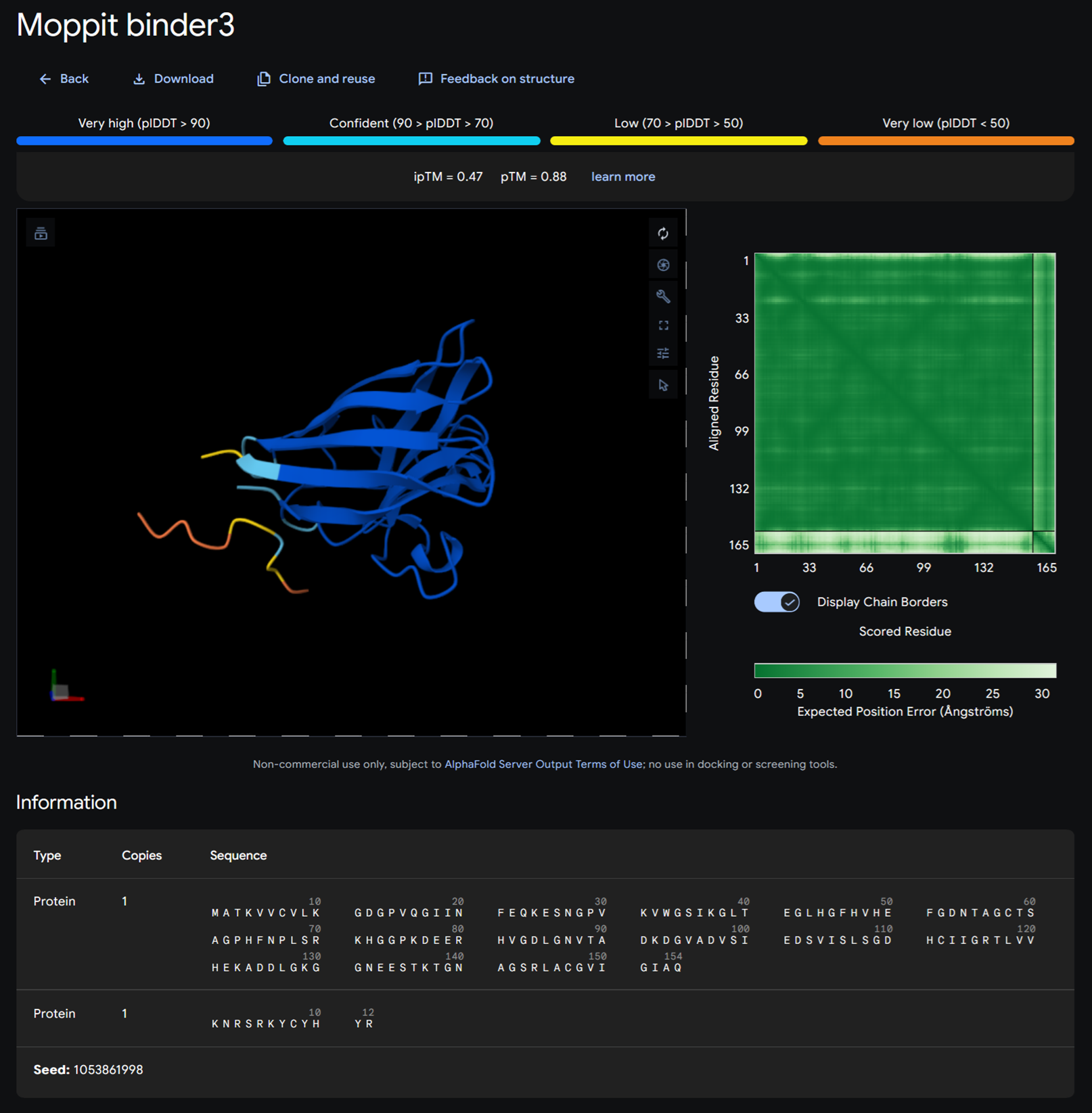
| KNRSRKYCYHYR | 0.9408624060451984 | 1.0 | 7.301994323730469 | 0.001087116077542305 | 0.9807692170143127 |
I then analysed these again on 1. peptiverse and finally I took the best binders and popped them up in alphafold. We focused on the binders that would bind to the N-terminus which was the motif we were interested in.
What is an N-terminus? The N-terminus (also amino-terminus) is the start of a protein or polypeptide chain, characterized by a free amino group on the first amino acid.
Photo from here!
Binder 1- KKSTYKSCLTKQ
Binder 2- KKKERCGGRSGK
Binder 3- KNRSRKYCYHYR
I then added KNRSRKYCYHYR and KKKERCGGRSGK on alphafold.
Moppit binder KKSTYKSCLTKQ on alphafold
Moppit binder KKKERCGGRSGK on alphafold
Moppit binder KNRSRKYCYHYR on alphafold
Part B: BRD4 Drug Discovery Platform Tutorial (Gabriele)- OPTIONAL
I will not be doing this part as I am really having a hard time with the protein weeks and need more time to digest this new knowledge.
Part C: Final project- L-Protein Mutants
High level summary: The objective of this assignment is to improve the stability and auto-folding of the lysis protein of a MS2-phage. This mechanism is key to the understanding of how phages can potentially solve antibiotic-resistance.
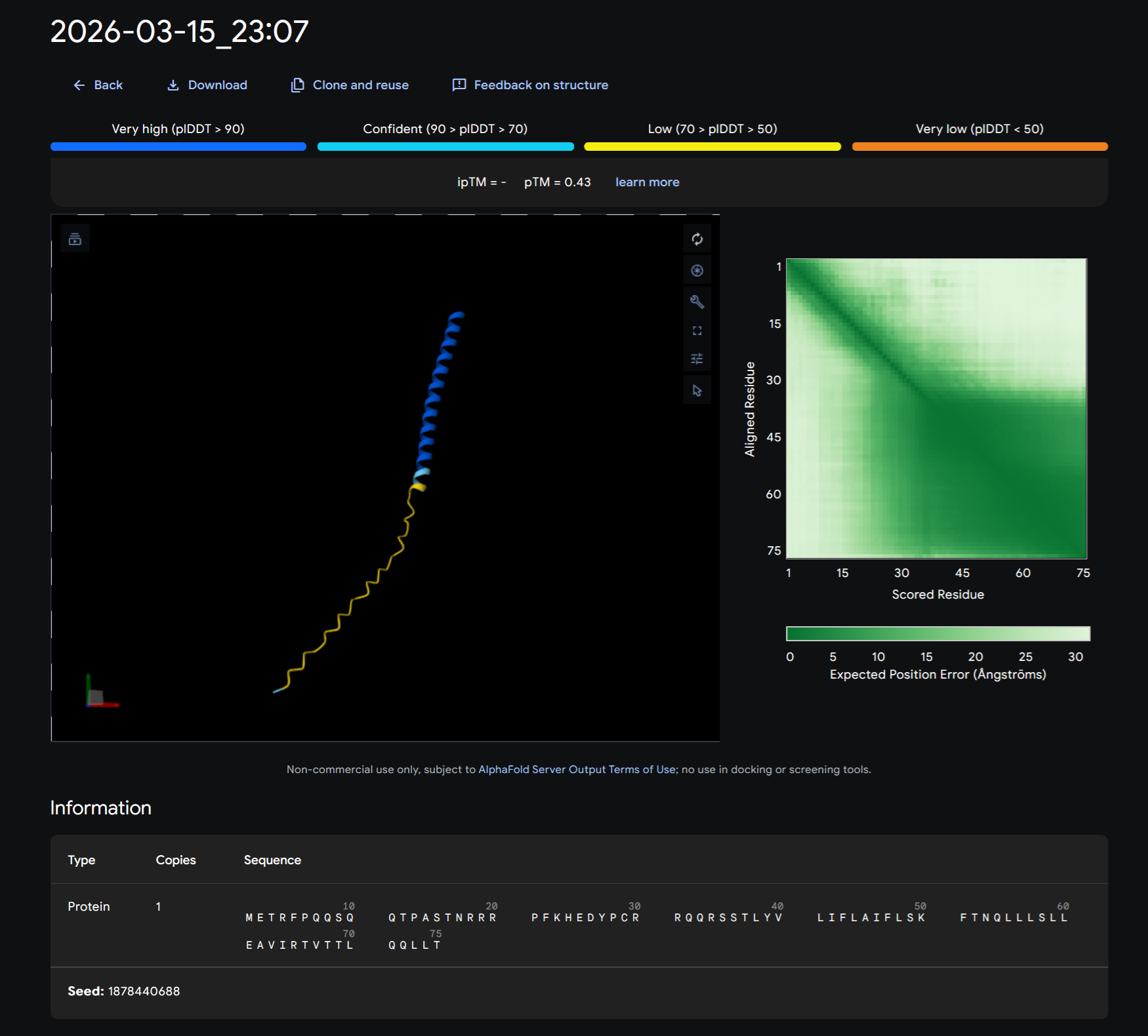
Ms2 Lysis Protein sequence
Note: Lysis protein contains a soluble N-terminal domain followed by a transmembrane protein (blue/last 35 residues). Transmembrane protein affects the lysis activity. The soluble domain (green) is the domain responsible for interaction with DnaJ.
After running the MS2 Lysis protein sequence on its own on Alphafold:
- Redered interaction of MS2 lysis protein with DnaJ
Dna J againts MS2 Lysis protein-> mutant
Dna J sequence

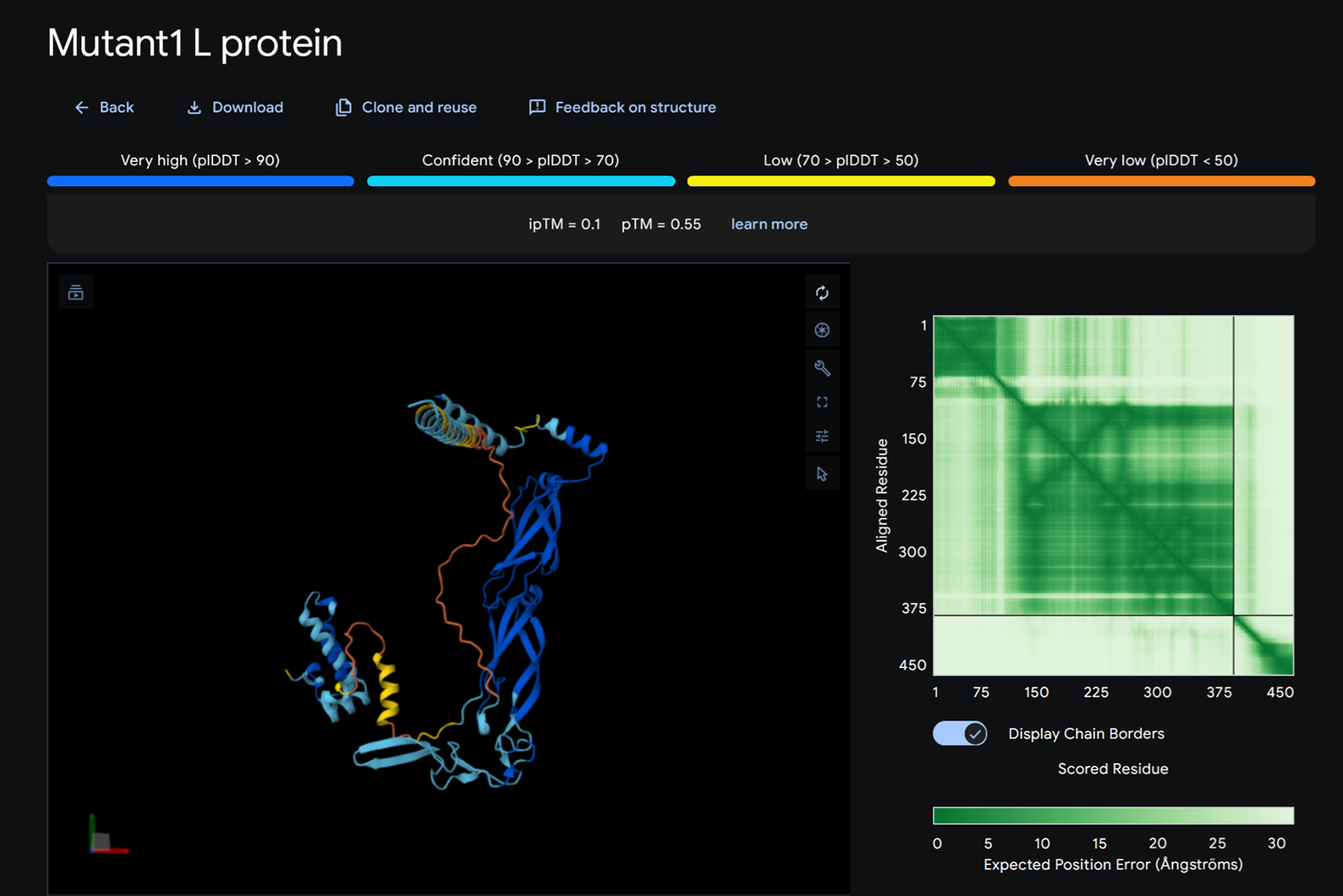
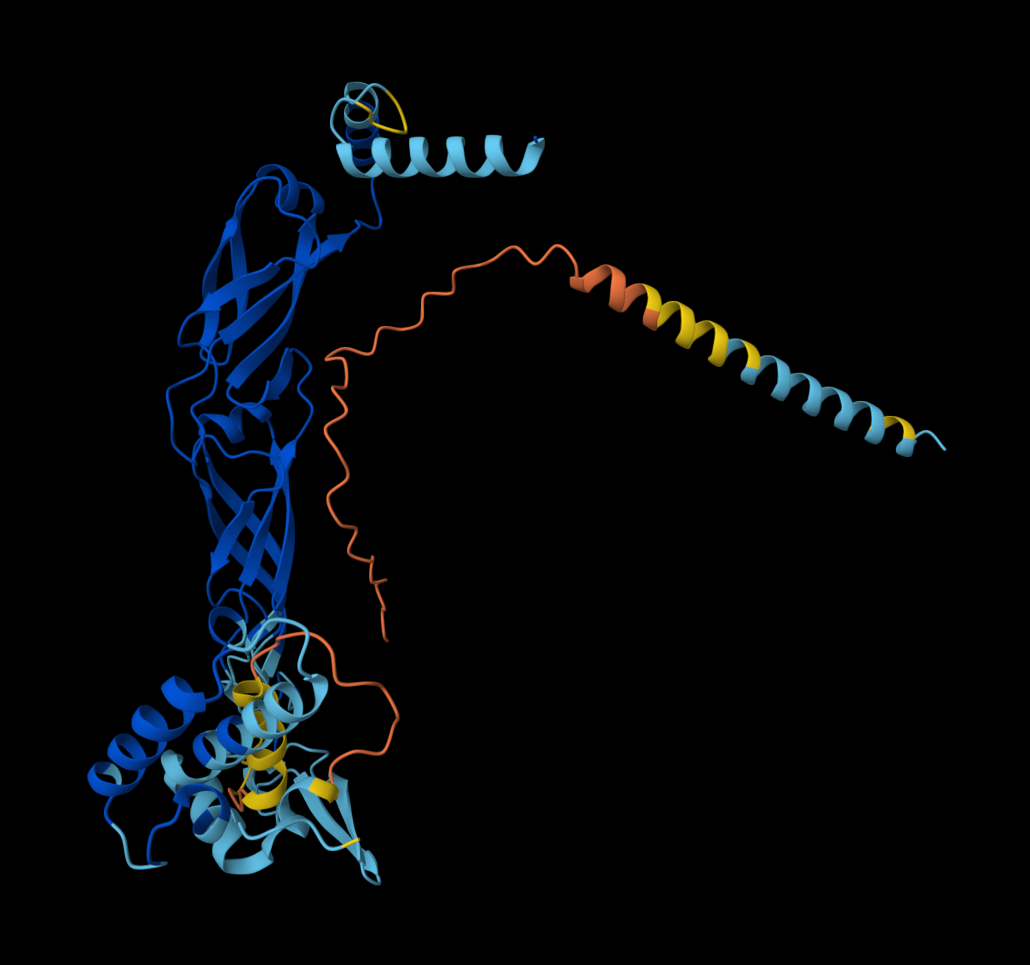
- Detail where we can see no bonds between DnaJ and L protein Mutant
- Multimer made up of 8 Mutant L proteins.