My final project Open Source body from fabricademy in 2022 <3
Final ideas presentation
My final project open source body from fabricademy in 2022 <3
Final project idea 1
Bacterial dyes from the human microbiome
Final project idea 2
Sweat collection device with PFA xenoestrogen biosensor.
Final project idea 3
A domestic diy lab for breaking down household single use plastics
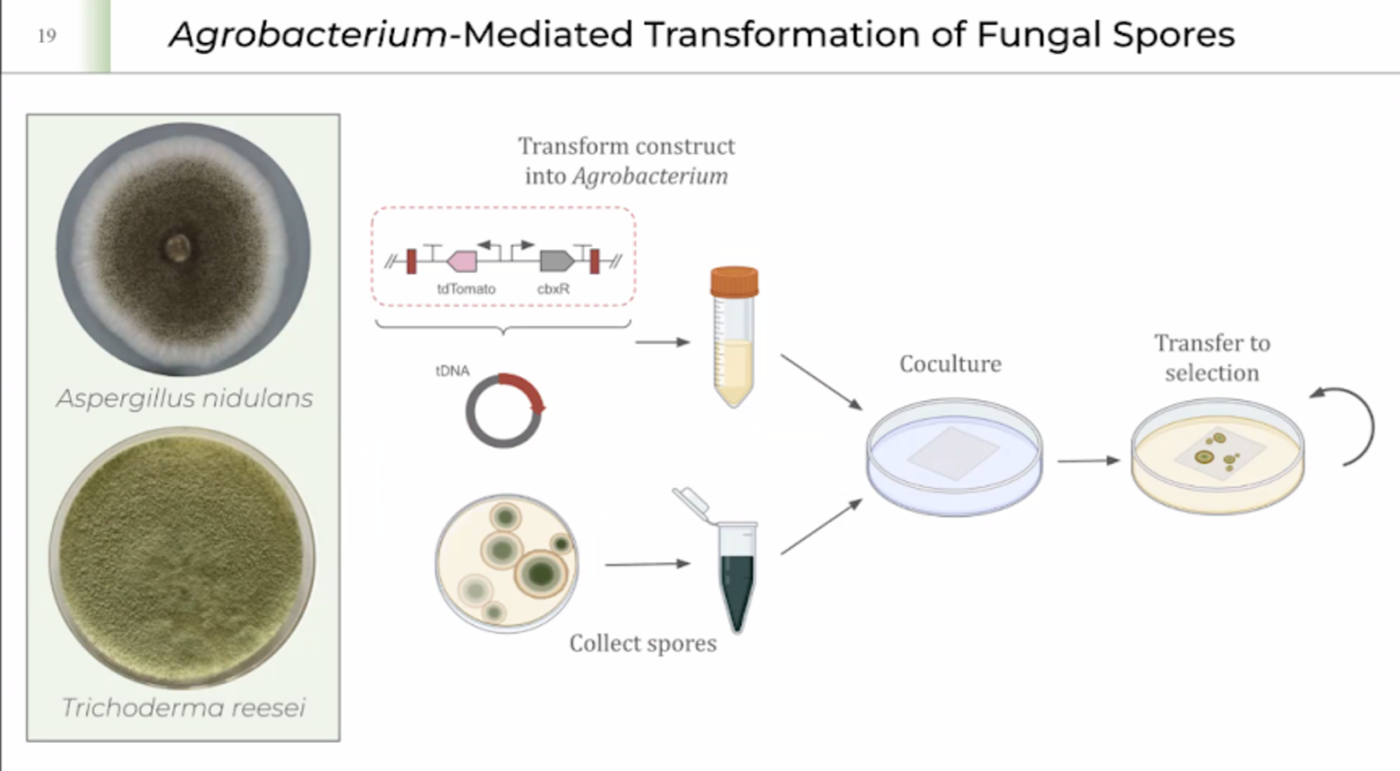
FINAL IDEA REVEAL!!!! HOW TO TRAIN YOUR MYCELIUM!!
I LOVE MUSHROOMS so guess which IDEA I AM GOING FOR :D In week 7 we got more information on fungal materials and I am inspired since I also stated my interest about mycoremediation and saprobionts in the first week homework. I took the last question of the 2nd part of week 7 homework below and used it as an opening question for my research into my final project!
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
This question right here is THE MOST IMPORTANT ONE FOR ME AND MY FINAL PROJECT!!!! I will try to answer this questions in more depth and add any more research HERE in this page during this week.
Fungi are biosensors and amazing bioremediation agents. They are extremely resilient and adaptable. I am interested in waste management, plastic degrading fungi and mycoremediation as well as solving issues that are universallike radiation exposure, microplastic pollution and pfa contamination of our bodies of water, soil and crops are posing huge threats and we are living in a damaged planet. I am also interested in researching the enzymes that break down plastics in saprophytic fungi and figure out how to train my mycelium/mushrooms naturally and how can I utilise synthetic biology to optimise the process. Certain molds and fungi have evolved to break down more than agricultural organic sources including plastic and radioactive waste. I wrote about the Chernobyl fungi in WEEK 01 here. The fungi in Chernobyl are radiotropic- they display radiotropism, another form of biosensing that melanin rich fungi posseses that demonstrates its ability to detect the presence of ionizing radiation, and to interact with it, favoring its growth and dissemination. This fungi carries out radiosynthesis, which means its using ionizing radiation as a main energy source to drive metabolization. ese melanized fungi are also called “black fungi” due to their coloration, usually darker than other fungi, since it has large amounts of melanin in their cell walls, which leads to changes in the color of the fungi and its hyphae, with these colors varying of shades of brown, gray and black, but can also exhibit lighter colors in more isolated cases (Chowdhary et al., 2014) in Radiotropic fungi and their use as bioremediation agents of areas affected by radiation.
In addition, yeast (saccharomyces cerevisiae) has also been used as a biosensor in modern-day biotechnology. It was the first eukaryote to have its genome sequenced in its entirety in 1996. Along with that advance was the concerted effort to assign functions to all 6000 open reading frames.
The benefits of yeast being used as a biosensor have opened new avenues for drug discovery, understanding molecular pathways involved in disease pathogenesis, protein–protein interaction studies, understanding of the molecular architecture of complex protein assemblies, identifying mutations in proteins that have significance in determining the functional differences, and detecting pollutants from the environment. Yeast has already proved its benefits in studying protein–protein interactions, drug screening against several diseases, including cancer, Alzheimer’s disease, Parkinson’s disease, and others, detection of pollutants, and diagnosis of diseases.
Yeast-based biosensors are engineered microorganisms, modified to detect and quantify target compounds, toxins, or environmental pollutants. These biosensors use genetic modifications—such as reporter genes (fluorescence/luminescence) and modified receptors—to produce measurable signals (colorimetry, electrical) upon interaction with specific molecules. They are widely used in environmental monitoring, pathogen detection, and pharmaceutical development due to their ease of culturing and genetic tractability.
I also virtually attended a workshop by artist Mary Maggic who were also on htgaa 2015 on Becoming with fungi where they used remazol blue (endorcrine disrupting chemical) to test the ability of the Schizophyllum commune mushroom for bioremediation. Marry Maggic have also worked with yeast biosensors.
YES-HER YEAST BIOSENSORS-DOES IT SAY (YES) TO (hER)?
Because endocrine disrupting compounds are usually found in minimal amounts (ng/L-1) in the water, one of the most common techniques for their detection is liquid chromatography-tandem mass spectrometry (LC-MS). But this approach is very expensive to perform on a routine basis, requiring both skilled personnel and a robust quality assurance/control program. Maybe biology is the answer... The YES-YEAST (yeast estrogen sensor) are a genetically modified strain of Saccharomyces cerevisiae (W303) that contain Human Estrogen Receptor (HER). They act as a biosensor: the input is estrogen and the output is a yellow color change. More importantly, the YES yeast are extensions of our bodies: what binds to their receptors also bind to ours, demonstrating a DIRECT biological response to xenoestrogens on our bodies. The same process of estrogen binding and activation is reproduced in the yeast. This bioassay detection method is more sensitive than the chemical approach either detecting estrogenic target compounds at lower concentrations, other non-target compounds, and synergistic effects that chemical methods and machines fail to detect.
This is both a "part two" of the Open Source Estrogen project as well as the Final Project for HTGAA, which combines lectures (1) "Synthetic Minimal Cells" with Kate Adamala, (2) "Bio-production" with Patrick Boyle, (3) "Computational Protein Design" with Srivatsan Raman, and (4) "Tools, Automation, and Open Hardware.
Another interest is studying radiotropic melanin producing fungi for protection from radiation and learning more about protein producing mechanisms by looking into their genome and editing it. I found this really cool radiation badge project made with freezer paper and modified yeast that inspired me.
A badge that helps hospital lab workers better track their daily radiation exposure, enabling a faster assessment of tissue damage that could lead to cancer. Rather than building portable cellars or ovens, Purdue University researchers have engineered yeast "microbreweries" within disposable badges made of freezer paper, aluminum, and tape. Simply adding a drop of water activates the yeast to show radiation exposure as read by an electronic device.
The success of the badge lies in the quick and measurable response of yeast to radiation: The higher the radiation dose, the higher the percentage of yeast cells that die. Wetting the badge activates the cells that are still alive to eat glucose and release carbon dioxide—the same fermentation process responsible for brewing beer and making bread rise.
All this research got me thinking-> Can I combine freeze dried cell free systems with mycelium instead of yeast?
What about fungi-based enzymatic degradation? How do I turn Waste to Resource? The resulting materials can be used as nutrient-rich soil or raw materials for chemical processes.
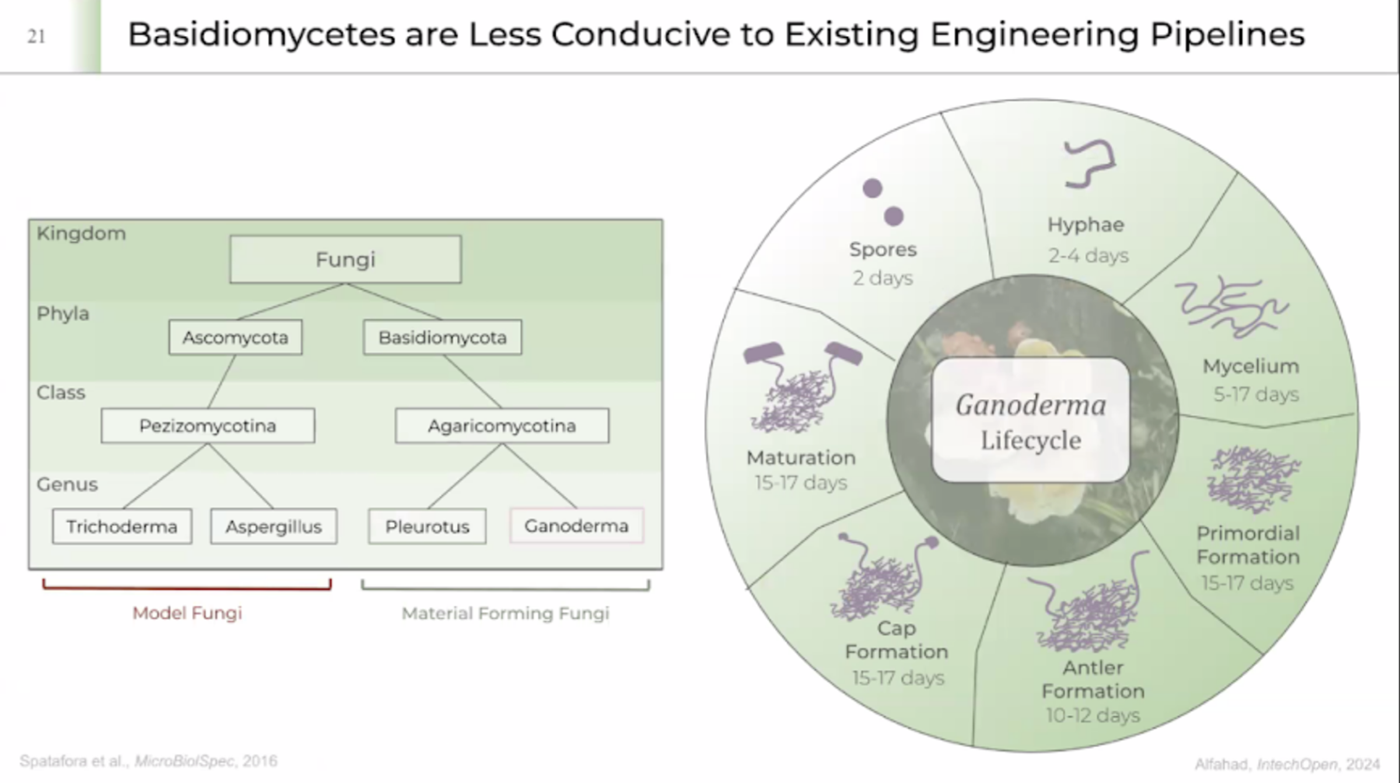
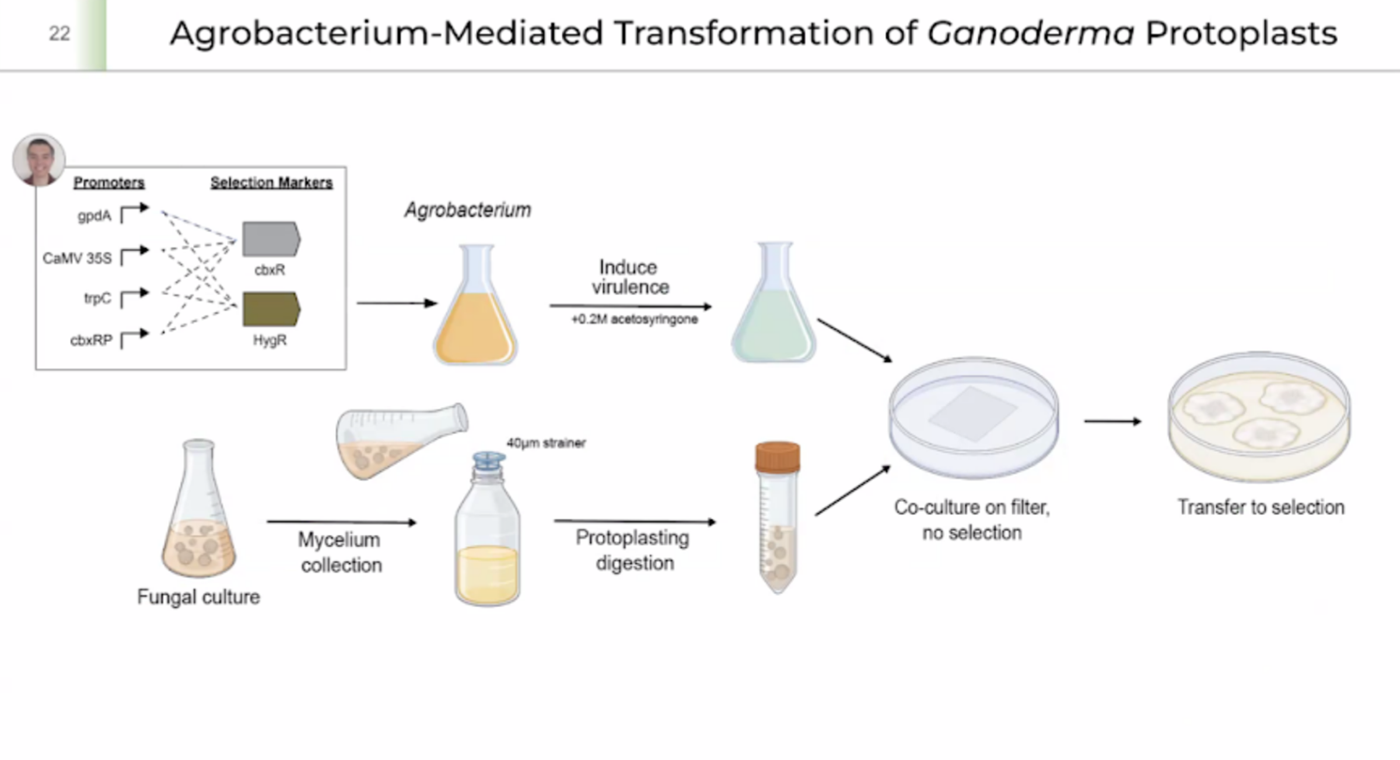
Genetically engineered fungi From Ren Ramlans presentation
Rens research-> working with Ascomycota and Basidiomycota are less conducive to existing engineering pipelines.
This is the question I wrote in the chat on that day of the presentation.
You 11:52 PM (Edited)
I am interested in plastic degrading fungi. How can I apply synthetic bio to this concept? I have a few ideas but it will be nice to get expert advice.
TA, Val Thompson, ChiTownBio, Chicago 11:57 PM
You could look at the pathways responsible for making the enzymes in mushrooms like oysters, that break down plastic, like lactase, manganese peroxidase, and lignin peroxidase, and maybe boost one of those pathways in some manner? The specifics beyond that are above me at this point in time.
Advantages of doing synthetic biology in fungi as opposed to bacteria
While fungi offer superior PTMs and secretion, they often have longer cell cycles (12–24 hours) compared to bacteria (20–60 minutes), and their genetic toolkit is often considered less developed compared to Escherichia coli.
Doing synthetic biology in fungi, particularly filamentous fungi and yeasts, offers several advantages over bacteria (such as E. coli) due to their eukaryotic nature, metabolic complexity, and specialized secretion systems. Key advantages include superior protein folding and secretion, a vast repertoire of secondary metabolites, and higher environmental robustness.
In Prospects of Fungal Biotechnologies for Livestock Volume 2. Fungal Biology. Springer, Cham., it is being mentioned that “engineered fungi like Aspergillus, Trichoderma, and Saccharomyces are increasingly used to produce valuable biomolecules such as enzymes, insulin, and antimicrobial peptides. These organisms naturally secrete large quantities of proteins, making them particularly attractive for industrial-scale applications”.
HTGAA 2026: Individual Final Project Documentation
HOW TO TRAIN YOUR MYCELIUM- BIOHYBRID EDITION
I realised that I wanted to work with mycelium no matter what. I made one of my 3 final ideas more specific because most of my ideas were super general- I enjoy designing circular systems and diy labs. I will post all my research about my final project and how I ended up choosing this project below! The question from fungal materials week above really helped me understand some ways in which mycelium can be genetically modified. I am still intrigued by the cell free freeze dried week and in the future I am definitely doing a mycelium project on that but for now this is the slide I added into the slide deck for the final ideas!
I will keep tweaking the idea and tuning the specifics until the final project presentation day! Below you can find my final 3 slides from the presentation day. I will document the process in more depth below :)
SECTION 1: ABSTRACT
Provide a concise, self-contained summary of your project (minimum 150 words). The abstract should allow a reader to understand the purpose, approach, and expected outcomes of the work without referring to other sections.
Your abstract should briefly address the following elements:
Significance: What problem or question does the project address, and why is it important?
Broad Objective: What is the overall goal of the project?
Hypothesis: What prediction or principle is the project testing or demonstrating?
Specific Aims: What key steps or milestones will be completed to achieve the objective?
Methods: What experimental or technical approaches will be used?
How can you train your mycelium to degrade synthetic petroleum derived polymers and environmental pollutants in order to harness its biocomputational capacity for soft robotic actuation and control? This project addresses the growing crisis of plastic pollution by exploring enzymatic degradation and mycoremediation processes as both an environmental and technological solution. How to train your mycelium investigates natural and engineered fungal systems as platforms for bioremediation and as living components in biohybrid robotics. The hypothesis is that enhancing or introducing plastic-degrading enzyme pathways—through selective exposure or through genetic modification will alter metabolic activity and produce distinct electrophysiological patterns that can be used as control inputs for actuating a soft robotic system. The overall objective is to train mycelium to metabolize synthetic polymers and pollutants in a controlled in vitro environment, while exploring and harnessing the biocomputational capacity of its electrical signaling as a biological interface for soft robotic control and actuation.
SECTION 2: PROJECT AIMS
Define three aims for your final project (minimum one sentence per aim).
Aim 1: Experimental Aim (this project):
“The first aim of my final project is to [achievable experimental goal] by utilizing [protocols, tools, or strategies].”
This aim should describe the core experimental objective you will attempt during this class. List or link any relevant methods or resources you plan to use (e.g., experimental protocols, automation workflows, DNA or protein designs, protein design tools, or Twist orders).
You will provide a detailed step-by-step experimental plan for Aim 1 in the Experimental Design section of this assignment.
The first aim of my final project is to explore the enzymatic degradation process of mycelium to break down different types of synthetic petroleum derived polymers (plastic) and other environmental pollutants while training my mycelium (Pleutotus Ostreatus) through exposure (naturally) vs synthetically through genetically optimising enzymatic degradation process in strains of mycelia that do not posses this enzymatic capability. I plan to do so by utilising mycelium cultivation techniques with a variety of scaffolds (substrates) containing plastics and pollutants as well as DNA sequencing, writing and editing.
Aim 2: Development Aim:
Describe the next step that would follow a successful Aim 1, extending the work beyond the scope of this course. This aim should represent a realistic progression of the project, such as executing additional experiments, solving a technical limitation, or developing the system or technology further.
The development aim of the project is to record enzymatic degradation capacity and analyse it via measuring bioelectrical activity via electrodes, and interfacing and translating these signals into actuation with microcontroller-driven soft robotic systems. The final developmental aim is to create a 3D printable or casted silicone soft robot like the ones below to combine with my mycelium brain.
Aim 3: Visionary Aim:
a) Describe the long-term vision for the project. Explain how the broader concept could have an impact if fully realized.
b) Examples include:
Challenging an existing paradigm or clinical practice.
Addressing a major barrier in a field.
Enabling a new experimental capability or research approach.
Harnessing sensing and mycoremediation capabilities of mycelium to produce and utilise regenerative forms of energy from the enzymatic degradation process. Enabling mycelium to be utilised as the living component in biohybrid robotics. Enabling future application experimentation around fungal materials through engineering additional sensing capabilities for microplastic biosensing and bioremediation.
AI final project interview- using the AI tutor
I ended up using the AI tutor on the forum and it helped me A LOT! The title for the project I was given is:
Pleurotus ostreatus as a Dual-Function Plastic-Degrading and Electrophysiological Biosensor Platform
This is my chat with the AI tutor that really helped me choose which type of plastic I will use for my project and how the dna design of the experiment will play out. I was super confused because I generated so many proposals and now I do not know which to follow.
Engineering Fungal Plastic Degrading Enzymes
Exported: 2026-04-27T12:41:58.054Z
Turns: 26
Turn 1
Question
I want to optimize plastic degrading enzymes in pleurotus ostreatus.
Answer
That's a fascinating direction — using oyster mushrooms as a chassis for plastic-degrading enzymes is genuinely exciting and has real-world relevance.
Before we dive into design, I want to make sure we're targeting the right problem. Which plastic(s) are you most interested in degrading? For example, PET, polyurethane, polyethylene, or something else?
Turn 2
Question
What is the easiest to do? I was thinking of LDPE but what do you think? I read that I have to pre treat the plastic in order to do that.
Answer
LDPE is notoriously hard — its C-C backbone is extremely stable and no well-characterized enzyme cleanly breaks it down yet. PET is the best starting point because PETase (and its improved variant FAST-PETase) is well-characterized, the reaction products are measurable, and there's a clear assay readout.
The pretreatment issue you read about is real for LDPE, but with PET you can skip harsh pretreatment and use amorphous PET film directly as a substrate.
So my recommendation: express a codon-optimized FAST-PETase in Pleurotus ostreatus and measure PET degradation by detecting the soluble product terephthalic acid (TPA) colorimetrically or by absorbance. Does that direction appeal to you, or do you have a strong reason to stick with LDPE?
Turn 3
Question
no PET would do too. The aim is to measure action potential through plastic degradation (mycoremediation process).
Answer
Great — PET + FAST-PETase in Pleurotus ostreatus it is. Measuring TPA release as a proxy for enzymatic activity is clean and automation-friendly.
Before we go further into construct design, I want to understand your expression strategy. Do you want to express FAST-PETase intracellularly, secreted extracellularly (so the enzyme is released into the growth medium to contact the plastic), or displayed on the fungal cell surface?
Turn 4
Question
what is the difference?
Answer
Great question. Here's the short version:
Intracellular: enzyme stays inside the cell — the plastic can't easily get in, so degradation is minimal. Not ideal here.
Secreted: enzyme is exported into the growth medium and contacts the plastic directly. This is the standard approach for fungal plastic degradation and gives you a clean, measurable TPA signal in the supernatant.
Surface display: enzyme is anchored to the outside of the cell membrane. Good for contact degradation but harder to engineer in Pleurotus.
For your mycoremediation goal, secreted expression is the most practical and well-precedented choice.
Does secreted expression sound right to you, or do you want to explore surface display?
Turn 5
Question
so let's go with secreted i think!
Answer
Secreted it is — great choice. To secrete FAST-PETase in Pleurotus ostreatus, we'll need to fuse it to a fungal signal peptide that directs the protein through the secretory pathway.
Do you have a preferred signal peptide in mind, or should we use a well-characterized one from Pleurotus itself (like the native glucoamylase signal peptide) or a broadly used fungal signal peptide such as the one from Trichoderma reesei cellobiohydrolase I?
Turn 6
Question
Let's use the well characterised one i think! I want to optimise the plastic degradation process and measure the action potential maybe with the optimisation and without? what do you think?
Answer
Great plan — using the native Pleurotus glucoamylase signal peptide keeps the secretion machinery familiar to the host, which should maximize export efficiency.
Your comparison idea is solid: a baseline construct (wild-type PETase + signal peptide) versus an optimized construct (FAST-PETase + signal peptide) gives you a clean experimental contrast, and TPA release measured by absorbance at 260 nm is your quantifiable readout.
To move into construct design, I need one more detail: what promoter do you want to drive expression in Pleurotus ostreatus — do you want a constitutive promoter (always on) or an inducible one (switched on by a specific signal, like a carbon source)?
Turn 7
Question
what is the difference? can you describe? The process will take place inside the petri dish and i have to add electrodes on the surface perhaps and measure.
Answer
Good clarification. A constitutive promoter drives expression continuously — the enzyme is always being made. An inducible promoter only activates when you add a specific trigger (e.g., a sugar or chemical), giving you an on/off switch.
For your setup, constitutive is simpler and more reliable — you don't need to manage an inducer, and the enzyme will be secreted steadily as the mycelium grows across the petri dish toward the plastic.
Which do you prefer: constitutive (simpler, always on) or inducible (more control, but more complexity)?
Turn 8
Question
Let's go for constitutive for now.
Answer
Constitutive it is — that keeps the design clean and experiment-ready.
For Pleurotus ostreatus, a strong constitutive promoter option is the native gpdA (glyceraldehyde-3-phosphate dehydrogenase) promoter, which is well-characterized and widely used for stable transgene expression in basidiomycetes.
So our construct backbone is taking shape: gpdA promoter → glucoamylase signal peptide → FAST-PETase coding sequence → terminator. Before we finalize the design, do you want to include a reporter tag (like a fluorescent protein or epitope tag) so you can also confirm the protein is being expressed and secreted, separate from the TPA degradation assay?
Turn 9
Question
hmmm.... I have not thought about that. Remind me what is an epitope tag?
Answer
An epitope tag is a short, well-known amino acid sequence (like FLAG, HA, or His-tag) fused to your protein. It lets you detect the protein using a standard antibody in a Western blot or dot blot — so you can confirm FAST-PETase is actually being made and secreted, independent of whether it's enzymatically active.
For your project, a small C-terminal His-tag would be the simplest choice — it adds minimal size, doesn't interfere with secretion, and lets you pull down the protein from the growth medium to confirm secretion.
Would you like to add a His-tag to the construct, or keep it tag-free and rely solely on the TPA assay to confirm activity?
Turn 10
Question
let's add a his-tag yes
Answer
Perfect — a C-terminal His-tag it is. Your construct is now fully defined:
gpdA promoter → glucoamylase signal peptide → FAST-PETase → His-tag → terminator
Now let's think about your assay. You mentioned measuring TPA release as the main readout of plastic degradation. How do you plan to measure TPA — do you have a specific instrument or method in mind, or should I suggest one that fits the available automation?
Turn 11
Question
I saw this project here https://www.science.org/doi/10.1126/scirobotics.adk8019.
Answer
That's a fascinating reference — the Science Robotics paper describes using electrophysiological signals from fungal mycelium as a readout of biological activity, which is a really creative angle.
Are you thinking of combining that approach with your PET degradation project — i.e., using mycelial action potentials as a real-time biosensor of FAST-PETase activity or plastic contact, alongside the TPA chemical assay?
Turn 12
Question
So this is the abstract of the project: How can you train your mycelium to degrade synthetic petroleum derived polymers and environmental pollutants in order to harness its biocomputational capacity for soft robotic actuation and control? This project addresses the growing crisis of plastic pollution by exploring enzymatic degradation and mycoremediation processes as both an environmental and technological solution. How to train your mycelium investigates natural and engineered fungal systems as platforms for bioremediation and as living components in biohybrid robotics.
The hypothesis is that enhancing or introducing plastic-degrading enzyme pathways—through selective exposure or through genetic modification will alter metabolic activity and produce distinct electrophysiological patterns that can be used as control inputs for actuating a soft robotic system. The overall objective is to train mycelium to decompose and metabolize synthetic polymers and pollutants in a controlled in vitro environment, while exploring and harnessing the biocomputational capacity of its electrical signaling from the mycoremediation process as a biological interface for soft robotic control and actuation.
Answer
This is a beautifully ambitious project — mycoremediation plus biocomputation plus soft robotics in one system. The hypothesis is clear and the vision is compelling.
Before we move into construct design and aims, I want to make sure the experimental scope is achievable for a single proposal. For Aim 1, would you like to focus specifically on engineering and validating FAST-PETase secretion in Pleurotus ostreatus (the construct we've been designing), with TPA release and electrophysiological signal recording as the dual readout — leaving the robotic actuation interface for Aim 3 as the visionary goal?
Turn 13
Question
Yes. I want to focus on aim 1 i think for now because there wont be enough time i think.
Answer
Perfect — Aim 1 is well-scoped and experimentally solid. Let's lock in the assay strategy.
You mentioned measuring TPA release (absorbance at 260 nm) and recording mycelial electrophysiological signals as your dual readout. For the electrophysiology, the setup is relatively simple: surface electrodes on the petri dish agar, connected to a multichannel amplifier, recording spike frequency before and after PET film contact.
For the TPA assay, we'll use the Spark Plate Reader (available in the lab) measuring absorbance at 260 nm from collected growth medium samples — fully automatable. Does that combination work for you, or do you want to adjust either readout?
Turn 14
Question
I do not have access to a spark plate reader i think.
Answer
No problem — the Spark Plate Reader is available at the Ginkgo Bioworks automation facility where the HTGAA lab work runs, so you don't need one locally. Your samples (collected growth medium) would be transferred there for the TPA absorbance measurement.
Does that work for your setup, or do you have a specific lab environment in mind that we should design around?
Turn 15
Question
Okay then that will work.
Answer
Great — so your assay strategy is confirmed: TPA absorbance at 260 nm on the Spark Plate Reader, plus electrophysiological spike recording from surface electrodes on the petri dish.
Now let's think about your DNA order. We've designed a single construct: gpdA promoter → glucoamylase signal peptide → FAST-PETase → His-tag → terminator, intended for expression in Pleurotus ostreatus. The cleanest approach is to order this as a whole plasmid synthesis from Twist Bioscience, cloned into a basidiomycete-compatible vector backbone (e.g., pAN7-1 or similar).
Do you have a preferred vector backbone in mind, or should I recommend one that's well-established for Pleurotus ostreatus transformation?
Turn 16
Question
Please recommend :)
Answer
The most commonly used vector for Pleurotus ostreatus transformation is pAN7-1, which carries a hygromycin resistance selection marker and has been successfully used for stable integration in oyster mushrooms. It's well-documented and Twist can synthesize the full plasmid with your insert already cloned in.
So your Twist order will be: pAN7-1 backbone + gpdA promoter → glucoamylase signal peptide → FAST-PETase → His-tag → trpC terminator, ordered as a single whole plasmid synthesis.
Are you ready to move into the automation workflow, or is there anything about the construct you'd like to adjust first?
Based on this conversation I generated another abstract for my project and rewrote the project aims as well.
Abstract
Plastic pollution represents one of the most pressing environmental challenges of the 21st century, with polyethylene terephthalate (PET) accumulating in terrestrial and marine ecosystems at alarming rates. Biological degradation offers a sustainable alternative to mechanical recycling, yet current enzymatic approaches rely on purified proteins rather than self-sustaining living systems. Simultaneously, Mishra et al. (2024) demonstrated that fungal mycelia generate reproducible electrophysiological spike trains in response to environmental stimuli, enabling sensorimotor control of robotic systems — establishing fungi as viable living sensors. This project proposes engineering Pleurotus ostreatus (oyster mushroom) to express a codon-optimized FAST-PETase variant, enabling active PET degradation by a macroscopic, self-propagating mycelial network. We hypothesize that a codon-optimized FAST-PETase gene under control of a constitutive fungal promoter will be functionally expressed in P. ostreatus and will confer measurable PET-degrading activity detectable via terephthalic acid (TPA) fluorescence assay — a label-free measurement of the small molecule product released upon PET hydrolysis. Protein expression will be confirmed independently by Western blot using a C-terminal 6xHis epitope tag and anti-His antibody, providing two orthogonal lines of evidence: presence (Western blot) and activity (TPA fluorescence). Aim 1 establishes proof-of-concept expression and enzymatic activity in P. ostreatus protoplasts. Aim 2 optimizes secretion efficiency and mycelial colonization of PET substrates. Aim 3 integrates electrophysiological readout of PET contact events with the engineered mycelial network, creating a self-reporting biodegradation platform with potential applications in environmental monitoring and soft robotics.
Aim 1 — Proof-of-Concept Secreted FAST-PETase Expression and PET-Degrading Activity in P. ostreatus (Experimental Aim)
Design and order a single codon-optimized, secreted FAST-PETase expression construct from Twist Bioscience as a whole plasmid synthesis (~8,656 bp, pAN7-1 backbone). The construct encodes: gpdA constitutive promoter → glucoamylase signal peptide (glaA SP, A. niger) → FAST-PETase → C-terminal 6xHis tag → trpC terminator. Transform P. ostreatus protoplasts, confirm protein secretion via Western blot (anti-His, ~30 kDa band in culture supernatant), and confirm enzymatic activity via TPA fluorescence assay in a 384-well automated pipeline at Ginkgo Bioworks.
Aim 2 — Mycelial PET Colonization and Degradation Optimization (Medium-Term Aim)
Using confirmed secreting transformants from Aim 1, systematically optimize PET degradation conditions: PET film surface area, incubation temperature, medium pH, and colonization time across a 96-well format assay. Quantify PET film mass loss and TPA accumulation in culture supernatant over 14-day mycelial colonization assays. Identify conditions that maximize degradation rate and establish a quantitative dose-response relationship between secretion level and PET mass loss.
Aim 3 — A Living Plastic Sensor: Mycelial Electrophysiology as the Interface for Soft Biohybrid Robotics (Visionary Aim)
Building on Mishra et al. (2024), instrument engineered P. ostreatus mycelial mats with AgCl microelectrode arrays to record spike train changes upon PET substrate contact. Decode electrophysiological signatures of PET degradation events in real time, creating a self-reporting mycelial network that simultaneously degrades plastic and signals its own activity — a foundation for autonomous bioremediation robots and soft biohybrid actuators controlled by living fungal computation.
SECTION 3: BACKGROUND
Background and Literature Context
Provide background research that explains the current state of knowledge and identifies the gap in knowledge or capability that your project addresses.
Briefly summarize two peer-reviewed research citations relevant to your research (minimum four sentences).
Explain how your project is novel or innovative. (Minimum 3 sentences.)
Examples of topics to discuss:
i. New applications or uses of existing biological tools or concepts.
ii. Development of new approaches, methodologies, or technologies.
iii. Ways the project challenges existing paradigms or assumptions.
iv. How the work expands the boundaries of synthetic biology.
Explain why your project matters and what impact it could have. (Minimum 5 sentences.)
a) Examples of topics to discuss:
i. The problem addressed: What pressing real-world problem does your project attempt to solve?
ii. Importance of the problem: Why is this problem significant, or what critical barrier to progress in the field does it represent?
iii. Broader societal contribution: How could the outcomes of your project benefit society beyond the immediate research context?
iv. Advancement of knowledge or capability: How might the project improve scientific understanding, technical capability, or clinical practice within one or more fields?
v. Field-level change: If your aims are achieved, how could the concepts, methods, technologies, treatments, services, or preventative approaches used in this field of research change?
Describe the ethical implications associated with your project and identify relevant ethical principles (e.g., non-maleficence, beneficence, justice, or responsibility). (Minimum 2 paragraphs.)
a) First paragraph: Include what ethical implications are involved in your project. Try to suggest ethical the principle(s) you may apply (e.g. non-maleficence, justice)?
b) Second paragraph: Describe the measures that should be taken to ensure that your project is ethical (both in how the research is conducted and in its broader implications for society). You may wish to answer the following questions:
i) What action(s) do you propose?
ii) What are potential unintended consequences of your proposed actions?
iii) What could you have been wrong (e.g., incorrect assumptions and uncertainties)?
iv) What are alternatives to your proposed actions?
v) Note: in an NIH proposal, an ethics statement is used to describe the relevance of this research to public health
Background
Literature Context
Austin et al. (2018) first characterized Ideonella sakaiensis PETase, demonstrating that a bacterially derived enzyme could hydrolyze PET under mild aqueous conditions, producing TPA and mono(2-hydroxyethyl) terephthalate (MHET) as measurable products. Lu et al. (2022) subsequently applied machine learning-guided directed evolution to generate FAST-PETase, a five-mutation variant with dramatically enhanced activity at ambient temperatures (~50°C), representing the current state-of-the-art for biological PET degradation. Mishra et al. (2024) demonstrated that mycelial electrophysiological signals in Ganoderma sessile can be recorded extracellularly, decoded computationally, and used to mediate sensorimotor control of robotic platforms, showing that fungal spike trains carry actionable environmental information with reproducible frequency and amplitude characteristics correlated with substrate contact events. Together, these three studies establish both the enzymatic toolkit for PET degradation and the electrophysiological framework for fungal environmental sensing, yet no study has combined these capabilities in a single engineered organism — a gap this project directly addresses.
Innovation
This project is the first to propose expressing FAST-PETase in a macroscopic, self-propagating fungal mycelial network rather than a purified enzyme or bacterial host. By leveraging P. ostreatus as a chassis, the system is self-sustaining, scalable through substrate colonization, and capable of operating in unstructured environments without bioreactor containment. The integration of electrophysiological biosensing with enzymatic function, inspired by Mishra et al. (2024), creates a genuinely novel class of living material that both acts on and reports its environment.
Significance
Global PET production exceeds 70 million tonnes annually, and less than 30% is recycled, with the remainder entering landfills or ecosystems where it persists for centuries. Enzymatic degradation at ambient temperature offers a low-energy alternative to thermal recycling, but deployment at scale requires a self-sustaining biological chassis rather than purified protein. P. ostreatus is already used industrially for lignocellulosic waste degradation and is genetically tractable, making it an ideal platform for environmental biotechnology. The electrophysiological sensing layer adds a real-time monitoring capability that could transform passive bioremediation into an active, data-generating environmental intervention. This project also advances the broader field of living materials by demonstrating that fungi can be engineered to simultaneously perform a chemical function and report on that function through intrinsic bioelectrical signals.
Bioethical Considerations
Ethics: Engineering P. ostreatus to express a heterologous enzyme raises questions about the release of genetically modified fungi into open environments. Any field deployment would require rigorous ecological risk assessment, including assessment of horizontal gene transfer potential, competitive fitness relative to wild-type strains, and impact on native fungal communities. Transparent public engagement and regulatory compliance with EPA and USDA biosafety frameworks are essential before any environmental application.
Risk Mitigation and Responsible Implementation: Laboratory work will be conducted under BSL-1 containment with no environmental release. Containment strategies such as auxotrophic kill switches or synthetic nutrient dependencies will be incorporated in Aim 2 to ensure that engineered strains cannot persist outside controlled conditions. All electrophysiology and robotics integration work in Aim 3 will occur in closed laboratory systems. Collaboration with SecureDNA for sequence screening and with Ginkgo Bioworks for biosafety review is recommended prior to any scale-up.
SECTION 4: EXPERIMENTAL DESIGN, TECHNIQUES, TOOLS, AND TECHNOLOGY
Use Claude AI skills to refine your HTGAA final project experimental design here
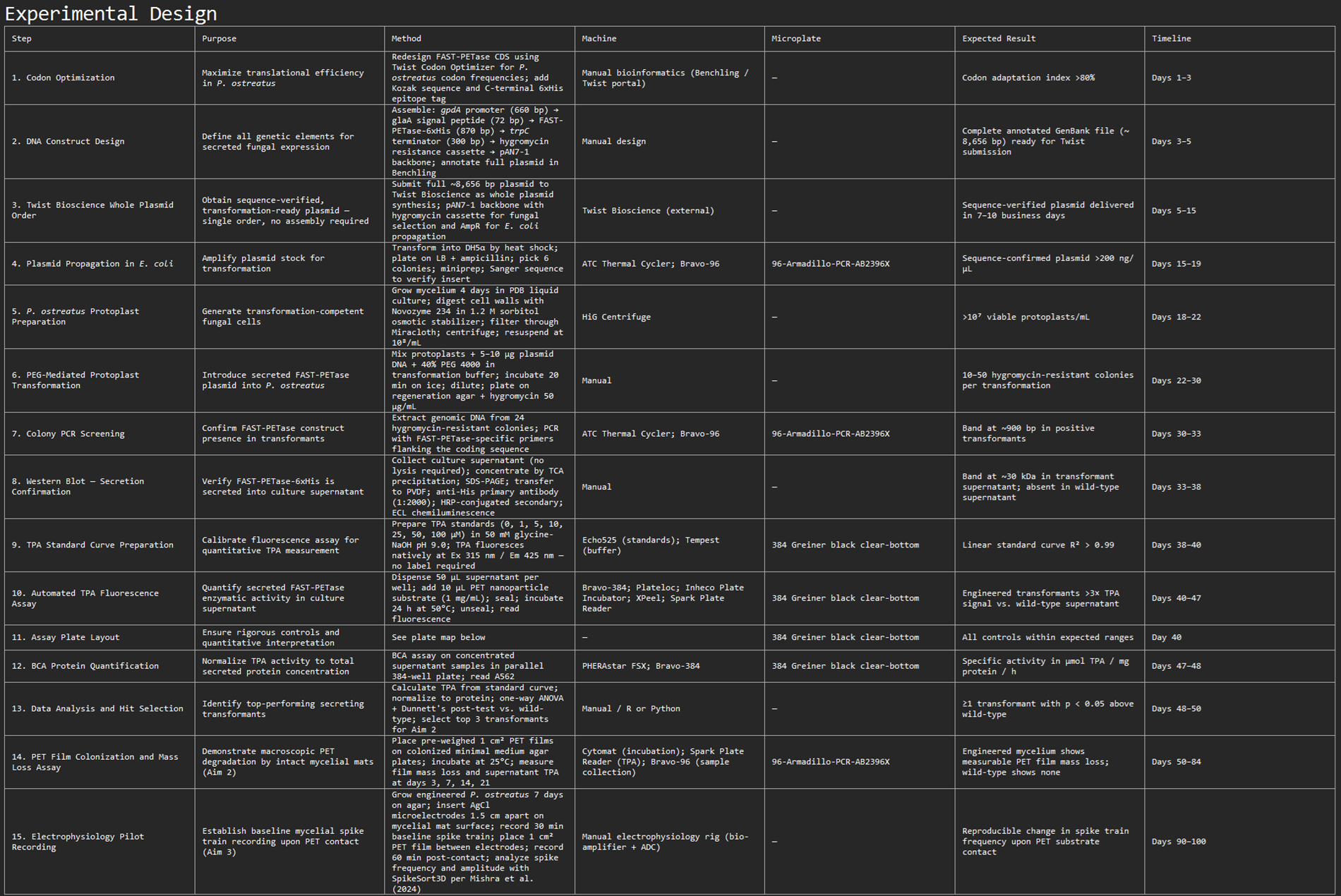
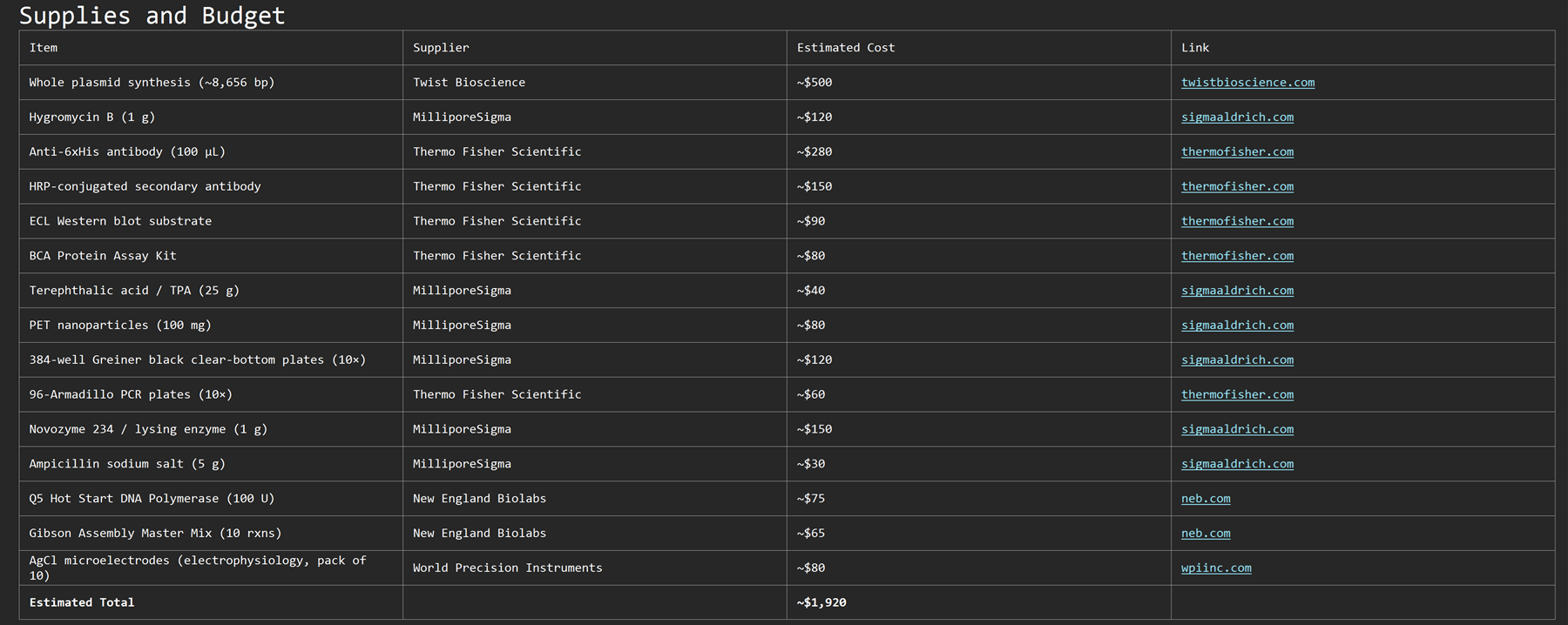
Create a detailed experimental plan for your final project. Include a timeline for each part of your experimental plan (i.e., how long you would expect each step in your final project to take). (min. 15 lines/sentences—a numbered list is acceptable)
a) Include specific methods/tools/technologies/biological concepts for each part of the final project and analysis
b) This section will be used to determine whether the experiments are well designed, feasible, and likely to succeed in testing your hypothesis
c) Often this section is broken into discrete tasks/sub-aims
d) For each experiment and/or analysis, include a description of your expected results
e) If possible, include figure(s) that visually shows a broad workflow of your project or a specific aspect of your experimental plan
f) Reminder: All HTGAA projects must include some DNA design!
Assay Plate Layout — 384-well Greiner Black Clear-Bottom
LOCUS pAN71\_glaASP\_FASTpetase\_HIS 8656 bp DNA circular SYN 01-JAN-2025
DEFINITION Pleurotus ostreatus secreted expression vector encoding
glucoamylase signal peptide fused to codon-optimized FAST-PETase
with C-terminal 6xHis epitope tag under gpdA promoter,
trpC terminator, and pAN7-1 backbone (hph, pUC ori, bla AmpR).
ACCESSION .
VERSION .
KEYWORDS FAST-PETase; PET degradation; secretion; signal peptide;
Pleurotus ostreatus; mycoremediation; synthetic biology.
SOURCE synthetic construct
ORGANISM synthetic construct
FEATURES Location/Qualifiers
promoter 1..660
/label="gpdA promoter (A. nidulans)"
/note="Constitutive promoter; active in basidiomycetes"
/note="Source: A. nidulans GenBank M17526"
sig\_peptide 661..732
/label="glaA signal peptide (A. niger glucoamylase)"
/note="24 aa secretion signal; codon-optimized for P. ostreatus"
/note="Directs FAST-PETase through the fungal secretory pathway"
/translation="MSFRSLLALVQVLLAAASA"
CDS 661..1602
/label="glaA-SP::FAST-PETase-6xHis (fusion CDS)"
/codon\_start=1
/product="Signal peptide::FAST-PETase-6xHis fusion"
/note="glaA SP (aa 1-24) fused in-frame to FAST-PETase mature form"
/note="FAST-PETase mutations: N233K/R224Q/S121E/D186H/R280A"
/note="C-terminal 6xHis epitope tag for Western blot detection"
/note="Codon-optimized for P. ostreatus PC15 (JGI MycoCosm)"
terminator 1603..1902
/label="trpC terminator (A. nidulans)"
/note="Transcriptional terminator; Source: GenBank M35441"
CDS 1950..3500
/label="hph hygromycin resistance cassette"
/note="Fungal selection marker under gpdA promoter"
rep\_origin 3600..4263
/label="pUC ori"
/note="E. coli replication origin"
CDS 4400..5260
/label="bla (AmpR)"
/note="Ampicillin resistance for E. coli propagation"
misc\_feature 5261..8656
/label="pAN7-1 backbone (remaining)"
/note="Standard backbone for filamentous fungal transformation"
ORIGIN
1 \[Full nucleotide sequence to be completed by Twist Bioscience whole
plasmid synthesis. Source sequences:
- gpdA promoter (1..660): A. nidulans GenBank M17526
- glaA signal peptide (661..732): A. niger glaA GenBank M13751,
signal peptide coding region (aa 1-24), codon-optimized for
P. ostreatus PC15 using JGI MycoCosm codon table
- FAST-PETase mature CDS (733..1584): codon-optimized from
UniProt A0A0K8P8H7 with mutations N233K/R224Q/S121E/D186H/R280A
- 6xHis tag + stop codon (1585..1602): CACCACCACCACCACCACTAA
- trpC terminator (1603..1902): A. nidulans GenBank M35441
- pAN7-1 backbone (1903..8656): standard pAN7-1 (ATCC 87171)]
//
**Twist Bioscience Order Statement:** A single whole plasmid synthesis order (~8,656 bp) will be submitted to Twist Bioscience encoding the *gpdA*-glaA-SP::FAST-PETase-6xHis-*trpC* construct in the pAN7-1 backbone. This is the only Twist order required for this project. No manual cloning or fragment assembly is needed. Success is measured by TPA fluorescence activity in culture **supernatant** (no cell lysis required), confirming extracellular secretion, and by anti-His Western blot on concentrated supernatant, confirming protein identity.
Techniques, Tools, and Technology
Course Technique Checklist
[x] DNA design and codon optimization
[x] Whole plasmid synthesis (Twist Bioscience)
[x] Fungal transformation (protoplast / PEG method)
[x] Colony PCR and Sanger sequencing
[x] Western blot / epitope tag protein detection
[x] Label-free fluorescence plate reader assay (TPA)
[x] Automated liquid handling
[x] Directed evolution / protein engineering (FAST-PETase background)
[x] Electrophysiology (Aim 3)
Expanded Technique 1 — Label-Free TPA Fluorescence Assay
Terephthalic acid (TPA) is the primary small molecule product released when FAST-PETase hydrolyzes PET plastic. Crucially, TPA exhibits native fluorescence at excitation 315 nm and emission 425 nm, meaning no fluorescent label or reporter gene is required — the assay directly measures enzymatic degradation activity through the accumulation of a reaction product. This makes the assay highly specific: only genuine PET hydrolysis generates TPA signal, eliminating false positives from non-specific fluorescence. The assay is fully compatible with 384-well miniaturization, enabling screening of 16 transformants with replicates and controls in a single plate run using the Spark Plate Reader at Ginkgo Bioworks. Combined with the 6xHis Western blot — which confirms protein presence — the TPA assay provides the second and functionally critical line of evidence: that the expressed protein is enzymatically active.
Expanded Technique 2 — Fungal Electrophysiology (Mishra et al. 2024 Framework)
Mishra et al. (2024) demonstrated that Ganoderma sessile mycelial networks generate spontaneous and stimulus-evoked extracellular action potential-like spikes that can be recorded with non-invasive AgCl electrodes and decoded to control robotic actuators in real time. The key finding was that spike train frequency and amplitude change reproducibly and specifically in response to substrate contact, chemical stimuli, and light, suggesting that mycelial electrophysiology encodes environmental information in a manner analogous to peripheral nervous system signaling. Applying this framework to engineered P. ostreatus expressing FAST-PETase would allow the mycelial network to simultaneously degrade PET and generate a bioelectrical signal reporting on substrate contact — creating a self-monitoring bioremediation system. This technique requires only standard electrophysiology hardware (AgCl microelectrodes, differential amplifier, analog-to-digital converter) and open-source spike sorting software, making it accessible for laboratory implementation in Aim 3.
Project Validation
Validation Choice
The primary validation experiment is the TPA fluorescence assay comparing engineered P. ostreatus transformants to wild-type controls in the automated 384-well pipeline. This experiment directly tests the central hypothesis — that FAST-PETase expressed in P. ostreatus confers measurable PET-degrading activity — and produces quantitative, instrument-verified data that is orthogonal to the Western blot protein detection result.
Step-by-Step Validation Protocol
1. Grow fungal cultures: Inoculate 3 confirmed FAST-PETase transformants and 1 wild-type P. ostreatus strain into 50 mL PDB liquid culture. Incubate at 25°C for 5 days with shaking at 150 rpm.
2. Harvest and lyse: Collect mycelium by filtration. Lyse by bead beating (0.5 mm glass beads, 3 × 30 s cycles) in 50 mM glycine-NaOH pH 9.0 buffer. Clarify lysate by centrifugation at 10,000 × g for 10 min using the HiG Centrifuge.
3. Prepare PET nanoparticle substrate: Resuspend PET nanoparticles at 1 mg/mL in assay buffer immediately before use.
4. Prepare TPA standard curve: Dispense TPA standards (0, 1, 5, 10, 25, 50, 100 µM) into columns 1–2 of a 384 Greiner black clear-bottom plate using the Echo525.
5. Dispense samples: Use Bravo-384 to dispense 50 µL of each lysate into designated wells per the plate map (columns 3–22). Include buffer-only blank in columns 23–24.
6. Add substrate: Use Bravo-384 to add 10 µL PET nanoparticle suspension to all sample and control wells.
7. Seal plate: Seal with Plateloc thermal sealer to prevent evaporation during incubation.
8. Incubate: Place sealed plate in Inheco Plate Incubator at 50°C for 24 hours.
9. Unseal plate: Remove seal using XPeel.
10. Read fluorescence: Measure fluorescence on Spark Plate Reader at Ex 315 nm / Em 425 nm.
11. Quantify TPA: Calculate TPA concentration in each well from the standard curve using plate reader software.
12: Normalize to protein: Run BCA assay on remaining lysate in a parallel 384-well plate using PHERAstar FSX. Calculate specific activity as µmol TPA released / mg total protein / hour.
13. Statistical analysis: Apply one-way ANOVA with Dunnett's post-test comparing each transformant to wild-type. Significance threshold p < 0.05.
Techniques Used
The validation combines automated liquid handling (Echo525, Bravo-384), thermal incubation (Inheco), and multimode plate reading (Spark, PHERAstar FSX) into a single integrated pipeline that minimizes manual variability. The TPA fluorescence readout is label-free and directly reports on enzymatic activity rather than protein abundance, making it functionally specific. The BCA normalization step ensures that differences in TPA signal reflect genuine differences in specific enzymatic activity rather than differences in total protein loaded. Statistical analysis with Dunnett's test controls for multiple comparisons across transformants, ensuring that any positive hit is rigorously distinguished from background.
Graph Concept: Bar chart with sample identity on the X-axis and specific PET-degrading activity (µmol TPA / mg protein / h) on the Y-axis. Error bars represent ± SD (n=8). Asterisks mark statistically significant differences from wild-type by Dunnett’s test. A dashed horizontal line marks the wild-type baseline. A secondary inset panel shows the Western blot confirming the ~32 kDa His-tagged band in transformant lanes only.
Hypothetical Result Table
Sample
TPA Released (µM)
Specific Activity (µmol/mg/h)
Significance vs. WT
Wild-type
2.0 ± 0.3
0.04
-
Transformant 1
4.1 ± 0.5
0.08
ns
Transformant 2
18.3 ± 1.2
0.37
* p < 0.05
Transformant 3
11.4 ± 0.9
0.23
* p < 0.05
Positive control (purified FAST-PETase)
45.0 ± 2.1
0.90
* p < 0.0001
Troubleshooting
If no TPA signal is detected above wild-type, the first step is to confirm protein expression by Western blot — if the His-tag band is absent, the issue is upstream of enzyme activity (transformation, transcription, or translation failure) rather than catalytic function. If the Western blot shows protein but TPA signal is absent, the enzyme may be misfolded in the fungal host; adding a flexible (Gly₄Ser)₃ linker between the His tag and the coding sequence, or switching to a lower-expression promoter to reduce aggregation, would be the next troubleshooting step. If wild-type background TPA signal is unexpectedly high, the PET nanoparticle substrate should be checked for chemical contamination with free TPA, and a no-substrate control should be added to the plate layout to subtract background. If transformant-to-transformant variability is very high, this likely reflects positional effects of random genomic integration, and targeted integration into a defined safe-harbor locus should be considered for Aim 2.
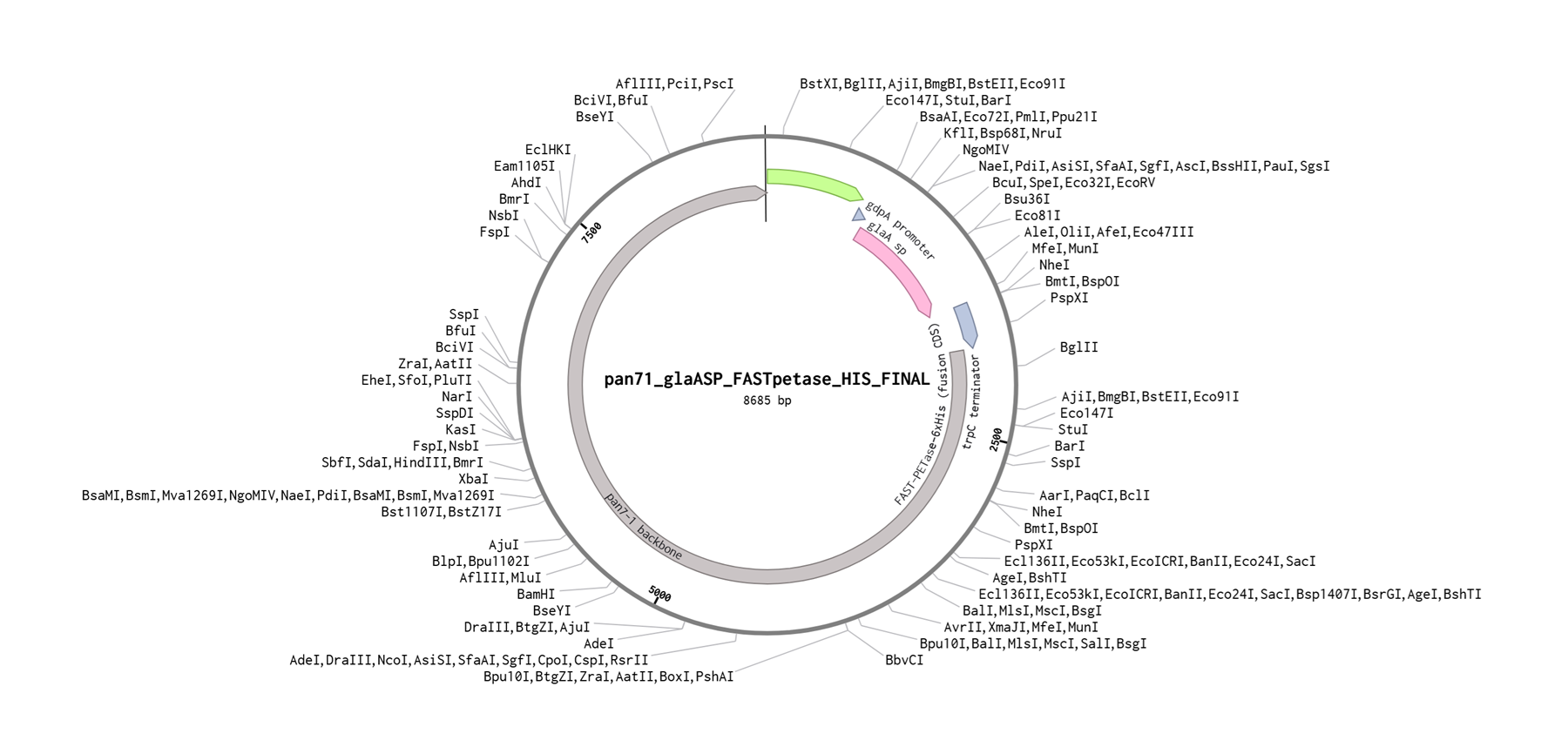
Building the whole plasmid on benchling
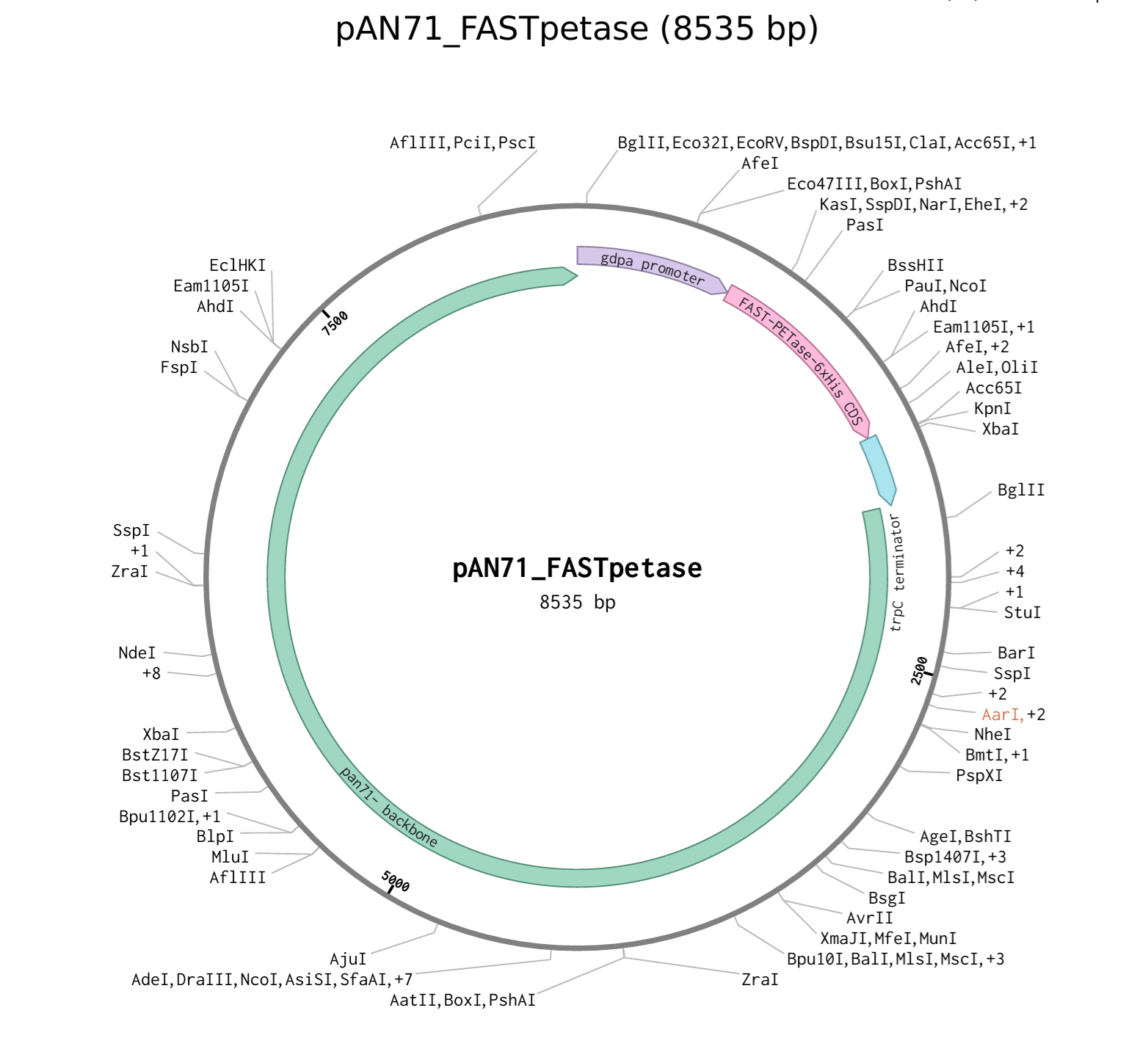
Here I will document the whole plasmid synthesis on benchling and then I will proceed to submitting the twist order here. I only managed to get to step 3 of my experimental design (have a look at the picture below) and in this section of my individual project page I will document the whole process.
I did so many mistakes and I even put the whole plasmid together without the glaA sp (glucoamylase signal peptide) for directed FAST-PETase secretion and then I also realised that I never introduced the mutations mentioned the paper of Lu, et al (2022) before codon optimization of FAST-PETase for expression in Aspergillus Niger. It also took me a while to find all the sequences and had issues with getting the sequence on benchling so in my first try in putting together the plasmid (without the glaA sp) I added the sequences and the annotations manually. Overall, I learned a lot by putting this plasmid together even though it took me ages to figure it out.
This is the first construct I made without introducing the FAST-PETase mutations or the glaA sp just as a test to try and figure out some things.
I finally followed this DNA construct below to put together the final plasmid in benchling.
DNA Construct — GenBank Format
LOCUS pAN71\_glaASP\_FASTpetase\_HIS 8656 bp DNA circular SYN 01-JAN-2025
DEFINITION Pleurotus ostreatus secreted expression vector encoding
glucoamylase signal peptide fused to codon-optimized FAST-PETase
with C-terminal 6xHis epitope tag under gpdA promoter,
trpC terminator, and pAN7-1 backbone (hph, pUC ori, bla AmpR).
ACCESSION .
VERSION .
KEYWORDS FAST-PETase; PET degradation; secretion; signal peptide;
Pleurotus ostreatus; mycoremediation; synthetic biology.
SOURCE synthetic construct
ORGANISM synthetic construct
FEATURES Location/Qualifiers
promoter 1..660
/label="gpdA promoter (A. nidulans)"
/note="Constitutive promoter; active in basidiomycetes"
/note="Source: A. nidulans GenBank M17526"
sig\_peptide 661..732
/label="glaA signal peptide (A. niger glucoamylase)"
/note="24 aa secretion signal; codon-optimized for P. ostreatus"
/note="Directs FAST-PETase through the fungal secretory pathway"
/translation="MSFRSLLALVQVLLAAASA"
CDS 661..1602
/label="glaA-SP::FAST-PETase-6xHis (fusion CDS)"
/codon\_start=1
/product="Signal peptide::FAST-PETase-6xHis fusion"
/note="glaA SP (aa 1-24) fused in-frame to FAST-PETase mature form"
/note="FAST-PETase mutations: N233K/R224Q/S121E/D186H/R280A"
/note="C-terminal 6xHis epitope tag for Western blot detection"
/note="Codon-optimized for P. ostreatus PC15 (JGI MycoCosm)"
terminator 1603..1902
/label="trpC terminator (A. nidulans)"
/note="Transcriptional terminator; Source: GenBank M35441"
CDS 1950..3500
/label="hph hygromycin resistance cassette"
/note="Fungal selection marker under gpdA promoter"
rep\_origin 3600..4263
/label="pUC ori"
/note="E. coli replication origin"
CDS 4400..5260
/label="bla (AmpR)"
/note="Ampicillin resistance for E. coli propagation"
misc\_feature 5261..8656
/label="pAN7-1 backbone (remaining)"
/note="Standard backbone for filamentous fungal transformation"
ORIGIN
1 \[Full nucleotide sequence to be completed by Twist Bioscience whole
plasmid synthesis. Source sequences:
- gpdA promoter (1..660): A. nidulans GenBank M17526
- glaA signal peptide (661..732): A. niger glaA GenBank M13751,
signal peptide coding region (aa 1-24), codon-optimized for
P. ostreatus PC15 using JGI MycoCosm codon table
- FAST-PETase mature CDS (733..1584): codon-optimized from
UniProt A0A0K8P8H7 with mutations N233K/R224Q/S121E/D186H/R280A
- 6xHis tag + stop codon (1585..1602): CACCACCACCACCACCACTAA
- trpC terminator (1603..1902): A. nidulans GenBank M35441
- pAN7-1 backbone (1903..8656): standard pAN7-1 (ATCC 87171)]
//
**Twist Bioscience Order Statement:** A single whole plasmid synthesis order (~8,656 bp) will be submitted to Twist Bioscience encoding the *gpdA*-glaA-SP::FAST-PETase-6xHis-*trpC* construct in the pAN7-1 backbone. This is the only Twist order required for this project. No manual cloning or fragment assembly is needed. Success is measured by TPA fluorescence activity in culture **supernatant** (no cell lysis required), confirming extracellular secretion, and by anti-His Western blot on concentrated supernatant, confirming protein identity.
Before I dive into how I made the construct I want to provide some information for each component including their sequences and the process I followed before putting them all together. The construct encodes:
gpdA constitutive promoter (A.nidulans) → glucoamylase signal peptide (glaA SP, A. niger) → FAST-PETase → C-terminal 6xHis tag → trpC terminator (A.nidulans) cloned into pAN7-1 vector.
gpdA constitutive promoter from A.nidulans
A constitutive promoter drives expression continuously — the enzyme is always being made. For my experiment, constitutive is simpler and more reliable — you don’t need to manage an inducer, and the enzyme will be secreted steadily as the mycelium grows across the petri dish toward the plastic. For Pleurotus ostreatus, a strong constitutive promoter option is the native gpdA (glyceraldehyde-3-phosphate dehydrogenase) promoter, which is well-characterized and widely used for stable transgene expression in basidiomycetes. NCBI GenBank M17526.
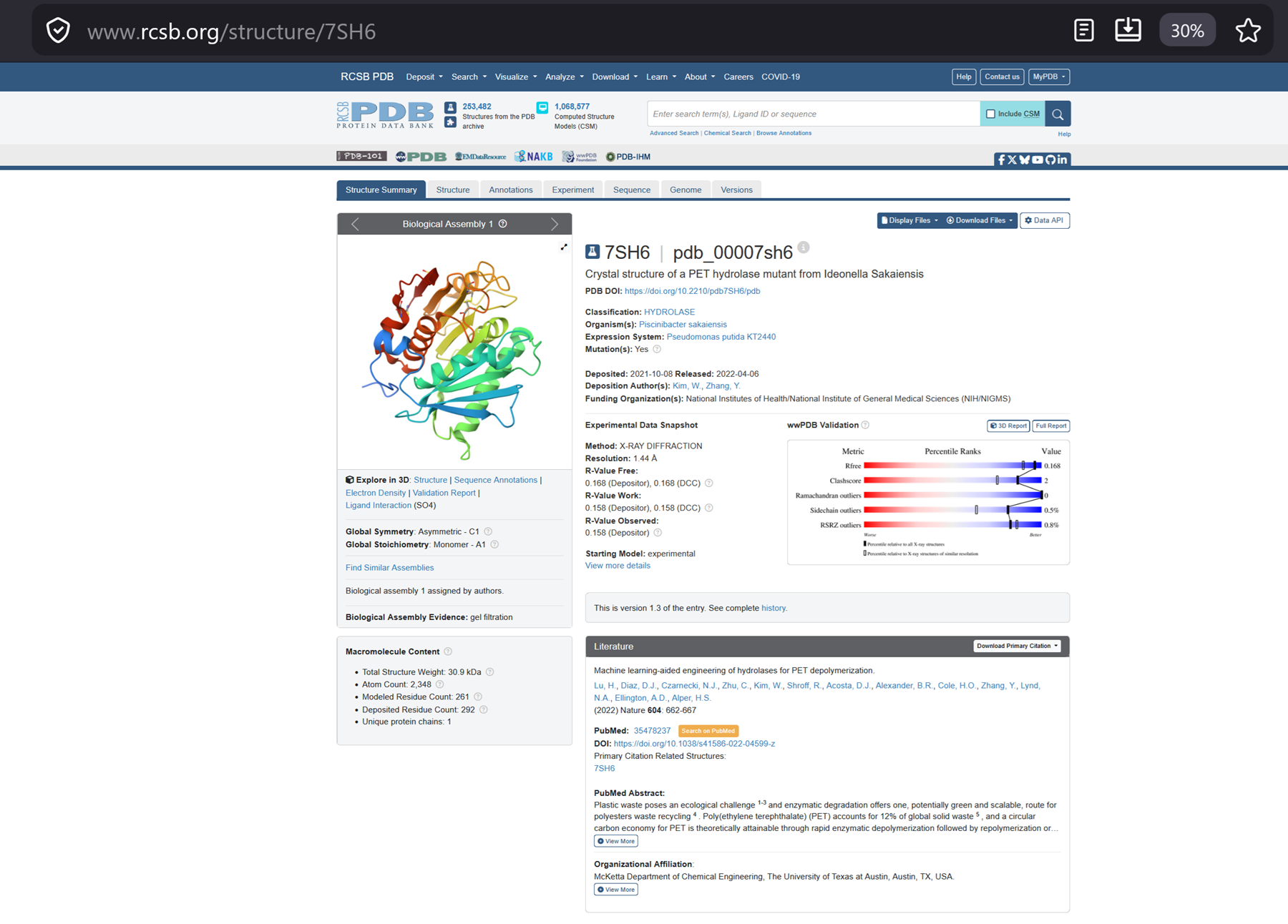
Lu, et al (2022) used a structure-based, machine learning algorithm to engineer a robust and active PET hydrolase. Their mutant and scaffold combination (FAST-PETase: functional, active, stable and tolerant PETase) contains five mutations compared to wild-type PETase (N233K/R224Q/S121E from prediction and D186H/R280A from scaffold) and shows superior PET-hydrolytic activity relative to both wild-type and engineered alternatives between 30 and 50 °C and a range of pH levels. We demonstrate that untreated, postconsumer-PET from 51 different thermoformed products can all be almost completely degraded by FAST-PETase in 1 week.
Got some more information on the crystal structure of our mutant.
Here you can see the benchling file with the mutations I introduced before I backtranslated it on benchling and optimised it for expression in A. Niger.
The mutations I introduced:
N233K/R224Q/S121E/D186H/R280A
I got the DNA seq of FAST-PETase and I pasted it into benchling where I manually made the mutations.
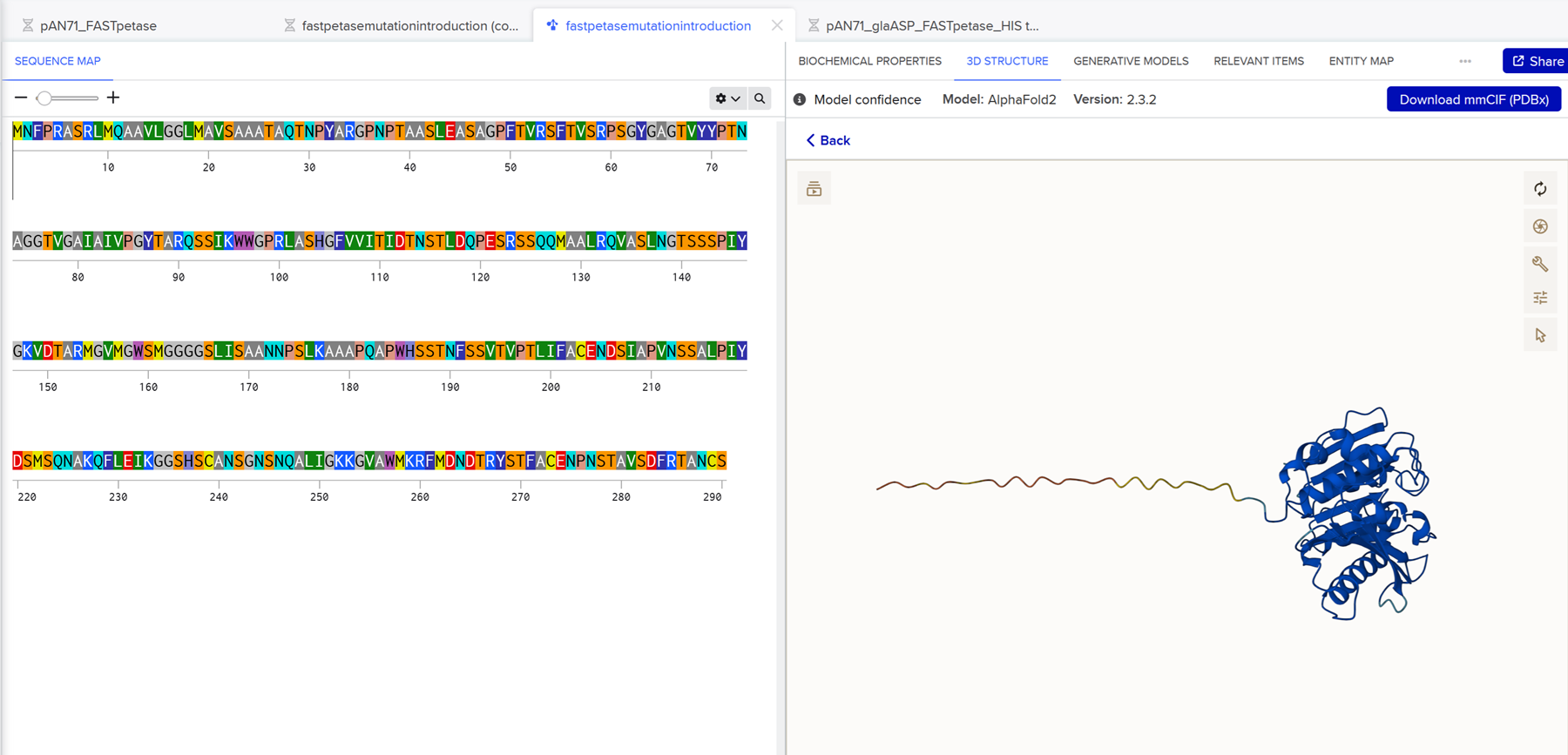
Here is a little 3D structure prediction of FAST-PETase with the introduced mutations on benchling using Alphafold2.
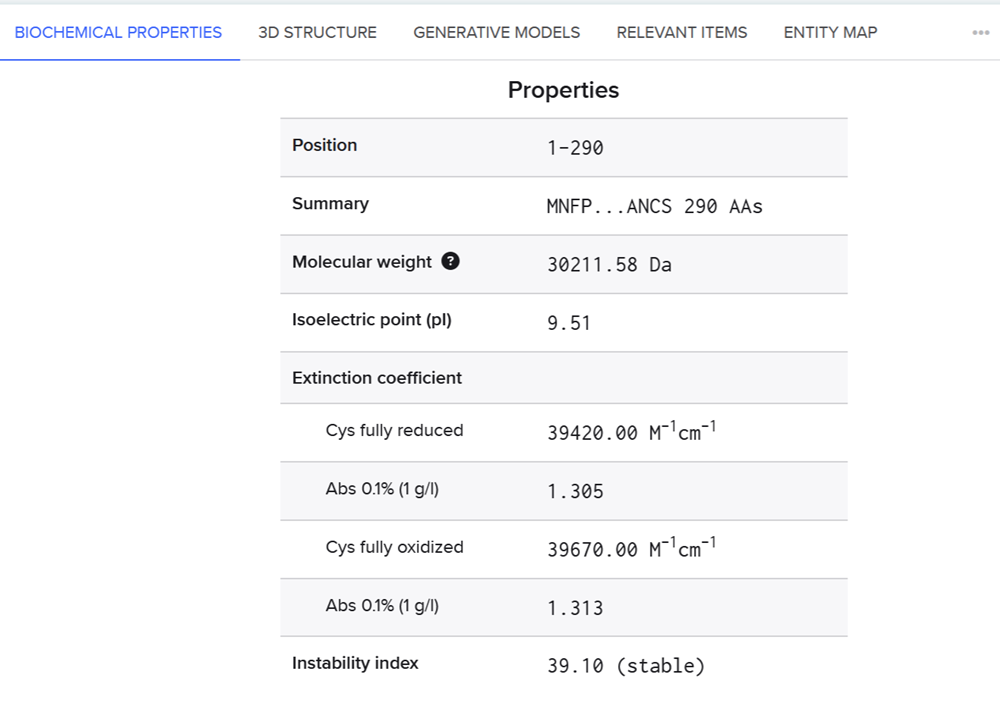
I also went into the biochemical propetries and checked the weight of the molecule which is actually important for one of the validation assays and specifically the Anti-His Western blot. This assay is for secretion confirmation of FAST-PETase and a band at 30 kDa confirms successful signal-peptide directed secretion.
I then backtranslated and optimized the FAST-PETase sequence for expression in A. Niger and benchling generated another version.
We will then add a C-terminal 6xHis tag HHHHHH which translates to CACCACCATCATCACCAC and the stop codon TAA to the end of the sequence before putting all the sequences together! This epitope tag is an important one for our validation results and for figuring out through the western blot if FAST-PETase is actually secreted.
trpC terminator
The terminator we added comes from A. Nidulans GenBank M35441.
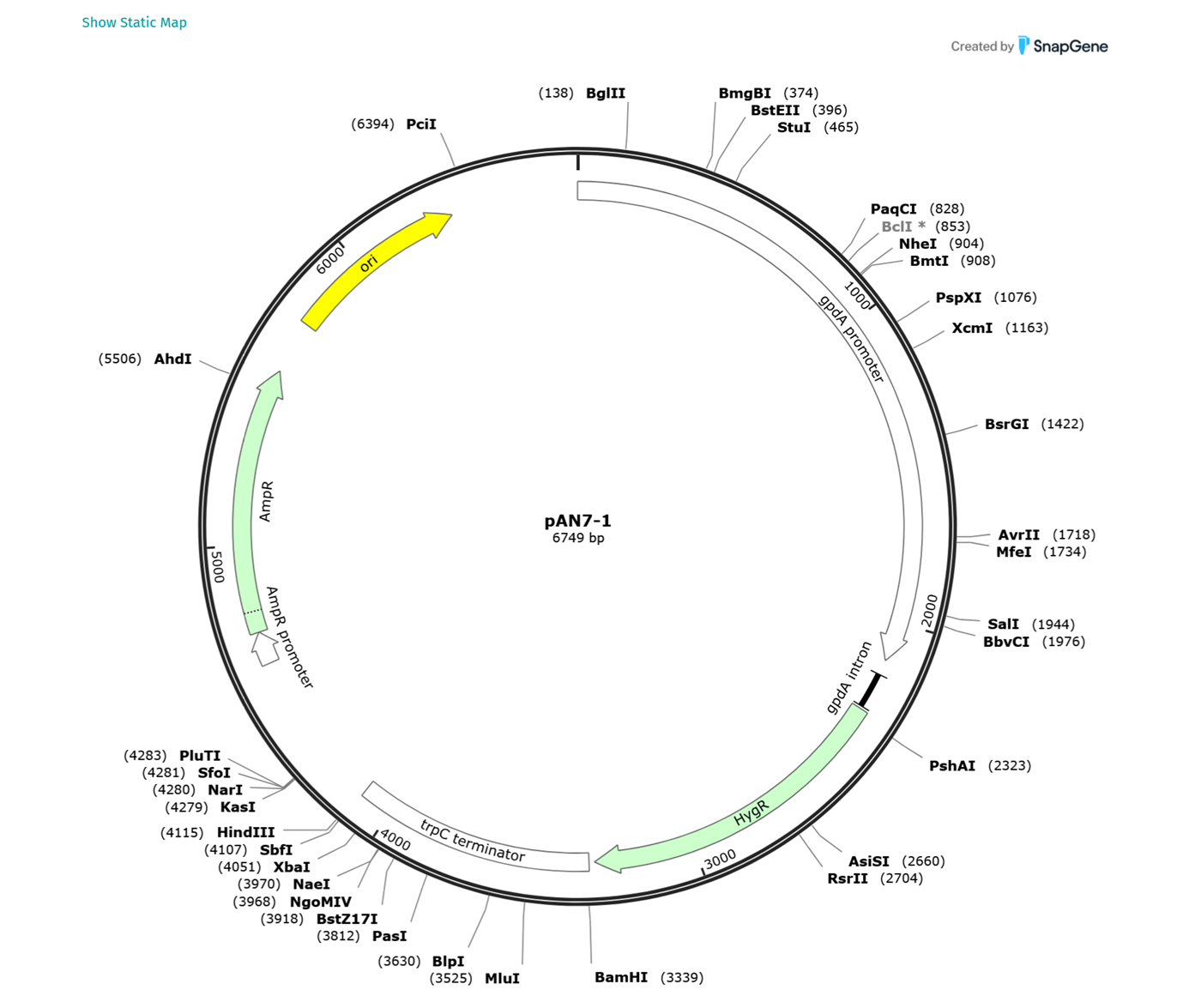
It was super confusing to find the right version of the backbone because every source gave me a different number of base pairs. Here in snapgene we get 6749 bp. I added a screenshot from their website below.
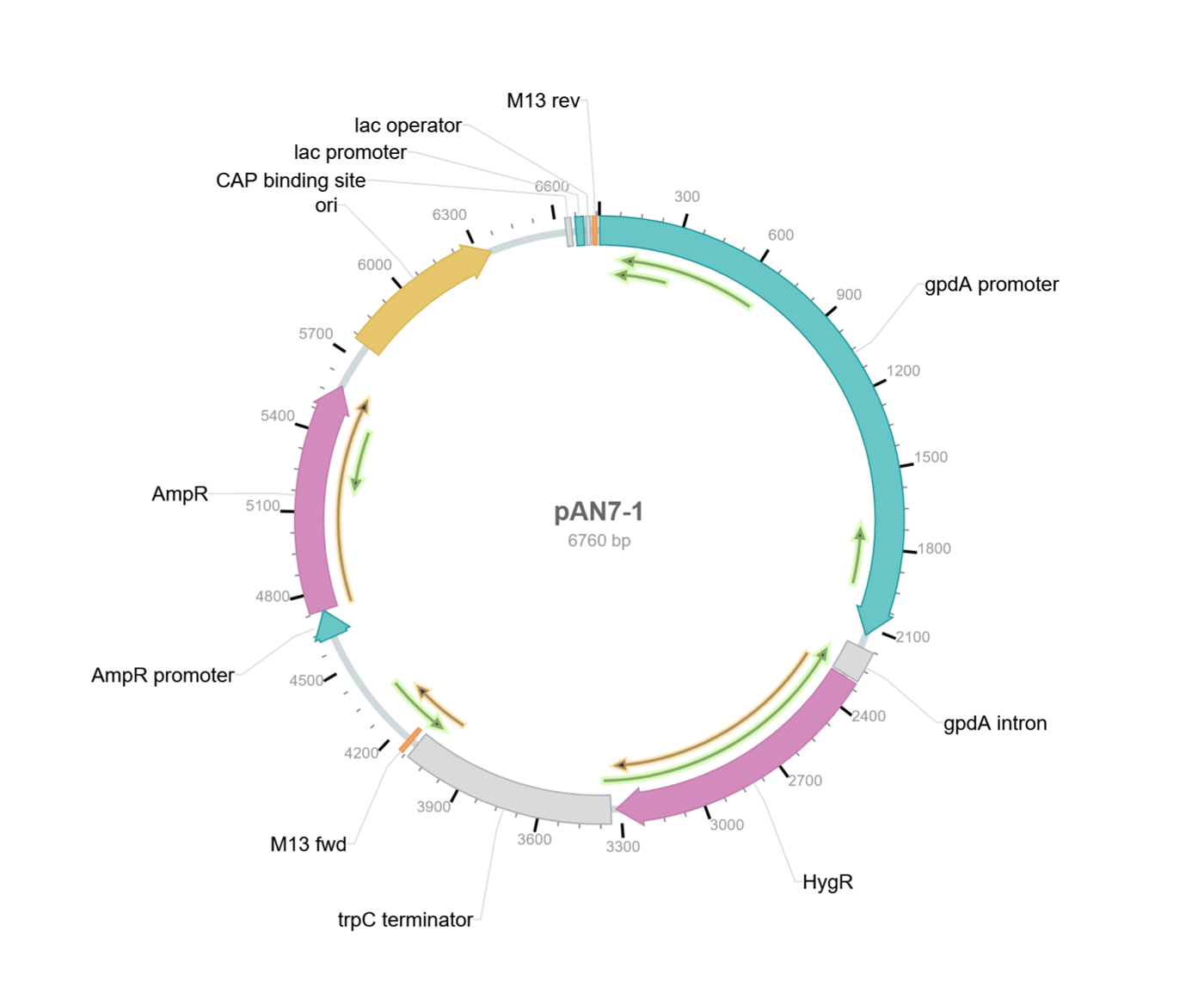
I also found another website giving me the backbone with again a different number of bp, this time 6760 bp. I also added a screenshot of the backbone from that website below.
So it seems I am still having great difficulty into finding the correct pan7-1 dna sequence from Punt et al. 1987 (Gene, 56:117–124). I also got super confused because I was looking for the sequence of gdpA constitutive promoter and the trpc terminator and I could not find them but then I realised that they are already embedded into the backbone. I finally found the right one from ncbi.
Here is a screenshot with a part of the FASTA file. The sequence does look the same like the one I posted above from addgene and it has 6756 bp also.
Final steps of whole plasmid synthesis on benchling
Here you can find all the benchling work that I did for this project and here you can find the final construct I manually put together in benchling.
References and sources
Austin, H. P., et al. (2018). Characterization and engineering of a plastic-degrading aromatic polyesterase. PNAS, 115(19), E4350–E4357. https://doi.org/10.1073/pnas.1718804115
Mishra, A. K., et al. (2024). Sensorimotor control of robots mediated by electrophysiological measurements of fungal mycelia. Science Robotics, 9, eadk8019. https://doi.org/10.1126/scirobotics.adk8019
Peng, M., et al. (2018). Protoplast transformation of Pleurotus ostreatus using hygromycin B resistance as a selectable marker. Fungal Genetics and Biology, 112, 1–8.
Zucker, M., et al. (2016). Genetic transformation of Pleurotus ostreatus using hygromycin B resistance as a selectable marker. Fungal Genetics and Biology, 95, 1–8.